

Abstract
Because the transcription factor activator protein-1 (AP-1) regulates a variety of protein-encoding genes, it is a participant in many cellular functions, including proliferation, transformation, epithelial mesenchymal transition (EMT), and apoptosis. Inhibitors targeting AP-1 have potential use in the treatment of cancer and other inflammatory diseases. Here, we identify veratramine as a potent natural modulator of AP-1, which selectively binds to a specific site (TRE 5′-TGACTCA-3′) of the AP-1 target DNA sequence and regulates AP-1-dependent gene transcription without interfering with cystosolic signaling cascades that might lead to AP-1 activation. Moreover, RNA-seq experiments demonstrate that veratramine does not act on the Hedgehog signaling pathway in contrast to its analogue, cyclopamine, and likely does not harbor the same teratogenicity and toxicity. Additionally, veratramine effectively suppresses EGF-induced AP-1 transactivation and transformation of JB6 P+ cells. Finally, we demonstrate that veratramine inhibits solar-ultraviolet-induced AP-1 activation in mice. The identification of veratramine and new findings in its specific regulation of AP-1 down stream genes pave ways to discovering and designing regulators to regulate transcription factor.
INTRODUCTION
Natural products have historically been invaluable as a source for the discovery and development of a variety of drugs (1). Veratramine, a known natural steroidal alkaloid isolated from plants of the lily family, such as the Veratrum species (2), has been shown to be effective in lowering blood pressure, antagonizing Na+ channel activity, and acting on serotonin (5-HT) with agonist activity (2–4). Importantly, veratramine is structurally similar to the Hedgehog (Hh) pathway modulator, cyclopamine, which prompted our interest in studying whether veratramine has similar pharmacological effects on the Hh pathway.
In this study, veratramine was identified as a downstream modulator of the activation of activator protein-1 (AP-1) by directly binding to the target DNA sequence of AP-1 instead of acting on the Hh signaling pathway. It could inhibit EGF-induced JB6 P+ cell transformation and EGF-induced AP-1 activation in a dose-dependent manner by specifically blocking the binding of AP-1 to its cognate DNA sequence. Furthermore, in an AP-1 transgenic mouse model, veratramine also blocked solar ultraviolet (UV)-induced AP-1 activation. These results suggest that veratramine might be a potential anticancer candidate acting through different pharmacological mechanisms.
The transcription factor AP-1 is a menagerie of dimeric basic region-leucine zipper (bZIP) proteins that belong to the Jun, Fos, Maf and ATF sub-families. AP-1 recognizes either 12-O-tetradecanoylphorbol-13-acetate (TPA) response elements (5′-TGAG/CTCA-3′) or cAMP response elements (CRE,5′-TGACGTCA-3′) (5) and regulates expression within the regulatory regions of various genes, thereby controlling cellular proliferation, transformation, differentiation, and apoptosis (6–8). Much evidence indicates that aberrant AP-1 activity is implicated in the pathogenesis of a number of diseases, including tumorigenesis (9–11), inflammation (12–15) and infection (16). Consequently, AP-1 is considered one of the most promising targets for therapeutic cancer intervention along with other transcription factors, such as c-Myc (17), HIF-1 (18), STAT3 (19) and NF-κB (20). Beyond that, the remarkable progress of the development of biomarkers which are used to diagnose and treat cancers (21,22), even provide information regarding the pathways or simulation responses that are involved in the activation of transcription factors, such as NF-κB (23), further pave the way for developing the AP-1 signalling pathway modulators. However, AP-1 is a double-edged sword in tumorigenesis. It can display anti-oncogenic characteristics by inducing apoptosis or it can exert oncogenic activity by signaling and promoting cell survival (24). The final outcome of AP-1 activity in cancer appears to depend on AP-1 dimer composition and on the cellular and genetic context in which it is studied (24). To achieve the aim of therapeutic intervention, many approaches have been explored for modulating various components of the transcription machinery using small molecules (25). Strategies are divided into two principle types: (i) directly targeting the DNA-binding domain or the cognate DNA sequence of a specific transcription factor (11) and (ii) indirectly targeting the co-activators or mediators of transcription (26). Of these two approaches, targeting specific transcription factors by blocking their binding to cognate DNA sequences seems to be more effective and offers several advantages. Specific DNA-binding agents could principally inhibit or affect the function of whole families of transcription factors that share highly conserved DNA sequence domains. This would allow the regulation of only a subset of genes controlled by the given transcription factor without affecting the expression of the entire set of genes. However, the advantages could also be considered drawbacks for DNA-binding drugs, due to the challenges in achieving selectivity for short nucleic acid sequences in the human genome (26). Synthetic polyamide-based inhibitors that selectively disrupt NF-κB binding by directly binding the DNA sequences, 5′-WGGWWW-3′ and 5′-GGGWWW-3′, have been recently reported (27). However, potent small molecules, especially potentially druggable natural products that can specifically and directly bind the AP-1 target DNA sequence, have not been reported. Consequently, to identify natural active DNA minor groove binders, we performed a virtual screen against a natural product database by specifically targeting the AP-1 target DNA sequence (TRE 5′-TGACTCA-3′). Results identified veratramine and several other active natural products, including compounds 1125 and 1126. Thus, to further understand the effect of veratramine and its active analogues, compounds 1125 and 1126, on AP-1-dependent transcription, RNA-seq was performed on JB6 P+ cells treated with these compounds to measure changes in gene expression. Furthermore, the in vivo and in vitro effects of these compounds on AP-1 activity were also demonstrated.
MATERIALS AND METHODS
Identification of veratramine by virtual screening
Structure-based virtual screening was conducted using our DNA specific molecular docking method, iDNAbinder, to examine an in-house natural product database of ∼2000 compounds. The compounds were first ionized and then their most probable tautomeric states were determined with Epik (version 2.0) (28) pH 7.0 ± 2.0. The prepared compounds were geometrically optimized using the OPLS2005 force field implemented in Maestro (29). The X-ray crystal structure of a heterodimer of the bZIP region of c-Fos and c-Jun bound to DNA (PDB code: 1FOS (30)) was used as the virtual-screening-based receptor structure. The c-Fos and c-Jun transcription factors were removed to expose the AP-1 target site on the DNA sequence. The DNA sequence was then prepared with Sybyl (31) to add hydrogens and partial charges. The related parameters were set as default, and only the best-scored binding pose for each compound was retained. The top ranked 500 compounds were kept for visual inspection analysis. Finally, 75 of the 500 compounds were selected as candidate compounds and 35 of the 75 compounds were eventually obtained to perform the bioassay experiments.
