

Abstract
Studies on the stoichiometric and catalytic reactivity of a geometrically constrained phosphorous triamide 1 with pinacolborane (HBpin) are reported. The addition of HBpin to phosphorous triamide 1 results in cleavage of the B-H bond of pinacolborane through addition across electrophilic phosphorus and nucleophilic N-methylanilide sites in a cooperative fashion. The kinetics of this process of were investigated by NMR spectroscopy, with the determined overall second order empirical rate law given by ν = − k[1][HBpin] where k = 4.76 × 10−5 M−1s−1 at 25 °C. The B–H bond activation process produces a P-hydrido-1,3,2-diazaphospholene intermediate 2, which exhibits hydridic reactivity capable of reacting with imines to give phosphorous triamide intermediates, as confirmed by independent synthesis. These phosphorous triamide intermediates are typically short-lived, evolving with elimination of the N-borylamine product of imine hydroboration with regeneration of the deformed phosphorous triamide 1. The kinetics of this latter process are shown to be first-order, indicative of a unimolecular mechanism. Consequently, catalytic hydroboration of a variety of imine substrates can be realized with 1 as catalyst and HBpin as terminal reagent. A mechanistic proposal implicating a P–N cooperative mechanism for catalysis that incorporates the various independently verified stoichiometric steps is presented and a comparison to related phosphorus-based systems is offered.
TOC GRAPHIC
1. Introduction
Bifunctional reactivity involving the cooperative action of donor and acceptor functionality juxtaposed within close molecular proximity is a potent strategy for the modification of chemical bonds. The power and breadth of this approach is well-exemplified in the many cases of metal-ligand cooperativity now evident in transition metal catalysis,1,2 where activation of a substrate covalent bond arises from the intimate participation and mutual modification of both a reactive metal center and supporting ligand. The documented scope of this reactivity mode across diverse transition metals and ligand structures3–7 is indicative of its generality.
Though less commonly described as such, aspects of ligand cooperativity are equally evident in chemistry of the main group; recent stoichiometric examples from Groups 2,8 13,9,10 14,11,12 and 1513 are representative. These observations regarding main group element-ligand cooperativity contribute to ongoing interest into the qualitative connections between reactivity of the transition metals and main group.14 In a compelling application of such stoichiometric reactivity to catalysis, Berben has described the cooperative metal-ligand reactivity of aluminum-bis(imino)pyridine compounds in catalytic amine15 and formic acid16 dehydrogenation.
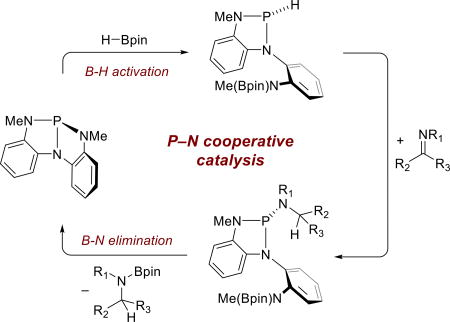
We have recently reported on the preparation and reactivity of geometrically deformed phosphorous triamide 1 (Figure 2).17 By virtue of the geometric constraint imposed by the triamide ligand, Cs-symmetric 1 exhibits an anomalously low nucleophilic reactivity, but instead demonstrates potent biphilic reactivity as exemplified in the reversible oxidative addition-reductive elimination of E–H bonds (E = -OR, -NHR) to the phosphorus center. Mechanistically, we have shown that the overall oxidative addition proceeds in stepwise phosphorus-ligand cooperative fashion, wherein the protic E–H substrate adds initially across one P–N linkage to give a σ3-P intermediate (A) in which a new P–E bond is formed and the resultant proton is accommodated on a methylanilide nitrogen (Figure 1, top). Subsequent intramolecular σ3→ σ5 ring-chain tautomerism then affords the final oxidative addition product B.
Figure 2.

Annotated partial two-dimensional 1H–1H NOESY spectrum of 2 showing correlations between protons that share dipolar coupling (C6D6, 298 K, 500 MHz). Hydrogen atom labels refer to numbering of 2 in Scheme 1. See SI for unabridged spectrum.
Figure 1.

Geometrically deformed phosphorous triamide 1 and its reactivity. (Top) Phosphorus-ligand cooperative addition of protic E–H substrates to 1, see ref. 17. (Bottom) Regioisomeric phosphorus-ligand cooperative addition of hydridic E–H to 1.
In view of this precedent, we considered the possible outcome from an analogous phosphorus-ligand cooperative activation but with formally hydridic E–H substrates (Figure 1, bottom). The difference in substrate polarization could be expected to invert the sense of E–H addition to 1, with hydride translocating to the phosphorus center and the attendant E+ fragment migrating to ligand nitrogen. This mode of activation would result in formation of a P-hydrido-1,3,2-diazaphospholene (C), a class of compound that Gudat18 has shown to exhibit significant hydridic reactivity and that Kinjo19 and most recently Speed20 have capitalized on in catalysis. We wished to ascertain whether interfacing these elementary reactivities with a subsequent group transfer from the pendant N-methylanilide with regeneration of 1 would permit an overall catalytic E–H transfer via a new P–N cooperative scheme.
We report here the realization of the foregoing postulated phosphorus-ligand cooperative scheme within the context of borane B–H addition to phosphorous triamide 1. We further show that this elementary mode of borane activation can be advanced to a catalytic B–H transfer application (imine hydroboration). Through spectroscopic and kinetic analyses, we validate the operation of the phosphorus-ligand cooperativity in which the reversible addition and elimination across a reactive P–N linkage drives catalysis. These results further expand the scope of ligand cooperative catalytic reactivity in the main group and portend a broader opportunity for the deliberate design of bifunctional, ligand cooperative catalysts based on inexpensive p-block elements.
2. Results
2.1 Stoichiometric Reactivity
2.1.1. Formation and Spectroscopic Characterization of P-hydrido-1,3,2-diazaphospholene 2
To test the hypothesis that 1 would exhibit bifunctional, phosphorus-ligand cooperative activation of B–H bonds, a solution of 1 (0.13 M in C6D6) was treated with one equivalent of pinacolborane (HBpin) (Scheme 1). Over the course of 12 h at 50 °C, 31P NMR spectra indicated the full consumption of 1 (δ 159.9 ppm) and formation of a major new phosphorus containing product (2) with a chemical shift δ 84.2 ppm (d, J= 146 Hz). Both the 31P chemical shift and multiplicity are suggestive of a tricoordinate phosphorus species with a direct P–H linkage. The presence of a P–H moiety is further supported by 1H NMR spectra of species 2, which display a new doublet at δ 6.88 ppm with complementary coupling (J = 146 Hz) to that observed in 31P NMR spectra. Additionally, the 1H NMR spectra show an apparent decrease in molecular symmetry of 2 relative to 1; the previously homotopic N-methyl groups from 1 are present in 2 as two distinct resonances (δ 3.05, 2.55 ppm). Only one of the N-methyl resonances (δ 2.55 ppm) displays 3JP–H coupling (J = 15 Hz), suggesting release of one N-methylanilide substituent from the phosphorus center of 1 upon reaction with HBpin. The 11B NMR signal of 2 was also identified at 24.2 ppm.
Scheme 1.

