


Abstract
Multi-protein DNA replication complexes called replisomes perform the essential process of copying cellular genetic information prior to cell division. Under ideal conditions, replisomes dissociate only after the entire genome has been duplicated. However, DNA replication rarely occurs without interruptions that can dislodge replisomes from DNA. Such events produce incompletely replicated chromosomes that, if left unrepaired, prevent the segregation of full genomes to daughter cells. To mitigate this threat, cells have evolved ‘DNA replication restart’ pathways that have been best defined in bacteria. Replication restart requires recognition and remodeling of abandoned replication forks by DNA replication restart proteins followed by reloading of the replicative DNA helicase, which subsequently directs assembly of the remaining replisome subunits. This review summarizes our current understanding of the mechanisms underlying replication restart and the proteins that drive the process in Escherichia coli (PriA, PriB, PriC and DnaT).
INTRODUCTION
The accurate transfer of genetic information from one cell to its progeny is an essential process for all organisms. The first step is to produce a copy of the genome. Genome duplication is catalyzed by protein complexes that mediate several key reactions that include origin recognition and replication initiation, double-stranded (ds) DNA unwinding, replication of template single-stranded (ss) DNA, and termination upon completion of replication (1). While these steps are carried out in all cells, variations exist in how diverse organisms regulate DNA replication, deal with unique chromosome structures and topologies, and overcome DNA damage or other events that require repair.
DNA replication in the model organism Escherichia coli has been extensively studied, providing a foundation for understanding the diverse mechanisms of genome duplication employed by all organisms. In E. coli, DNA replication is initiated at oriC, a unique origin locus within the ∼5-million base pair circular chromosome (Figure 1A). oriC is ‘melted’ by the action of the DnaA initiator protein to expose two template ssDNA strands that act as platforms for loading the replicative DnaB helicase (2–4). One full DnaB hexamer is loaded onto each ssDNA strand with the aid of the helicase loader, DnaC (5–8). Additional exposed ssDNA is quickly coated by the ssDNA-binding protein (SSB), which protects DNA and blocks additional DnaB helicase loading (9). Each DnaB hexamer recruits primase (DnaG), which synthesizes RNA primers used to initiate DNA synthesis, along with the subunits that comprise the replicative DNA polymerase III holoenzyme (PolIII HE) (10–13). These proteins form the core replisomes that copy the E. coli genome. Once assembled, replisomes replicate bi-directionally away from oriC until, ideally, they undergo programmed disassembly at the termination region, where they encounter ter sites bound by Tus proteins that create ‘replication fork traps’ (14–16). After completion of DNA replication, the newly synthesized genomes are separated and segregated to daughter cells.
Figure 1.
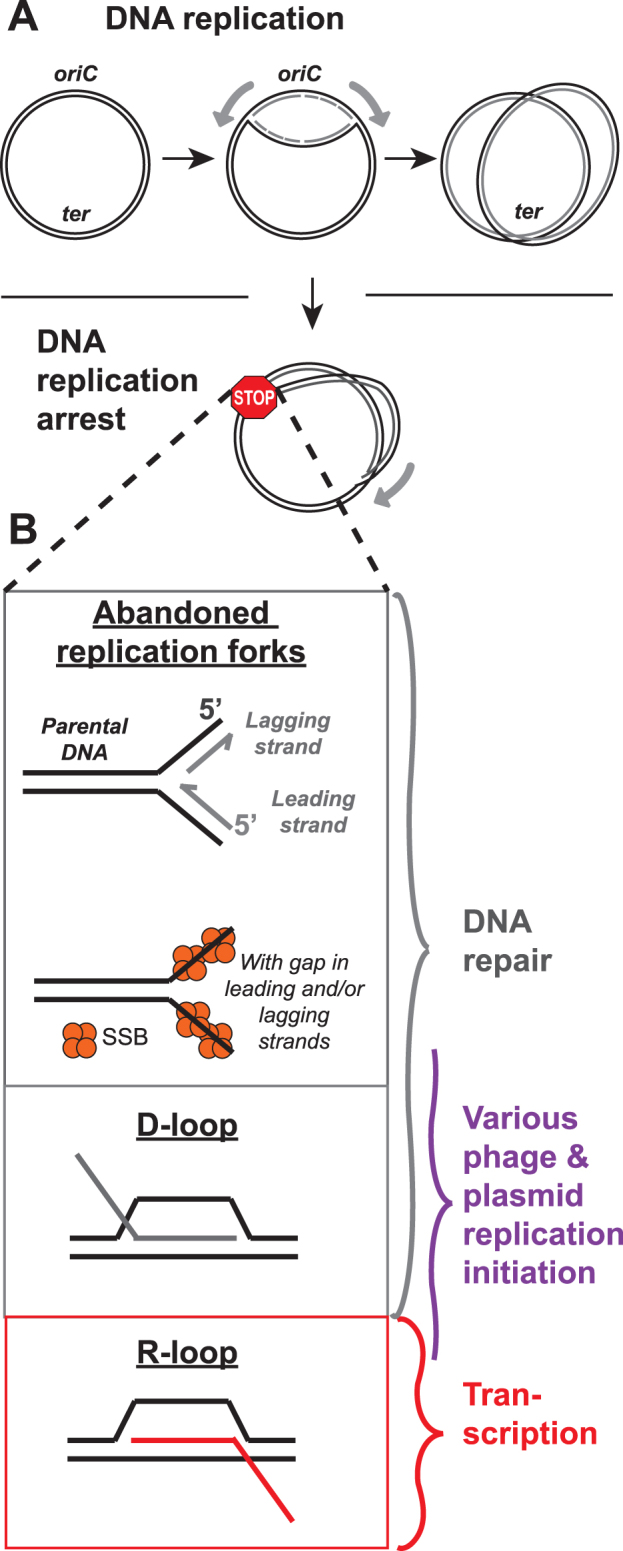
DNA replication and forked DNA structures that can be recognized by the replication restart machinery. (A) Cartoon representation of DNA replication in E. coli from the origin of replication (oriC) to the terminus (ter), depicting replication fork arrest and collapse that often occurs during this process. (B) Schematic of the DNA fork structures present at abandoned replication forks and other fork types that replication restart can recognize. These additional fork types are found during DNA repair processes, certain phage and plasmid DNA replication initiation, and during/after transcription. DNA is indicated in black (parental) and grey (nascent) with RNA shown in red. SSB is shown in orange.
While this description may give the impression that replication occurs without challenges, the reality is that replisomes face numerous barriers including collisions with damage in the template and with transcription complexes or other genome-bound protein factors (17–21). Such obstacles can cause the replisome to stall or dissociate. The latter premature termination event leaves collapsed/abandoned replication forks that cannot be reinitiated by the same sequence-specific process that functions at oriC. Premature termination of the replication fork is potentially lethal, as double-strand DNA breaks (DSBs) will be created upon the next origin firing event and chromosome partitioning and cell division will be impaired. The frequency of replication failure is difficult to measure. However, one estimate based on multiple lines of evidence suggests that replisome dissociation occurs roughly once per cell cycle in E. coli (22), whereas a more recent study suggests that replisome disassembly occurs multiple times per cell cycle and that replication/transcription collision is a significant source of these events (20). Accumulating studies have emphasized the prevalent nature of transcription-replication conflicts and the severity of their consequences in all organisms (blocking replication progression, causing replisome disassembly, creating DSBs, leading to cell death, shaping the landscape of the bacterial genome, and resulting in cancer and disease in eukaryotes) (21,23–25). In vitro studies have shown that the E. coli replisome can withstand and even bypass complications related to replication fork stalling/collapse, repriming DNA synthesis downstream (26). However the frequency with which these complications occur within the cell appears to be high enough that full replisome disassembly (replicative helicase disassembly) and replication restart remains a relatively common event.
How do E. coli and other related bacteria survive this possible calamity? Bacteria have evolved DNA replication restart mechanisms that detect abandoned replication forks and, in a structure-dependent and sequence-independent manner, reload the replicative helicase onto sites far removed from oriC. In this review, we summarize our current understanding of the structural mechanisms that govern DNA replication restart processes in E. coli and briefly address the differences found in other organisms.
DNA REPLICATION RESTART: WHEN AND WHERE
Dissociation of the replicative helicase, one of the most stable components of the bacterial replisome (27), is the step at which a replication fork can be considered ‘abandoned’, and helicase reloading appears to be the primary objective of bacterial DNA replication restart pathways. Under normal growth, DNA replication restart is regularly required to resolve and restore abandoned replication fork structures. Replication restart events, including those resulting from replication-transcription conflicts, proceed either directly (without the need for repair) or following DNA repair (28–30). Requirements for replication restart become enhanced under stressed conditions that affect DNA replication or genome integrity (31,32). On forks that require repair, restart proteins likely recognize the fork after repair proteins have recognized and acted on the lesion. This review focuses on the mechanisms of proteins that function most directly in replicative helicase loading; discussions of the roles of DNA repair proteins in DNA replication restart are reviewed elsewhere (33).
Abandoned replication forks can take on various forms, with gapped (SSB-coated ssDNA) or dsDNA on the leading and/or lagging strands (Figure 1B). Either of these fork types may be physiologically relevant substrates under the various conditions that lead to replication failure, and our current understanding of replication restart substrates has largely come from the DNA preferences of restart proteins in reconstituted in vitro reactions. Additional forked substrates recognized by replication restart include D-loops (homologous recombination products) and R-loops (transcription products) (Figure 1B), and restart from each of these structures occurs in processes termed induced or constitutive stable DNA replication (iSDR and cSDR), respectively. iSDR is an SOS DNA damage response-induced process that utilizes D-loops, whereas cSDR is a process in which replication initiation occurs on R-loops that are stabilized in bacterial strains lacking RNase HI (see review (34)). Replication restart pathways also initiate replication of certain plasmid and phage DNA that encode regions in their chromosomes that form replication fork-like structures that can be acted on by the cellular/host restart proteins. Hfr conjugation and P1 transduction use a 3′ end invasion site to form D-loops whereas bacteriophage ΦX174 DNA replication, bacteriophage Mu replicative transposition, and ColE1-like plasmids use a replication fork-like structure (31,32,35–37).
The involvement of the DNA replication restart proteins (RRPs) in phage replication is also important historically, since the E. coli RRPs were originally defined as host factors required for replicating the bacteriophage ΦX174 genome in vitro (35,38). This led to the proteins being named ‘primosome assembly proteins’ since they were found to catalyze priming of DNA synthesis. The RRPs were purified using ΦX174 phage DNA replication as a measure of activity (39–42). In these classic experiments, PriA, PriB, PriC and DnaT (collectively referred to as the pre-primosome) were shown to load DnaB onto SSB-coated ΦX174 DNA at its origin of replication, called the ‘primosome assembly site’ (pas). This site forms a replication fork-like hairpin structure that would be recognized as an abandoned replication fork in E. coli. Subsequent studies revealed that PriA, PriB, PriC and DnaT are dispensable for the core DNA replication process in E. coli, and it is clear now that these proteins are specialized for DNA replication restart. The next section describes what is known about each of these proteins.
THE REPLICATION RESTART PROTEINS
PriA
Originally called ‘protein n' ’ or ‘replication factor Y,’ PriA is a ∼80kDa 3′-5′ superfamily II (SF2) DNA helicase that can interact with a range of DNA structures (dsDNA, ssDNA and forked DNA) and proteins (SSB, PriB, and DnaT). PriA acts as a master orchestrator of replication restart by recognizing and remodeling abandoned DNA replication forks (43). Consistent with its fundamental role in replication restart, PriA is also the only RRP that is conserved across all bacterial phyla.