Isothermal titration calorimetry (ITC)
Three sequences of oligonucleotides (the cognate DNA of AP-1 and the random designed decoys DNA) were synthesized and annealed to form double-stranded DNA. The study about the interactions of compound-DNA was performed using Isothermal titration calorimetry (MicroCal iTC200). For the titration of 1125 to DNA, the prepared double-stranded DNA and the compound 1125 were diluted using the buffer (10 mM HEPES, 100 mM NaCl, pH 7.2 and 7.1% DMSO). The concentration of DNA in the cell was 100 μM, and the concentration of 1125 in the syring was 3.57 mM. Similarly, for the interaction study about 1529-DNA, each sequence of DNA was diluted to 10 μM and the compound 1529 was diluted to 1 mM using the buffer (10 mM HEPES, 100 mM NaCl, pH 7.2 and 10% DMSO). All titration experiments were performed by adding the compound in steps of 2 μl at 25°C. The data were analyzed using Microcal origin software by fitting to a one-site binding model.
Electrophoretic mobility shift assay (EMSA)
EMSA assays were conducted using the Lightshift chemiluminescent EMSA kit (Pierce, Shanghai, China). The specific DNAs corresponding to the cognate DNA of AP-1 were synthesized with 5′-biotin labels and annealed to form double-stranded DNA. Reaction solutions contained 2 μl of 10× binding buffer (100 mM Tris–HCl pH 7.5, 500 mM KCl, 10 mM MgCl2, 50 mM DTT, 5 mM EDTA, 50% glycerol), 2.5 μg poly(dI-dC), 100 fmol biotin-labeled probes, different concentrations of the specific compound and ultra-pure water to a total volume of 20 μl. These solutions were incubated at room temperature for 10 min and then 2 μl of 5 mg/ml HeLa nuclear extract (Promega, Beijing, China) were added. The mixtures were then incubated for another 20 min at room temperature. Samples were electrophoresed on a 6% non-denaturing polyacrylamide gel and then transferred to nylon membranes, UV cross-linked, probed with streptavidin–HRP conjugate and incubated with the respective substrate. The results were detected by a chemiluminescence imaging system.
Luciferase assay to determine AP-1 or NF-κB transactivation
Confluent monolayers of JB6 P+ cells stably transfected with an AP-1 or NF-κB plasmid were trypsinized and 8 × 103 viable cells suspended in 100 μl of 5% FBS-MEM were added to each well of a 96-well plate. Plates were incubated at 37°C in a humidified atmosphere of 5% CO2. When cells reached 80 to 90% confluence, they were cultured in 0.1% FBS-MEM for another 24 h. The cells were treated with the respective compound for 60 min before they were exposed to 10 ng/ml EGF for 18 h. After treatment, cells were disrupted with 100 μl lysis buffer (0.1 M potassium phosphate pH 7.8, 1% Triton X-100, 1 mM DTT and 2 mM EDTA), and the luciferase activity was measured using a luminometer (Luinoskan Ascent, Labsystems, MD, USA).
Anchorage-independent cell transformation assay
The effects of compounds 586, 1134, 1451, 1460, 1512, 1474 and 1529 on EGF-induced transformation were investigated in JB6 C141 cells as described by Colburn et al. (32). Colonies were counted under a microscope using the Image-Pro Plus software program (Version 6, Media Cybernetics, Silver Spring, MD, USA). Data are shown as means ± S.D. of values obtained from triplicate experiments. The asterisk (*) indicates a significant (P < 0.05) change in the number of colonies as indicated.
Cell lines and culture
JB6 P+ cells alone and JB6 P+ cells stably transfected with an AP-1 or NF-κB plasmid were maintained in 5% FBS/MEM at 37°C in a humidified atmosphere of 5% CO2. Cells were passaged when they reached 80–90% confluence.
Transcription microarray experiments
Total RNA was isolated using the TRIzol Reagent (Invitrogen, Shanghai, China) following the manufacturer's instructions. Synthesis of the cDNA target, its hybridization to microarrays and scanning of those arrays was performed using Illumina Whole-Genome Gene Expression Bead Chips (MouseWG-6) and reagents according to the product recommendations (Genergy Biotechnology (Shanghai) Co., Ltd., Shanghai, China). Each treatment was repeated in triplicate.
Solar-ultraviolet-induced AP-1 luciferase activity in vivo
AP-1 luciferase transgenic mice were identified, grouped, and housed as described previously (33). The mice were developed, propagated and maintained at The Hormel Institute. Mice were maintained under conditions according to guidelines established by Research Animal Resources and the Institutional Care and Use Committee, University of Minnesota. Two weeks after grouping, the basal level of AP-1 luciferase activity was measured by punch skin biopsy. AP-1 luciferase reporter-bearing male and female mice were randomly divided into groups (12 mice/group). Two weeks after the punch biopsy, the mice were treated with veratramine by painting on the back of each mouse (10 μg of veratramine in 200 μl of acetone/mouse). The mice were treated with solar UV (60 kJ/m2 UVA/3.6 kJ/m2 UVB) using a UVA-340 sunlamp (Q-Lab Corporation, Cleveland, OH, USA) 3 h after veratramine treatment. Skin biopsies of equal weight were harvested in 100 μl of lysis buffer (0.1 M potassium phosphate buffer at pH 7.8, 1% Triton X-100, 1 mM DTT and 2 mM EDTA) for measurement of luciferase activity at the times indicated. AP-1-dependent luciferase activity in the tissue extract was determined using the luciferase assay reagent from Promega and a luminometer (Luinoskan Ascent, Labsystems).