Activation of pinacolborane with 1 and resulting P-hydrido-1,3,2-diazaphospholene 2.
Additional information regarding the overall composition of 2 emerges from two-dimensional NOESY spectra (Figure 2). Cross peaks indicate 1H–1H NOE correlation signals between H1 (i.e. P–H) and both H6 and H7 (N–methyl C–Hs), confirming the open structure of the ligand framework. Also, the correlation between H8 (pinacol methyl C–Hs) with both H1 and H7 confirm the presence of the Bpin moiety on the pendant N-methylanilide arm. Taken together, these spectral data are consistent with the formulation of 2 as a P-hydrido-1,3,2-diazaphospholene arising from P–N cooperative activation of the B–H bond of HBpin. The atropisomeric diastereomers, which were previously observed in the analogous compounds A, are not resolved spectroscopically for 2 presumably as function of the small size of the P–H moiety. Compound 2 is solution stable (0.12 M, 95 % NMR yield), further evolving to σ5–P hydridophosphoranes upon prolonged standing in the presence of excess HBpin. We have not succeeded to date in isolating single crystals of 2 for x-ray analysis despite repeated attempts.
2.1.2. Kinetic Study of the B-H Activation Reaction
Kinetic analyses were conducted to elucidate the reaction rate and molecularity of the phosphorus-ligand cooperative B-H activation. The reaction of 1 (78 mM in C6D6) with excess HBpin (15 equiv) was monitored via 1H NMR as a function of time (25 °C, 1,3,5-(MeO)3C6H3 internal standard), which displayed pseudo-first order dependence on 1 (Figure 3A). Plots of kobs vs. [HBpin]0 (Figure 3B) demonstrate a linear relationship over the concentration range 0.78 M ≤ [HBpin] ≤ 2.73 M, thus establishing an overall second order reaction rate law for addition of HBpin to 1 (eq 1, k = 4.76 × 10−5 M−1s−1 at 25 °C) in accord with the notion of direct bimolecular phosphorus-ligand cooperative activation of the B–H bond.
| (1) |
Figure 3.

Kinetics experiments of the cooperative HBpin activation with 1. (A) Plot of [1] vs time monitored ([1]o = 78 mM, [HBpin]o = 117 mM, C6D6, 297 K). (Inset) Plot of ln [1] vs time with linear least-squares fit. (B) Plot of kobs vs [HBpin]0 with linear least-squares fit.
2.1.3. Hydridic Reactivity of 2 – Alkylhalide Reduction
In line with the rationale from Gudat regarding the impact of hyperconjugative effects on 1,3,2-diazaphospholene reactivity,21 we wished to evaluate the hydridic behavior of 2. Indeed, NBO charge analysis implicates a hydridic nature of the P–H moiety in 2 (atomic charge of −0.11). In agreement with this analysis, the addition of chloroform to a solution of 2 (78 mM in C6D6) resulted in conversion over the course of 6 h to a new compound (31P NMR δ 146.4 ppm) consistent with the assignment as P-chloro-1,3,2-diazaphospholene 3 (Scheme 2). In an alternative preparation, compound 3 may be accessed directly from 1 by treatment with HBpin in chloroform solution. Surprisingly in this latter reaction, compound 2 is not observed to accumulate in solution as a persistent intermediate. This observation suggests that the reaction consuming 2 (with the halogenated substrate) is more rapid than the reaction generating 2 (from 1 by HBpin activation), consistent with the potent hydricity of P-hydrido-1,3,2-diazaphospholenes, such as 2.
Scheme 2.

Hydrodechlorination of chloroform by 2 and fomation of 3.
Crystalline samples of 3 suitable for x-ray diffraction were prepared by crystallization from CH2Cl2/pentane solution. The molecular structure (Figure 4) confirms the tricoordinate phosphorus local environment with pendant N(Bpin) moiety, supporting the analogous structural assignment for 2. Compound 3 displays a small displacement (0.251 Å) of phosphorus from the C2N2 plane and a rather long P–Cl bond length (2.276(1) Å, compare 2.180(4) Å for (Me2N)2PCl22), indicative of hyperconjugative n(π)→σ* overlap as has been noted in related compounds by Gudat. It is reasonable to assume that compound 2 experiences similar electronic interactions that underpin its hydridic reactivity.
Figure 4.