PriA genetics
Escherichia coli priA-null mutants are very slow growing and rich media sensitive (31,44,45), a phenotype that contributed to the original rationale that replisomes require restarting at least once per cell cycle (38,44,46). In accordance with their rich-media sensitivity and the prevalent nature of replication-transcription conflicts, priA-null cells are extremely sensitive to inhibition of Rho-dependent transcription termination by bicyclomycin (19). These cells are induced for the SOS DNA-damage response and a portion are filamented (with or without sulA or sulB mutations that suppress cell division) and are defective in proper chromosomal partitioning (31,44,47,48). Additionally, priA-null (priA2::kan) strains are as recombination deficient and sensitive to UV irradiation as recB21 recC22 mutants (49,50). PriA is critical for repair of DSB, UV-induced restriction alleviation, and resumption of DNA replication after UV-irradiation (51–55). Indeed, recombination in the absence of PriA is harmful: an rdgC deletion, which removes RdgC control of RecA loading, is synthetically lethal with PriA inactivation, and the viability of a priA-null mutation is improved in a recA recB mutant background (47,56). PriA mutant cells are unable to properly replicate certain plasmids and phages or undergo primed adaptive CRISPR-Cas E. coli Type 1 immunity, as most of these processes proceed or initiate through forked DNA structures and require other RRPs (31,32,35–37,57). PriA mutant cells also cannot perform iSDR and cSDR (described above) (31,34,45).
Many mutations are synthetically lethal with priA-null mutations. These mutations reveal related pathways that accomplish similar functions in the cell or processes that in the absence of one function increases the frequency at which other functions are required. Many genes with synthetically lethal relationships with priA-null cells, ruvABC, recG, ftsK, gyrB, holD and rep, are most easily explained by the latter category, since replication fork arrest is elevated (gyrBts, holD, rep), or predicted to be elevated (ruvABC, recG, ftsK) in these backgrounds. Certain synthetic lethalities can be overcome by the dnaC809 suppressor of priA2::kan, whereas others cannot (holD, rep (58,59)). In fact, the increase in arrested replication forks from a priA-null mutation can elevate the rate of integration of transposon types that target ‘slow’ replication forks (60).
Beyond the impact of priA-null mutations, many priA genetic studies have examined point mutations within priA. Of these, some were designed to test the roles of specific activities (e.g. ATPase function) whereas others were found as suppressors of separate mutations in E. coli. Specifically, a few point mutations that affect PriA helicase activity have revealed an interplay between RecG and PriA at DNA forks. PriA and RecG substrate preferences overlap, and missense mutants of priA that affect helicase activity have been isolated as suppressors of the UV-sensitive phenotype of recG-null mutants (61–63). These priA suppressors (srgA for suppressor to recG) retain the ability to unwind the lagging strand when the leading strand is present but have reduced helicase activity when the leading strand is absent (62). The reason for this could be rationalized with in vitro data indicating that the latter is the preferred substrate for RecG and the former is thought to be the preferred substrate for PriA (61,64,65). A more straight-forward helicase dead variant, priA300, is also able to rescue the UV sensitivity of a recG mutant strain, leading to the conclusion that PriA helicase activity is harmful in the absence of RecG (63,66). Given their interplay and the relatively high affinity that PriA has for all fork types, RecG is proposed to promote formation of productive fork conformations for PriA binding and to prevent PriA from unwinding parental DNA or duplex regions that join DNA molecules during DSB repair (67,68). Tus/ter sites, at which replication forks meet and create 3′ flaps, are potentially detrimental PriA substrates when RecG is absent (69). Interestingly, the primed adaptive immunity of the E. coli Type 1 CRISPR-Cas system requires both RecG and PriA (although not PriA helicase activity) and has an enhancement of spacer acquisition from ter sites relative to the rest of the chromosome (57,70). However, in the interplay of PriA and RecG at replication forks, a lack of PriA ATPase activity (priA300) cannot fully replicate srgA phenotypes and reduced PriB levels also partially suppresses some recG-null phenotypes (71).
A recent study looked at PriA distribution across the E. coli genome as a measure for stalled replication fork distribution under normal and nucleotide-depleted conditions using ChIP-chip (72). The authors found PriA distribution under non-stressed and UV-exposed conditions to be too low and/or disperse to detect. This may be the result of fork sites being randomly dispersed through the genome as well as low levels of PriA (1–2 molecules per fork (73)). Following endogenous levels of PriA via microscopy has also proven difficult, perhaps due to some of these properties of PriA (74,75). However, the PriA ChIP-chip study did find that upon nucleotide depletion, PriA association was enhanced in a 600 kb segment around oriC in asynchronous cells. It was proposed that the forks in this area around oriC may be proceeding more slowly or arresting more easily under nucleotide depletion (72).
PriA biochemistry
Given the importance of PriA as a well-conserved driver of DNA replication restart, multiple labs have investigated the structural mechanisms by which PriA proteins operate. Proteolysis experiments showed the E. coli PriA protein to be comprised of two major domains: an N-terminal DNA-binding domain (DBD, residues 1–181) and a C-terminal helicase domain (HD, residues 182–732) (Figure 2A) (76–78). The first high-resolution structures of a domain from PriA were of the N-terminal-most 105 residues of the E. coli PriA DBD, a region referred to as the 3′ binding domain (3′ BD) (Figure 2A: pale red with the terminal nucleotide in black) (79–81). The 3′ BD structures helped to explain how PriA can recognize the 3′ hydroxyl end of leading strand DNA in D-loop and abandoned replication fork structures. The structures identified a 3′ BD pocket that interacts specifically with the 3′ end in a non-base-selective manner. Asp17 hydrogen bonds with a ring atom present in any base type, Tyr18 stacks against the deoxyribose ring, and Lys61 and Gly37 interact with the 5′ phosphodiester group of the terminal base (80). The remaining domain of the PriA DBD (residues 109–181; now known as the winged helix domain) also binds DNA and facilitates the DBD interaction with forks without a 3′ end, although these interactions require cooperation with the rest of PriA for high DNA affinity (81).
Figure 2.

Structures of the bacterial DNA replication restart proteins. (A) Domain architecture and crystal structure of Klebsiella pneumoniae PriA (pdb: 4NL4; ADP in red sticks and Zn2+ in gray spheres (43)), with each structural domain individually colored, overlayed with the structure of the E. coli 3′ BD cocrystalized with dinucleotide (pale red and black respectively; pdb: 2D7G, (80)). (B) Structure of the E. coli PriB dimer (pdb: 1WOC, (183)) shown in magenta, overlayed with the structure of E. coli PriB dimer cocrystalized with ssDNA (grey and black respectively; pdb: 2CCZ, (119)). Inset: two DNA-binding domains of E. coli SSB bound to ssDNA (grey and black respectively, pdb: 1EYG, (184)). (C) Left: The solution NMR structure of the 98 N-terminal residues of E. coli PriC shown in wheat (pdb: 2RT6, (129)). Right: The solution NMR structure of full-length Cronobacter sakazakii PriC protein shown in brown (pdb: 2NCJ,(127)). (D) The solution NMR structure of the C-terminal 90 residues of the E. coli DnaT monomer shown in dark blue (middle; pdb: 2RU8, (96)) overlayed with the crystal structure of E. coli DnaT 84–153 bound to dT10 (the five subunits in the oligomer shown in cyan, where the middle one has been overlayed with 2RU8 and the outer two are shown with transparent grey surface, and DNA in black; pdb: 4OU7, (98)). Domain architecture shown to left of each structure, where grey sections indicated the protein sections not included and/or resolved in the structure(s) at right.
More recently, crystal structures of the full-length Klebsiella pneumoniae PriA protein (88/98% sequence identity/similarity with E. coli PriA) have revealed a multi-domain structure that provides additional insights into the physical mechanisms of PriA functions (Figure 2A) (82). The DBD within these structures contains the 3′ BD as expected and identifies the second DNA-binding element within the PriA DBD as a circularly-permutated winged helix (WH) fold. Within the PriA HD, which comprises the majority of the protein, several domains were observed: two RecA-like lobes that contained the expected SF2 helicase motifs, a cysteine-rich region (CRR) subdomain coordinating two zinc atoms, and a C-terminal domain (CTD) that shares structural homology with the S10 ribosomal subunit (82). In contrast to the 3′ BD, which is tightly packed against the PriA helicase domain (HD), the WH domain is loosely tethered to the rest of PriA through linkers and the domain is packed against symmetrically-related molecules within the crystal lattice. Related to the flexibility of the WH linker, a priA-null mutation suppressor was recently found from an inactivating antibiotic resistance insertion within the WH at residue 154 (83). The suppressor introduced a start codon at residue 157 that yields an almost fully-functional PriA when both PriA segments are present.
Previous priA mutations provided information on the function of some of the helicase core elements. Extensive studies have been performed on PriA variants in which the conserved lysine (K230) residue of motif I (Walker A) has been altered. The mutant priA300 (K230R) abolishes ATPase, helicase and translocase activities of PriA. Despite loss of enzymatic function, priA300 E. coli behave much like wild type cells in vivo and the PriA variant is active in in vitro replication restart assays (32,84,85). This suggests that ATPase, helicase, and/or translocase activities of PriA are not required for its replication restart function. However, PriA helicase activity is well-conserved across bacteria (except in Deinococcus (86)) and is required in certain restart pathways (described below). When priA300 is coupled with a priB-null mutation, the double mutant behaves like a priA2::kan mutant, indicating that PriA helicase activity is required for the PriA–PriC pathway but not the PriA–PriB pathway (Figure 3) (48). Additionally, PriA helicase activity is required for Mu bacteriophage replicative transposition in vivo and in vitro (48,64,87), normal iSDR levels (88), and to restore the viability of holD mutants with priA2::kan or priA300 (58). PriA K230A and PriA K230D variants have also been studied in vitro and in vivo, and both are similar to K230R, except that priA K230D may have a specific defect in replication of ColEI plasmids (84,89). Several studies have suggested the presence of a second, weaker nucleotide binding site (90–93) and studies with a small-molecule inhibitor propose that it binds at this second location (94) although high resolution structures of PriA have yet to identify the location of this binding site (79–82). The helicase core also contains the other helicase sequence motifs of a SF2 helicase. Similar to the motif IIa/aromatic-rich loop (ARL) of some SF2 helicases, the full-length PriA structure revealed an aromatic and basic residue-rich loop in helicase lobe 1. This loop was found to crosslink to DNA at the DNA fork junction and couple DNA-binding to ATP hydrolysis (95). The HD also contains the SSB-binding site (between helicase lobe 1, 3′ BD and CTD) and the PriB interaction site (likely near the CRR—see below) (78,82). SSB and PriB interactions each stimulate PriA helicase activity (77,96–98). The DnaT interaction site on PriA is unknown but hypothesized to be near the PriA–PriB site.
Figure 3.

DNA replication restart pathways in Escherichia coli. E. coli and related bacteria possess three functional pathways for DNA replication restart, with the fourth column likely only occurring in vitro. All pathways serve to reload the replicative helicase DnaB on sites far removed from the origin of replication in a DNA structure-dependent manner. Key steps in the process are separated by row. Abandoned replication forks (various forms depicted in top row, depending on whether the strands are dsDNA or SSB-coated ssDNA gaps) are recognized by either by PriA or PriC (second row; SSB-interaction/remodeling not shown). Remodeling of the fork through helicase activity (third row) or SSB-interaction may allow for or/and proceed subsequent protein-protein interactions (fourth row). Replication restart ends with DnaB loading (bottom). Note that for simplicity PriA and PriC recognition of a select/preferred fork type is shown. PriA helicase activity is not required on many fork types and two PriA molecules could be shown on a fork. Rep helicase may function before PriC recognition on the first/left fork type. Helicase activity likely does not function exactly in this order and could occur during all steps.
The CRR element within the PriA HD (Figure 2A) contains eight cysteines that coordinate two zinc ions within a CXXCXnCXXCXnCXXCXnCXXC motif. Genetic studies have uncovered a range of phenotypes associated with cysteine sequence changes in this region that have led to the conclusion that the CRR is involved in helicase function and PriB interaction. Three cysteine variants (C445G, C448G and C476G) display wild type DNA-dependent ATPase activities levels but greatly reduced helicase activity that could be stimulated with addition of ZnCl2 (99). A PriA variant with two cysteine residues altered (C445A/C448A) can bind D-loop DNA in gel mobility shift assays, but the shift is to a species of different mobility than wild type PriA (possibly indicating a protein fold disruption) and the variant cannot complement a priA-null mutation (89). Other cysteine variants (C439Y and C445Y) are able to bind ssDNA, but are unable to interact with PriB and therefore cannot assemble a complex suitable for replication restart (100). The requirement for PriB can be eliminated if excess DnaT is supplemented in the reaction, an observation that was also found to be true for wild type PriA. Lastly, priA301 (C479Y), like priA300, has little or no phenotype as a single mutant except for a slightly lower plating efficiency for Mu bacteriophage (48) and is non-functional in the PriA-PriC pathway (48). Unlike priA300, priA301 is unable to facilitate cSDR when combined with an rnhA mutant and has increased basal levels of SOS expression when combined with a priC mutation (48,101). Taken together, these studies support the idea that the CRR is important for coupling helicase with ATPase activities and for interactions with PriB.