Kinase phosphorylation assay
JB6 cells were cultured with 0.1% FBS-MEM and starved for 24 h. The cells were treated with 20 μM veratramine for 2 h and then stimulated with 10 ng/ml EGF for 30 min. At 30 min after treatment, protein extracts (500 mg) were used for phospho-MAPK array analysis. Array spots were visualized using an ECL kit (Shanghai ChemPartner Co., Ltd., Shanghai, China). The density of each duplicated array spot was measured using the ImageJ software program (NIH). The normalized changes in EGF-induced phosphorylation of MAPKs were compared with the untreated control (i.e. a value of 1). Data are shown as an average of duplicate samples.
Kinase activity profiling
The kinase profiling study was conducted using the Caliper assay screening platform (Shanghai ChemPartner Co., Ltd., Shanghai, China). All kinases and other materials were purchased from commercial sources. The compounds were screened in vitro against the different kinases, and staurosporine and PI103 were used as reference compounds. Two concentrations (3 and 10 μM) of the compounds were tested in duplicate on each kinase.
Statistical analysis
All quantitative data are expressed as mean values ± S.E. or S.D. as indicated. One-way ANOVA was used for statistical analysis. A probability of P < 0.05 was used as the criterion for statistical significance.
RESULTS
Identification of veratramine from a natural compound database
To identify specific molecules that bind to the AP-1 target DNA sequence (TRE 5′-TGACTCA-3′), virtual screening analysis was performed by searching an in-house natural product database of approximately 2,000 compounds. The virtual screening protocol was assembled based on iDNAbinder (http://lilab.ecust.edu.cn/), which is specifically designed to recognize distinct target DNA sequences and identify potential small molecules that bind to this sequence built on a base-sequence-sensitive scoring function (details in Supplemental Information). The top 500 compounds identified by our specific scoring function from the screening solution were inspected visually by considering the following criteria: (i) the binding pose of the candidate compound has to be positioned well into the site of interest (5′-TGACTCA-3′) and (ii) the predicted binding pose of the compound has to form favorable molecular interactions, including hydrogen bonds, hydrophobic interactions, and ionic bonds, with site bases. As a result of the screening, 75 compounds were selected as ‘hits’ and 35 of those compounds were prepared for in vitro testing.
These compounds were evaluated for their effect on AP-1 activity in JB6 P+ cells transfected with an AP-1 reporter plasmid and 18 of the 35 compounds inhibited AP-1 activity (Supplementary Table S1). Additionally, these compounds were evaluated for their effect against NF-κB, the most thoroughly studied transcription factor, to study their binding specificity. The assays were performed with JB6 P+ cells transfected with an NF-κB reporter plasmid. Notably, the compounds had almost no effect on NF-κB activity in vitro (Supplementary Table S1). This result suggests that the 18 active compounds are potentially selective inhibitors of transcription factor AP-1 with little effect on NF-κB.
Specific recognition of AP-1 target DNA sequence
To further investigate the binding specificity and the interactions of compounds with the cognate AP-1 DNA (top panel in Figure 2A), isothermal titration calorimetry (ITC) experiments were performed and two decoy sequences were designed as controls. The first decoy sequence (TRE 5′-CGCTTGATGACTTGGCCGGAA-3′, 21 bp; middle panel in Figure 2A) is a reported mutant target DNA sequence of AP-1, whose promoter activity was reduced to a small fraction compared with the wildtype (34). The second decoy sequence (5′-GCGGATCCCATAACAATGAC AGACT-3′, 25 bp; bottom panel in Figure 2A), relative to the cognate DNA, was a random sequence generated by our group. The mutant AP-1 DNA and the random designed DNA were used to illustrate the binding specificity of the compounds with the DNA. The ITC results indicated that veratramine (1529) clearly showed binding specificity with the cognate DNA sequence of AP-1 over the two decoy DNA sequences (Figure 1). Compound 1125 interacted with the cognate DNA sequence of AP-1, but showed no binding with the mutant AP-1 DNA or the random designed DNA.
Figure 2.

Veratramine (compound 1529) selectively binds and inhibits AP-1-dependent activity, but does not affect NF-κB. (A) The three sequence of DNA templates used for the EMSA and the luciferase assays, they are: cognate sequence of AP-1(top panel, the AP-1 binding site is highlighted in red color), reported mutant target DNA sequence of AP-1(middle panel, mutation is highlighted in italic and AP-1 binding site is in red), and randomly designed decoy sequence (bottom panel); (B) computational analysis shows that veratramine inserts into the minor groove of the AP-1 DNA sequence. (C) EMSA results show the inhibition of the transcription factor/DNA complex formation by veratramine or compound 1125. A 100-fold concentration of specific or nonspecific (NS) non-labeled probe (relative to the biotin-labeled specific DNA probe) was used to illustrate the specificity of the protein/DNA complexes. Oligonucleotides were incubated alone (lanes labeled ‘AP-1 probe’), with HeLa nuclear extracts in the absence (lanes labeled ‘DMSO’) or the presence of increasing concentrations of the compound as specified at the top of the lane.
Figure 1.

Binding isotherm obtained from ITC show the specific binding interactions of AP-1 cognate DNA with veratramine (compound 1529, upper three subfigures) and compound 1125 (lower three subfigures). (A) The chemical structures of veratramine and compound 1125. (B) ITC data for the titration of veratramine (upper panels) and compound 1125 (lower panels) into three types of DNA sequences (from left to right, AP-1 cognate DNA; AP-1 mutant DNA; and randomly-designed DNA). The upper portion in each panel shows the change in enthalpy per injection of veratramine (compound 1529) or 1125 into a corresponding DNA sequence. The lower portions indicate the concentration normalized heat form titration at the molar ratio of compounds.
In silico and EMSA characterization of the veratramine binding mode
To characterize the site-specific mode of veratramine binding to the AP-1 target DNA sequence, virtual screening was used to predict the binding mode of veratramine. Results indicated that veratramine could fit well into the minor groove of the AP-1 DNA sequence with a well-shaped complementary conformation (Figure 2B). Veratramine formed hydrophobic contacts with bases A35 and C34 and hydrogen bonds between its hydroxyl groups and bases A35, T7 and C34. Thus, veratramine binds to the AP-1 DNA cognate sequence by specifically recognizing the -AC- subsequence.