Molecular structure of 3 with 50% probability level rendered thermal ellipsoid plot. Selected bond lengths [Å] and angles [°]: P(1)–Cl(1) 2.276(1), P(1)–N(1) 1.681(2), P(1)–N(2) 1.665(2), N(3)–B(1) 1.418(4), O(1)–B(1) 1.377(4), O(2)–B(1) 1.371(3), N(1)-C(12) 1.401(4), N(2)-C(7) 1.400(7), N(1)-C(1) 1.428(3), N(2)-C(13) 1.459(4); N(1)-P(1)–Cl(1) 103.20(8), N(2)-P(1)–Cl(1) 98.64(9), N(1)-P(1)–N(2) 90.40(1).
2.1.4. Hydridic Reactivity of 2 – Imine Reduction
To further evaluate its hydricity with respect to organic substrates, compound 2 was treated with N-tosylbenzaldimine at rt in benzene solution (Scheme 3) resulting in complete conversion to 4 (31P NMR δ 92.6 ppm). X-ray crystallography confirms the incorporation of the imine fragment through a formal hydrophosphination giving a triamide local environment about tricoordinate phosphorus with the Bpin moiety on a pendant N-methylanilide arm (Figure 5). As above, compound 4 may also be accessed directly from 1 by treatment with PhHC=NTs and HBpin at ambient temperature, where 2 is a presumed but unobservable intermediate. Compound 4 is a stable species in both the solid state and solution; no decomposition is observed after heating a benzene solution at 60 °C for 48 h.
Scheme 3.

Reduction of N-tosyl-1-phenylmethanimine and formation of the triazaphospholene 4.
Figure 5.

Molecular structure of 4. The unit cell contains two crystallographically independent molecules, only one is shown for clarity. Thermal ellipsoid plot rendered at the 50% probability level. Selected bond lengths [Å] and angles [°]: P(1)–N(1) 1.712(4), P(1)–N(2) 1.681(4), P(1)–N(3) 1.770(4), N(3)-S(1) 1.652(5), N(1)-C(2) 1.388(7), N(2)-C(7) 1.390(7), N(2)-C(8) 1.439(6), N(1)-C(1) 1.469(6); N(1)-P(1)-N(3) 103.7(2), N(2)-P(1)-N(3) 101.4(2), N(2)-P(1)-N(1) 88.2(2).
2.1.5. Evidence for Cooperative B-N Elimination
In contrast to the simple hydrophosphination of N-tosylbenzaldimine, reaction of 2 with an N-alkyl benzaldimine results in a markedly different outcome. Treatment of a benzene solution of 2 with N-benzyl p-methoxybenzaldimine (5) at ambient temperature resulted in the consumption of 2 and, unexpectedly, the formation of 1 (Scheme 4). 1H NMR spectra confirm the concomitant formation of hydroboration product 7 under these conditions.
Scheme 4.

Reaction of 2 with imine 5 and the resulting regeneration of 1 via intermediate 6. PMP = p-methoxyphenyl.
In situ 31P NMR monitoring of the above reaction progress provides a window into this transformation (Figure 6). An initial solution of 2, prepared in situ from reaction of 1 with HBpin in C6D6,23 was treated with imine 5. As the reaction progresses (Figure 6b,c), the concentration of 2 was observed to decrease while the concentration of 1 was simultaneously observed to increase. Also evident in these spectra is the appearance of a transient minor peak δ 105.1 ppm. In view of the chemical shift of this signal, we considered that this species might correspond to the phosphorous triamide 6 from hydrophosphination of 5 with 2. Indeed, independent synthesis of 6 from treatment of 3 with N-benzyl-N-(p-methoxyphenyl)methanamine (Scheme 5) gives the same 31P NMR signal δ 105.1 ppm. Consistent with the above observations, 6 is metastable in solution but not isolable. Instead, 6 decomposes cleanly to 1, proceeding to completion within 60 h at ambient temperature (31P NMR spectra, Figure 7). Correspondingly, 1H NMR spectra confirm the formation of the N-borylamine product 7 by B–N elimination from 6.
Figure 6.

Representative time-stacked 31P NMR spectra (C6D6, 145.79 MHz, 25 °C) for the reaction of 2 (δ 84.2 ppm) with imine 5 to give 1 (δ 159.9 ppm) via 6 (δ 105.1 ppm) at time points: (A) 10 min, (B) 30 min, (C) 12 h. Asterisk marks unidentified by-product (δ 140 ppm).
Scheme 5.

The independent synthesis of 6 and subsequent elimination of the N-borylamine 7. PMP=p-methoxyphenyl.
Figure 7.

Time-stacked 31P NMR spectra (C6D6, 145.79 MHz, 25 °C) for the conversion of 6 to 1 at time points (A) 10 min, (B) 3 h, (C) 12 h, (D) 36 h, (E) 60 h.
Although 6 is not isolable itself, a related phosphorous triamide 8 (δ 91.3 ppm) can be prepared by treatment of 3 with benzylamine as depicted in Scheme 6. The composition of this compound was unambiguous proven by X-ray diffraction analysis of single crystalline samples (Figure 8). The similarity in 31P NMR chemical shifts between isolable 8 and transient 6 supports the assignment of the latter as the phosphorous triamide depicted in Scheme 4. In a further analogy to 6, compound 8 can be directly converted to 1 upon heating (50 °C, 48 h) with liberation of N-boryl benzylamine 9. Taken together, the totality of these experiments demonstrates that the N-boryl benzylamines are generated from the phosphorous triamide intermediates via a ligand-phosphorus cooperative pathway.
Scheme 6.

Synthesis of 8 and its conversion to 1.
Figure 8.

Molecular structure of 8. Thermal ellipsoid plot rendered at the 50% probability level. Selected bond lengths [Å] and angles [°]: P(1)-N(1) 1.664(2), P(1)-N(2) 1.732(2), P(1)-N(3) 1.716(2), N(2)-C(8) 1.408(2), N(2)-C(14) 1.417(2), N(3)-C(13) 1.392(3), N(3)-C(20) 1.452(3); N(1)-P(1)-N(2) 105.36(9), N(1)-P(1)-N(3) 105.25(9), N(2)-P(1)-N(3) 87.57(8).
2.1.6. Kinetic Study of the B-N Elimination Reaction
The rate of the conversion from 6 to 1 is suitable for NMR monitoring of kinetic reaction progress. Compound 6 was prepared in solution as depicted in Scheme 5, with purification by rapid filtration over celite to eliminate triethylammonium chloride. The decay of 6 was then monitored at ambient temperature by 1H NMR spectroscopy to greater than 75% conversion (Figure 9). The linear relationship between ln[6] and time (Figure 9, inset) demonstrates first-order behavior, as would be expected for a unimolecular decomposition of 6. The extracted rate constant (k = 0.04 s−1) is independent of absolute concentration within the range 0.58 mM < [6] < 3.26 mM. These results are fully consistent with a ligand cooperative intramolecular transfer of the boryl unit from the N-methylanilide arm to the N-benzyl-N-(p-methoxyphenyl)methanamine fragment, elimination of N-borylamine 7, and reconstitution of a P–N bond in the conversion of 6 to 1.
Figure 9.