The CTD (Figure 2A) remained largely unstudied until the recent publication of the full-length PriA structure. A recG-null suppressor has been identified in which an insertion in priA removed the four terminal residues (66). These residues sit at the junction between the CTD and the helicase lobes and likely form a key packing/stabilization element. The CTD as a whole shares structural homology to the ribosomal S10 subunit, which binds to branched rRNA. The isolated CTD was found to bind ssDNA, dsDNA and forked DNA (82). Additionally, the interface between the CTD with the 3′ BD and helicase lobe 1 forms the SSB tail-binding site on the PriA face opposite the DNA-binding face, indicating that PriA may be able to interact with DNA and SSB simultaneously (82).
PriA–DNA interaction
PriA–DNA replication fork interactions have been the focus of many studies but our understanding of the complex remains limited. The 3′ BD-nucleotide structures are the only PriA/DNA structures and experiments to probe the PriA/DNA fork interaction are complicated by the high affinity that PriA has to ssDNA, dsDNA and forked DNA of various forms, as well as the fact that all six domains/subdomains have been implicated in binding DNA. Despite the high affinity of PriA for ssDNA and dsDNA, the replication restart activity of PriA is restricted to replication fork structures, although the biochemical mechanism for this selectivity is not known. When bound to a replication fork structure, a conformational change in PriA and/or in the DNA facilitates loading of PriB and DnaT in the PriA/PriB pathway (78,90). The 3′ BD interacts with the 3’OH end of the nascent leading strand and the presence of that strand or SSB orients the PriA helicase core onto the lagging strand (80–82,98). It was thought that PriA prefers forked substrates without a gap in the leading strand, although PriA was recently shown to be more active in an in vitro restart assay on forks with a gap size of >5 nucleotides (102). This size may allow a second PriA molecule to bind, increasing restart efficiency. On the lagging strand, PriA is highly active on replication forks with a 5–7 nucleotide gap (in the presence or absence of SSB), however PriA is active as a helicase on forks with gaps >1 nucleotide and on forks without a gap in the presence of SSB (98,103). Footprinting on forks with a nascent leading strand (to mimic physiological conditions where at least SSB would be present to orient PriA similarly) have shown that PriA protects strands around the fork junction. This protection (–22 to +10 on the lagging strand and –26 to –4 and +1 to +7 on the leading strand) is retained in the presence of ATP, supporting the idea that PriA remains associated with fork junctions even during helicase activity (67,81,104). In vitro, multiple PriA molecules can bind a given DNA substrate. PriA is thought to function as a monomer, although two PriA molecules can bind to forks with ssDNA on both arms (e.g. pas) (73,105). Recent protein–DNA crosslinking studies have expanded upon footprinting information. When phenyldiazirine-dU was placed at select sites within a DNA fork, PriA was found to crosslink to all sites proximal to the fork junction (106). PriA crosslinked to probes in the parental duplex (–3), ssDNA lagging template (+3), leading strand gap (+6 in leading template), and to the penultimate base in the nascent leading strand before a gap (+12 from fork junction). When benzylphenylalanine (Bpa) is placed at PriA residues of interest and the site of the crosslinked partner (nucleotide) mapped, a protein-region specific map of PriA binding was observed (95). Bpa-incorporated within motif IIa/ARL within helicase lobe 1 was found to crosslink to both strands at the fork junction (–1) to +3 within the lagging strand, and Bpa incorporated at a residue within helicase lobe 2 was found to crosslink to bases +5/+6 within the lagging strand. In the presence of ATP analogs, the helicase lobe 2 crosslinks shifted closer to the helicase lobe 1 (ARL) crosslinks, consistent with the current model of SF1/SF2 helicase lobe movement with respect to each other during helicase/ATP hydrolysis cycles (95,107). Additionally, the altering affinities of PriA for DNA in the presence of ADP and ATP analogs have been investigated (93,103). It has not been shown what part of PriA interacts with the parental duplex, and the roles of the WH domain and the CTD in the model of PriA on a DNA fork remain unclear.
PriB
Originally called ‘protein n ’, PriB is an ∼11 kDa protein that homodimerizes (73,108,109) and interacts with PriA, DnaT, DNA and SSB (100,110). PriB appears to stabilize protein–protein interactions required for replication restart and can stimulate PriA helicase activity (73,78,96,105).
PriB genetics
Deleting priB has little effect alone since the PriA–PriB and PriA–PriC pathways (see below) are redundant, whereas a priB priC double mutant is barely viable (worse than a priA2::kan mutant) (111). However, the PriA–PriB pathway (Figure 3) is thought to be the dominant pathway of replication restart, as there are some situations where PriB is necessary and PriC is dispensable. Aside from the requirement for PriB in the absence of PriA helicase activity, PriB appears to be more important for restart following DNA recombination than PriC (111). Consistent with this, priB-null mutations are more deleterious than priC-null mutations in a holD mutant strain that has increased instances of fork arrest and increased fork reversal DNA repair processes (58). PriB is also important for restart following DNA repair after UV-induced DNA damage while PriC is not (112). PriB may be particularly important for replication restart that occurs after repair of DSBs. Two lines of evidence support this. First, priB mutations are synthetically lethal with dam mutations, which are known to have more DSBs than wild-type cells (113–115). Second, this lethality is suppressed by the introduction of a mutH mutation that fails to incise DNA during mismatch repair (113). PriB is also important for DNA replication restart that occurs independently of DNA recombination. For example, priB-null mutations impact gyrBts mutants with lowered gyrase activity at semi-permissive temperatures whereas priC-null mutations do not (116). Additionally, recG null suppressors that partially reduce or completely eliminate PriB expression levels have been found, and mutations in RNA polymerase can further improve this suppression by alleviating some of the negative phenotypes of a priB deletion (71). These results implicate the importance of restart during replication-transcription conflicts, especially with the over-replication that occurs in recG-null strains (71). One case where PriB seems to be important for a restart function in the absence of PriA is during cSDR: A functional priB gene is required with a dnaC809,820 mutant that suppresses the need for PriA, DnaT and PriC during this process (101). It is possible that during the assembly of the replisome during cSDR, the role of PriB may be different than the role it plays in replication restart because the replisome is being loaded at an R-loop. Alternatively, PriB may differentially interact with DnaC809,820 to facilitate functional interaction of the DnaC variant with DnaB. The complex interaction between priB and dnaC is further substantiated by studies showing that a novel mutation, dnaC1331, causes a decrease in plasmid copy number and an increase in SOS expression and cell filamentation in a priA300 mutant similar to a priB mutation (113,117).
PriB biochemistry
Biochemical and structural data indicate that PriB forms a dimer of oligonucleotide-oligosaccharide binding (OB) folds that are structurally similar to SSB (Figure 2B) (108,118). PriB binds ssDNA and a crystal structure of the complex has been determined (Figure 2B: grey and black (119)). However, unlike SSB, dsDNA binding by PriB has also been observed (118) and PriB is thought to have only one ssDNA-binding site (119,120). The PriB ssDNA-binding site recognizes an 8–12 nucleotide stretch, with a preference for homo-pyrimidine over homo-purine ssDNA (120,121), as has been seen for PriA (122). However, recently two modes of PriB-ssDNA binding have been proposed with longer ssDNA (dT35), with the ssDNA-interacting residues of the second mode being distinct from those previously observed with ssDNA and involving histidines (121). These longer dT substrates (≥24–30 nucleotides) are also required for positive cooperativity of PriB-ssDNA binding (119,120). In a crosslinking study, PriB interactions were observed across ssDNA, dsDNA and forked DNA (106). However, upon inclusion of SSB and PriA, these crosslinks were largely lost. Thus PriB may bind DNA in a specific manner in the context of a PriA/DNA complex and in a less specific manner in isolation. The PriA and DnaT-interacting residues of PriB have also been studied and were found to overlap with the PriB DNA-interacting site (78).
PriC
Originally called ‘protein n’ ‘, PriC is a ∼24 kDa protein that interacts with SSB, ssDNA, DnaT and DnaB (123–126). PriC is a single-domain, alpha-helical bundle that readily oligomerizes on DNA (73,124,127).
PriC genetics
The priC-null mutation (priC303::kan) has no measurable phenotype by itself, but it is synthetically lethal when combined with a priA mutation and barely viable with a priB mutation (111). However, there is at least one situation where priC is required without a requirement for priB: priC mutations are synthetically lethal with certain temperature-sensitive dnaA mutants (128). This lethality can be suppressed by either making the strain dnaA-independent (with an rnhA mutation) or by a dnaC809,820 mutation.
PriC biochemistry
Following determination of the structure of an N-terminal portion of E. coli PriC, the full-length Cronobacter sakazakii PriC (41/55% identical/similar to E. coli PriC) structure was recently reported, revealing a bundle of five alpha helices, with a 20-residue loop connecting α-helix 1 and α-helix 2 (127,129) (Figure 2C). PriC has been shown to bind DNA primarily through the C-terminal portion of the protein, with a conserved region of positively charged residues across two faces of the protein being the predicted ssDNA binding site (124,127,130). Perturbation of DNA binding by mutation of residues within this site leads to a decrease in in vitro DnaB loading, but not in vivo functionality, suggesting that modest reductions in PriC DNA-binding affinity are not detrimental to PriC-mediated DNA replication restart (127,130). While it is able to bind both ds- and ssDNA, PriC preferentially binds to ssDNA with a site size of 7–9 nucleotides (124). This fits well with in vitro data that suggest PriC is most active on stalled forks with ≥7 base gaps between the nascent leading strand and the fork junction (64). DNA binding also appears to induce oligomerization that is mediated by the N-terminal region of the protein, although the role of this oligomerization in replication restart has not been determined (129). The SSB interaction site is adjacent to the ssDNA-binding site. SSB binding is stabilized by residues R121 and R155, and abolishing this interaction by altering either of these residues or by altering the C-terminus of SSB (to which PriC binds) eliminates DnaB loading in vitro and PriC-mediated replication restart in vivo (123).
A direct interaction between PriC and DnaB and with the DnaB/DnaC complex has recently been observed (127). Using isothermal titration calorimetry, a 1:1 stoichiometry of PriC:DnaB/DnaC was measured, suggesting that six PriC molecules can interact with the DnaB hexamer within the DnaB/DnaC complex. It is furthermore predicted that interaction between PriC and DnaB, especially in the context of PriC oligomerization, could play a role in recruiting the helicase to the stalled fork (127).
DnaT
Originally known as ‘protein i,’, DnaT is a ∼19 kDa protein that binds ssDNA, the PriA–PriB–DNA complex, and PriC (126). DnaT is a two domain protein, with the structure of the CTD comprising a bundle of alpha-helices (Figure 2D), and it is thought to function as either a monomer or a homotrimer at replication restart sites (73,131).
DnaT genetics
The dnaT gene was first identified by the dnaT1 (R152C) mutation in E. coli 15T− (132,133). This is a dominant, temperature sensitive mutation that arrests DNA replication under non-permissive conditions and thus DnaT was thought to be involved with termination of DNA replication (hence the designation ‘T’). However no such role has been demonstrated. When this mutant allele was cloned into a pACYC184 derivative and tested for functionality, it was found that dnaT1 is only temperature sensitive and dominant negative in E. coli 15T (132). In E. coli K12, dnaT822, an in-frame deletion of codons 87–92 that is thought to be a dnaT-null mutation, causes a phenotype that is similar to priA2::kan and can be suppressed by dnaC809 (like priA2::kan), suggesting that, like PriA, DnaT is only required for the PriA-PriB and PriA-PriC pathways and not for the PriA-independent PriC(-Rep) pathway (Figure 3) (132). The dnaT gene is directly upstream of dnaC (134,135), and it is tempting to speculate that the two proteins interact. However, no specific interaction has been documented.