To further examine the binding of AP-1 to its consensus recognition site, we conducted electrophoretic mobility shift assays using HeLa cell nuclear extracts. Two bands were detected by chemiluminescence (Figure 2C). The uppermost band was the AP-1/specific DNA complex and the lower band corresponded to the free biotin-labeled specific DNA probe. A 100-fold concentration of specific or nonspecific (NS) unlabeled probe was used to illustrate the binding specificity of AP-1 with its cognate DNA sequence. The formation of the AP-1/DNA complex was effectively inhibited by increasing the concentration of veratramine (compound 1529; left panel) or compound 1125 (right panel).
Veratramine suppresses EGF induced AP-1 transactivation and EGF-induced JB6 P+ cell transformation
To investigate the effect of veratramine on AP-1 in cells, we performed an AP-1 transactivation assay using JB6 P+ cells stably transfected with a luciferase reporter gene or cells stably transfected with the NF-κB luciferase reporter gene as a control. Approximately 70% of the EGF-induced AP-1 transactivation activity was inhibited by 2.5 μM veratramine in a dose-dependent manner (Figure 3A). NF-κB transactivation was not significantly affected by any concentration of veratramine. These results indicated that veratramine might exert its anticancer effects by selectively inhibiting AP-1. Blocking tumor promoter-induced AP-1 activation can inhibit neoplastic transformation in JB6 cells. (35) We therefore investigated whether inhibiting AP-1 with veratramine could affect EGF-induced cell transformation. Results indicated that veratramine inhibited EGF-induced JB6 cell transformation in a dose-dependent manner (Figure 3B).
Figure 3.

Veratramine (compound 1529) suppresses AP-1 activity and EGF-induced JB6 P+ cell transformation. (A) Veratramine selectively suppresses AP-1-dependent transactivation activity, but not NF-κB transactivation. (B) JB6 P+ cells were treated with EGF and increasing concentrations of veratramine and incubated for one week in a 37°C, 5% CO2 incubator. Cells were harvested and colonies counted using a microscope and the Image-Pro Plus software program (Version 6, Media Cybernetics, Silver Spring, MD) software program.
Genome-wide effects of veratramine on EGF-induced gene expression
Because veratramine effectively inhibited EGF-induced AP-1 transactivation and JB6 P+ cell transformation, RNA-seq experiments were conducted to establish the global modulating effects of veratramine (1529) or its analogues (compounds 1125 or 1126) on the EGF-inducible JB6 P+ cell transcriptome. Genes with a log2 normalized change of >2-fold and adjusted P-values of 0.05 or less were considered to be significantly and differentially expressed. Results revealed a total of 529 out of 22,310 genes with a significant change in expression after treatment with 10 μM veratramine or 20 μM compound 1125 or 1126 compared with the control (DMSO). These genes represent ∼2.4% of the entire JB6 P+ genome. Hierarchical clustering analysis (i.e. Euclidian distance and average linkage) was performed on all genes (Figure 4A), including the 529 genes with significant changes in expression (Figure 4B). Consistent with the results of the binding validation experiments, changes induced by compound 1125 and veratramine (1529), which directly target the AP-1 DNA sequence, are clustered into the same sub-group. From the 529 significantly expressed genes, 39 are commonly up-regulated and 10 are commonly down-regulated by all 3 compounds (Figure 4A, lower panels). Genes that overlapped between compound 1125 and veratramine (1529) included 20 that were up-regulated and 15 that were down-regulated and were unaffected by compound 1126. Additionally, a subset of 75 genes was up-regulated and 66 genes were down-regulated only by veratramine.
Figure 4.

Profiling of the AP-1 pathway using the RNA-seq global transcriptome to assess changes in gene expression in JB6 P+ cells treated with veratramine (1529) or the analogue compound 1125 or 1126. All ratios are normalized to the EGF-induced state. (A) Changes in the gene expression profile of the whole genome set (top left). Significant differences in gene expression JB6 P+ cells treated with veratramine or its analogues exposed to veratramine or its analogues (top right; log2 expression change > 2.0 fold after treatment with any compound). Venn diagrams showing the overlap among the 529 genes activated (bottom left) or repressed (bottom right) by treatment with compounds. (B) Profiling of the changes in gene expression in the AP-1 pathway. (C, D) Individual normalized changes in MMP and CDK gene expression. (E) Profiling of changes in gene expression in the MAPK signaling pathway. Data were analyzed by hierarchical clustering (Euclidian distance and average linkage).
Veratramine inhibits the downstream signalling pathway of AP-1
To examine the mechanisms of veratramine's effect on AP-1, most genes from this signaling pathway were collected, and the magnitude of their change in expression induced by each inhibitor was examined by gene profiling (Figure 4B). Expression changes were substantially below 2-fold in most cases for all three compounds, excluding a significant induction of Il16 and some genes in the clustered group 100 and the repression of Mettl7a1 and Il11ra1 and Tesk1 in group 117 (Figure 4B). With the competitive binding of veratramine on the AP-1 site, the expression of AP-1 family proteins, including Myc, Jun, FosB and Fos clustered in groups 99 and 100, were significantly (∼3.0-fold) or relatively (∼1.5-fold) increased, as expected for positive feedback. These changes in expression led to T cell activation by inducing the expression of its modulator, Il16 (Figure 4B). Alternatively, the stimulation of Il16 can lead to the phosphorylation of c-Jun and p38 MAPK (36). Thus, the increasing expression of Il16 is likely the result of positive feedback of the AP-1 signaling pathway. Additionally, another transcription factor, Egr1, was up-regulated. Although the AP-1 proteins were activated, their downstream target genes, including CDKs, Ils and AP-1 inducible MMPs, such as Mmp3, whose overexpression correlates with carcinogenesis, remain unaffected or even suppressed (Figure 4C and D), indicating that the downstream genes in the AP-1 signaling pathway were inhibited by veratramine. However, other MMPs genes, such as Mmp2, Mmp24 and Mmp28, which do not contain TATA box, are not affected by AP-1 and do not belong to the AP-1 downstream, therefore, their expression should not be affected by the veratramine, but may be altered by their upstream genes. We also performed significance-calculating analyses by using gene set enrichment analysis (GSEA) (37,38) and the results are shown in Supplementary Figure S1. The enrichment plot (Supplementary Figure S1A) and the relative expression heat map (Supplementary Figure S1B) again confirmed the repression of AP-1 downstream genes, such as Cd14, Mmp9, Il6, Nudt1, Ets1, Timp1 (Metalloproteinases1), Nr4a2, Maf1, etc., while the AP-1 genes, such as, Fos, Jun and Myc etc. are activated by the modulation of veratramine.