Kinetics experiments of the cooperative B–N elimination from 6. (Main) Plot of [6] vs time monitored ([6]o = 3.3 mM, C6D6, 297 K). (Inset) Plot of ln [6] vs time with linear least-squares fit.
2.2 Catalytic Reactivity
In view of the foregoing stoichiometric reactivities, we pursued investigations of hydroboration reactions catalyzed by 1. In an initial reaction, a benzene solution of N-benzyl p-anisaldimine 5 (1 equiv) and HBpin (1 equiv) was exposed to 10 mol% of 1 at 50 °C. Under these conditions, imine 5 was completely converted to the hydroboration product 7 over the course of 24 h (eq 2). Control reactions between 5 and HBpin under otherwise identical conditions but omitting catalytic 1 show less than 15% conversion (eq 3), consistent with a transformation that is indeed under catalyst control.
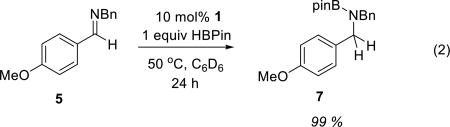 |
(2) |
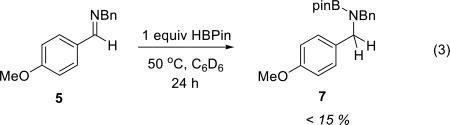 |
(3) |
In situ spectral monitoring experiments of the catalytic reaction was conducted to further substantiate the action of 1 during catalysis. Following mixing of 5, HBpin, and 10 mol% 1 in C6D6 at 50 °C in a thermostatted probe, a 31P NMR spectrum indicates the presence of small quantities of 2, formed at the expense of the initial concentration of 1. At subsequent intermediate time points (t = 0.5–12 h), the concentrations of 2 showed no significant change. As the transformation progressed over the next 24 h, continued production of the corresponding hydroboration product 7 could be observed by 1H NMR spectra. Catalyst 1, although diminished, was never completely consumed. The existence of 1 during the entire course of catalytic reaction as opposed to its total conversion to 2 in stoichiometric reaction suggested again that the reduction of the imine by 2 leads to an ephemeral intermediate 5 (see Scheme 4) which decomposes to afford 1 and the hydroboration product.
Under the aforementioned catalytic conditions, imines displaying an array of structural variations are reduced catalytically and produce the corresponding hydroboration products with good to excellent yields. As depicted in eq 4, a range of aldimines are tolerated, including those with N-aryl (10) and N-alkyl (11) substitution, and electronically diverse (14 – 18) and sterically demanding (19) C-substituents. Ketimines are similarly reduced under catalytic conditions (21 – 23). Lower yields in the reduction of ketimines are probably caused by a steric effect.
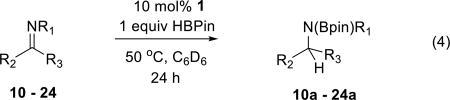 |
(4) |
| 10 | R1=Ph | R2=Ph | R3=H | 99 % y |
| 11 | R1=Bn | R2=p-BrC6H4 | R3=H | 73 % y |
| 12 | R1=p-MeC6H4 | R2=p-MeC6H4 | R3=H | 88 % y |
| 13 | R1=p-OMeC6H4 | R2=Ph | R3=H | 76 % y |
| 14 | R1=p-OMeC6H4 | R2=p-FC6H4 | R3=H | 94 % y |
| 15 | R1=p-OMeC6H4 | R2=1-naphthyl | R3=H | 99 % y |
| 16 | R1=p-OMeC6H4 | R2=m-CF3C6H4 | R3=H | 96 % y |
| 17 | R1=p-OMeC6H4 | R2=p-OMeC6H4 | R3=H | 64 % y |
| 18 | R1=p-CF3C6H4 | R2=p-OMeC6H4 | R3=H | 99 % y |
| 19 | R1=p-OMeC6H4 | R2=mesityl | R3=H | 77 % y |
| 20 | R1=p-FC6H4 | R2=Ph | R3=H | 91 % y |
| 21 | R1=Ph | R2=Ph | R3=Me | 48 % y |
| 22 | R1=Bn | R2=Ph | R3=Ph | 64 % y |
| 23 | R1=Bn | R2=Ph | R3=Me | 94 % y |
| 24 | R1=Ph | R2=p-Ts | R3=H | <5%y |
Consistent with the observed thermal stability of N-sulfonyl derivative 4 (Sect. 2.1.4), N-p-toluenesulfonyl aldimines (24) are not suitable substrates for catalytic reduction. In this circumstance, presumably catalytic turnover is prohibited by the failure of an intermediate 4 to undergo cooperative elimination of N-borylated product with regeneration of catalyst 1. This negative result supports the notion that the P–N cooperativity reactivity demonstrated in stoichiometric transformations (Sect. 2.1) is both necessary and operative in catalytic reactions.
3. Discussion
In line with ongoing investigations into the catalytic function of nontrigonal tricoordinate phosphorus compounds, we find that compound 1 is a competent nonmetal reactant for the activation of the B–H bond in HBpin, and for subsequent catalytic hydroboration of imines. The sum of the experimentally validated stoichiometric transformations establishes the basis for the proposed P–N cooperative mechanism of catalytic hydroboration by 1, as depicted in Scheme 7: (1) addition of HBpin to 1 generates intermediate 2 via a bimolecular cooperative mechanism, (2) formal insertion of imine substrate to the P–H of 2 giving a phosphorous triamide 25, and (3) intramolecular boryl transfer to eliminate N-borylamine 26 with regeneration of catalyst 1 and closure of the catalytic cycle.
Scheme 7.