DnaT biochemistry
DnaT binds to the PriB–PriA–ssDNA complex and appears to recruit the DnaB–DnaC complex to the primosome (73,78,100,136). Excess levels of DnaT allow bypass of the requirement for PriB in ΦX174 complementary strand DNA replication in vitro, which could indicate that DnaT directly binds to the PriA/DNA complex and that this association is assisted by PriB (100). Like PriB and PriC, DnaT lacks enzymatic activity and is not found in all bacterial species containing PriA. Both monomer and trimer DnaT states have been implicated in functioning in primosome assembly (73,137–139). DnaT homotrimerization is cooperative, with no dimer observed in solution (137–139). In E. coli, DnaT residues 1–161 form a dimer, which has led to a trimerization model in which a third DnaT subunit associates through contacts between the C-terminal domains (137). A recent solution structure of the C-terminal domain of E. coli DnaT (residues 89–179) demonstrates that it forms a three-helix bundle with a highly flexible C-terminal 20-residue tail (140) (Figure 2D:center cyan). This domain is monomeric, highlighting the importance of the N-terminal domain in trimerization (140). The N-terminal domain of K. pneumoniae DnaT (residues 1–83) has been proposed to be important in PriB binding and trimerization, but not ssDNA-binding (139). Recent work on the E. coli DnaT protein has shown that residues 42–66 within the N-terminal domain mediate trimerization and binding to PriB (140).
DnaT from K. pneumoniae has been shown to bind ∼25 nucleotides of ssDNA as a trimer through the highly conserved C-terminal domain (141). A crystal structure of E. coli DnaT 84–153 in complex with dT10 ssDNA as well as electron microscopy studies indicate that DnaT is capable of forming spiral filaments on ssDNA (142) (Figure 2D: shades of blue with black DNA). The ssDNA binds through base stacking and electrostatic interactions with two nucleotides per DnaT monomer (142). Additional fragment analysis has shown that an acidic linker (residues 70–88) plays a role in causing dissociation of ssDNA from a PriB/ssDNA complex (140). The ssDNA and PriB interaction sites on DnaT have been investigated, but the DnaT–PriA and DnaT–PriC interaction sites remain unknown.
PROPOSED MODELS OF DNA REPLICATION RESTART
To carry out their essential functions, cellular replication restart pathways must meet three requirements. First, RRPs must recognize bona fide abandoned DNA replication forks in a structure selective manner. As noted above, legitimate replication restart substrates are forked DNA of diverse types that result from a wide variety of processes, so RRPs must maintain sufficient structural plasticity to accommodate a variety of structures without being overly promiscuous and facilitating restart at inappropriate sites. Second, remodeling of the DNA at abandoned replication forks is required to expose ssDNA on the lagging strand for replicative helicase loading. Lastly, RRPs must aid in recruitment and loading of the replicative helicase. Genetic and biochemical studies have delineated three replication restart pathways in E. coli that can be classified as either PriA-mediated (two pathways) or PriC-mediated (one pathway) based on the protein that first binds to the replication fork (Figure 3). The functional redundancy of these pathways may allow cells to confront a vast array of situations that might arrest DNA replication and highlights the essential nature of restart for bacterial survival. Interactions between pathways in this model are still being discovered in efforts to determine the physiological roles of each.
PriA/PriB pathway
Based upon genetic observations, the PriA/PriB pathway appears to be the dominant restart pathway in E. coli. These observations include the following: (1) the priA-null phenotype is severe, (2) the priC-null mutation has no phenotype in an otherwise wild type background, and (3) the priB-null mutation can be more detrimental than a priC-null mutation (see PriB section) (31,44,50,111). Besides its ability to bind ssDNA and dsDNA, PriA interacts with forked DNA with high affinity (35,43,61,143,144). This forked DNA can take on a variety of forms, with and without gaps on leading and/or lagging strands (all fork types in Figure 3) that include D-loops formed during homologous recombination (98,104,145). PriA recognition of forked structures also includes R-loops, formed by an RNA strand invading duplex DNA to displace one of the DNA strands (45,88). On forked DNA substrates, PriA has higher affinity for those containing a fork-proximal nascent leading strand 3’OH, which are hypothesized to be common products of collapsed forks and of DNA recombination pathways (43,61,67,76,81,98,102,146). Upon binding, PriA can remodel forks with duplex lagging strands through its preferential unwinding of the nascent lagging strand. On forks with ssDNA, PriA can remodel the SSB/ssDNA complex in a manner that exposes ssDNA through a direct interaction with SSB (82,97,98,106). The PriA-SSB interaction may also function to enhance binding of PriA to forked DNA to promote the efficiency and/or specificity with which PriA recognizes DNA forks (75,147). The PriA–SSB interaction additionally stabilizes a PriA orientation that positions the HD to unwind the lagging strand and stimulates PriA helicase activity (96,98). Once bound, PriA serves as a protein block that prevents utilization of the fork prior to DnaB reloading (and PriA removal) by competing with Pol III holoenzyme for the leading strand 3’end at forks (102,148,149). This latter function is likely important for maintaining the integrity of replication fork structures and preventing DNA chain growth prior to the completion of replication restart.
Once PriA is bound to a replication restart substrate, it recruits additional RRPs to the replication re-initiation site. DnaT is required for DnaB recruitment and loading and PriB appears to assist DnaT recruitment to the PriA/DNA complex. The coordinated effort of PriA, PriB and DnaT in reloading DnaB onto a replication fork appears to take place via a ‘hand-off mechanism’ (78). First, PriB binds PriA and ssDNA, preferentially in the PriA–DNA complex. It is hypothesized that binding of PriA to DNA leads to a conformational change in PriA that exposes the PriB binding site on the PriA HD (78,96). PriB can also interact with PriA in the absence of DNA, increasing the affinity of PriA for forked DNA over ssDNA or dsDNA (105). Second, PriB stimulates PriA helicase activity and facilitates formation of a ternary complex with DnaT (78,96,105). In fact, the requirement for PriB can be alleviated by excess DnaT in vitro and PriB binding sites for ssDNA, PriA and DnaT appear to partially overlap (100). Third, binding of DnaT leads to release of PriB from the complex, and it is thought that DnaT exposes ssDNA for DnaB loading by DnaC (73,78,135,140,142). This ternary complex is then competent to load DnaB from the DnaB/DnaC complex, although the exact mechanisms by which this occurs remain undefined. Whether DnaT within the ternary complex plays a role in directly recruiting DnaB by protein-protein interactions with DnaB or the DnaB/DnaC complex remains unknown. This ‘hand-off mechanism’ of the fork-proximal ssDNA by these RRPs (and DnaC) to DnaB was supported and expanded upon in a recent study, where photo-crosslinkable probes at unique sites across a DNA fork were used to map RRP contacts within and out of the primosome complex (106). This work showed that PriA crosslinks with all DNA strands/sites proximal to the fork junction and this is unchanged upon the addition of SSB, PriB, and DnaT, supporting a peripheral association of these proteins.
PriA/PriC and PriC-mediated pathways
PriC is involved in two of the three replication restart pathways in E. coli and can mediate its own independent restart reaction in vitro (Figure 3). PriC binds ssDNA, preferentially associating with replication forks that include ≥7 nucleotide gaps between the nascent leading strand and replication fork (64,124). Such substrates could form as a result of blocked nascent leading strand replication coupled with continued fork unwinding. PriC might also bind ssDNA present on the lagging strand in the PriA/PriC pathway where PriA helicase activity exposes ssDNA on the lagging strand and PriC may serve a similar role to that of PriB (129). Like PriA, PriC can remodel SSB-coated forks by directly binding SSB which could be important for exposing ssDNA DnaB loading sites (123).
On replication forks lacking nascent leading and lagging strands in in vitro reactions (Figure 3, right column), PriC is sufficient to load DnaB from DnaB/DnaC complexes in the absence of any other restart proteins (64). One simple way this could occur is by PriC interacting with DnaB/DnaC, which has recently been observed (127). However, this minimal PriC pathway may not be significant in vivo, as forks of this type would presumably re-anneal until a nascent strand abuts the fork junction, stabilizing the structure. Nonetheless, the PriC pathway serves as a useful tool to define the minimal requirements for restart in vitro.
When a nascent lagging strand abuts the fork junction, PriC requires PriA or another helicase for either of two reasons. First, PriC may associate with ssDNA on the leading strand and require PriA (or Rep) helicase activity to create ssDNA on the lagging strand for restart pathway progression (64,113,145). Second, PriC may require initial PriA recognition and processing of replication forks that are entirely duplex to create a ssDNA PriC binding site on the lagging strand to facilitiate the PriA/PriC pathway (43,61,64,76,145,146). Direct interaction between PriA and PriC is hypothesized but has not been observed.
Besides its activity on DNA forks without nascent strands in vitro, the PriA-independent PriC-mediated pathway is active in vivo but requires Rep helicase in priA-null or PriA helicase-deficient strains (59,113,150). In this PriC/Rep pathway, the role of Rep may simply be to replace PriA in removing nascent lagging strand DNA to enable PriC-mediated loading of DnaB. PriC stimulates the helicase activity of Rep in vitro (151). Rep also interacts with DnaB and could act as an intermediary in DnaB recruitment (152). Our understanding of the PriC/Rep pathway is complicated by the fact that Rep also has a role in stabilizing the intact replication fork. Rep is able to remove proteins bound to DNA and helps resolve DNA replication/transcription conflicts (153–157). Moreover, rep deletion strains exhibit slower DNA replication rates than wild type cells (155). Slower replication rates and poor resolution of transcription/replication conflicts could lead to an increase in DNA damage and the need for replication restart processes, thus complicating analysis of Rep functions in fork progression and in DNA replication restart.
Although PriA helicase activity is highly conserved and thought to be significant in replication restart, selective removal of PriA ATPase activity (priA300) alone fails to confer a phenotype in vivo. Rep helicase is thought to fill-in for this role (59,145). However, the PriA-PriC pathway requires PriA helicase activity (a priA300 priB-null double mutation copies a priA-null phenotype). Interestingly, PriB stimulates PriA helicase activity but PriA–PriB pathways are functional without PriA helicase activity (priA300 priC-null cells are viable) (48,96). This leads to the question of whether Rep helicase activity fills in for PriA helicase activity in these strains or whether most abandoned replication forks simply do not require helicase activity for restart function? Could PriA binding alone expose ssDNA, since PriA (in the presence of SSB) can function on forks without a ssDNA gap on the lagging strand (62,87)? Future experiments will be required to answer these questions.
From synthetic lethality experiments, the PriC-dependent/PriA-independent pathway does not appear to require other restart proteins. However, DnaT is thought to function in all PriA pathways, including the PriA/PriC pathway (since priA dnaT double mutants have the same viability as single null mutants (132)). This suggests that the PriC–DnaB(DnaC) interaction cannot load DnaB/DnaC when PriA is present. An interaction between PriC and DnaT is anticipated from the requirement for DnaT in all PriA pathways and the observation that an RRP is needed for DnaT association with replication forks (only excess DnaT in vitro has been shown to be able to bypass the need for PriB). Interestingly, PriC can interact with DnaT in the absence of PriA and DNA (126). However, the synthetic lethal relationships described above indicate that this interaction is not required in the PriA-independent PriC-mediated pathway.
DnaB/DnaC interactions
DnaC is thought to facilitate DnaB loading by associating with the C-terminal domain of DnaB and stabilizing an opening in the hexameric DnaB ring through which DNA passes (4,158,159). DnaC dissociation is then triggered, along with DnaB ring closure and DnaB translocation/helicase initiation. In oriC-dependent initiation, DnaA interacts directly with the N-terminal domain of DnaB and mediates localization and binding of DnaB (6,160,161). PriC also interacts with DnaB in the DnaB/DnaC complex and it may serve to recruit DnaB to abandoned replication forks (127). It is possible that various interactions between the RRPs and DnaB/DnaC may have allosteric effects on DnaB (or DnaC) and therefore alter DnaB interactions with other partners that control the steps in some of these processes. For example, DnaC interaction with DnaB appears to have allosteric effects on DnaB that occlude DnaB-primase interactions (162). It remains unclear whether the PriC–DnaB interaction (and inferred RRP-DnaB interaction in the other restart pathways) functions to recruit DnaB/DnaC and/or facilitate reloading of DnaB.