Veratramine may not affect the MAPK pathways or other kinases
To determine whether AP-1 activity is involved in the regulation of MAPKs expression in response to various stimuli, we analyzed the activity of MAPKs and expression of genes in the MAPKs signaling pathways (Figure 4E). Except for the AP-1 family genes, only three genes (Serac1, Map4k1 and Pla2g5) in groups 175 and 179 were up-regulated >2.0-fold after treatment with veratramine or compound 1125; whereas the expression of the majority, including MPKs, was not affected (Supplementary Table S2). The increased expression level of Map4k1 is likely correlated with the activation of c-Myc (39). In contrast to veratramine and compound 1125, compound 1126 affected the expression of genes in groups 169, 170 and 173 differently, resulting in easily detectable up- or down-regulation. Overall, the EGF-induced cytosolic MAPKs signal cascades are not affected by veratramine or compound 1125. The activation of AP-1 by veratramine probably occurs through positive feedback for the downstream regulation of the AP-1 signaling pathway.
To further investigate the effect of veratramine on other AP-1-activation-related upstream kinases, we performed kinase assays either with EGF alone (10 ng/ml) or EGF plus veratramine (1529, 20 μM) treatment. The results indicate that the AP-1 related upstream kinases, including JNKs, ERKs, p38, Msk and Hsp27 (Figure 5A and B), were not affected by veratramine (1529) treatment. Furthermore, Western blot analysis revealed that AP-1-activation- related kinases, including ERKs, Msk, Rsk2, c-Fos, c-Jun and CREB, were not affected by treatment with different concentrations of veratramine (1529; Figure 5C). These results further verify that blocking AP-1 activity by veratramine does not occur through its upstream kinases. In addition, the inhibitory effects of veratramine against critical AP-1 related up-stream kinases were investigated by kinase activity profiling. Results indicated that these kinases may not be obviously affected by veratramine (Figure 5D), which again indicates that veratramine is a modulator that acts on the downstream signaling of AP-1.
Figure 5.

Veratramine (compound 1529) has no effect on EGF-induced MAPK pathways. (A) JB6 cells were cultured with 0.1% FBS/MEM and starved for 24 h. The cells were treated or not treated with compound 1529 (20 μM) for 2 h and then cells were stimulated or not stimulated with 10 ng/ml of EGF for 30 min. Protein extracts (500 μg) were prepared and used for phospho-MAPK array analysis. Array spots were visualized using an ECL detection kit. (B) The density of each duplicated array spot in ‘a’ was measured as described in Materials and Methods. The graph shows the normalized change in EGF-induced phosphorylation of JNKs, ERKs, p38, MSK and HSP27 compared with untreated control (i.e. a value of 1). Data are shown as an average of duplicate samples. (C) The important protein components related to AP-1 were verified by Western blotting. The phosphorylation of c-Fos and c-Jun and their upstream kinases were not affected by veratramine. Each experiment was repeated three times and representative blots are shown. (D) Percent of inhibition by veratramine (10 or 3 μM) against 17 kinases.
Veratramine does not act on the Hh signaling pathway
The Hh pathway is an essential signaling pathway in embryonic development and is critical for maintaining tissue polarity and stem cell populations (40,41). Deregulated Hh pathway activity can consequently result in the hereditary developmental disorder, holoprosencephaly (42). In contrast, hyperactivation of this pathway is linked to many extracutaneous cancers (43). In the absence of Hh ligands, the unbound Pathced 1 (Ptch1) protein acts catalytically to suppress the activity of Smoothened (Smo), a key player in Hh signal transduction, by preventing its localization to the cell surface. Once Ptch1 is bound to active Hh ligands, the inhibition of Smo by Ptch1 is released, allowing Smo to signal downstream, eventually leading to activation of Gli family transcription factors (Gli2 and Gli3) to regulate target gene expression (44,45). The first Hh pathway inhibitor to be identified was the plant-derived steroidal alkaloid, cyclopamine, which binds directly to the transmembrane helices of Smo and blocks cellular responses to Hh signaling (46,47). The discovery of cyclopamine has created exciting new prospects for molecular targeted therapies and prevention of human cancers associated with Hh signaling (48). However, cyclopamine has limited potential as an orally bioactive therapeutic agent due to its instability and concentration sensitivity in modulating Hh signaling (41). A relatively low concentration of cyclopamine can specifically inhibit Hh signaling, but a high dose can cause cell death without affecting Hh target gene expression (5).
Cyclopamine is a known inhibitor of the Hh signaling pathway (49). To determine whether veratramine interacts with Smo and affects the Hh signaling pathway in the same manner as cyclopamine (Figure 6A), gene expression profiling of the Hh signaling pathway after treatment with veratramine (1529) or its analogues was conducted (Figure 6B). Results indicated no obvious perturbation in gene expression of this signaling pathway, except for the enhancement of the Myc and Boc transcription factors. This result supports the hypothesis that veratramine acts as a downstream modulator of AP-1 and does not interfere with the cytosolic signaling cascade that can lead to the activation of the Hh signaling pathway. Moreover, this result could indicate that veratramine might not cause side effects, unlike the Smo inhibitor, cyclopamine which could lead to fetal deformities.
Figure 6.