Proposed phosphorus-ligand cooperative mechanism for the catalytic imine hydroboration.
Borane B–H activation is, of course, well-known with transition metals both via oxidative addition24 and metal-ligand cooperativity25; a great many catalytic hydroboration applications are based on these fundamental bond activation reactions.26 Main group B–H activation is also precedented, for instance 1,1-oxidative addition by carbenes.27 More common, though, are formally heterolytic reactivity modes, either FLP-type activation28 in the presence of Lewis bases or formally metathetical transformations across polarized σ-bonds,29 where catalytic applications are known.30 Also, although not rigorously involving B–H activation, the Corey-Bakshi-Shibata protocol warrants mention in conjunction with a discussion of bifunctional main group catalyzed hydroboration.31 Nikonov32 and Kinjo33 have recently reviewed various aspects of main group catalyzed reduction chemistry.
A theme emerging from our study of 1 and related distorted structures concerns the extent to which descent from local threefold symmetry about tricoordinate phosphorus results in increased electrophilic character at P. Enthalpic (strain) arguments have been advanced in related systems that account for this observation,34 although electronic effects are also very likely to play an equally decisive role. In the case of 1, this mode of reactivity is accentuated as a function of the geometric constraint imposed by the triamide support framework. Whereas we had previously probed the increased electrophilic reactivity at phosphorus by reaction of 1 with protic E–H substrates (E = OR, NHR),35 here we access a regioreversal by reaction with a hydridic E–H (E = Bpin) compound. In this event, the acceptance of a hydride at phosphorus results in boryl transfer to the pendant N-methylanilide with P–N scission. Kinetics experiments demonstrate that the B–H reaction occurs in a bimolecular fashion, effectively a net σ-bond metathesis. In view of the ubiquity of phosphine-borane dative interactions, the regioselectivity of the B–H activation bears special mention; presumably both the weak P-nucleophilicity of 1 and weak B-electrophilicity of HBpin disfavor the common P→B interaction.
The P-hydrido-1,3,2-diazaphospholene intermediate 2 arising from P–N cooperative activation of the B–H bond in HBpin serves as a point of intersection with previous studies by Gudat, Kinjo, and Speed. Gudat has demonstrated the controlling role of hyperconjugative effects and π-σ* aromaticity in the structure and reactivity of P-substituted diazaphos-pholenes, phenomena that are manifest in a marked reactivity of the P–H moiety toward formal imine insertion as in 2. Similar electronic effects presumably also underpin the subsequent intramolecular boryl transfer reaction that leads to elimination of N-borylamine products 26 and reforms 1. This latter step is rapid under catalytic conditions as a function of the enforced intramolecularity; consequently, throughput via intermediates 2 and 25 is fast and 1 represents the resting state of the catalytic cycle. By way of contrast, recently reported diazaphospholene catalysts are devoid of possible ligand cooperativity, forcing the formation of reactive P-hydrido-1,3,2-diazaphospholenes to be turnover limiting. The P-N cooperative function of 1 is therefore a distinctive and essential aspect of its catalytic performance. Overall, the proposed mechanistic framework provides an entry point for next-generation optimization of P–N cooperative catalysis with 1 by deliberate targeting of individual steps for acceleration through rational modification and substituent effects.
4. Conclusions
Distorted phosphorous triamide 1 was shown to be a catalyst for the hydroboration of imines, and the mechanistic details underpinning this observation have been eludicated. The reactivity arises from the cooperative action of an electrophilic tricoordinate phosphorus center with a proximal basic N-methylanilide nitrogen within the distorted molecular environment imposed by the triamide supporting structure. The phosphorus-ligand cooperative bimolecular pathway is involved in both the initial B–H activation and the restoration of distorted phosphabicyclic structure of 1 following substrate reduction. The ability of distorted phosphorous triamide 1 to catalyze B–H transfer through a P-N ligand cooperative mechanism suggests further opportunity for targeted design of novel main group catalysts. On the basis of the described mechanistic model, new bifunctional P-based catalysts are being developed with potential applications across a range of E–H transfer chemistries and will be reported in due course.
