


Abstract
Influenza polymerase uses short capped primers snatched from nascent Pol II transcripts to initiate transcription of viral mRNAs. Here we describe crystal structures of influenza A and B polymerase bound to a capped primer in a configuration consistent with transcription initiation (’priming state’) and show by functional assays that conserved residues from both the PB2 midlink and cap-binding domains are important for positioning the capped RNA. In particular, mutation of PB2 Arg264, which interacts with the triphosphate linkage in the cap, significantly and specifically decreases cap-dependent transcription. We also compare the configuration of the midlink and cap-binding domains in the priming state with their very different relative arrangement (called the ‘apo’ state) in structures where the potent cap-binding inhibitor VX-787, or a close analogue, is bound. In the ‘apo’ state the inhibitor makes additional interactions to the midlink domain that increases its affinity beyond that to the cap-binding domain alone. The comparison suggests that the mechanism of resistance of certain mutations that allow virus to escape from VX-787, notably PB2 N510T, can only be rationalized if VX-787 has a dual mode of action, direct inhibition of capped RNA binding as well as stabilization of the transcriptionally inactive ‘apo’ state.
INTRODUCTION
Influenza RNA-dependent RNA polymerase is a heterotrimer with subunits PA, PB1 and PB2. It binds the conserved 3′ and 5′ ends of each of the eight negative-sense RNA genome segments and is responsible for transcription and replication of the genomic RNA in the nucleus of infected cells. The polymerase synthesizes translation competent viral mRNA in a unique manner. Transcription is initiated by short capped primers that are pirated from nascent host Pol II transcripts so that 10–14 nucleotides of host sequence precede the virally encoded sequences in the resultant chimeric viral mRNA. Transcription termination is achieved by a stuttering mechanism resulting in poly(A)-tail formation. The recent high resolution structures of influenza polymerase suggest a plausible mechanistic model for cap-dependent transcription and polyadenylation (1,2). The four key steps in the structure-based model for synthesis of poly-adenylated viral mRNA are as follows (2). (a) Binding of the donor host nascent Pol II transcript to the PB2 cap-binding domain followed by cleavage 10–14 nucleotides downstream of the cap by the endonuclease at the N-terminus of the PA subunit. (b) Rotation by ∼60° of the cap-binding domain from the ‘cleavage’ configuration to the ‘priming’ configuration to direct the short capped primer down into the RNA synthesis catalytic centre of the PB1 subunit (1). It is thought that those primers with 3′ ends complementary to the 3′ extremity of the template are preferentially selected for transcription initiation (3,4). (c) Progression into processive elongation with the translocating template and product extruding through separate tunnels. (d) Formation of the poly(A)-tail by reiterative copying of the 5′ proximal oligo-U stretch of the template corresponding to nucleotides 17–22 from the 5′ end (5,6).
Transcription of viral mRNAs is essential for viral replication and therefore inhibitors that target the transcriptional activity of influenza polymerase are potential anti-viral drugs. Novel anti-influenza drugs as a therapeutic option complementary to vaccination are urgently needed, since influenza is a recurrent, unpredictable and potentially very serious threat to public health worldwide with enormous social and economic costs. In recent years potent inhibitors that target the three active sites on the polymerase relevant for transcription, the PB2 cap-binding, the PA endonuclease and the PB1 RNA synthesis site have been developed and are entering clinical trials (2,7). One of these, VX-787 (JNJ63623872, Pimodivir), is a potent cap-binding inhibitor (8–11).
In this work, we focus on step (a) of the cap-dependent transcription model and present the first structural data showing how a capped oligomer is bound to the heterotrimeric influenza A or B polymerase when in the ‘priming’ configuration. The mode of binding of cap analogues such as m7GTP to the isolated PB2 cap-binding domain for both influenza A (12,13) and influenza B polymerases (14,15) is well understood (12,14), but not how the physiological substrate, capped RNA, binds to the full polymerase. The structure of the complete polymerase with capped RNA shows that capped primer binding induces a highly ordered structure. The cap-binding (PB2 residues 320–489) and the midlink domain (PB2 residues 251–319 (mid) + 490–536 (link), into which the cap-binding domain is inserted (16)), intimately contact each other and both domains contribute conserved residues that interact with the capped RNA. The importance of these residues is assessed by functional studies in vitro and in cells. We then compare the ‘priming’ configuration of the cap-binding and midlink domains (denoted cap-midlink double domain) as observed in the capped RNA complex with the quite different alternative configuration of these domains observed previously (denoted ‘apo’ configuration) (16). Crystal structures of a close analogue of the cap-binding inhibitor VX-787 bound to influenza A or B constructs containing only the PB2 cap-midlink double domain show that in the ‘apo’ configuration the compound makes additional interactions with the midlink domain. Taking into account the location of reported resistance mutants to this compound (9), our analysis suggests that VX-787 may act by both direct inhibition of capped-RNA binding as well as by stabilizing the transcriptionally inactive ‘apo’ configuration of the cap-binding and midlink domains.
MATERIALS AND METHODS
Purification, crystallization and structure determination of bat A/H17N10 polymerase complexes
Expression and purification of the influenza A/little yellow-shouldered bat/Guatemala/164/2009(H17N10) polymerase was as described (17), but eliminating the final gel filtration step. After elution from the Heparin column, the sample was in 50 mM HEPES/NaOH pH 7.5, 5% (v/v) glycerol, 2 mM tris(2-carboxyethyl)phosphine (TCEP) and 500–700 mM NaCl. In this buffer, the protein was concentrated to about 7–9 mg/ml, flash frozen and submitted to crystallization trials. Co-crystals of bat polymerase with m7GTP were grown as described (17) but with addition of 5 mM of the cap analogue. For crystallization with capped RNA, the protein was mixed with equimolar amounts of a capped RNA primer (m7GpppAAUCUAUAAUAG) and vRNA promoter comprising nucleotides 1–16 from the 5′ end (5′-pAGUAGUAACAAGAGGG-3′) and nucleotides 1–18 or 3–18 from the 3′ end (3′-UCGUCUUCGUCUCCAUAU-5′). The complex was crystallized at 4°C by vapour diffusion in 0.1 M bicine pH 9.0 and 17% (v/v) MPD. Well solution supplemented with 15% (v/v) glycerol was employed as cryo-protectant for flash freezing the crystals. Diffraction data were collected on beamline ID23–1 at the European Synchrotron Radiation Facility (ESRF) and integrated with XDS (18). Structures were solved by molecular replacement with PHASER (19) using the previously determined bat polymerase structure (PDB: 4WSB) and refined using REFMAC (20) and BUSTER (https://www.globalphasing.com/buster/). For the m7GTP bound structure, the endonuclease is poorly ordered as observed previously for this crystal form (17). For the capped RNA bound structure, local NCS restraints were used for the two complexes in the asymmetric unit. See Table 1 for crystallographic statistics.
Table 1. Crystallographic data processing and refinement statistics.