Suppressors of priA-null phenotypes and RRP synthetic lethalities are commonly found in dnaC. Interestingly there are separate dnaC suppressors of PriA and PriC pathways that may suggest that the PriC-DnaB/DnaC interaction occurs in a similar but distinct location/mechanism than potential DnaB/DnaC interactions with PriA complexes. priA-null phenotypes are suppressed by dnaC809 (E176G). However PriC and Rep are required in this suppressor pathway as in normal priA-null strains (48,111). This requirement is relieved by dnaC809,820, which bypasses the requirement for PriC through the addition of the dnaC820 mutation (K172N) (59,111).
Regulation of replication restart proteins
Very little is known about the regulation of the RRP genes expression. The genes are spread throughout the chromosome in separate operons. The priA gene exists by itself, dnaT and dnaC genes are in an operon along with two genes of unknown function, and priC is in an operon with ybaM (also of unknown function). Interestingly, priB is the second of four genes in an operon with ribosomal subunit proteins: rpsF – priB – rpsR – rplI. None of the RRPs are SOS-induced (163), underlying their general importance in DNA maintenance. PriA levels are estimated in E. coli to be ∼70 molecules/cell (164) and estimates from B. subtilis are ∼50–100 molecules/cell (165). Ribosomal profiling in E. coli estimates PriA to be present at 60–105 molecules of protein per cell per generation (166). Other early estimates from RRP levels in E. coli indicated that PriB is present at ∼80 molecules/cell (167) and DnaT is at ∼50 molecules/cell (131). Escherichia coli ribosomal profiling suggests that PriB would exist at 250–1150 molecules/cell, DnaT at 90–605 molecules/cell, and PriC at 100–160 molecules/cell (166).
How are RRPs regulated at the protein level to ensure that they act exclusively on ‘warranted’ DNA forks rather than on other DNA substrates? This is a pertinent question considering that the RRPs restrict their activity to forked DNA yet all RRPs have affinity for ssDNA. It is thought that this control results from other proteins present on the DNA. For the targeting of RRPs to forked DNA over ssDNA, PriA has specific increased affinity for forked DNA when tested with PriB (100,105) and PriB–ssDNA binding outside of the PriA complex may be inhibited by competition with SSB (102). For RRP restriction to DNA forks warranted for replication restart, DNA forks may become warranted if they become abandoned by other proteins (the replisome components, DNA repair proteins upon the completion of homologous recombination, or DNA replication termination components) and leave an exposed DNA fork for RRP recognition.
REPLICATION RESTART IN OTHER ORGANISMS
The DNA replication restart pathways described above are those found in E. coli, however PriA is the only RRP conserved across all bacterial phyla. PriC is found in proteobacteria, with PriB conservation extending across a few more gram-negative species than PriC. No PriA-independent restart pathways (excluding PriA suppressor mutations) have been reported in other bacteria. It remains unclear whether PriA is the only replication restart-specific factor in other bacteria or if, without strong sequence conservation, additional factors have simply not yet been identified. In Neisseria gonorrhoeae, which has PriB and no recognized PriC, priA-null mutations have a similarly severe phenotype to inactivation of PriA in E. coli (168). This raises the question of how DNA replication restart is facilitated to keep priA-null N. gonorrhoeae cells alive. PriB-ssDNA and PriB-PriA interactions are retained in N. gonorrhoeae but with significantly altered affinities, suggesting a somewhat altered mechanism of action (169). A recent study has observed a direct interaction of Deinococcus radiodurans PriA with DnaB in vitro, without other factors present, including DNA (86). This study also showed that D. radiodurans PriA retains DNA fork binding but is not an active helicase (86). Does PriA alone serve to recruit/load the replicative helicase/loader complex in bacteria such as D. radiodurans?
In the Gram-positive bacteria Bacillus subtilis and Staphylococcus aureus, a multi-protein restart complex is employed: PriA, DnaD (no clear homolog in E. coli), DnaB (unrelated to the E. coli DnaB), and DnaI (homolog of E. coli DnaC) function to load the replicative helicase, DnaC (homolog of DnaB in E. coli). As with E. coli PriB, gram-positive DnaD binds ssDNA and PriA and stimulates PriA helicase/ATPase activities (170,171). Although DnaB and DnaD (and DnaI) are also part of DnaA-mediated helicase loading during replication initiation in Gram-positive bacteria, DnaD may treat DnaA and PriA pathways distinctly by having different interaction locations on its surface for PriA and DnaA (170). A direct interaction between DnaB and PriA from Geobacillus stearothermophilus has also recently been observed (172). Additionally, the PriA–SSB interaction, including the resulting stimulation of PriA helicase activity, may be altered in the gram-positive system (173).
Unlike bacterial systems, the presence of multiple licensed origins in eukaryotes allows for the rescue of arrested replication forks by adjacent replisomes, potentially bypassing the need for origin-independent loading of a new replisome. Furthermore, many of the components at the replication fork that contribute to DNA synthesis and fork stability differ from those in bacteria and eukaryotic replisomes appear to be less likely to dissociate than prokaryotic replisomes (174). Therefore, the term ‘DNA replication restart’ in eukaryotes does not often refer to helicase reloading, as it does in prokaryotes, but to restart of a paused replisome. Despite this replisome stability, the presence of adjacent replisomes, and late origin firing, stalled forks and rare collapsed forks require attention to ensure complete duplication of the region. Failure to resolve and repair arrested DNA replication forks in eukaryotes is a potentially dangerous situation that can lead to genomic instability (disease) and cell death. The pathways by which replication forks can be stabilized and restarted in eukaryotes (using the term restarted generally here) have been the recent focus of much of the DNA maintenance field (175–179). No PriA or any RRP homologs have been found outside of bacteria. Interestingly though, the PriA 3’BD was found to share structural (and functional) homology to the HIRAN domain of HLTF, a eukaryotic DNA damage tolerance pathway fork reversal protein, providing one example of where the mechanisms of bacterial DNA replication restart may shed light on the mechanisms of eukaryotic fork maintenance (180–182).
FUTURE OF DNA REPLICATION RESTART
After more than 40 years of research, the study of DNA replication restart stands at an exciting point. Biochemical, genetic, and structural studies have combined to provide novel mechanistic insights into this essential process in bacteria. Despite these advances, several key questions remain in DNA replication restart. How is the replicative helicase/helicase loader complex recruited and the replicative helicase loaded by the replication restart machinery? How are the RRPs regulated and removed from the replication fork? Are there functional homologs to PriC/PriB/DnaT (or gram-positive DnaB/DnaD) in bacteria where only PriA has been identified or does PriA act alone to restart DNA replication in these bacteria? Understanding diverse mechanisms of DNA replication restart will provide insights into eukaryotic systems that may employ related mechanisms.
FUNDING
DNA replication restart studies in the Keck lab are supported by a grant from the National Institutes of Health [R01 GM098885]; National Science Foundation Graduate Research Fellowship [DGE-1256259 to T.A.W.); National Institutes of Health training grant in Molecular Biosciences [T32 GM07215 to S.R.W. and B.B.] (in part). The open access publication charge for this paper has been waived by Oxford University Press - NAR.
Conflict of interest statement. None declared.
REFERENCES
- 1. Kornberg A., Baker T.A.. Freeman WH. DNA Replication. 1992; 2nd edn, NY. [Google Scholar]
- 2. Bramhill D., Kornberg A.. Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell. 1988; 52:743–755. [DOI] [PubMed] [Google Scholar]
- 3. Grimwade J.E., Torgue J.J.-C., McGarry K.C., Rozgaja T., Enloe S.T., Leonard A.C.. Mutational analysis reveals Escherichia coli oriC interacts with both DnaA-ATP and DnaA-ADP during pre-RC assembly. Mol. Microbiol. 2007; 66:428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bleichert F., Botchan M.R., Berger J.M.. Mechanisms for initiating cellular DNA replication. Science. 2017; 355:eaah6317. [DOI] [PubMed] [Google Scholar]
- 5. Ludlam A.V., McNatt M.W., Carr K.M., Kaguni J.M.. Essential amino acids of Escherichia coli DnaC protein in an N-terminal domain interact with DnaB helicase. J. Biol. Chem. 2001; 276:27345–27353. [DOI] [PubMed] [Google Scholar]
- 6. Marszalek J., Kaguni J.M.. DnaA protein directs the binding of DnaB protein in initiation of DNA replication in Escherichia coli. J. Biol. Chem. 1994; 269:4883–4890. [PubMed] [Google Scholar]
- 7. Wahle E., Lasken R.S., Kornberg A.. The dnaB-dnaC replication protein complex of Escherichia coli. I. Formation and properties. J. Biol. Chem. 1989; 264:2463–2468. [PubMed] [Google Scholar]
- 8. Wahle E., Lasken R.S., Kornberg A.. The dnaB-dnaC replication protein complex of Escherichia coli. II. Role of the complex in mobilizing dnaB functions. J. Biol. Chem. 1989; 264:2469–2475. [PubMed] [Google Scholar]
- 9. Mok M., Marians K.J.. The Escherichia coli preprimosome and DNA B helicase can form replication forks that move at the same rate. J. Biol. Chem. 1987; 262:16644–16654. [PubMed] [Google Scholar]
- 10. Kim S., Dallmann H.G., McHenry C.S., Marians K.J.. Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell. 1996; 84:643–650. [DOI] [PubMed] [Google Scholar]
- 11. Kim S., Dallmann H.G., McHenry C.S., Marians K.J.. Tau protects beta in the leading-strand polymerase complex at the replication fork. J. Biol. Chem. 1996; 271:4315–4318. [DOI] [PubMed] [Google Scholar]
- 12. Gao D., McHenry C.S.. tau binds and organizes Escherichia coli replication proteins through distinct domains. Domain IV, located within the unique C terminus of tau, binds the replication fork, helicase, DnaB. J. Biol. Chem. 2001; 276:4441–4446. [DOI] [PubMed] [Google Scholar]
- 13. Costa A., Hood I.V., Berger J.M.. Mechanisms for initiating cellular DNA replication. Annu. Rev. Biochem. 2013; 82:25–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Neylon C., Kralicek A.V., Hill T.M., Dixon N.E.. Replication termination in Escherichia coli: structure and antihelicase activity of the Tus-Ter complex. Microbiol. Mol. Biol. Rev. 2005; 69:501–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duggin I.G., Wake R.G., Bell S.D., Hill T.M.. The replication fork trap and termination of chromosome replication. Mol. Microbiol. 2008; 70:1323–1333. [DOI] [PubMed] [Google Scholar]
- 16. Hill T.M., Marians K.J.. Escherichia coli Tus protein acts to arrest the progression of DNA replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:2481–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGlynn P., Lloyd R.G.. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 2002; 3:859–870. [DOI] [PubMed] [Google Scholar]
- 18. Gupta M.K., Guy C.P., Yeeles J.T., Atkinson J., Bell H., Lloyd R.G., Marians K.J., McGlynn P.. Protein-DNA complexes are the primary sources of replication fork pausing in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:7252–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Washburn R.S., Gottesman M.E.. Transcription termination maintains chromosome integrity. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:792–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mangiameli S.M., Merrikh C.N., Wiggins P.A., Merrikh H.. Transcription leads to pervasive replisome instability in bacteria. Elife. 2017; 6:e19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Merrikh H., Zhang Y., Grossman A.D., Wang J.D.. Replication-transcription conflicts in bacteria. Nat. Rev. Microbiol. 2012; 10:449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cox M.M., Goodman M.F., Kreuzer K.N., Sherratt D.J., Sandler S.J., Marians K.J.. The importance of repairing stalled replication forks. Nature. 2000; 404:37–41. [DOI] [PubMed] [Google Scholar]