5. Experimental Section
A full description of the general experimental methods, including preparation and purification of starting materials, can be found in the Supporting Information.
Synthesis of 2
Under nitrogen atmosphere, pinacolborane (15 mg, 0.12 mmol) was mixed with a C6D6 solution (1 mL) of compound 1 (30 mg, 0.12 mmol) in a sealable threaded NMR tube. The mixture was stirred at 50 °C for 12 h to afford crude 2 (estimated purity 95 % by 1H NMR). 1H NMR (C6D6, 500 MHz): δ 7.29 (d, 1H, J = 7.8 Hz), 7.20 (d, 1H, J = 7.8 Hz), 7.07 (d, 1H, J = 7.7 Hz), 6.92-6.77 (m, 4H), 6.86 (d, 1H, 1 J P-H = 146.3 Hz), 6.52 (d, 1H, J = 7.8 Hz), 3.05 (s, 3H), 2.85 (d, 3H, J = 12.8 Hz), 1.05 (s, 12H) ppm. 31P NMR (C6D6, 202 MHz): δ 84.2 (d, J= 146 Hz) ppm. MS (ESI) C20H27BN3O2P (M+) calc’d: 383.1934; found: 383.1938.
Synthesis of 3
Under nitrogen atmosphere, pinacolborane (50 mg, 0.39 mmol) was added into a CHCl3 solution (2 mL) of compound 1 (100 mg, 0.39 mmol) and the solution was stirred under room temperature. After 12 h, all volatiles were removed in vacuo and the remaining white solid was recrystallized in pentane to afford 3 as a white crystals (80 mg, 70 %). 1H NMR (CDCl3, 400 MHz): δ 7.64 (d, 1H, J = 8.0 Hz), 7.37-7.26 (m, 3H), 7.08 (t, 1H, J = 8.0 Hz), 7.01-6.97 (m, 2H), 3.39 (d, 3H, J = 16.0 Hz), 2.85 (s, 3H), 0.94 (broad s, 12H) ppm. 13C NMR (CDCl3, 100 MHz): δ 145.0 (d, 2 J PC = 4.0 Hz), 136.9 (d, 2JPC = 12.0 Hz), 132.0 (d, 2JPC = 11.0 Hz), 129.5, 129.5, 128.8, 128.3, 125.7, 121.5, 121.1, 111.9, 110.0, 82.7, 37.2, 29.4 (d, 2JPC = 18.0 Hz), 24.2 ppm. 31P NMR (CDCl3, 145 MHz): δ 146.4 ppm. MS (ESI) C20H26BN3O2P (M+-Cl) calc’d: 382.1856; found: 382.1859.
Synthesis of 4
Under nitrogen atmosphere, a C6D6 solution of in situ generated 2 (1 ml, 0.24 M) was added N-tosyl-1-phenylmethanimine (64 mg, 0.24 mmol). The mixture was stirred at room temperature for 16 h, all volatiles were removed in vacuo and the resulting white solid was washed with pentane (60 mg, 63%). The solid was then recrystallized in a 10:1 pentane/CH2Cl2 solution to obtain the samples for X-ray crystallography. 1H NMR (C6D6, 400 MHz): δ 7.80 (d, 2H, J = 7.9 Hz), 7.53-7.50 (m, 2H), 7.37 (d, 1H, J = 8.0 Hz), 7.20-7.07 (m, 5H), 6.93-6.69 (m, 5H), 6.65 (d, 1H, J = 7.4 Hz), 6.05 (d, 1H, J = 7.5 Hz), 4.51 (d, 1H, J = 15.9 Hz), 3.69 (d, 1H, J = 15.9 Hz), 2.93 (s, 3H), 2.38 (d, 3H, J = 11.8 Hz), 2.07 (s, 3H), 1.12 (s, 12H) ppm. 13C NMR (C6D6, 126 MHz): δ 145.1, 141.6, 139.5, 137.9, 137.7, 137.7, 137.0 (d, 2JPC = 10.1 Hz), 134.1 (d, 2JPC = 12.6 Hz), 129.8, 129.3, 129.1, 128.1, 126.8, 126.8, 125.2, 124.2, 119.9, 118.5, 108.6, 107.8, 82.8, 47.9, 36.2, 36.2, 28.6, 28.4, 24.6, 24.4 ppm. 31P NMR (C6D6, 145 MHz): δ 92.6 ppm. MS (ESI) C34H41BN4O4PS (M++H) calc’d: 643.2679; found: 643.2685.
Compound 6 and NMR monitoring of its decay
Under nitrogen atmosphere, a 1:1 THF/ether solution of 3 (30 mg, 70 mM) and an ether solution of N-benzyl-1-(4-methoxyphenyl)methanamine (33 mg, 140 mM) were both cooled to −30 °C and then mixed together. The resulting cloudy mixture was quickly filtered through Celite and all volatiles were removed in vacuo to afford crude 5. (NMR yield 70 %). The product was then dissolved in C6D6 and the solution was monitored by 31P and 1H NMR1H NMR (C6D6, 500 MHz): δ 7.56 (d, 1H, J = 11.0 Hz), 7.40 (d, 1H, J = 11.0 Hz), 7.23-6.80 (m, 12H), 6.80 (d, 2H, J = 11.5 Hz), 6.67 (d, 1H, J = 10.5 Hz), 4.25 (broad s, 2H), 3.70 (broad s, 2H), 3.45 (s, 3H), 3.20 (s, 3H), 3.04 (d, 3H, 3JPH = 13.8 Hz), 1.16 (broad s, 12H) ppm. 31P NMR (C6D6, 145 MHz): δ 105.2 ppm.
Synthesis of 8
Under nitrogen atmosphere, benzyl amine (26 mg, 0.24 mmol) was added into a CH2Cl2 solution of 3 (50 mg, 120 mM). The resulting cloudy solution was quickly filtered through Celite and all volatiles were removed in vacuo to afford crude 8. The crude product was recrystallized in a 1:5 dichloromethane:pentane solution to afford 8 as a white crystals (40 mg, 71 %). 1H NMR (CD2Cl2, 360 MHz): δ 7.40-7.21 (m, 3H), 7.08 (d, 1H, J = 7.0 Hz), 6.83 (t, 1H, J = 7.3 Hz), 6.68 (t, 1H, J = 7.6 Hz), 6.61 (d, 1H, J = 7.5 Hz), 6.56 (d, 1H, J = 7.5 Hz), 3.07 (d, 3H, J = 10.3 Hz), 2.75 (s, 3H) ppm, 1.17 (s, 12H). 31P NMR (CD2Cl2, 145 MHz): δ 91.34 ppm. 13C NMR (CD2Cl2, 125 MHz): δ 144.5, 143.9, 140.9, 129.3, 129.0, 128.3 (d, 2JPC = 11.3 Hz), 127.4, 127.0, 126.8, 126.5, 126.2, 125.2, 118.7, 117.3, 106.0, 82.6, 46.4, 44.8, 35.9, 29.0 (d, 2JPC = 21.2 Hz), 24.3 ppm. MS (ESI) C27H35BN4O2P (M++H) calc’d: 489.2591; found 489.2595.