| Crystal | Bat A/H17N10 polymerase +3′ vRNA 3–18 +5′ vRNA 1–16 +m7GTP | Bat A/H17N10 polymerase +3′ vRNA 1–18 +5′ vRNA 1–16 +12-mer capped RNA primer | A/Victoria/H3N2 PB2 cap-midlink double-domain +VX-787N | B/Memphis PB2 cap-midlink double-domain +VX-787N |
|---|---|---|---|---|
| Diffraction data | ||||
| Space group | C2 | C2 | P2221 | P3121 |
| Cell dimensions (Å) | a = 269.34, b = 148.70, c = 88.51 β = 98.17 | a = 185.46, b = 286.17, c = 171.93 β = 93.27 | a = 49.46, b = 112.32, c = 201.09 α = β = γ = 90 | a = b = 73.20 c = 103.74 α = β = 90 γ = 120 |
| Wavelength (Å) | 0.979 | 0.979 | 0.966 | 0.966 |
| Beamline (ESRF) | ID23–1 | ID23–1 | ID30A1 | ID30A1 |
| No. Crystals | 1 | 1 | 1 | 1 |
| Resolution range of data (Å) | 50.0–2.90 | 50.0–3.90 | 50.0–3.00 | 50.0–2.05 |
| (last shell) | (2.98–2.90) | (4.00–3.90) | (3.18–3.00) | (2.15–2.05) |
| Completeness (%) (last shell) | 99.6 (99.8) | 99.7 (98.9) | 98.2 (97.7) | 99.7 (99.7) |
| R-sym (last shell) | 0.102 (1.22) | 0.118 (1.24) | 0.065 (0.63) | 0.119 (1.10) |
| R-meas (last shell) | 0.115 (1.38) | 0.137 (1.44) | 0.082 (0.80) | 0.131 (1.21) |
| I/σI (last shell) | 10.75 (1.08) | 8.28 (0.93) | 9.36 (1.45) | 10.46 (1.75) |
| CC(1/2) (%)(last shell) | 99.8 (59.7) | 99.8 (53.9) | 99.7 (79.4) | 99.7 (64.4) |
| Redundancy (last shell) | 4.63 (4.65) | 3.87 (3.87) | 4.8 (4.7) | 5.70 (5.78) |
| Refinement | ||||
| Reflections used in refinement: work (free) | 72389 (3755) | 77007 (4018) | 22122 (1118) | 19649 (1022) |
| R-work (last shell) | 0.234 (0.436) | 0.207 (0.381) | 0.233 (0.401) | 0.229 (0.327) |
| R-free (last shell) | 0.273 (0.471) | 0.245 (0.376) | 0.263 (0.551) | 0.254 (0.306) |
| No. of non-hydrogen atoms | 18166 | 36717 | 6595 | 1987 |
| Protein | 17435 | 35378 | 6478 | 1856 |
| RNA | 600 | 1153 | - | - |
| Ligand | 33 (m7GTP) | 186 (capped RNA) | 87 (3 x VX-787N) | 29 (VX-787N) |
| Solvent | 98 (12 phosphate + water) | - | 30 (6 sulphate) | 102 (water) |
| Geometry and B-factors | ||||
| RMSD bond lengths (Å) | 0.008 | 0.007 | 0.008 | 0.007 |
| RMSD bond angles (°) | 1.14 | 0.96 | 1.28 | 1.07 |
| Average B-factor ALL (Å2) | 117.9 | 186.3 | 122.4 | 50.9 |
| Protein (Å2) | 118.7 | 185.9 | 122.7 | 51.5 |
| vRNA 5′, 3′ (Å2) | 94.6 | 190.9 | - | - |
| Ligand (Å2) | 105.3 (m7GTP) | 235.4 (capped RNA) | 79.8 (3 x VX-787N) | 34.1 (VX-787N) |
| Solvent (Å2) | 108.2 | - | 153.2 | 45.4 |
| Ramachandran favoured (%) | 92.3 | 95.1 | 94.9 | 97.2 |
| Ramachandran outliers (%) | 0.7 | 0.5 | 0.4 | 0.0 |
| Molprobity score | 1.57 | 1.12 | 1.65 | 0.83 |
| Clash score | 1.14 | 0.76 | 1.29 | 0.26 |
Mutation of bat A/H17N10 polymerase
The mutations I260A, R264A and Y432A were inserted into the expression constructs via site-directed mutagenesis employing Phusion High-Fidelity DNA Polymerase (NEB) and the following primer pairs (with mutated nucleotides in lower case): BF_PB2_I260A_fwd (5′-ACCCT CgccA TTGCC TCTAG AAACA TAGTC CG-3′) x BF_PB2_I260A_rev (5′-GGCAA TggcG AGGGT CTGAT CCAAG TCGTC G-3′),
BF_PB2_R264A_fwd (5′-GCCTC TgcaA ACATA GTCCG AAGGA GCATC G-3′) x BF_PB2_R264A_rev (5′-TATGT TtgcA GAGGC AATGA TGAGG GTCTG-3′),
BF_PB2_Y432A_fwd (5′-CCCAT GgctC AGCTGC TGAG ACATT TCCAG-3′) x BF_PB2_Y432A_rev (5′-CAGCT GagcC ATGGG GTTGA GTCGCT GG-3′).
Since polymerase chain reactions failed when using the expression construct of the entire H17N10 polymerase complex as template (a 12.8 kb plasmid), the strategy was changed in favour of a three-step approach. First, the mutagenesis was carried out in a construct containing only the coding sequence of PB2, a plasmid of size 7.1 kb. Then 1500 bp fragments, containing the desired point mutations, were amplified from these vectors with primers batPB2_mega_fwd (5′-GTCGT TCATT ACCCT CGGGT G-3′) x batPB2_mega_rev (5′-GTTGC CGCGA ATGAG AAGCC-3′), the product DNA gel-purified and used as megaprimer in a third PCR to amplify the pKLPBac expression vector carrying all three polymerase subunits.
Minigenome assays
Site-directed mutagenesis was used to replace influenza A/Victoria/3/75(H3N2) PB2 residues I260, R264, Q306, R423 and H432 with alanines in the corresponding pcDNA3 plasmid for minigenome experiments. The modified constructs were generated with Phusion High-Fidelity DNA Polymerase (NEB) and following primer pairs (with mutated nucleotides in lower case):
H3-PB2_I260A_fwd (5′-AAGCC TAgct ATTGC AGCCA GGAAC ATAGT G-3′) x H3-PB2_I260A_rev (5′-CTGCA ATagc TAGGC TTTGG TCAAT ATCGT CATTC-3′),
H3-PB2_R264A_fwd (5′-TGCAG CCgcg AACAT AGTGA GAAGA GCATC AG-3′) x H3-PB2_R264A_rev (5′-TATGT TcgcG GCTGC AATAA TTAGG CTTTG GTC-3′),
H3-PB2_Q306A_fwd (5′-GGAAG AAgca GCTGT GGATA TATGC AAGGC TG-3′) x H3-PB2_Q306A_rev (5′-CCACA GCtgc TTCTT CCGTC GGGTT CTGTC TAAG-3′),
H3-PB2_R423A_fwd (5′-CGTTA ACgcg GCAAA TCAGC GGTTG AATCC C-3′) x H3-PB2_R423A_rev (5′-GATTT GCcgc GTTAA CGAAA TTCAG GTCAC CTCTA AC-3′),
H3-PB2_H432A_fwd (5′-AATCC CATGg ctCAA CTTTT AAGGC ATTTT CAGAA AG-3′) x H3-PB2_H432A_rev (5′-TAAAA GTTGa gcCAT GGGAT TCAAC CGCTG ATT-3′).
HEK293T cells were seeded in 12-well plates and transfection was performed with LipoD293™ transfection reagent (Tebu-bio). Each well was transfected with 100 ng pcDNA3 expressing nucleoprotein (NP), 10 ng pcDNA3 plasmid expressing PB2 or corresponding mutants, 10 ng pcDNA3 plasmids expressing PB1 and 5ng pcDNA3 plasmids expressing PA subunits of the influenza A/Victoria/3/75(H3N2) polymerase and 50 ng pPolI-NP-Luc, encoding a Firefly luciferase reporter gene in negative polarity, flanked by the 5′ and 3′ regions of the NP segment (described in (21)). Wild-type plasmids were donated by J. Ortin (22), as well as the antibody used for verification of PB2 expression. Transfection mix without PB2 was used as a negative control. pRenilla-TK plasmid (Promega) was used to correct for the transfection efficiencies. Cells were lysed 20 hours post-transfection and Firefly and Renilla luciferase activities were measured with a dual-luciferase reporter assay system (Promega) using a Clariostar microplate reader (BMG) according to the manufacturer's protocol. All experiments were performed in biological triplicates.
Viral RNA quantification
For RNA detection using primer extension assay HEK293T cells were transfected with 300 ng pcDNA3-PA, 600 ng pcDNA3-PB1 and 600 ng pcDNA3-PB2 (wild-type or variants), 6 μg pcDNA3-NP and 60 ng pPolI-NP-Luc. One day post-transfection, RNAs were extracted with TRIzol reagent (Ambion) and mixed with primers for negative-stranded RNA (5′-GGATTACGTCGCCAGTCAAG-3′), positive-stranded RNA (5′-GCTCTCCAGCGGTTCCATC-3′) and 5S rRNA (5′-TCCCAGGCGGTCTCCCATCC-3′). Primer extension was performed as described (23). Transcription products were analysed on 6% (w/v) polyacrylamide gels containing 7 M urea in Trisborate-EDTA (TBE) buffer, detected by autoradiography, and quantified by densitometry of phospho-images using ImageJ 1.51p software.