Changes in gene expression induced by cyclopamine. (A) Chemical structure of the Hedgehog (Hh) inhibitor cyclopamine. (B) Changes in the gene expression profile of the Hh signaling pathway after treatment with veratramine (compound 1529) or its analogues, compound 1125 or 1126.
Veratramine suppresses solar UV-induced AP-1 activation in vivo
Ultraviolet (UV) irradiation activates AP-1 through MAPK signaling pathways, which play an important role in UV-induced skin inflammation and carcinogenesis. Agents that block AP-1 activation show promise for use in protecting against UV-induced inflammation and skin carcinogenesis. To further examine the effect of veratramine (1529) on AP-1 activity in vivo, AP-1 activities were measured in solar UV-irradiated AP-1-luciferase transgenic mice. The results indicate that pretreatment of mouse dorsal skin with veratramine (1529) before UV exposure effectively and markedly suppressed solar UV induced AP-1 activation at 36 h after solar UV exposure (Figure 7).
Figure 7.
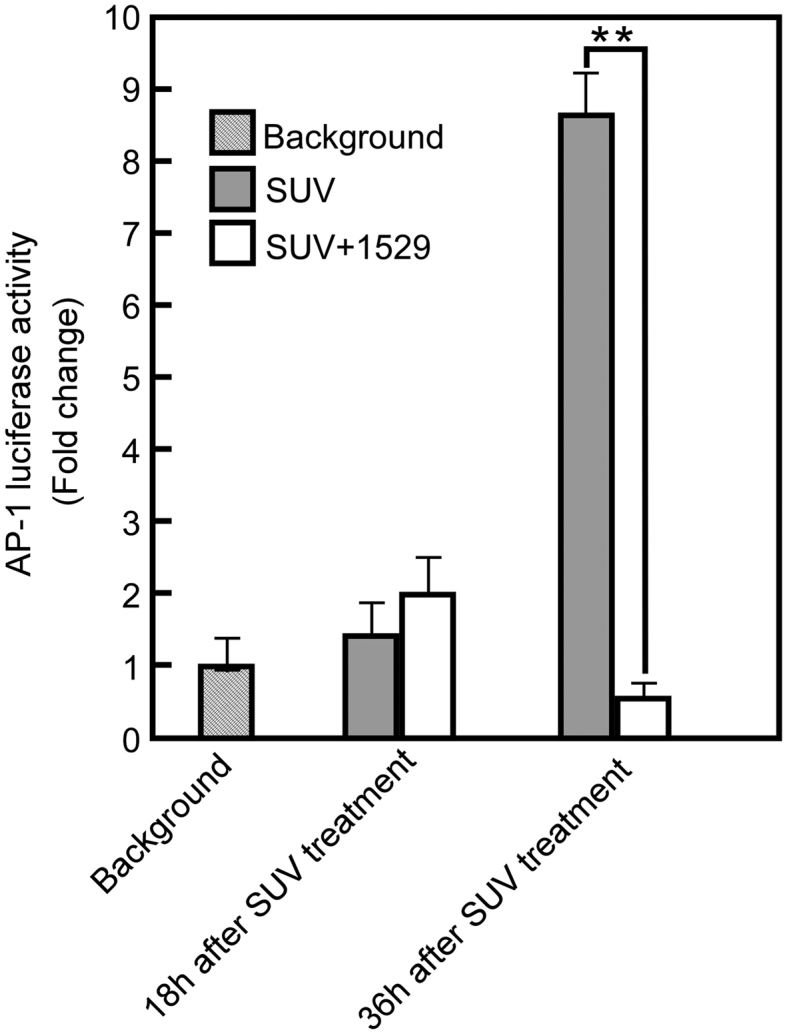
Veratramine suppresses solar UV-induced AP-1 activity in vivo. AP-1 luciferase reporter-bearing male and female mice (12 mice/group) were treated with veratramine solvent by painting on the back of each mouse (10 μg of veratramine in 200 μl of acetone/mouse). The mice were treated with solar UV 3 h after veratramine treatment. Skin biopsies of equal weight were harvested in 100 μl of lysis buffer for measurement of luciferase activity at the times indicated. AP-1-dependent luciferase activity in the tissue extract was determined using the luciferase assay reagent (Promega) and a luminometer.
DISCUSSION
Because the gene promoters recognized by any particular transcription factor could have different sequences, targeting a specific promoter sequence by a small molecule might allow the regulation of only a subset of genes controlled by the given transcription factor and might at least partially avoid any unexpected side effects. The current study introduces a binding-site-specific downstream modulator of AP-1, veratramine, which was identified in a virtual screening of an in-house natural product database. Veratramine, long considered a natural product of interest, has been reported to have anti-thrombotic activity and serotonin-agonist activity (3). Moreover, this compound has been reported to be a potential signal transduction inhibitor for treating cancers4, but its direct binding target was not determined until now. An important aspect of this work is the discovery of veratramine as a specific natural small molecule inhibitor, which selectively recognizes and binds the AP-1 DNA target sequence (5′-TGACTCA-3′). This discovery exemplifies the efficiency of our iDNAbinder protocol in the design of specific DNA-binding agents, which challenges the selectivity for short nucleic acid sequences that may be abundant in the human genome. Furthermore, a series of in vitro experiments indicates that veratramine acts on the downstream signaling of AP-1, but does not interfere with the EGF-induced cytosolic MAPK signaling cascades or other AP-1 activation-related kinases. This indicates that the gene modulation occurs only on a subset of genes controlled by AP-1.
To explore the anti-cancer effects of veratramine, its ability to suppress EGF-induced AP-1 transactivation and transformation was evaluated in JB6 P+ cells. As expected, the AP-1 dependent transactivation could be effectively suppressed by veratramine in a dose-dependent manner. However, the NF-κB dependent transactivation was not affected by treatment with veratramine. Moreover, cell transformation also could be effectively inhibited by veratramine. These findings demonstrate a selective inhibition of the AP-1-dependent signaling pathway by veratramine.