Supplementary Material
Acknowledgments
The stoichiometric reactivity of 1 with HBpin was supported under NSF award CHE-1724505. The catalytic hydroboration studies were supported under NIH grant GM114547. A.T.R. gratefully acknowledges additional support from the Alfred P. Sloan Foundation and Amgen. We thank Dr. Nicole L. Dunn (PSU) for pre-liminary experiments.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Full experimental procedures; 1H, 13C, and 31P NMR spectra for all synthetic compounds; NOESY spectra for 2; crystallographic details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Khusnutdinova JR, Milstein D. Angew. Chem. Int. Ed. 2015;54:12236–12273. doi: 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]
- 2.Trincado M, Grützmacher H. In: Cooperative Catalysis. Peters R, editor. Wiley-VCH Verlag GmbH & Co. KGaA; 2015. pp. 67–110. [Google Scholar]
- 3.Gunanathan C, Milstein D. Acc. Chem. Res. 2011;44:588–602. doi: 10.1021/ar2000265. [DOI] [PubMed] [Google Scholar]
- 4.Zell T, Milstein D. Acc. Chem. Res. 2015;48:1979–1994. doi: 10.1021/acs.accounts.5b00027. [DOI] [PubMed] [Google Scholar]
- 5.Koren-Selfridge L, Londino HN, Vellucci JK, Simmons BJ, Casey CP, Clark TB. Organometallics. 2009;28:2085–2090. [Google Scholar]
- 6.Schneider S, Meiners J, Askevold B. Eur. J. Inorg. Chem. 2012;2012:412–429. [Google Scholar]
- 7.(a) Hashiguchi S, Fujii A, Takehara J, Ikariya T, Noyori R. J. Am. Chem. Soc. 1995;117:7562–7563. [Google Scholar]; (b) Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R. J. Am. Chem. Soc. 1996;118:2521–2522. [Google Scholar]; (c) Uematsu N, Fujii A, Hashiguchi S, Ikariya T, Noyori R. J. Am. Chem. Soc. 1996;118:4916–4917. [Google Scholar]
- 8.(a) Arrowsmith M, Hadlington TJ, Hill MS, Kociok-Köhn G. Chem. Commun. 2012;48:4567–4569. doi: 10.1039/c2cc30565h. [DOI] [PubMed] [Google Scholar]; (b) Arrowsmith M, Hill MS, Kociok-Köhn G. Chem. Eur. J. 2013;19:2776–2783. doi: 10.1002/chem.201203190. [DOI] [PubMed] [Google Scholar]; (c) Arrowsmith M, Hill MS, Hadlington T, Kociok-Köhn G, Weetman C. Organometallics. 2011;30:5556–5559. [Google Scholar]
- 9.(a) Dureen MA, Stephan DW. J. Am. Chem. Soc. 2010;132:13559–13568. doi: 10.1021/ja1064153. [DOI] [PubMed] [Google Scholar]; (b) R Cabrera A, S Rojas R, Valderrama M, Plüss P, Berke H, G Daniliuc C, Kehr G, Erker G. Dalton Trans. 2015;44:19606–19614. doi: 10.1039/c5dt01966d. [DOI] [PubMed] [Google Scholar]; (c) Theuergarten E, Schlösser J, Schlüns D, Freytag M, G Daniliuc C, G Jones P, Tamm M. Dalton Trans. 2012;41:9101–9110. doi: 10.1039/c2dt30448a. [DOI] [PubMed] [Google Scholar]
- 10.(a) Gellrich U, Diskin-Posner Y, Shimon LJW, Milstein D. J. Am. Chem. Soc. 2016;138:13307–13313. doi: 10.1021/jacs.6b07454. [DOI] [PubMed] [Google Scholar]; (b) Appelt C, Westenberg H, Bertini F, Ehlers AW, Slootweg JC, Lammertsma K, Uhl W. Angew. Chem. Int. Ed. 2011;50(17):3925–3928. doi: 10.1002/anie.201006901. [DOI] [PubMed] [Google Scholar]
- 11.(a) Li J, Li B, Liu R, Jiang L, Zhu H, Roesky HW, Dutta S, Koley D, Liu W, Ye Q. Chem. Eur. J. 2016;22:14499–14503. doi: 10.1002/chem.201603544. [DOI] [PubMed] [Google Scholar]; (b) Yu Y, Li J, Liu W, Ye Q, Zhu H. Dalton Trans. 2016;45:6259–6268. doi: 10.1039/c5dt03873a. [DOI] [PubMed] [Google Scholar]; (c) Freitag S, Henning J, Schubert H, Wesemann L. Angew. Chem. Int. Ed. 2013;52:5640–5643. doi: 10.1002/anie.201301153. [DOI] [PubMed] [Google Scholar]
- 12.(a) Xiong Y, Yao S, Driess M. Organometallics. 2009;28:1927–1933. [Google Scholar]; (b) Gau D, Rodriguez R, Kato T, Saffon-Merceron N, de Cózar A, Cossío FP, Baceiredo A. Angew. Chem. Int. Ed. 2011;50:1092–1096. doi: 10.1002/anie.201006265. [DOI] [PubMed] [Google Scholar]; (c) Delawar M, Gehrhus B, B Hitchcock P. Dalton Transactions. 2005;0(17):2945–2953. doi: 10.1039/b507462b. [DOI] [PubMed] [Google Scholar]
- 13.Nifantiev EE, Grachev MK, Burmistrov SY. Chem. Rev. 2000;100:3755–3800. doi: 10.1021/cr9601371. [DOI] [PubMed] [Google Scholar]
- 14.Power PP. Nature. 2010;463:171–177. doi: 10.1038/nature08634. [DOI] [PubMed] [Google Scholar]
- 15.Myers TW, Berben LA. J. Am. Chem. Soc. 2013;135:9988–9990. doi: 10.1021/ja4032874. [DOI] [PubMed] [Google Scholar]
- 16.W Myers T, A Berben L. Chem. Sci. 2014;5(7):2771–2777. [Google Scholar]