In vitro RNA synthesis assay
The RNA synthesis activity of wild-type A/H17N10 and PB2-variant influenza polymerases was analyzed by the fluorescence polarization (FP) based assay as described (24). Briefly, RNA synthesis was initiated by adding 0.25 μM polymerase (pre-incubated by a 1.2-fold molar excess of 5′ RNA nt 1–14) to 0.15 μM fluorescently-labelled template RNA and 0.2 mM NTPs (each) in assay buffer (50 mM HEPES/NaOH, 150 mM NaCl, 10% (v/v) glycerol, 5 mM MgCl2, 2 mM TCEP, pH 7.4) at 24°C. To determine the KM (capped RNA primers), reactions were supplemented by the indicated concentrations of the capped RNA primer (5′-m7GpppAAUCUAUAAUAG-3′; TriLink BioTechnologies, San Diego, USA). To determine the IC50-values of VX-787N (for structural formula, see Supplementary Figure 5), ApG primed (0.5 mM; IBA, Germany) or capped RNA primed (0.5 μM) RNA synthesis reactions from the vRNA promoter were quantified at the indicated concentrations of VX-787N (Chembiotek, India and a gift from Savira pharmaceuticals). At indicated reaction times, 5 μl aliquots were transferred to 80 μl 4.5 M NaCl and the FP recorded as described (24). Progress curves were fitted to single- or double-exponentials according to pseudo-first order kinetics. The initial rate (rate constant multiplied by the corresponding amplitude) was plotted against the respective concentration of capped RNA primer or VX-787N and fitted to the quadratic velocity equation or hyperbolically, respectively, yielding the MICHAELIS constant of the capped RNA primer (KM (capped RNA primer)) and the maximal reaction rate (Vmax) or the IC50-value. Vmax (mP/min) was converted to the turnover number kcat (min–1) by division by the maximal signal amplitude of 80 mP that correlated to all template RNA being converted from initially single-stranded to dsRNA. Catalytic efficiency is expressed as kcat/KM (min–1 μM–1). All constants are reported as mean values with standard deviation, derived from at least duplicate experiments. The vRNA promoter was purchased from IBA (v5′ nt 1–14: 5′-pAGUAGAAACAAGGC-3′; v3′ nt 1–18: 5′-(FAM-Ex-5)-AGUUUGCCUGCUUCUGCU-3′).
Figure 5.

Binding and inhibitory function of VX-787N or VX-787 to influenza A and B polymerase. (A) Effect of VX-787N on bat A/H17N10 influenza polymerase RNA synthesis activity. While VX-787N shows no effect on ApG primed RNA synthesis from the vRNA promoter (black), at an excess of VX-787N over polymerase and capped RNA primer, VX-787N completely inhibits capped RNA primed RNA synthesis from the vRNA promoter (orange) as expected. Squares and circles of identical color correspond to duplicate experiments. (B) Quantification of VX-787N-FAM binding to A/H5N1 influenza polymerase PB2 cap-binding domain or cap-midlink double domain. Interaction of A/H5N1 PB2 cap-binding domain (black) and cap-midlink double domain (red) with the fluorescently labelled VX-787N results in increased fluorescence polarization that was used to determine KDs of respectively 0.017 ± 0.004 μM and 0.007 ± 0.001 μM (error of the fitting routine of mean values of duplicate experiments; standard deviation of the mean is indicated). (C) Representative ITC data and curve fit to derive the affinity of VX-787 to A/H3N2 cap-midlink double domain. The derived KD is 2.4 ± 1.3 nM (N = 4). (D) Comparison of VX-787N-FAM binding to bat A/H17N10 polymerase or B/Memphis polymerase by fluorescence anisotropy assay giving KDs of respectively 0.0061 ±0.00021 μM (mean and standard deviation of duplicates; black and pink circles correspond to independent binding isotherms) and 1.2 ± 0.02 μM (representative binding isotherm ± error of the fitting routine; red squares). (E) Representative ITC data and curve fit to derive the affinity of VX-787 to B/Memphis cap-midlink double domain. The derived KD is 1.2 ± 0.3 μM (N = 5). (F) Representative ITC data and curve fit to derive the affinity of VX-787 to A/H3N2 (N501T) cap-midlink double domain. The derived KD is 10.7 ± 4.8 nM (N = 6).
Inhibitor binding to heterotrimeric polymerase
Quantification of A/H17N10 influenza polymerase binding to VX-787N was performed using a competition assay using a fluorescently labelled VX-787N (VX-787N-FAM, ChemPartner, Shanghai, China). Briefly, protein was successively added to 0.01 μM VX-787N analogue in binding buffer (50 mM HEPES/NaOH, 0.15 M NaCl, 10% (v/v) glycerol, 2 mM TCEP, 1% (v/v) DMSO, pH 7.4) at the concentrations indicated. After attaining equilibrium, total fluorescence intensity and fluorescence anisotropy were measured at 24 °C with a fluorescence spectrometer (Photon Technology International) equipped with polarizers with excitation and emission wavelengths set to 494 and 520 nm, respectively. Total fluorescence intensity was corrected for the volume change due to addition of protein and the observed fluorescence anisotropy corrected for the change in fluorescence intensity according to Equation 1 (25).
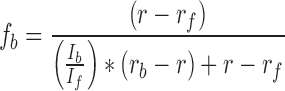 |
(1) |
where fb represents the fractional concentration of bound probe (VX-787N-FAM), r the observed anisotropy, rf and rb the anisotropy of free and bound probe and If and Ib the fluorescence intensity of free and bound probe, respectively. Using KaleidaGraph (Synergy Software), binding isotherms at the indicated concentrations of VX-787N were fitted to the HILL-equation (26), derived microscopic dissociation constants (ligand concentration occupying half of the binding sites) converted to association constants and analysed by Equation (2) (quadratic velocity equation):
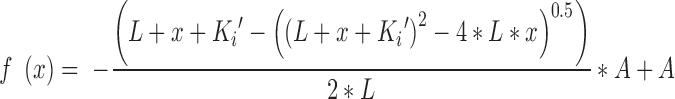 |
(2) |
with L being the concentration of spectroscopic probe VX-787N-FAM (L = 0.01 μM), Ki′ the apparent inhibition constant, x the concentration of competing VX-787N and A, the association constant of polymerase and VX-787N analogue in the absence of competitor. Ki′ was converted to the true inhibition constant Ki (Pol+VX-787N) according to Equation (3) (competitive inhibition):
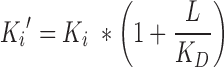 |
(3) |
with L equaling the concentration of VX-787N-FAM (L = 0.01 μM) and KD being the dissociation constant of polymerase and VX-787N-FAM in the absence of VX-787N (KD (Pol+VX-787N analogue) = 0.0061 ±0.00021 μM; mean and corresponding standard deviation of duplicate experiments).
Isothermal titration calorimetry (ITC)
ITC measurements were performed at 25°C using a MicroCal ITC200 system (Malvern). Proteins were dialyzed overnight in a buffer containing 20 mM Tris pH 7.5, 150 mM NaCl, 0.25 mM TCEP and the ligand VX-787 was dissolved in the same buffer. Ligand was injected at intervals of 120 s into the protein contained in the calorimetric cell at a stirring speed of 800 rpm. Control experiments were performed using the same set up but with only buffer in the cell. The heat effects for the control run were small and constant so the average heat of injection was subtracted from the results of the sample run. The experimental data were analysed with MicroCal ITC Origin and were fitted to a single-site binding equation.
Expression, purification and crystallization of influenza A and B cap-midlink double domains
Residues 247–536 from A/Victoria/3/1975(H3N2) were cloned in pETM11 (EMBL) and expressed in Escherichia coli Rosetta 2 (Novagen) at 16°C overnight after induction with 0.25 mM IPTG. Cells were collected by centrifugation at 6000 g, for 15 min at 4°C. Pellets were re-suspended and sonicated in lysis buffer (20 mM Tris pH 8, 300 mM NaCl, 5% (v/v) glycerol, 5 mM beta-mercapto-ethanol) containing CompleteTM protease inhibitor cocktail (Roche). The protein was purified at room temperature by Ni2+ affinity chromatography (Ni-NTA, Qiagen). Nickel elutions were dialyzed against 20 mM Tris pH 8, 200 mM NaCl, 5% (v/v) glycerol, 2 mM beta-mercapto-ethanol and the histidine tag removed by Tobacco Etch Virus (TEV) protease. The resulting untagged proteins were further purified by a second passage on Ni2+ affinity chromatography and gel filtration chromatography using a Superdex 75 column (GE-Healthcare) (20 mM Tris pH 8, 200 mM NaCl, 5% (v/v) glycerol, 0.5 mM beta-mercapto-ethanol). The N510T mutation was introduced into the A/H3N2 double domain expression construct via site-directed mutagenesis using the primer pair (with mutated nucleotides in lower case): N510T_H3N2_fwd (5′- GATCA GCGTG GTAcT GTTCT GCTGA GC -3′) x N510T_H3N2_rev (5′-GCTCA GCAGA ACAgT ACCAC GCTGA TC-3′). The protein was expressed and purified as described above.