- 23. Helmrich A., Ballarino M., Nudler E., Tora L.. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013; 20:412–418. [DOI] [PubMed] [Google Scholar]
- 24. Hamperl S., Cimprich K.A.. Conflict resolution in the genome: how transcription and replication make it work. Cell. 2016; 167:1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang E.Y., Stirling P.C.. Replication fork protection factors controlling R-loop bypass and suppression. Genes. 2017; 8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yeeles J.T.P., Poli J., Marians K.J., Pasero P.. Rescuing stalled or damaged replication forks. Cold Spring Harbor Perspect. Biol. 2013; 5:a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beattie T.R., Kapadia N., Nicolas E., Uphoff S., Wollman A.J., Leake M.C., Reyes-Lamothe R.. Frequent exchange of the DNA polymerase during bacterial chromosome replication. Elife. 2017; 6:e21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Merrikh H., Machon C., Grainger W.H., Grossman A.D., Soultanas P.. Co-directional replication-transcription conflicts lead to replication restart. Nature. 2011; 470:554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Septenville A.L., Duigou S., Boubakri H., Michel B.. Replication fork reversal after replication-transcription collision. PLoS Genet. 2012; 8:e1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Million-Weaver S., Samadpour A.N., Merrikh H.. Replication restart after replication-transcription conflicts requires RecA in Bacillus subtilis. J. Bacteriol. 2015; 197:2374–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee E.H., Kornberg A.. Replication deficiencies in priA mutants of Escherichia coli lacking the primosomal replication n' protein. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:3029–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kogoma T., Cadwell G.W., Barnard K.G., Asai T.. The DNA replication priming protein, PriA, is required for homologous recombination and double-strand break repair. J. Bacteriol. 1996; 178:1258–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michel B., Sandler S.J.. Replication restart in bacteria. J. Bacteriol. 2017; 199:e00102–00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 1997; 61:212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shlomai J., Kornberg A.. A prepriming DNA replication enzyme of Escherichia coli. II. Actions of protein n': a sequence-specific, DNA-dependent ATPase. J. Biol. Chem. 1980; 255:6794–6798. [PubMed] [Google Scholar]
- 36. Jones J.M., Nakai H.. The phiX174-type primosome promotes replisome assembly at the site of recombination in bacteriophage Mu transposition. EMBO J. 1997; 16:6886–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Minden J.S., Marians K.J.. Replication of pBR322 DNA in vitro with purified proteins. Requirement for topoisomerase I in the maintenance of template specificity. J. Biol. Chem. 1985; 260:9316–9325. [PubMed] [Google Scholar]
- 38. Marians K.J. Prokaryotic DNA replication. Annu. Rev. Biochem. 1992; 61:673–719. [DOI] [PubMed] [Google Scholar]
- 39. Schekman R., Weiner A., Kornberg A.. Multienzyme systems of DNA replication. Science (New York, N.Y.). 1974; 186:987–993. [DOI] [PubMed] [Google Scholar]
- 40. Schekman R., Weiner J.H., Weiner A., Kornberg A.. Ten proteins required for conversion of phiX174 single-stranded DNA to duplex form in vitro. Resolution and reconstitution. J. Biol. Chem. 1975; 250:5859–5865. [PubMed] [Google Scholar]
- 41. Wickner S., Hurwitz J.. Association of phiX174 DNA-dependent ATPase activity with an Escherichia coli protein, replication factor Y, required for in vitro synthesis of phiX174 DNA. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wickner S., Hurwitz J.. Conversion of phiX174 viral DNA to double-stranded form by purified Escherichia coli proteins. Proc. Natl. Acad. Sci. U.S.A. 1974; 71:4120–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nurse P., Liu J., Marians K.J.. Two modes of PriA binding to DNA. J. Biol. Chem. 1999; 274:25026–25032. [DOI] [PubMed] [Google Scholar]
- 44. Nurse P., Zavitz K.H., Marians K.J.. Inactivation of the Escherichia coli priA DNA replication protein induces the SOS response. J. Bacteriol. 1991; 173:6686–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Masai H., Asai T., Kubota Y., Arai K., Kogoma T.. Escherichia coli PriA protein is essential for inducible and constitutive stable DNA replication. EMBO J. 1994; 13:5338–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Seufert W., Messer W.. Initiation of Escherichia coli minichromosome replication at oriC and at protein n' recognition sites. Two modes for initiating DNA synthesis in vitro. EMBO J. 1986; 5:3401–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCool J.D., Sandler S.J.. Effects of mutations involving cell division, recombination, and chromosome dimer resolution on a priA2::kan mutant. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8203–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sandler S.J., McCool J.D., Do T.T., Johansen R.U.. PriA mutations that affect PriA-PriC function during replication restart. Mol. Microbiol. 2001; 41:697–704. [DOI] [PubMed] [Google Scholar]
- 49. Kushner S.R., Nagaishi H., Templin A., Clark A.J.. Genetic recombination in Escherichia coli: the role of exonuclease I. Proc. Natl. Acad. Sci. U.S.A. 1971; 68:824–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sandler S.J. Overlapping functions for recF and priA in cell viability and UV-inducible SOS expression are distinguished by dnaC809 in Escherichia coli K-12. Mol. Microbiol. 1996; 19:871–880. [DOI] [PubMed] [Google Scholar]
- 51. Eykelenboom J.K., Blackwood J.K., Okely E., Leach D.R.F.. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol. Cell. 2008; 29:644–651. [DOI] [PubMed] [Google Scholar]
- 52. Meddows T.R., Savory A.P., Lloyd R.G.. RecG helicase promotes DNA double-strand break repair. Mol. Microbiol. 2004; 52:119–132. [DOI] [PubMed] [Google Scholar]
- 53. Ivancić-Bacće I., Vlasić I., Cogelja-Cajo G., Brcić-Kostić K., Salaj-Smic E.. Roles of PriA protein and double-strand DNA break repair functions in UV-induced restriction alleviation in Escherichia coli. Genetics. 2006; 174:2137–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Courcelle J., Belle J.J., Courcelle C.T.. When replication travels on damaged templates: bumps and blocks in the road. Res. Microbiol. 2004; 155:231–237. [DOI] [PubMed] [Google Scholar]
- 55. Rangarajan S., Woodgate R., Goodman M.F.. Replication restart in UV-irradiated Escherichia coli involving pols II, III, V, PriA, RecA and RecFOR proteins. Mol. Microbiol. 2002; 43:617–628. [DOI] [PubMed] [Google Scholar]
- 56. Moore T., McGlynn P., Ngo H.P., Sharples G.J., Lloyd R.G.. The RdgC protein of Escherichia coli binds DNA and counters a toxic effect of RecFOR in strains lacking the replication restart protein PriA. EMBO J. 2003; 22:735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ivancic-Bace I., Cass S.D., Wearne S.J., Bolt E.L.. Different genome stability proteins underpin primed and naive adaptation in E. coli CRISPR-Cas immunity. Nucleic Acids Res. 2015; 43:10821–10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Flores M.J., Ehrlich S.D., Michel B.. Primosome assembly requirement for replication restart in the Escherichia coli holDG10 replication mutant. Mol. Microbiol. 2002; 44:783–792. [DOI] [PubMed] [Google Scholar]
- 59. Sandler S.J. Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics. 2000; 155:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shi Q., Parks A.R., Potter B.D., Safir I.J., Luo Y., Forster B.M., Peters J.E.. DNA damage differentially activates regional chromosomal loci for Tn7 transposition in Escherichia coli. Genetics. 2008; 179:1237–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McGlynn P., Al-Deib A.A., Liu J., Marians K.J., Lloyd R.G.. The DNA replication protein PriA and the recombination protein RecG bind D-loops. J. Mol. Biol. 1997; 270:212–221. [DOI] [PubMed] [Google Scholar]
- 62. Gregg A.V., McGlynn P., Jaktaji R.P., Lloyd R.G.. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol. Cell. 2002; 9:241–251. [DOI] [PubMed] [Google Scholar]
- 63. Al-Deib A.A., Mahdi A.A., Lloyd R.G.. Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K-12. J. Bacteriol. 1996; 178:6782–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Heller R.C., Marians K.J.. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol. Cell. 2005; 17:733–743. [DOI] [PubMed] [Google Scholar]
- 65. McGlynn P., Lloyd R.G.. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8227–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jaktaji R.P., Lloyd R.G.. PriA supports two distinct pathways for replication restart in UV-irradiated Escherichia coli cells. Mol. Microbiol. 2003; 47:1091–1100. [DOI] [PubMed] [Google Scholar]
- 67. Tanaka T., Masai H.. Stabilization of a stalled replication fork by concerted actions of two helicases. J. Biol. Chem. 2006; 281:3484–3493. [DOI] [PubMed] [Google Scholar]
- 68. Azeroglu B., Mawer J.S., Cockram C.A., White M.A., Hasan A.M., Filatenkova M., Leach D.R.. RecG directs DNA synthesis during double-strand break repair. PLoS Genet. 2016; 12:e1005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rudolph C.J., Upton A.L., Stockum A., Nieduszynski C.A., Lloyd R.G.. Avoiding chromosome pathology when replication forks collide. Nature. 2013; 500:608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Levy A., Goren M.G., Yosef I., Auster O., Manor M., Amitai G., Edgar R., Qimron U., Sorek R.. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015; 520:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mahdi A.A., Briggs G.S., Lloyd R.G.. Modulation of DNA damage tolerance in Escherichia coli recG and ruv strains by mutations affecting PriB, the ribosome and RNA polymerase. Mol. Microbiol. 2012; 86:675–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tanaka T., Nishito Y., Masai H.. Fork restart protein, PriA, binds around oriC after depletion of nucleotide precursors: replication fork arrest near the replication origin. Biochem. Biophys. Res. Commun. 2016; 470:546–551. [DOI] [PubMed] [Google Scholar]
- 73. Ng J.Y., Marians K.J.. The ordered assembly of the phiX174-type primosome. II. Preservation of primosome composition from assembly through replication. J. Biol. Chem. 1996; 271:15649–15655. [DOI] [PubMed] [Google Scholar]
- 74. Bentchikou E., Chagneau C., Long E., Matelot M., Allemand J.F., Michel B.. Are the SSB-interacting proteins RecO, RecG, PriA and the DnaB-interacting protein Rep bound to progressing replication forks in Escherichia coli. PLoS One. 2015; 10:e0134892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lecointe F., Sérèna C., Velten M., Costes A., McGovern S., Meile J.-C., Errington J., Ehrlich S.D., Noirot P., Polard P.. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO J. 2007; 26:4239–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tanaka T., Mizukoshi T., Taniyama C., Kohda D., Arai K., Masai H.. DNA binding of PriA protein requires cooperation of the N-terminal D-loop/arrested-fork binding and C-terminal helicase domains. J. Biol. Chem. 2002; 277:38062–38071. [DOI] [PubMed] [Google Scholar]
- 77. Chen H.W., North S.H., Nakai H.. Properties of the PriA helicase domain and its role in binding PriA to specific DNA structures. J. Biol. Chem. 2004; 279:38503–38512. [DOI] [PubMed] [Google Scholar]