Although veratramine has the same scaffold as the Smo inhibitor cyclopamine, a completely different pharmacological mechanism was confirmed in this study. Veratramine selectively and directly binds with a specific site of the target DNA sequence of AP-1 instead of acting on the Hh signaling pathway; thus, it may not have the same teratogenicity and toxicity as cyclopamine. Finally, our study clearly demonstrated that veratramine effectively suppresses SUV-induced AP-1 activation in vivo. All these findings indicate that veratramine is a potentially valuable anticancer agent that should be further explored and developed.
AVAILABILITY
DNA-specific docking program iDNABinder is an open source and can be requested by email to Prof. Honglin Li (hlli@ecust.edu.cn).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Yongjun Dang at the Fudan University, Prof. Jingyan Zhang at East China University of Science and Technology for their technological assistance and helpful suggestions.
Author Contributions: The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint First Authors. F.B. performed the computational experiments and analysed gene expression data, K.D.L., J.W.W, J.S.Z., L.L.Z. and W.Y.M. performed the cellular, biochemical and animal experiments and analysed data. H.L., S.D.Z., L.Z. isolated and synthesized the natural compounds. P.H. contributed to transcription microarray analysis. K.D.L., W.D.Z, H.L.L. and Z.G.D. designed the project. F.B., K.D.L. A.M.B. and H.L.L. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
National Key Research and Development Program [2016YFA0502304 to H.L.L.]; Professor of Chang Jiang Scholars Program (to W.D.Z); National Natural Science Foundation of China [81372269 to K.D.L., 81230090, 81520108030, 21472238 to W.D.Z.]; Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products [16DZ2280200 to W.D.Z.]; Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase) [U1501501]; Honglin Li is also sponsored National Program for Support of Top-notch Young Professionals. Funding for open access charge: National Key Research and Development Program [2016YFA0502304].
Conflict of interest statement. None declared.
REFERENCES
- 1. Koehn F.E., Carter G.T.. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005; 4:206–220. [DOI] [PubMed] [Google Scholar]
- 2. Tremblay M., McGovern K. Xie J. Cyclopamine and Its Derivatives for Cancer Therapeutics. Hedgehog Signaling Activation in Human Cancer and its Clinical Implications. 2011; NY: Springer; 187–212. [Google Scholar]
- 3. El Sayed K.A. Microbial biotransformation of veratramine. J. Nat. Prod. 1998; 61:149–151. [DOI] [PubMed] [Google Scholar]
- 4. Khanfar M., El Sayed K.. The Veratrum alkaloids jervine, veratramine, and their analogues as prostate cancer migration and proliferation inhibitors: biological evaluation and pharmacophore modeling. Med. Chem. Res. 2013; 22:1–12. [Google Scholar]
- 5. Chinenov Y., Kerppola T.K.. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001; 20:2438–2452. [DOI] [PubMed] [Google Scholar]
- 6. Sankpal N.V., Mayfield J.D., Willman M.W., Fleming T.P., Gillanders W.E.. Activator protein 1 (AP-1) contributes to EpCAM-dependent breast cancer invasion. Breast Cancer Res. 2011; 13:R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Milde-Langosch K., Roder H., Andritzky B., Aslan B., Hemminger G., Brinkmann A., Bamberger C.M., Loning T., Bamberger A.M.. The role of the AP-1 transcription factors c-Fos, FosB, Fra-1 and Fra-2 in the invasion process of mammary carcinomas. Breast Cancer Res. Trans. 2004; 86:139–152. [DOI] [PubMed] [Google Scholar]
- 8. Shaulian E., Karin M.. AP-1 as a regulator of cell life and death. Nat. Cell. Biol. 2002; 4:E131–E136. [DOI] [PubMed] [Google Scholar]
- 9. Matthews C.P., Colburn N.H., Young M.R.. AP-1 a target for cancer prevention. Curr. Cancer Drug Tar. 2007; 7:317–324. [DOI] [PubMed] [Google Scholar]
- 10. Zhao C.Y., Qiao Y.C., Jonsson P., Wang J., Xu L., Rouhi P., Sinha I., Cao Y.H., Williams C., Dahlman-Wright K.. Genome-wide profiling of AP-1-regulated transcription provides insights into the invasiveness of triple-negative breast cancer. Cancer Res. 2014; 74:3983–3994. [DOI] [PubMed] [Google Scholar]
- 11. Kamide D., Yamashita T., Araki K., Tomifuji M., Tanaka Y., Tanaka S., Shiozawa S., Shiotani A.. Selective activator protein-1 inhibitor T-5224 prevents lymph node metastasis in an oral cancer model. Cancer Sci. 2016; 107:666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schonthaler H.B., Guinea-Viniegra J., Wagner E.F.. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann. Rheum. Dis. 2011; 70(Suppl. 1):i109–i112. [DOI] [PubMed] [Google Scholar]
- 13. Zenz R., Eferl R., Kenner L., Florin L., Hummerich L., Mehic D., Scheuch H., Angel P., Tschachler E., Wagner E.F.. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005; 437:369–375. [DOI] [PubMed] [Google Scholar]
- 14. Wang A., Al-Kuhlani M., Johnston S.C., Ojcius D.M., Chou J., Dean D.. Transcription factor complex AP-1 mediates inflammation initiated by Chlamydia pneumoniae infection. Cell. Microbiol. 2013; 15:779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aikawa Y., Morimoto K., Yamamoto T., Chaki H., Hashiramoto A., Narita H., Hirono S., Shiozawa S.. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotech. 2008; 26:817–823. [DOI] [PubMed] [Google Scholar]
- 16. Duverger A., Wolschendorf F., Zhang M., Wagner F., Hatcher B., Jones J., Cron R.Q., van der Sluis R.M., Jeeninga R.E., Berkhout B. et al. An AP-1 binding site in the enhancer/core element of the HIV-1 promoter controls the ability of HIV-1 to establish latent infection. J. Virol. 2012; 87:2264–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adhikary S., Eilers M.. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005; 6:635–645. [DOI] [PubMed] [Google Scholar]