Crystallization trials were performed with a Cartesian robot. Crystals (space-group P2221) were obtained of VX-787N with A/H3N2 double domain at 15 mg/ml protein mixed with 5 mM VX-787N in 0.2 M ammonium sulphate, 20% PEG3350. The crystals grew in 3 days at 20°C and were frozen with mother liquor plus 20% (v/v) glycerol. Crystals (space-group P1) were obtained of VX-787 (Pimodivir, purchased from MedChemExpress) with A/H3N2 double domain at 16 mg/ml protein mixed with 2 mM VX-787 in 0.7 M sodium formate and 0.1 M MES pH 6. The crystals grew in 3 days at 20°C and were frozen with mother liquor supplemented by 35% (v/v) glycerol. Crystals of VX-787N with A/H5N1 cap-binding domain (424-loop truncation variant purified as described (13)) were grown by mixing 1 μl of protein solution at 13 mg/ml and 2 mM VX-787N with an equal volume of solution containing 0.2 M ammonium phosphate bibasic (pH 8) and 20% PEG 3350. Crystals were frozen in liquid nitrogen in the presence of 20% (v/v) glycerol as cryoprotectant.
The equivalent double-domain construct was made from residues 249–540 of influenza B/Memphis/13/03 PB2 subunit and purified in a similar way but using buffers containing 50 mM Tris/HCl pH 7.5 and 10% (v/v) glycerol. Crystals were obtained with influenza B double domain at 15 mg/ml mixed with 5 mM of VX-787N in 0.1 M PTCP and 25% (w/v) PEG1500 at 4°C and were cryoprotected with 20% (v/v) glycerol. Diffraction data were collected on ESRF beamline ID30A1 and integrated with XDS (18).
The structures were solved by molecular replacement with PHASER (19) using previously determined influenza A or B cap-binding and midlink domain structures and refined using REFMAC (20) and PHENIX (27). See Table 1 and Supplementary Table S1 for crystallographic statistics and Supp. Figures 7A and B for omit electron density for the VX-787N ligand for both structures. For the A/Victoria/H3N2 structure one of the three molecules in the asymmetric unit is less well-ordered and refinement was done using local NCS and TLS parameters.
RESULTS
Structure determination of influenza polymerase-cap complexes
The original crystal form of the promoter bound bat influenza A/H17N10 polymerase exhibited an orientation of the cap-binding domain compatible with the first step of cap-snatching where capped RNA bound in the PB2 cap-binding domain can bridge across to the PA endonuclease for cleavage (‘cleavage’ conformation) (17). Co-crystallization with m7GTP yielded a structure at 2.9 Å resolution (Figure 1A, Table 1) with clear difference density for the ligand (Supplementary Figure S1A). The cap-analogue binds as to the isolated cap-binding domain (12) with no contacts being made to the midlink domain. To gain more insight into capped RNA binding, we next attempted to determine a crystal structure of the cap-dependent transcription initiation state. This was done by co-crystallizing bat influenza A polymerase with the vRNA promoter (5′ nt 1–16, 3′ nt 1–18) and a capped 12-mer primer ending in AG-3′, which could in principle make two base-pairs with the terminal 3′-UC of the template (Supplementary Figure S1D). A new monoclinic crystal form, diffracting to 3.9 Å resolution, was obtained, with two polymerase complexes in the asymmetric unit (Table 1). Similar to the original bat influenza A polymerase crystal form, the new crystals were stabilized by intermolecular vRNA–vRNA contacts made by the distal base-paired regions of the promoter (17). The novel feature of this structure is that the cap-binding domain has rotated ∼60° into the presumed ‘priming’ configuration (Figure 1B), whereas this position of the cap-binding domain had only previously been observed with promoter bound influenza B/Memphis polymerase (1). The cap-proximal part of the primer (m7GpppAA) is clearly visible bound to the cap-binding and midlink domains (Supplementary Figure S1B) although the rest of the primer has no electron density, due to either disorder or degradation. As in the original bat influenza A polymerase structure, the template 3′ end is only visible beyond nucleotide 5 and consequently the anticipated base-pairing of the 3′ end of the primer to the 3′ extremity of the template is not observed.
Figure 1.

Structure of bat influenza A polymerase with bound m7GTP or capped RNA. (A) Overview of the structure of bat A/H17N10 polymerase with bound m7GTP (yellow spheres). Subunits and domains are colored and annotated. The bound vRNA promoter is also indicated. (B) Structure of the bat A/H17N10 polymerase with rotated cap-binding domain and bound capped RNA (m7GTP moiety as yellow spheres, subsequent bases as blue spheres). Color scheme is as in A. (C) Close up of capped RNA binding site showing key interactions with conserved residues in the cap-binding and midlink domains (yellow sticks). Putative hydrogen bonds are shown as green dotted lines. Colors of domains as in A.
In parallel, co-crystallization attempts with influenza B polymerase and a capped 13-mer primer ending in AGC-3′ yielded the structure of a very similar complex at 3.8 Å resolution (24). This not only showed the cap-proximal part of the primer (m7GpppAAU, Supplementary Figure S1C) but also the template 3′ end entering the active site. A previous report of this influenza B polymerase structure (PDB:5MSG, (24)) focussed on the path of the template 3′ end through the template entrance tunnel, whereas here we focus on the very similar mode of binding observed for the capped primer to both bat influenza A or influenza B polymerase. Although both structures are only of moderate resolution, the electron density after map sharpening is reasonably good, particularly for the bat influenza A polymerase (Supplementary Figure S1B).
Figure 1C shows the observed cap-proximal part of the capped primer and its environment when bound to heterotrimeric bat influenza A polymerase. Both the cap-binding domain (PB2 residues 318–484) and the midlink domain (PB2 residues 251–322 and 493–536) are well ordered. This includes the 424-loop (PB2 residues 420–427) and residues 485–492 which connect the cap-binding domain to the second half of the midlink domain, regions that were disordered in the previous structures of the bat heterotrimer in the ‘cleavage’ conformation. The 424-loop is stabilized in a particular conformation with Arg423 at its extremity making a salt bridge with PB1 residue Glu277 (both conserved residues in all A/B strains) and Asn425 with main-chain of PB1 Glu267. The m7G moiety of the cap interacts only with the cap-binding domain as previously observed (12) with Phe323, Phe357 (a histidine in human/avian A strains) and Phe404 making stacking interactions and Glu361 and Lys376 making base-specific interactions. Asn429 hydrogen bonds to both the alpha- and gamma-phosphates of the triphosphate. Moreover, new interactions are observed between the ribose and tri-phosphate moiety and the adjacent mid domain, notably with residues from its central long helix (α14). Gln306 (conserved in all influenza A strains), contacts the m7G ribose hydroxyls and Arg264 (conserved in all influenza strains) as well as Ser519 (conserved in all A/B strains) contact the beta-phosphate. The first two bases of the capped RNA, which are both adenines in the RNA used here, are splayed out in separate pockets and stack respectively with Ile260 and Tyr432. Residue 432 is a conserved aromatic residue (tyrosine in bat A/H17N10 and influenza B and histidine in human/avian A strains) whereas Ile260 is conserved in all A/B strains.
A very similar conformation of the capped RNA is observed in the previously published equivalent structure with influenza B polymerase (PDB: 5MSG, (24)). Between influenza A and B the protein-RNA interactions involving Ile262 (Ile260 in influenza A), Arg266 (Arg264), Tyr434 (Tyr432) and S519 (S520) are conserved. However other interactions are not conserved (e.g. Gln306 becomes Val308 in influenza B, Asn429 becomes Ser431) but are compensated by alternative interactions (e.g. Gln259 and Ser524 interact with the phosphate and ribose of the second base). The similar and distinct interactions between bat A/H17N10 and influenza B polymerases with the capped RNA are shown in Figures 1C and Supplementary Figure S2. These results, combined with the high sequence identity, particularly in functional regions, of bat A/H17N10 and avian/human A strains suggests that all influenza strains bind capped RNA in the same fashion.
Functional analysis of conserved residues interacting with the capped RNA
We selected a number of the newly identified and functionally conserved PB2 residues interacting with the capped RNA for mutational analysis by in vitro transcription assays, using bat A/H17N10 polymerase and/or minigenome assays in cells using human A/Victoria/3/75(H3N2) polymerase. Residues chosen were the base-stacking aromatic residues Tyr/His432 and Ile260, ribose interacting Gln306, triphosphate interacting Arg264, and Arg423, which stabilizes the conformation of the 424-loop. Mutation of these might be expected to affect capped RNA in the priming configuration and hence transcription efficiency.