- 17.Zhao W, McCarthy SM, Lai TY, Yennawar HP, Radosevich AT. J. Am. Chem. Soc. 2014;136(50):17634–17644. doi: 10.1021/ja510558d. [DOI] [PubMed] [Google Scholar]
- 18.Burck S, Gudat D, Nieger M, Du Mont W-W. J. Am. Chem. Soc. 2006;128:3946–3955. doi: 10.1021/ja057827j. [DOI] [PubMed] [Google Scholar]
- 19.(a) Chong CC, Hirao H, Kinjo R. Angew. Chem. Int. Ed. 2014;53:3342–3346. doi: 10.1002/anie.201400099. [DOI] [PubMed] [Google Scholar]; (b) Chong CC, Hirao H, Kinjo R. Angew. Chem. Int. Ed. 2015;54:190–194. doi: 10.1002/anie.201408760. [DOI] [PubMed] [Google Scholar]
- 20.Adams MR, Tien C-H, Huchenski BSN, Ferguson MJ, Speed AWH. Angew. Chem. Int. Ed. 2017 doi: 10.1002/anie.201611570. [DOI] [PubMed] [Google Scholar]
- 21.Gudat D. Acc. Chem. Res. 2010;43:1307–1316. doi: 10.1021/ar100041j. [DOI] [PubMed] [Google Scholar]
- 22.Snow SS, Jiang DX, Parry RW. Inorg. Chem. 1985;24:1460–1463. [Google Scholar]
- 23.A unidentified trace impurity from the in situ preparation of 2, whose concentration does not change with time, is also observed in 31P NMR at δ 140.1 ppm.
- 24.(a) Keaton RJ, Blacquiere JM, Baker RT. J. Am. Chem. Soc. 2007;129:1844–1845. doi: 10.1021/ja066860i. [DOI] [PubMed] [Google Scholar]; (b) Pun D, Lobkovsky E, J Chirik P. Chem. Commun. 2007;0:3297–3299. doi: 10.1039/b704941b. [DOI] [PubMed] [Google Scholar]; (c) Chaplin AB, Weller AS. Angew. Chem. Int. Ed. 2010;49:581–584. doi: 10.1002/anie.200905185. [DOI] [PubMed] [Google Scholar]
- 25.(a) Stahl T, Müther K, Ohki Y, Tatsumi K, Oestreich M. J. Am. Chem. Soc. 2013;135:10978–10981. doi: 10.1021/ja405925w. [DOI] [PubMed] [Google Scholar]; (b) Anaby A, Butschke B, Ben-David Y, Shimon LJW, Leitus G, Feller M, Milstein D. Organometallics. 2014;33:3716–3726. [Google Scholar]
- 26.(a) Wilczynski R, Sneddon LG. J. Am. Chem. Soc. 1980;102:2857–2858. [Google Scholar]; (b) Wilczynski R, Sneddon LG. Inorg. Chem. 1981;20:3955–3962. [Google Scholar]; (c) Wilczynski R, Sneddon LG. Inorg. Chem. 1982;21:506–514. [Google Scholar]; (d) Davan T, Corcoran EW, Sneddon LG. Organometallics. 1983;2:1693–1694. [Google Scholar]; (e) Pender MJ, Carroll PJ, Sneddon LG. J. Am. Chem. Soc. 2001;123:12222–12231. doi: 10.1021/ja011432s. [DOI] [PubMed] [Google Scholar]; (f) Gridnev ID, Miyaura N, Suzuki A. Organometallics. 1993;12:589–592. [Google Scholar]; (g) Kaithal A, Chatterjee B, Gunanathan C. Org. Lett. 2015;17:4790–4793. doi: 10.1021/acs.orglett.5b02352. [DOI] [PubMed] [Google Scholar]; (h) Burgess K, Jaspars M. Tetrahedron Lett. 1993;34:6813–6816. [Google Scholar]; (i) Burgess K, Jaspars M. Organometallics. 1993;12:4197–4200. [Google Scholar]; (j) Yamamoto Y, Fujikawa R, Umemoto T, Miyaura N. Tetrahedron. 2004;60:10695–10700. [Google Scholar]; (k) Satoh M, Nomoto Y, Miyaura N, Suzuki A. Tetrahedron Lett. 1989;30:3789–3792. [Google Scholar]; (l) Männig D, Nöth H. Angew. Chem. Int. Ed. Engl. 1985;24:878–879. [Google Scholar]
- 27.(a) Frey GD, Masuda JD, Donnadieu B, Bertrand G. Angew. Chem. Int. Ed. 2010;49:9444–9447. doi: 10.1002/anie.201005698. [DOI] [PubMed] [Google Scholar]; (b) Lastovickova DN, Bielawski CW. Organometallics. 2016;35:706–712. [Google Scholar]
- 28.(a) Dureen MA, Lough A, Gilbert TM, Stephan DW. Chem. Commun. 2008:4303–4305. doi: 10.1039/b808348g. [DOI] [PubMed] [Google Scholar]; (b) Eisenberger P, Bailey AM, Crudden CM. J. Am. Chem. Soc. 2012;134:17384–17387. doi: 10.1021/ja307374j. [DOI] [PubMed] [Google Scholar]
- 29.Waterman R. Organometallics. 2013;32:7249–7263. [Google Scholar]
- 30.Hill MS, Liptrot DJ, Weetman C. Chem. Soc. Rev. 2016;45:972–988. doi: 10.1039/c5cs00880h. [DOI] [PubMed] [Google Scholar]
- 31.(a) Corey EJ, Bakshi RK, Shibata S, Chen CP, Singh VK. J. Am. Chem. Soc. 1987;109:7925–7926. [Google Scholar]; (b) Corey EJ, Bakshi RK, Shibata S. J. Am. Chem. Soc. 1987;109:5551–5553. [Google Scholar]; (c) Corey EJ, Helal CJ. Angew. Chem. Int. Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 32.Revunova K, Nikonov GI. Dalton Trans. 2014;44:840–866. doi: 10.1039/c4dt02024c. [DOI] [PubMed] [Google Scholar]
- 33.Chong CC, Kinjo R. ACS Catal. 2015;5:3238–3259. [Google Scholar]
- 34.(a) Greenhalgh R, Newbery JE, Woodcock R, Hudson RF. Chem. Commun. 1969:22. [Google Scholar]; (b) Hudson RF, Brown C. Acc. Chem. Res. 1972;5:204–211. [Google Scholar]
- 35.McCarthy SM, Lin Y-C, Devarajan D, Chang JW, Yennawar HP, Rioux RM, Ess DH, Radosevich AT. J. Am. Chem. Soc. 2014;136:4640–4650. doi: 10.1021/ja412469e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.