- 18. Ke Q., Costa M.. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006; 70:1469–1480. [DOI] [PubMed] [Google Scholar]
- 19. Michaud E.J., Yoder B.K.. The primary cilium in cell signaling and cancer. Cancer Res. 2006; 66:6463–6467. [DOI] [PubMed] [Google Scholar]
- 20. Sen R., Baltimore D.. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986; 46:705–716. [PubMed] [Google Scholar]
- 21. Liu C., Yang C., Lu L., Wang W., Tan W., Leung C.-H., Ma D.-L.. Luminescent iridium(iii) complexes as COX-2-specific imaging agents in cancer cells. Chem. Commun. 2017; 53:2822–2825. [DOI] [PubMed] [Google Scholar]
- 22. Li X., Kim C.Y., Lee S., Lee D., Chung H.-M., Kim G., Heo S.-H., Kim C., Hong K.-S., Yoon J.. Nanostructured phthalocyanine assemblies with protein-driven switchable photoactivities for biophotonic imaging and therapy. J. Am. Chem. Soc. 2017; 139:10880–10886. [DOI] [PubMed] [Google Scholar]
- 23. Wang W., Yang C., Lin S., Vellaisamy K., Li G., Tan W., Leung C.-H., Ma D.-L.. First synthesis of an oridonin-conjugated iridium(III) complex for the intracellular tracking of NF-κB in living cells. Chem. Eur. J. 2017; 23:4929–4935. [DOI] [PubMed] [Google Scholar]
- 24. Eferl R., Wagner E.F.. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer. 2003; 3:859–868. [DOI] [PubMed] [Google Scholar]
- 25. Ye N., Ding Y., Wild C., Shen Q., Zhou J.. Small molecule inhibitors targeting activator protein 1 (AP-1). J. Med. Chem. 2014; 57:6930–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leung C.-H., Chan D.S.-H., Ma V.P.-Y., Ma D.-L.. DNA-binding small molecules as inhibitors of transcription factors. Med. Res. Rev. 2013; 33:823–846. [DOI] [PubMed] [Google Scholar]
- 27. Raskatov J.A., Meier J.L., Puckett J.W., Yang F., Ramakrishnan P., Dervan P.B.. Modulation of NF-κB-dependent gene transcription using programmable DNA minor groove binders. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shelley J., Cholleti A., Frye L., Greenwood J., Timlin M., Uchimaya M.. Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided-Mol. Des. 2007; 21:681–691. [DOI] [PubMed] [Google Scholar]
- 29. Suite 2010: Maestro, v., Schrödinger, LLC, New York, NY, 2011.
- 30. Glover J.N.M., Harrison S.C.. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature. 1995; 373:257–261. [DOI] [PubMed] [Google Scholar]
- 31. Tripos 2001; SYBYL (Tripos, S.L., MO), Version 6.8.
- 32. Colburn N.H., Wendel E.J., Abruzzo G.. Dissociation of mitogenesis and late-stage promotion of tumor cell phenotype by phorbol esters: mitogen-resistant variants are sensitive to promotion. Proc. Natl. Acad. Sci. U.S.A. 1981; 78:6912–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhong S., Quealy J.A., Bode A.M., Nomura M., Kaji A., Ma W.Y., Dong Z.. Organ-specific activation of activator protein-1 in transgenic mice by 12-o-tetradecanoylphorbol-13-acetate with different administration methods. Cancer Res. 2001; 61:4084–4091. [PubMed] [Google Scholar]
- 34. Muralikrishna B., Parnaik V.K.. SP3 and AP-1 mediate transcriptional activation of the lamin A proximal promoter. Eur. J. Biochem. 2001; 268:3736–3743. [DOI] [PubMed] [Google Scholar]
- 35. Dong Z., Birrer M.J., Watts R.G., Matrisian L.M., Colburn N.H.. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:609–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krautwald S. IL-16 activates the SAPK signaling pathway in CD4+ macrophages. J. Immunol. 1998; 160:5874–5879. [PubMed] [Google Scholar]
- 37. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mootha V.K., Lindgren C.M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E. et al. PGC-1[alpha]-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003; 34:267–273. [DOI] [PubMed] [Google Scholar]
- 39. Wang Q., Zhang Y., Yang H.-S.. Pdcd4 knockdown up-regulates MAP4K1 expression and activation of AP-1 dependent transcription through c-Myc. Biochim. Biophys. Acta (BBA) - Mol. Cell Res. 2012; 1823:1807–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jiang J., Hui C.-C.. Hedgehog signaling in development and cancer. Dev. Cell. 2008; 15:801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie J., Epstein E.. Xie J. Activation of hedgehog signaling in human cancer. Hedgehog Signaling Activation in Human Cancer and its Clinical Implications. 2011; NY: Springer; 85–104. [Google Scholar]
- 42. Roessler E., Belloni E., Gaudenz K., Jay P., Berta P., Scherer S.W., Tsui L.-C., Muenke M.. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 1996; 14:357–360. [DOI] [PubMed] [Google Scholar]
- 43. Xie J. Hedgehog signaling pathway: development of antagonists for cancer therapy. Curr. Oncol. Rep. 2008; 10:107–113. [DOI] [PubMed] [Google Scholar]
- 44. Lu Y., Li J.L., Cheng J.L., Lubahn D.B.. Genes targeted by the Hedgehog-signaling pathway can be regulated by Estrogen related receptor beta. BMC Mol. Biol. 2015; 16:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang L., Xie G., Fan Q., Xie J.. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010; 29:469–481. [DOI] [PubMed] [Google Scholar]
- 46. Gupta S., Takebe N., Lorusso P.. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010; 2:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Firestone A.J., Chen J.K.. Xie J. Small-molecule inhibitors of the hedgehog pathway. Hedgehog Signaling Activation in Human Cancer and its Clinical Implications. 2011; NY: Springer; 163–186. [Google Scholar]
- 48. Yauch R.L., Gould S.E., Scales S.J., Tang T., Tian H., Ahn C.P., Marshall D., Fu L., Januario T., Kallop D. et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008; 455:406–410. [DOI] [PubMed] [Google Scholar]
- 49. Sheikh A., Alvi A.A., Aslam H.M., Haseeb A.. Hedgehog pathway inhibitors – current status and future prospects. Infect. Agents Cancer. 2012; 7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.