Mutations I260A, R264A, Q306A, R423A and H432A were introduced singly into plasmids encoding the PB2 subunit of A/Victoria(H3N2) and used, together with plasmids for PA, PB1 and NP, in minigenome assays in HEK293T mammalian cells, with a luciferase reporter that can only be expressed by an actively transcribing viral ribonucleoprotein (RNP) complex (Figure 2). The results show that the R264A mutation completely abolished RNP biological activity, the I260A and H432A mutations have slightly reduced or comparable to wild-type polymerase activity and the Q306A and R423A mutations consistently exhibit increased polymerase activity (Figure 2A). We confirmed by Western blot that the PB2 mutant subunits were well expressed (Figure 2A). To verify and extend these results we used primer extension to quantify the amounts of viral mRNA, vRNA and cRNA in the minigenome system (Figure 2B–E). The results for viral mRNA levels confirm the absence of viral mRNA for the R264A mutation and show decreases for I260A and H432A (Figure 2D) but do not confirm the enhanced activity of the Q306A and R423A mutants. The vRNA levels for each mutant were little changed compared to wild-type (Figure 2C), whereas the cRNA levels, which are low in these experiments, show slight decreases paralleling the mRNA levels (Figure 2E), suggesting some effect of the mutations on replication.
Figure 2.

Biological activity of wild-type and PB2 mutated recombinant RNPs. HEK293T cells were co-transfected with plasmids expressing PB1, PA, NP and either WT or mutant PB2, a plasmid encoding Renilla luciferase and a plasmid encoding Firefly luciferase in negative polarity, flanked by the non-translated regions of influenza NP segment. Plasmid expressing PB2 was omitted as a negative control. (A) At 20 h post-transfection, luciferase accumulation was determined. Values were normalized to Renilla expression, and the activity of the WT RNP was set to 100% (mean ± SEM; N = 9–18; **** P < 0.0001 by Student's t test compared to WT RNP activity). At the bottom is shown confirmation of WT or mutant PB2 expression in human HEK cells by western blotting using β-actin as a loading control. (B) Representative gel of primer extension assay showing the different RNA levels produced by WT and mutant polymerases in the minigenome assay. (C-E). Viral RNA accumulation measured by primer extension assay. 24 h post-transfection, total RNA was isolated and analysed by luciferase gene-specific primer extension. A primer specific for 5S rRNA was used for normalization (mean ± SEM; N = 6; ***P < 0.001 by Student's t-test compared to WT RNP activity).
The single point mutants of PB2 showing significant decrease of mRNA synthesis in cells, I260A, R264A and H432A were expressed in the context of the bat A/H17N10 polymerase heterotrimer in insect cells and purified (Y432A in the case of A/H17N10). They were analysed using a recently reported fluorescence based RNA synthesis assay, which can monitor the rate of synthesis of full-length transcripts in a capped RNA primed reaction (24). Briefly, this assay uses a model template comprising the 3′ vRNA with a fluorescent tag at its 5′ end. Incubation with nucleotide triphosphates and a capped 12-mer RNA as primer (which does not require cleavage by the endonuclease) leads to cap-dependent transcription. The reaction is quenched in high salt to dissociate the RNA from the protein, whereupon the transcript can anneal with the complementary template leading to a double-stranded environment of the fluorophore and an enhanced fluorescence anisotropy signal. Mutations which affect the binding of the capped RNA in the priming configuration might be expected to correspondingly affect the KM for capped RNA and the kcat/KM in the overall transcription assay. These kinetic parameters were derived from fitting RNA synthesis time courses measured at variable capped RNA concentrations (Figure 3, Supplementary Figure S3). The three mutants Y432A, I260A, R264A show respectively increases of 4, 8 and 17-fold in the KM compared to wild-type, whereas Vmax is less affected (Figure 3B). The most detrimental mutation was R264A with a 17-fold increase of KM and 21-fold decrease of kcat/KM.
Figure 3.

In vitro cap-dependent transcription assay. (A) Vmax of capped RNA primed RNA synthesis of A/H17N10 wild-type influenza polymerase (black) and PB2-variants R264A (pink), I260A (purple) and Y432A (orange) at varying concentrations of capped RNA primer (see Materials and Methods and Supplementary Figure S3). (B) Quantification of derived kinetic parameters for capped RNA primed transcription. Mean values and corresponding standard deviation of duplicate experiments are indicated.
Comparison of different configurations of the cap-binding and midlink domains
The particular juxtaposition of the cap-binding and midlink domains we observe in the full polymerase complex with capped primer (‘priming’ configuration, Figure 4A) is quite different from the alternative ‘apo’ configuration of these two domains (Figure 4B). The latter configuration is found in the context of the isolated C-terminal two-thirds of A/H5N1 PB2 (residues 247–736, PDB:5FMM, (16)), the cRNA 5′ end bound influenza B polymerase structure (PDB:5EPI, (16)) and the RNA-free influenza C structure (PDB:5D98, (28)). In the ‘priming’ configuration the midlink domain is rotated by ∼77° compared to the ‘apo’ configuration (after superposition by the cap-binding domain) and the 424-loop of the cap-binding domain is stabilized in different configurations in the two structures (Figure 4A,B). For instance, PB2 Arg423 interacts with PB1 Glu277 (Figure 1C) or with PB2 Glu520 (Figure 4B) in respectively the ‘priming’ and ‘apo’ configurations. Distinct again is the cap-binding and midlink domain arrangement seen in the putative ‘cleavage’ configuration as observed in the original bat polymerase structure (PDB:4WSB) and the equivalent m7GTP bound structure described here. Figure 4C shows a comparison of the three cap-midlink double domain configurations. To further investigate the preferred configurations we made a minimal two domain construct comprising only the cap-binding and midlink domains (PB2 residues 248–536) for the A/H3N2 and A/H5N1 strains. We were unable to obtain crystals of this construct with capped RNA, either in the ‘priming’ configuration or the ‘apo configuration’. This suggests firstly that the ‘priming’ configuration with bound capped RNA is only stable in the context of the full polymerase. Secondly, as discussed previously (16), the ‘apo’ configuration allows m7GTP to bind, albeit in an unusual bent conformation, but not the physiological ligand which is capped RNA. This is because beyond the m7G cap moiety there would be a steric clash of the capped RNA with residues 511–515 of the midlink domain. For this reason the ‘apo’ configuration appears to be not directly relevant for cap-snatching or transcription (16,28).
Figure 4.

Three different configurations of the cap-midlink double domain. (A) ‘Priming’ conformation. The juxtaposition of the midlink (magenta) and cap-binding (orange) domains as observed in the H17N10 capped RNA (blue sticks) complex (Figure 1B). The location of the three VX-787 resistance mutations, Gln306, Ser324 and Asn510 is shown as well as Arg423 in the 424-loop which makes a salt bridge with PB1 Glu277 (not shown)(compare (B) and see text). (B) ‘Apo’ conformation of A/H3N2 midlink (pink) and cap-binding (orange) domains as observed in the double domain structure with bound VX-787N (red sticks). The location of the three VX-787 resistance mutations, Gln306, Ser324 and Asn510 is shown as well as the salt-bridge of Arg423 in the 424-loop with Glu520 (compare (A) and see text). A very similar configuration of the two domains has already been reported for A/H5N1 with bound m7GTP (PDB: 5FMM, root-mean-square deviation of 0.95 Å for 276 superposed Cα atoms) and for influenza B polymerase (PDB: 5EPI) (16). (C) Superposition by means of the cap-binding domain (orange) of the ‘cleavage’, ‘priming’ and ‘apo’ configurations of the midlink (respectively wheat, magenta and pink) and cap-binding double domain with the respective rotation angles marked.
Mechanism of inhibition by cap-binding inhibitors
Recently 7-azaindole substituted compounds have been shown to protect mice against lethal influenza A infection with high potency, the most well-studied being VX-787 (now known as JNJ63623872 or Pimodivir, Supplementary Figure S5A) (8–11). VX-787 binds the isolated influenza A PB2 cap-binding domain with an affinity of around 24 nM, some 60 times tighter than for m7GTP, and the EC50 against various influenza A strains varies from 0.1 to 3.2 nM (9). Much of the potency of VX-787 derives from the excellent pi-stacking of the pyrimidine ring together with its exocyclic fluorine on Phe323 in the cap-binding site, as well as that of the azaindole between His357 and Phe404 and the packing of the cyclohexyl group with Phe325, as observed in a co-crystal structure (PDB: 4P1U) (8,9). Based on these observations, the mechanism of inhibition of VX-787 has been hitherto presumed to be direct competition with capped RNA for the PB2 cap-binding site.