- 78. Lopper M., Boonsombat R., Sandler S.J., Keck J.L.. A hand-off mechanism for primosome assembly in replication restart. Mol. Cell. 2007; 26:781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sasaki K., Ose T., Tanaka T., Mizukoshi T., Ishigaki T., Maenaka K., Masai H., Kohda D.. Crystallization and preliminary crystallographic analysis of the N-terminal domain of PriA from Escherichia coli. Biochim. Biophys. Acta. 2006; 1764:157–160. [DOI] [PubMed] [Google Scholar]
- 80. Sasaki K., Ose T., Okamoto N., Maenaka K., Tanaka T., Masai H., Saito M., Shirai T., Kohda D.. Structural basis of the 3′-end recognition of a leading strand in stalled replication forks by PriA. EMBO J. 2007; 26:2584–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tanaka T., Mizukoshi T., Sasaki K., Kohda D., Masai H.. Escherichia coli PriA protein, two modes of DNA binding and activation of ATP hydrolysis. J. Biol. Chem. 2007; 282:19917–19927. [DOI] [PubMed] [Google Scholar]
- 82. Bhattacharyya B., George N.P., Thurmes T.M., Zhou R., Jani N., Wessel S.R., Sandler S.J., Ha T., Keck J.L.. Structural mechanisms of PriA-mediated DNA replication restart. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:1373–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Leroux M., Jani N., Sandler S.J.. A priA mutant expressed in two pieces has almost full activity in E. coli K-12. J. Bacteriol. 2017; 199:e00267–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zavitz K.H., Marians K.J.. ATPase-deficient mutants of the Escherichia coli DNA replication protein PriA are capable of catalyzing the assembly of active primosomes. J. Biol. Chem. 1992; 267:6933–6940. [PubMed] [Google Scholar]
- 85. Sandler S.J., Samra H.S., Clark A.J.. Differential suppression of priA2::kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA, and dnaC. Genetics. 1996; 143:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lopper M.E., Boone J., Morrow C.. Deinococcus radiodurans PriA is a Pseudohelicase. PLoS One. 2015; 10:e0133419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jones J.M., Nakai H.. Duplex opening by primosome protein PriA for replisome assembly on a recombination intermediate. J. Mol. Biol. 1999; 289:503–516. [DOI] [PubMed] [Google Scholar]
- 88. Tanaka T., Taniyama C., Arai K., Masai H.. ATPase/helicase motif mutants of Escherichia coli PriA protein essential for recombination-dependent DNA replication. Genes Cells. 2003; 8:251–261. [DOI] [PubMed] [Google Scholar]
- 89. Masai H., Deneke J., Furui Y., Tanaka T., Arai K.I.. Escherichia coli and Bacillus subtilis PriA proteins essential for recombination-dependent DNA replication: involvement of ATPase/helicase activity of PriA for inducible stable DNA replication. Biochimie. 1999; 81:847–857. [DOI] [PubMed] [Google Scholar]
- 90. Lucius A.L., Jezewska M.J., Bujalowski W.. Allosteric interactions between the nucleotide-binding sites and the ssDNA-binding site in the PriA helicase-ssDNA complex. 3. Biochemistry. 2006; 45:7237–7255. [DOI] [PubMed] [Google Scholar]
- 91. Lucius A.L., Jezewska M.J., Roychowdhury A., Bujalowski W.. Kinetic mechanisms of the nucleotide cofactor binding to the strong and weak nucleotide-binding site of the Escherichia coli PriA helicase. 2. Biochemistry. 2006; 45:7217–7236. [DOI] [PubMed] [Google Scholar]
- 92. Lucius A.L., Jezewska M.J., Bujalowski W.. The Escherichia coli PriA helicase has two nucleotide-binding sites differing dramatically in their affinities for nucleotide cofactors. 1. Intrinsic affinities, cooperativities, and base specificity of nucleotide cofactor binding. Biochemistry. 2006; 45:7202–7216. [DOI] [PubMed] [Google Scholar]
- 93. Szymanski M.R., Jezewska M.J., Bujalowski W.. The Escherichia coli PriA helicase-double-stranded DNA complex: location of the strong DNA-binding subsite on the helicase domain of the protein and the affinity control by the two nucleotide-binding sites of the enzyme. J. Mol. Biol. 2010; 402:344–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sunchu B., Berg L., Ward H.E., Lopper M.E.. Identification of a small molecule PriA helicase inhibitor. Biochemistry. 2012; 51:10137–10146. [DOI] [PubMed] [Google Scholar]
- 95. Windgassen T.A., Keck J.L.. An aromatic-rich loop couples DNA binding and ATP hydrolysis in the PriA DNA helicase. Nucleic Acids Res. 2016; 44:9745–9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cadman C.J., Lopper M., Moon P.B., Keck J.L., McGlynn P.. PriB stimulates PriA helicase via an interaction with single-stranded DNA. J. Biol. Chem. 2005; 280:39693–39700. [DOI] [PubMed] [Google Scholar]
- 97. Cadman C.J., McGlynn P.. PriA helicase and SSB interact physically and functionally. Nucleic Acids Res. 2004; 32:6378–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jones J.M., Nakai H.. Escherichia coli PriA helicase: fork binding orients the helicase to unwind the lagging strand side of arrested replication forks. J. Mol. Biol. 2001; 312:935–947. [DOI] [PubMed] [Google Scholar]
- 99. Zavitz K.H., Marians K.J.. Helicase-deficient cysteine to glycine substitution mutants of Escherichia coli replication protein PriA retain single-stranded DNA-dependent ATPase activity. Zn2+ stimulation of mutant PriA helicase and primosome assembly activities. J. Biol. Chem. 1993; 268:4337–4346. [PubMed] [Google Scholar]
- 100. Liu J., Nurse P., Marians K.J.. The ordered assembly of the phiX174-type primosome. III. PriB facilitates complex formation between PriA and DnaT. J. Biol. Chem. 1996; 271:15656–15661. [DOI] [PubMed] [Google Scholar]
- 101. Sandler S.J. Requirements for replication restart proteins during constitutive stable DNA replication in Escherichia coli K-12. Genetics. 2005; 169:1799–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Manhart C.M., McHenry C.S.. The PriA replication restart protein blocks replicase access prior to helicase assembly and directs template specificity through its ATPase activity. J. Biol. Chem. 2013; 288:3989–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Szymanski M.R., Jezewska M.J., Bujalowski W.. The Escherichia coli PriA helicase specifically recognizes gapped DNA substrates: effect of the two nucleotide-binding sites of the enzyme on the recognition process. J. Biol. Chem. 2010; 285:9683–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liu J., Marians K.J.. PriA-directed assembly of a primosome on D loop DNA. J. Biol. Chem. 1999; 274:25033–25041. [DOI] [PubMed] [Google Scholar]
- 105. Szymanski M.R., Jezewska M.J., Bujalowski W.. Binding of two PriA-PriB complexes to the primosome assembly site initiates primosome formation. J. Mol. Biol. 2011; 411:123–142. [DOI] [PubMed] [Google Scholar]
- 106. Manhart C.M., McHenry C.S.. Identification of subunit binding positions on a model fork and displacements that occur during sequential assembly of the Escherichia coli primosome. J. Biol. Chem. 2015; 290:10828–10839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Singleton M.R., Dillingham M.S., Wigley D.B.. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007; 76:23–50. [DOI] [PubMed] [Google Scholar]
- 108. Lopper M., Holton J.M., Keck J.L.. Crystal structure of PriB, a component of the Escherichia coli replication restart primosome. Structure. 2004; 12:1967–1975. [DOI] [PubMed] [Google Scholar]
- 109. Liu J.H., Chang T.W., Huang C.Y., Chen S.U., Wu H.N., Chang M.C., Hsiao C.D.. Crystal structure of PriB, a primosomal DNA replication protein of Escherichia coli. J. Biol. Chem. 2004; 279:50465–50471. [DOI] [PubMed] [Google Scholar]
- 110. Huang Y.H., Lin M.J., Huang C.Y.. Yeast two-hybrid analysis of PriB-interacting proteins in replication restart primosome: a proposed PriB-SSB interaction model. Protein J. 2013; 32:477–483. [DOI] [PubMed] [Google Scholar]
- 111. Sandler S.J., Marians K.J., Zavitz K.H., Coutu J., Parent M.A., Clark A.J.. dnaC mutations suppress defects in DNA replication- and recombination-associated functions in priB and priC double mutants in Escherichia coli K-12. Mol. Microbiol. 1999; 34:91–101. [DOI] [PubMed] [Google Scholar]
- 112. Courcelle C.T., Landstrom A.J., Anderson B., Courcelle J.. Cellular characterization of the primosome and rep helicase in processing and restoration of replication following arrest by UV-induced DNA damage in Escherichia coli. J. Bacteriol. 2012; 194:3977–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Boonsombat R., Yeh S.-P., Milne A., Sandler S.J.. A novel dnaC mutation that suppresses priB rep mutant phenotypes in Escherichia coli K-12. Mol. Microbiol. 2006; 60:973–983. [DOI] [PubMed] [Google Scholar]
- 114. Marinus M.G. Recombination is essential for viability of an Escherichia coli dam (DNA adenine methyltransferase) mutant. J. Bacteriol. 2000; 182:463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Nowosielska A., Marinus M.G.. Cisplatin induces DNA double-strand break formation in Escherichia coli dam mutants. DNA Repair. 2005; 4:773–781. [DOI] [PubMed] [Google Scholar]
- 116. Grompone G., Ehrlich S.D., Michel B.. Replication restart in gyrB Escherichia coli mutants. Mol. Microbiol. 2003; 48:845–854. [DOI] [PubMed] [Google Scholar]
- 117. Harinarayanan R., Gowrishankar J.. A dnaC mutation in Escherichia coli that affects copy number of ColE1-like plasmids and the PriA-PriB (but not Rep-PriC) pathway of chromosomal replication restart. Genetics. 2004; 166:1165–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Huang Y.H., Lo Y.H., Huang W., Huang C.Y.. Crystal structure and DNA-binding mode of Klebsiella pneumoniae primosomal PriB protein. Genes Cells. 2012; 17:837–849. [DOI] [PubMed] [Google Scholar]
- 119. Huang C.-Y., Hsu C.-H., Sun Y.-J., Wu H.-N., Hsiao C.-D.. Complexed crystal structure of replication restart primosome protein PriB reveals a novel single-stranded DNA-binding mode. Nucleic Acids Res. 2006; 34:3878–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Szymanski M.R., Jezewska M.J., Bujalowski W.. Interactions of the Escherichia coli primosomal PriB protein with the single-stranded DNA. Stoichiometries, intrinsic affinities, cooperativities, and base specificities. J. Mol. Biol. 2010; 398:8–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fujiyama S., Abe Y., Takenawa T., Aramaki T., Shioi S., Katayama T., Ueda T.. Involvement of histidine in complex formation of PriB and single-stranded DNA. Biochim. Biophys. Acta. 2014; 1844:299–307. [DOI] [PubMed] [Google Scholar]
- 122. Jezewska M.J., Bujalowski W.. Interactions of Escherichia coli replicative helicase PriA protein with single-stranded DNA. Biochemistry. 2000; 39:10454–10467. [DOI] [PubMed] [Google Scholar]
- 123. Wessel S.R., Marceau A.H., Massoni S.C., Zhou R., Ha T., Sandler S.J., Keck J.L.. PriC-mediated DNA replication restart requires PriC complex formation with the single-stranded DNA-binding protein. J. Biol. Chem. 2013; 288:17569–17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Aramaki T., Abe Y., Ohkuri T., Mishima T., Yamashita S., Katayama T., Ueda T.. Domain separation and characterization of PriC, a replication restart primosome factor in Escherichia coli. Genes Cells. 2013; 18:723–732. [DOI] [PubMed] [Google Scholar]
- 125. Butland G., Peregrin-Alvarez J.M., Li J., Yang W., Yang X., Canadien V., Starostine A., Richards D., Beattie B., Krogan N. et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005; 433:531–537. [DOI] [PubMed] [Google Scholar]
- 126. Huang C.C., Huang C.Y.. DnaT is a PriC-binding protein. Biochem. Biophys. Res. Commun. 2016; 477:988–992. [DOI] [PubMed] [Google Scholar]
- 127. Wessel S.R., Cornilescu C.C., Cornilescu G., Metz A., Leroux M., Hu K., Sandler S.J., Markley J.L., Keck J.L.. Structure and function of the PriC DNA replication restart protein. J. Biol. Chem. 2016; 291:18384–18396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hinds T., Sandler S.J.. Allele specific synthetic lethality between priC and dnaAts alleles at the permissive temperature of 30 degrees C in E. coli K-12. BMC Microbiol. 2004; 4:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Aramaki T., Abe Y., Katayama T., Ueda T.. Solution structure of the N-terminal domain of a replication restart primosome factor, PriC, in Escherichia coli. Protein Sci. 2013; 22:1279–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Aramaki T., Abe Y., Furutani K., Katayama T., Ueda T.. Basic and aromatic residues in the C-terminal domain of PriC are involved in ssDNA and SSB binding. J. Biochem. 2015; 157:529–537. [DOI] [PubMed] [Google Scholar]