Here we examine further the mechanism of action of VX-787, but have used a close analogue denoted VX-787N. VX-787N is nearly identical to VX-787 but is an azaindazol instead of an azaindole, thus having an extra nitrogen instead of a carbon in the head group (Supplementary Figure S5B). A 1 Å resolution structure of VX-787N bound to A/H5N1 cap-binding domain (Supplementary Figure S5C, D and Supplementary Table S1) shows that the mode of binding of VX-787 and VX-787N are virtually identical except for a perturbation in the position of Arg332 to exploit the enhanced hydrogen-bonding capacity of the azaindazol (Supplementary Figure S5D-F). Consistent with this, VX-787N was found to have an IC50 of 0.66 nM in a cytopathic effect (CPE) reduction assay performed as described (29).
In agreement with the proposed mechanism of action, VX-787N did not interfere with ApG-primed RNA synthesis from the vRNA promoter, which is independent of capped RNA primer binding (Figure 5A) suggesting that VX-787N does not target intrinsic RNA synthesis activity. In contrast and as expected, VX-787N strongly inhibited capped RNA primed RNA synthesis at otherwise identical conditions (Figure 5A), although the experimental design did not allow accurate quantification of this inhibition. To quantify binding of VX-787N to the full-length heterotrimeric bat A/H17N10 polymerase we performed a competition experiment using a fluorescent derivative of the compound with the fluorophore FAM linked to the terminal carboxyl group ((VX-787N-FAM, Supplementary Figure S6A). We used the fluorescence anisotropy signal to measure the fraction of bound VX-787N-FAM as a function of polymerase concentration for a series of VX-787N concentrations (Supplementary Figure S6B). This allowed estimation of the Ki for VX-787N as 13 nM for bat A/H17N10 polymerase (see Materials and Methods and Supplementary Figure S6C), comparable to the reported IC50 for inhibition of viral growth (9,30).
These results are consistent with VX-787/VX-787N inhibiting cap-dependent transcription by binding tightly in the cap-binding site. However as emphasized above, the cap-binding domain is in at least three different configurations with respect to the midlink domain (Figure 4C) and the question arises whether VX-787/VX-787N preferentially binds to either of these and which is most relevant for inhibition. To investigate this point, we first considered the reported resistance mutants generated upon passaging influenza A/PR/8/34/H1N1 virus in the presence of VX-787 (9). Mutations at four sites in PB2 were found to induce resistance: F404Y (257 fold increase in EC50 compared to wild-type), S324I/N/R (respectively 157-, 127- and 63-fold), Q306H (186-fold) and N510T (133-fold) (9), although the fitness of the mutant viruses remains to be assessed. The F404Y mutation can be rationalized by the predicted steric clash of the additional OH with the ligand and will not be considered further. However Ser324, whilst in the cap-binding domain, is solvent exposed and orientated away from the cap-binding site (and hence not in contact with the inhibitor) in the co-crystal structures with the cap-binding domain and, intriguingly, Gln306 and Asn510 are not even in the cap-binding domain but in the mid and link domains respectively. In the bat A/H17N10 capped RNA structure in the ‘priming’ configuration, as discussed above, Q306 interacts with the ribose of the m7G (even though this interaction is not essential for transcription as shown by the Q306A mutation), whereas Glu510, which is the bat equivalent of Asn510 in avian/human influenza A, is remote (∼16 Å) from the cap-binding site (Figure 4A).
Structural analysis of inhibitor binding to the influenza A and B cap-midlink double domain
To investigate further the structural basis for resistance, we co-crystallized VX-787N with the A/Victoria(H3N2) cap-midlink double domain (residues 247–536, henceforth called the double-domain), yielding crystals which diffracted to 3 Å resolution, with three complexes in the asymmetric unit (Table 1, Supplementary Figure S7A). The H3N2 double-domain conformation (Figure 4B) is very similar to that seen previously in the larger A/H5N1 PB2 construct (residues 247–736, PDB:5FMM, (16) and the interactions of VX-787N with the cap-binding domain are as described above. However, additional interactions of VX-787N to the midlink domain effectively occlude the otherwise solvent exposed part of the compound. These interactions include hydrogen bonds to the main-chain carbonyl and amide of Val511 by respectively the amide linking the pyrimidine and cyclohexyl groups and the carboxyl-group of the compound as well as van der Waals contacts with Val511 and Asn510 (Figure 6A and B). We also determined the equivalent A/Victoria(H3N2) double domain crystal structure with VX-787 (Pimodivir) instead of VX-787N. The crystals diffracted to 2.7 Å resolution, with four complexes in the asymmetric unit (Supplementary Table S1). As expected, the structure is overall very similar to that with VX-787N (RMSD for all Cα atoms 0.8 Å). By ITC we measured a KD of 2.4 ± 1.3 nM for VX-787 to the A/H3N2 cap-midlink double domain (Figure 5B), compared to 24 nM reported previously for the cap-binding domain alone (9).
Figure 6.

Structure of VX-787N bound to influenza A and B cap-midlink double domains. (A) Close up of the interactions in the vicinity of VX-787N (red) bound at the interface between the A/H3N2 cap-binding (orange) and midlink (magenta) domains. Hydrogen bonds are represented by green dotted lines and the red-dotted line highlights the close contact (3.2 Å) of the Cβ atom of Asn510 with the exocyclic fluorine F24 of the compound. In the domain interface, Glu517 plays a key role, hydrogen bonding to Ser324*, Gly326, Gly327 and His 432. Starred residues correspond to sites of resistant mutants. Other interacting residues from the cap-binding domain are omitted for clarity but shown in (B). (B) Schematic diagram derived from (A) showing hydrogen bonds (green) and van der Waals contacts (red arcs) with bound VX-787N as calculated using LIGPLOT+ (31). Note that the main chain of Val511 makes two hydrogen bonds to the compound. (C) Close up of the interactions in the vicinity of VX-787N (red) bound at the interface between the B/Memphis cap-binding (light orange) and midlink (light violet) domains. For clarity, the orientation is slightly different from in (A). Hydrogen bonds are represented by green dotted lines and the red-dotted line highlights the close contacts of the exocyclic fluorine F24 of the compound. In the domain interface, Asp518 (equivalent to Glu517 in influenza A) plays a key role, hydrogen bonding to Gly328 (Gly326) and Tyr434 (His432). Other interacting residues from the cap-binding domain are omitted for clarity but shown in (B). (D) Schematic diagram derived from on the B/Memphis cap-midlink structure showing hydrogen bonds (green) and van der Waals contacts (red arcs) with bound VX-787N as calculated using LIGPLOT+ (31).
It has been reported that VX-787 has no potency against influenza B strains (8,9). This is consistent with the 200-fold increase in KD of VX-787N-FAM measured for the B/Memphis polymerase (∼1.2 μM) compared to the bat A/H17N10 polymerase (∼ 6 nM) (Figure 5C). This is consistent with a KD of 1.2 μM ± 0.3 for VX-787 to the B/Memphis cap-midlink double domain measured by ITC (Figure 5D). To investigate the structural basis for the reduced affinity to influenza B polymerase, we co-crystallized the inhibitor with the B/Memphis double-domain construct (PB2 residues 249–540). Co-crystallization was successful despite the lower affinity of VX-787N for influenza B polymerase yielding, for the double-domain, crystals diffracting to 2.05 Å resolution (Table 1, Supp. Figure 7B). In this crystal form, the cap-binding domain, the inhibitor and part of the midlink domain interfacing them are well ordered but the rest of the midlink domain is poorly visible.
VX-787N binds to influenza B cap-binding domain similarly to influenza A, with the azaindazole moiety stacking between Trp359 (His 357 in influenza A) and Phe406 (Phe404) and hydrogen bonding to conserved Glu363 (Glu361), Lys378 (Lys376) and via a water to Arg334 (Arg332) (Figure 6C and D). The carboxyl-group of VX-787N hydrogen bonds to the amide in the Trp359 side-chain and makes a salt-bridge with Lys341. In the influenza B structure, the midlink domain is slightly shifted compared to influenza A (Supplementary Figure S7C). Consequently, the exocyclic fluorine of VX-787N is in contact with the carbonyl oxygen of Val512, whereas the equivalent oxygen hydrogen bonds to the VX-787N amide in influenza A (Figure 6). There is a similar, but less extensive, inter-domain interaction network mediated by Asp518 (Glu517 in A/H3N2), Gly328 (Gly326) and Tyr434 (His432). However, the most significant difference between VX-787N interactions with A and B polymerases is that in influenza B, Gln325 replaces Phe323 thus eliminating the strong pi stacking with the pyrimidine ring of the inhibitor. Indeed this substitution leaves a void under this part of the compound enclosed by Gln325, Leu332, Phe365 and Phe406 (Supplementary Figure S7C).
DISCUSSION
Based on our original structures of bat influenza A and human influenza B polymerases we hypothesized that the cap-binding domain could first be orientated to direct a capped RNA towards the endonuclease active site for cleavage and then rotate by around 60° to direct the capped primer into the polymerase active site to initiate transcription (1). Here, we confirm by co-crystallization of bat influenza A polymerase with either m7GTP or a capped 12-mer RNA, that both ‘cleavage’ and ‘priming’ states are indeed accessible to one and the same polymerase. In the ‘priming’ state, the capped primer binds not only to the cap-binding domain but also makes significant interactions with the midlink domain. Furthermore, both bat influenza A and influenza B polymerases bind capped RNA in essentially the same way (compare Figure 1C and Supplementary Figure S2A). We had hoped to visualize the putative transcription initiation state, with the 3′ end of the primer base-pairing with the template in the polymerase active site (Supplementary Figure S1B). However in both the A/H17N10 and B/Memphis structures, only the cap and 2–3 cap-proximal bases are visible in the electron density and whereas in the B/Memphis structure the 3′ end of the template partially occupies the active site (24), this is not the case in the bat A/H17N10 structure. There are several possible reasons for this including degradation of the capped RNA by the endonuclease, or, as discussed previously, the need for a conformational change to accommodate base-pairs in the active site, which should be correlated with the breaking of the distal base-paired region of the promoter (24). However, in both the A/H17N10 and B/Memphis co-crystals with capped RNA, the base-paired region of the promoter is involved in critical crystal contacts.
We tested the importance of several of the newly observed interactions with the capped-primer by in cell and in vitro functional assays. These confirm a critical role in cap-dependent transcription of the highly conserved PB2 residue Arg264, which interacts with the triphosphate of the cap in both the bat influenza A and B polymerase complexes. This highlights the fact that the triphosphate conformations observed in various co-crystal structures of the cap-binding domain alone with the cap-analogue m7GTP do not necessarily reflect the physiological relevant situation when capped RNA is bound to the full-length polymerase. Individual mutation of the residues upon which the first two bases of the capped RNA stack, Ile260 and Tyr/His432 respectively, diminishes mRNA synthesis in the minigenome assay (Figure 2D) and decreases the catalytic efficiency of cap-dependent transcription in vitro (Figure 3), confirming the importance of these interactions. In the luciferase assay, the Q306A and R423A mutations appear to have increased polymerase activity although this is not reflected in the primer extension experiments. The reason for this discrepancy is not clear.
In addition to the ‘cleavage’ and ‘priming’ configurations of the polymerase, which differ principally by the rotational orientation of the cap-binding domain, a significantly different third conformation has been described for both influenza B (16) and C (28) polymerases (Figure 4). In these structures, there is a radical repacking of the flexibly linked midlink, cap-binding, 627 and NLS domains of PB2 (16) and as the promoter is only partially present or absent altogether, we denote this conformation as ‘apo’. We found that for influenza A/H5N1 the isolated C-terminal region of PB2 (including the cap-binding, midlink and 627 domains) preferentially takes up the ‘apo’ configuration and small-angle scattering and cross-link mass-spectroscopy suggests that in solution there is an equilibrium between different states of the polymerase (16). Here we show that the isolated A/Victoria(H3N2) cap-midlink double domain, when co-crystallized with the potent cap-binding inhibitors VX-787 or VX-787N, also takes up the ‘apo’ configuration (Figure 4B). In this configuration, the midlink and cap-binding domains are juxtaposed in a more closed configuration that is stabilized by several inter-domain interactions. For instance, Gln306 and Glu517 of the midlink domain make a hydrogen bond network with side-chains of Ser324, and His432 (which in the ‘priming’ configuration is stacked on the second RNA base) and main-chain amides of Gly326 and Gly327 (Figure 6A) (an identical interaction network is observed in the A/H5N1 PB2-C structure (PDB:5FMM)). This VX-787N/VX-787 bound double domain structures also reveal that the inhibitor not only makes the previously described interactions with cap-binding domain but additional interactions with the main-chain of the midlink domain in the region of Val511 as well as a close contact with Asn510 (Figure 6A and B). ITC measurements confirm that VX-787 binds with considerably higher affinity to the cap-midlink double domain than to the cap-binding domain alone (Figure 5).
The above observations lead to the suggestion that VX-787/VX-787N could partially act by stabilizing the polymerase in the ‘apo’ conformation. This mode of inhibition would not involve a direct competition with capped RNA, which cannot bind to the ‘apo’ conformation, but a shift of equilibrium towards a transcriptionally inactive state of the polymerase. Support for this hypothesis is found in the location and role of some of the residues whose mutation was selected to impart resistance to VX-787, S324I/N/R, Q306H (186-fold) and N510T (133-fold) (9). As just discussed, each of Ser324, Gln306 and Asn510 plays a role in stabilizing the inhibitor bound ‘apo’ conformation of the cap-midlink domain. Since the Cβ of Asn510 is in direct contact with the exocyclic fluorine of the VX-787 pyrimidine ring (red-dotted line in Figure 6A), the substitution of Asn510 by Thr510, which is branched at the Cβ atom, would cause a steric clash and thus weaken the affinity to VX-787. In contrast, in the ‘priming’ configuration, residue 510 (Glu510 in bat polymerase) is solvent exposed and remote (∼16 Å) from the cap-binding site and its mutation is thus unlikely to affect binding of either capped RNA or VX-787 in the cap-binding site (Figure 4A). That the mutation N510T allows the virus to escape VX-787 inhibition thus suggests that the indirect mode of action whereby the compound stabilizes a transcriptionally inactive state of the polymerase is significant. This is supported by a four-fold higher KD of VX-787 to the N510T mutant compared to the wild-type H3N2 cap-midlink double domain (Figure 5B and E). Does this explanation also hold for the other escape mutants Q306H and S324I/N/R? In the cap-midlink inter-domain hydrogen bond network involving Gln306 and Ser324, substitution of the buried Ser324 by more bulky side-chains, particularly isoleucine or arginine, would likely disrupt the interface. This would destabilize the close interaction between the two domains in the ‘apo’ conformation and disfavour formation of the extra interactions provided by Asn510 and Val511 to VX-787. In the capped RNA structure with bat polymerase, Ser324 is on the other side of the first base of the capped RNA from Ile260, solvent exposed and orientated away from the cap-binding site (Figure 4A). Indeed superposing VX-787N bound in the cap-binding domain into the ‘priming’ configuration shows that it is at closest ∼7 Å from the side-chain of Ser324 (Supplementary Figure S7D). Thus in the ‘priming’ configuration substitutions of Ser324 to Ile, Asn or Arg would not appear to affect either capped RNA or inhibitor binding. Indeed Ser324 is equivalent to Arg326 in influenza B polymerase, and therefore the influenza B complex with capped RNA directly shows how an arginine can be accommodated at this position without affecting capped RNA binding. The Q306H escape mutant is less straightforward to explain since a histidine is of a similar size and can make similar interactions as a glutamine and would not be expected to significantly disrupt the interface between the cap-binding and midlink domains (Figure 6A). However if VX-787 is modelled into the ‘priming’ conformation there would be a close contact between the side-chain of Gln306 (which normally interacts with the ribose of the cap, Figures 1C and 4A) and the exocyclic fluorine of the VX-787N pyrimidine ring which protrudes further out than the ribose (Supplementary Figure S7D). Perhaps mutation of flexible glutamine to the more constrained histidine side-chain disfavours VX-787N binding without significantly affecting capped RNA binding.
The preceding discussion suggests that VX-787, by binding tightly in the influenza A cap-binding site, can compete directly with capped RNA binding in the ‘priming’ or ‘cleavage’ configurations, or compete indirectly by stabilizing the transcriptionally inactive ‘apo’ configuration of the cap-midlink domains, to which the inhibitor binds with higher affinity than to the cap-binding domain alone. Supporting evidence for the latter hypothesis comes from a comparison of the different roles in the ‘priming’ and ‘apo’ states of certain residues whose mutation imparts resistance. In the case of influenza B polymerase, which is naturally resistant to VX-787 due to a 200 fold reduction in affinity compared to influenza A polymerase (Figure 5C and D), we also obtained a structure with the compound bound to the influenza B cap-midlink double domain. This resembles that with influenza A, although the two domains are juxtaposed slightly differently (Figure 6), but suggests that the major loss in affinity derives from the substitution of Gln325 in influenza B instead of Phe323 in influenza A, thus removing the strong pi-stacking with the pyrimidine ring of the compound. Indeed this substitution leaves a void under this part of the compound (Supplementary Figure S7C) which could perhaps be filled with appropriately orientated modifications to the original compound to enhance affinity to influenza B polymerase.
AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures are deposited with the wwPDB under accession numbers:
6EVK A/H17N10 polymerase with vRNA promoter and m7GTP
6EVJ A/H17N10 polymerase with vRNA promoter and capped RNA primer
6EUY A/Victoria(H3N2) PB2 cap-midlink double domain with VX-787N
6EUV A/Victoria(H3N2) PB2 cap-midlink double domain with VX-787
6EUX B/Memphis PB2 cap-midlink double domain with VX-787N
6EUW A/H5N1 PB2 cap-binding domain with VX-787N
Supplementary Material
ACKNOWLEDGEMENTS
We thank the staff of the ESRF-EMBL Joint Structural Biology Group, in particular Matthew Bowler, for access to and help on ESRF beamlines. We also acknowledge the EMBL Eukaryotic Expression Facility, the EMBL high-throughput crystallization facility (HTX) and the Partnership for Structural Biology (PSB) biophysical platform, for access and help. We thank Helmut Buschmann, Oliver Szolar and Ulrike Uhrig for help in acquiring the compounds VX-787N, VX-787N-FAM and VX-787.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council Advanced Grant V-RNA [322586 to S.C.]; Grenoble Instruct Center [ISBG: UMS 3518 CNRS-CEA-UJF-EMBL] within the Grenoble Partnership for Structural Biology (PSB), with support from FRISBI [ANR-10-INSB-05-02]; GRAL [ANR-10-LABX-49-01]. Funding for open access charge: EMBL.
Conflict of interest statement. None declared.
REFERENCES
- 1. Reich S., Guilligay D., Pflug A., Malet H., Berger I., Crepin T., Hart D., Lunardi T., Nanao M., Ruigrok R.W. et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature. 2014; 516:361–366. [DOI] [PubMed] [Google Scholar]
- 2. Pflug A., Lukarska M., Resa-Infante P., Reich S., Cusack S.. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017; 234:103–117. [DOI] [PubMed] [Google Scholar]
- 3. Hagen M., Tiley L., Chung T.D., Krystal M.. The role of template-primer interactions in cleavage and initiation by the influenza virus polymerase. J. Gen. Virol. 1995; 76:603–611. [DOI] [PubMed] [Google Scholar]
- 4. Geerts-Dimitriadou C., Zwart M.P., Goldbach R., Kormelink R.. Base-pairing promotes leader selection to prime in vitro influenza genome transcription. Virology. 2011; 409:17–26. [DOI] [PubMed] [Google Scholar]
- 5. Robertson J.S., Schubert M., Lazzarini R.A.. Polyadenylation sites for influenza virus mRNA. J. Virol. 1981; 38:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poon L.L., Pritlove D.C., Fodor E., Brownlee G.G.. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J. Virol. 1999; 73:3473–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stevaert A., Naesens L.. The influenza virus polymerase complex: an update on its structure, functions, and significance for antiviral drug design. Med. Res. Rev. 2016; 36:1127–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clark M.P., Ledeboer M.W., Davies I., Byrn R.A., Jones S.M., Perola E., Tsai A., Jacobs M., Nti-Addae K., Bandarage U.K. et al. Discovery of a novel, first-in-class, orally bioavailable azaindole inhibitor (VX-787) of influenza PB2. J. Med. Chem. 2014; 57:6668–6678. [DOI] [PubMed] [Google Scholar]
- 9. Byrn R.A., Jones S.M., Bennett H.B., Bral C., Clark M.P., Jacobs M.D., Kwong A.D., Ledeboer M.W., Leeman J.R., McNeil C.F. et al. Preclinical activity of VX-787, a first-in-class, orally bioavailable inhibitor of the influenza virus polymerase PB2 subunit. Antimicrob. Agents Chemother. 2015; 59:1569–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Farmer L.J., Clark M.P., Boyd M.J., Perola E., Jones S.M., Tsai A., Jacobs M.D., Bandarage U.K., Ledeboer M.W., Wang T. et al. Discovery of novel, orally bioavailable beta-amino acid azaindole inhibitors of influenza PB2. ACS Med. Chem. Lett. 2017; 8:256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bandarage U.K., Clark M.P., Perola E., Gao H., Jacobs M.D., Tsai A., Gillespie J., Kennedy J.M., Maltais F., Ledeboer M.W. et al. Novel 2-substituted 7-azaindole and 7-azaindazole analogues as potential antiviral agents for the treatment of influenza. ACS Med. Chem. Lett. 2017; 8:261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guilligay D., Tarendeau F., Resa-Infante P., Coloma R., Crepin T., Sehr P., Lewis J., Ruigrok R.W., Ortin J., Hart D.J. et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008; 15:500–506. [DOI] [PubMed] [Google Scholar]
- 13. Pautus S., Sehr P., Lewis J., Fortune A., Wolkerstorfer A., Szolar O., Guilligay D., Lunardi T., Decout J.L., Cusack S.. New 7-methylguanine derivatives targeting the influenza polymerase PB2 cap-binding domain. J. Med. Chem. 2013; 56:8915–8930. [DOI] [PubMed] [Google Scholar]
- 14. Xie L., Wartchow C., Shia S., Uehara K., Steffek M., Warne R., Sutton J., Muiru G.T., Leonard V.H., Bussiere D.E. et al. Molecular Basis of mRNA Cap Recognition by Influenza B Polymerase PB2 Subunit. J. Biol. Chem. 2016; 291:363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Y., Yang Y., Fan J., He R., Luo M., Zheng X.. The crystal structure of the PB2 cap-binding domain of influenza B virus reveals a novel cap recognition mechanism. J. Biol. Chem. 2015; 290:9141–9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thierry E., Guilligay D., Kosinski J., Bock T., Gaudon S., Round A., Pflug A., Hengrung N., El Omari K., Baudin F. et al. Influenza polymerase can adopt an alternative configuration involving a radical repacking of PB2 domains. Mol. Cell. 2016; 61:125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pflug A., Guilligay D., Reich S., Cusack S.. Structure of influenza A polymerase bound to the viral RNA promoter. Nature. 2014; 516:355–360. [DOI] [PubMed] [Google Scholar]
- 18. Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2011; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murshudov G.N. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997; 53:240–255. [DOI] [PubMed] [Google Scholar]
- 21. Gabriel G., Dauber B., Wolff T., Planz O., Klenk H.D., Stech J.. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:18590–18595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Resa-Infante P., Jorba N., Zamarreno N., Fernandez Y., Juarez S., Ortin J.. The host-dependent interaction of alpha-importins with influenza PB2 polymerase subunit is required for virus RNA replication. PLoS One. 2008; 3:e3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robb N.C., Smith M., Vreede F.T., Fodor E.. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J. Gen. Virol. 2009; 90:1398–1407. [DOI] [PubMed] [Google Scholar]
- 24. Reich S., Guilligay D., Cusack S.. An in vitro fluorescence based study of initiation of RNA synthesis by influenza B polymerase. Nucleic Acids Res. 2017; 45:3353–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lakowicz J.R. Principles of Fluorescence Spectroscopy. 2006; 3rd edn, Springer Science + Business Media. [Google Scholar]
- 26. Hill A.V. A new mathematical treatment of changes of ionic concentration in muscle and nerve under the action of electric currents, with a theory as to their mode of excitation. J. Physiol. 1910; 40:190–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D.. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012; 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hengrung N., El Omari K., Serna Martin I., Vreede F.T., Cusack S., Rambo R.P., Vonrhein C., Bricogne G., Stuart D.I., Grimes J.M. et al. Crystal structure of the RNA-dependent RNA polymerase from influenza C virus. Nature. 2015; 527:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kowalinski E., Zubieta C., Wolkerstorfer A., Szolar O.H., Ruigrok R.W., Cusack S.. Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza pH1N1 (2009) polymerase. PLoS Pathogens. 2012; 8:e1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fu Y., Gaelings L., Soderholm S., Belanov S., Nandania J., Nyman T.A., Matikainen S., Anders S., Velagapudi V., Kainov D.E.. JNJ872 inhibits influenza A virus replication without altering cellular antiviral responses. Antiviral Res. 2016; 133:23–31. [DOI] [PubMed] [Google Scholar]
- 31. Laskowski R.A., Swindells M.B.. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inform. Model. 2011; 51:2778–2786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.