- 131. Arai K., McMacken R., Yasuda S., Kornberg A.. Purification and properties of Escherichia coli protein i, a prepriming protein in phi X174 DNA replication. J. Biol. Chem. 1981; 256:5281–5286. [PubMed] [Google Scholar]
- 132. McCool J.D., Ford C.C., Sandler S.J.. A dnaT Mutant With Phenotypes Similar to Those of a priA2::kan Mutant in Escherichia coli K-12. Genetics. 2004; 167:569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lark C.A., Riazi J., Lark K.G.. dnaT, dominant conditional-lethal mutation affecting DNA replication in Escherichia coli. J. Bacteriol. 1978; 136:1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Masai H., Arai K.. Operon structure of dnaT and dnaC genes essential for normal and stable DNA replication of Escherichia coli chromosome. J. Biol. Chem. 1988; 263:15083–15093. [PubMed] [Google Scholar]
- 135. Nakayama N., Bond M.W., Miyajima A., Kobori J., Arai K.. Structure of Escherichia coli dnaC. Identification of a cysteine residue possibly involved in association with dnaB protein. J. Biol. Chem. 1987; 262:10475–10480. [PubMed] [Google Scholar]
- 136. Ng J.Y., Marians K.J.. The ordered assembly of the phiX174-type primosome. I. Isolation and identification of intermediate protein-DNA complexes. J. Biol. Chem. 1996; 271:15642–15648. [DOI] [PubMed] [Google Scholar]
- 137. Szymanski M.R., Jezewska M.J., Bujalowski W.. Energetics of the Escherichia coli DnaT protein trimerization reaction. Biochemistry. 2013; 52:1858–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Szymanski M.R., Jezewska M.J., Bujalowski W.. The Escherichia coli primosomal DnaT protein exists in solution as a monomer-trimer equilibrium system. Biochemistry. 2013; 52:1845–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Huang Y.-H., Huang C.-Y.. The N-terminal domain of DnaT, a primosomal DNA replication protein, is crucial for PriB binding and self-trimerization. Biochem. Biophys. Res. Commun. 2013; 442:147–152. [DOI] [PubMed] [Google Scholar]
- 140. Fujiyama S., Abe Y., Tani J., Urabe M., Sato K., Aramaki T., Katayama T., Ueda T.. Structure and mechanism of the primosome protein DnaT-functional structures for homotrimerization, dissociation of ssDNA from the PriB·ssDNA complex, and formation of the DnaT·ssDNA complex. FEBS J. 2014; 281:5356–5370. [DOI] [PubMed] [Google Scholar]
- 141. Huang Y.-H., Lin M.-J., Huang C.-Y.. DnaT is a single-stranded DNA binding protein. Genes Cells. 2013; 18:1007–1019. [DOI] [PubMed] [Google Scholar]
- 142. Liu Z., Chen P., Wang X., Cai G., Niu L., Teng M., Li X.. Crystal structure of DnaT84-153-dT10 ssDNA complex reveals a novel single-stranded DNA binding mode. Nucleic Acids Res. 2014; 42:9470–9483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Shlomai J., Kornberg A.. An Escherichia coli replication protein that recognizes a unique sequence within a hairpin region in phi X174 DNA. Proc. Natl. Acad. Sci. U.S.A. 1980; 77:799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Lee M.S., Marians K.J.. Escherichia coli replication factor Y, a component of the primosome, can act as a DNA helicase. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:8345–8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Heller R.C., Marians K.J.. Unwinding of the nascent lagging strand by Rep and PriA enables the direct restart of stalled replication forks. J. Biol. Chem. 2005; 280:34143–34151. [DOI] [PubMed] [Google Scholar]
- 146. Mizukoshi T., Tanaka T., Arai K., Kohda D., Masai H.. A critical role of the 3′ terminus of nascent DNA chains in recognition of stalled replication forks. J. Biol. Chem. 2003; 278:42234–42239. [DOI] [PubMed] [Google Scholar]
- 147. Yu C., Tan H.Y., Choi M., Stanenas A.J., Byrd A.K., K D.R., Cohan C.S., Bianco P.R.. SSB binds to the RecG and PriA helicases in vivo in the absence of DNA. Genes Cells. 2016; 21:163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Xu L., Marians K.J.. PriA mediates DNA replication pathway choice at recombination intermediates. Mol. Cell. 2003; 11:817–826. [DOI] [PubMed] [Google Scholar]
- 149. Yuan Q., McHenry C.S.. Strand displacement by DNA polymerase III occurs through a tau-psi-chi link to single-stranded DNA-binding protein coating the lagging strand template. J. Biol. Chem. 2009; 284:31672–31679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Seigneur M., Bidnenko V., Ehrlich S.D., Michel B.. RuvAB acts at arrested replication forks. Cell. 1998; 95:419–430. [DOI] [PubMed] [Google Scholar]
- 151. Heller R.C., Marians K.J.. Non-replicative helicases at the replication fork. DNA Repair (Amst). 2007; 6:945–952. [DOI] [PubMed] [Google Scholar]
- 152. Atkinson J., Gupta M.K., McGlynn P.. Interaction of Rep and DnaB on DNA. Nucleic Acids Res. 2011; 39:1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Yancey-Wrona J.E., Wood E.R., George J.W., Smith K.R., Matson S.W.. Escherichia coli Rep protein and helicase IV. Distributive single-stranded DNA-dependent ATPases that catalyze a limited unwinding reaction in vitro. Eur. J. Biochem. 1992; 207:479–485. [DOI] [PubMed] [Google Scholar]
- 154. Boubakri H., de Septenville A.L., Viguera E., Michel B.. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010; 29:145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Lane H.E., Denhardt D.T.. The rep mutation. IV. Slower movement of replication forks in Escherichia coli rep strains. J. Mol. Biol. 1975; 97:99–112. [DOI] [PubMed] [Google Scholar]
- 156. Colasanti J., Denhardt D.T.. The Escherichia coli rep mutation. X. Consequences of increased and decreased Rep protein levels. Mol. Gen. Genet.: MGG. 1987; 209:382–390. [DOI] [PubMed] [Google Scholar]
- 157. Guy C.P., Atkinson J., Gupta M.K., Mahdi A.A., Gwynn E.J., Rudolph C.J., Moon P.B., van Knippenberg I.C., Cadman C.J., Dillingham M.S. et al. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol. Cell. 2009; 36:654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. O'Shea V.L., Berger J.M.. Loading strategies of ring-shaped nucleic acid translocases and helicases. Curr. Opin. Struct. Biol. 2014; 25:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Arias-Palomo E., O'Shea V.L., Hood I.V., Berger J.M.. The bacterial DnaC helicase loader is a DnaB ring breaker. Cell. 2013; 153:438–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Sutton M.D., Carr K.M., Vicente M., Kaguni J.M.. Escherichia coli DnaA protein. The N-terminal domain and loading of DnaB helicase at the E. coli chromosomal origin. J. Biol. Chem. 1998; 273:34255–34262. [DOI] [PubMed] [Google Scholar]
- 161. Seitz H., Weigel C., Messer W.. The interaction domains of the DnaA and DnaB replication proteins of Escherichia coli. Mol. Microbiol. 2000; 37:1270–1279. [DOI] [PubMed] [Google Scholar]
- 162. Chodavarapu S., Jones A.D., Feig M., Kaguni J.M.. DnaC traps DnaB as an open ring and remodels the domain that binds primase. Nucleic Acids Res. 2016; 44:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Courcelle J., Khodursky A., Peter B., Brown P.O., Hanawalt P.C.. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics. 2001; 158:41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Shlomai J., Kornberg A.. A prepriming DNA replication enzyme of Escherichia coli. I. Purification of protein n': a sequence-specific, DNA-dependent ATPase. J. Biol. Chem. 1980; 255:6789–6793. [PubMed] [Google Scholar]
- 165. Polard P., Marsin S., McGovern S., Velten M., Wigley D.B., Ehrlich S.D., Bruand C.. Restart of DNA replication in Gram-positive bacteria: functional characterisation of the Bacillus subtilis PriA initiator. Nucleic Acids Res. 2002; 30:1593–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Li G.W., Burkhardt D., Gross C., Weissman J.S.. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014; 157:624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Low R.L., Shlomai J., Kornberg A.. Protein n, a primosomal DNA replication protein of Escherichia coli. Purification and characterization. J. Biol. Chem. 1982; 257:6242–6250. [PubMed] [Google Scholar]
- 168. Kline K.A., Seifert H.S.. Mutation of the priA gene of Neisseria gonorrhoeae affects DNA transformation and DNA repair. J. Bacteriol. 2005; 187:5347–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Dong J., George N.P., Duckett K.L., DeBeer M.A., Lopper M.E.. The crystal structure of Neisseria gonorrhoeae PriB reveals mechanistic differences among bacterial DNA replication restart pathways. Nucleic Acids Res. 2010; 38:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Huang Y.H., Lien Y., Huang C.C., Huang C.Y.. Characterization of Staphylococcus aureus primosomal DnaD protein: highly conserved C-terminal region is crucial for ssDNA and PriA helicase binding but not for DnaA protein-binding and self-tetramerization. PLoS One. 2016; 11:e0157593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Bruand C., Velten M., McGovern S., Marsin S., Serena C., Ehrlich S.D., Polard P.. Functional interplay between the Bacillus subtilis DnaD and DnaB proteins essential for initiation and re-initiation of DNA replication. Mol. Microbiol. 2005; 55:1138–1150. [DOI] [PubMed] [Google Scholar]
- 172. Li Y.C., Naveen V., Lin M.G., Hsiao C.D.. Structural analyses of the bacterial primosomal protein DnaB reveal that it is a tetramer and forms a complex with a primosomal re-initiation protein. J. Biol. Chem. 2017; 292:15744–15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Huang Y.H., Guan H.H., Chen C.J., Huang C.Y.. Staphylococcus aureus single-stranded DNA-binding protein SsbA can bind but cannot stimulate PriA helicase. PLoS One. 2017; 12:e0182060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Yao N., O’Donnell M.. Bacterial and eukaryotic replisome machines. JSM Biochem. Mol. Biol. 2016; 3:1013. [PMC free article] [PubMed] [Google Scholar]
- 175. Berti M., Vindigni A.. Replication stress: getting back on track. Nat. Struct. Mol. Biol. 2016; 23:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Techer H., Koundrioukoff S., Nicolas A., Debatisse M.. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017; 18:535–550. [DOI] [PubMed] [Google Scholar]
- 177. Saldivar J.C., Cortez D., Cimprich K.A.. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017; 18:622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Branzei D., Szakal B.. Building up and breaking down: mechanisms controlling recombination during replication. Crit. Rev. Biochem. Mol. Biol. 2017; 52:381–394. [DOI] [PubMed] [Google Scholar]
- 179. Gao Y., Mutter-Rottmayer E., Zlatanou A., Vaziri C., Yang Y.. Mechanisms of post-replication DNA repair. Genes. 2017; 8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Kile A.C., Chavez D.A., Bacal J., Eldirany S., Korzhnev D.M., Bezsonova I., Eichman B.F., Cimprich K.A.. HLTF’s ancient HIRAN domain binds 3′ DNA ends to drive replication fork reversal. Mol. Cell. 2015; 58:1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Achar Y.J., Balogh D., Neculai D., Juhasz S., Morocz M., Gali H., Dhe-Paganon S., Venclovas C., Haracska L.. Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res. 2015; 43:10277–10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Hishiki A., Hara K., Ikegaya Y., Yokoyama H., Shimizu T., Sato M., Hashimoto H.. Structure of a novel DNA-binding domain of helicase-like transcription factor (HLTF) and its functional implication in DNA damage tolerance. J. Biol. Chem. 2015; 290:13215–13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Shioi S., Ose T., Maenaka K., Shiroishi M., Abe Y., Kohda D., Katayama T., Ueda T.. Crystal structure of a biologically functional form of PriB from Escherichia coli reveals a potential single-stranded DNA-binding site. Biochem. Biophys. Res. Commun. 2005; 326:766–776. [DOI] [PubMed] [Google Scholar]
- 184. Raghunathan S., Kozlov A.G., Lohman T.M., Waksman G.. Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat. Struct. Biol. 2000; 7:648–652. [DOI] [PubMed] [Google Scholar]