

Abstract
Purpose:
The purpose of the study was to report aperture of retinal pigment epithelium (RPE) as a late complication and an unreported finding during the natural course of adult-onset foveomacular vitelliform dystrophy (AFVD).
Methods:
Four diagnosed cases of AFVD followed for a period ranging from 4 to 8 years. All patients had documented records of clinical examination, fundus autofluorescence and fluorescein angiography, and spectral domain-optical coherence tomography at regular intervals.
Results:
Besides the known stages in the natural course of AFVD, RPE aperture was noted as an additional finding during the vitelliruptive stage of the disease. The vitelliform material was noted beneath the disrupted RPE before disappearance. Accumulation of vitelliform material continued even after the vitelliruptive stage.
Conclusion:
RPE aperture may represent an ongoing process in the natural course of AFVD.
Keywords: Adult-onset foveomacular vitelliform dystrophy, aperture, fundus autofluorescence, retinal pigment epithelium, spectral domain-optical coherence tomography
Adult-onset foveomacular vitelliform dystrophy (AFVD) is characterized by bilateral subretinal yellowish macular lesions usually seen between fourth and fifth decades of life.[1,2] Although the long-term follow-up of AFVD reveals progressive stages (vitelliform, pseudohypopyon, vitelliruptive, and atrophic) during its natural course, the exact underlying mechanism into its pathogenesis is not well understood.[3] It is still not known how the vitelliform material gets reabsorbed.[3]
The imaging modalities such as fundus autofluorescence (FAF) and spectral domain-optical coherence tomography (SD-OCT) show characteristic findings as the lesions evolve through various stages.[2,3,4,5] We observed aperture of retinal pigment epithelium (RPE) as a new finding on SD-OCT during the natural course of AFVD that has not been reported so far.
Methods
We performed a retrospective analysis of a case series of four patients (age range: 45–82 years) with AFVD who presented in the retina services between 2000 and 2008. Institute Ethics Committee approval was obtained. The study adhered to the tenets of the Declaration of Helsinki. The inclusion criteria were the presence of AFVD and absence of dry age-related macular degeneration (AMD)/choroidal neovascular membrane (CNVM) by clinical records and multimodal image analysis, minimum 1-year follow up, and complete documented records of ophthalmic examination including best-corrected visual acuity (BCVA), slit-lamp biomicroscopic examination, fundus photography, fundus fluorescein angiography (FFA), FAF imaging, and SD-OCT (Cirrus HD-OCT [Carl Zeiss Meditec, Dublin, CA, USA] or Spectralis HRA + OCT [Heidelberg Engineering, Heidelberg, Germany]) done at baseline and follow-up visits. Eyes with additional retinal pathologies (such as diabetic retinopathy, vascular occlusion, macular hole, and other vitreoretinal interface disorders) were excluded from the study.
Case examples
Case example 1
A 45-year-old female presented with complaints of metamorphopsia. Her BCVA was 20/20 in the right eye and 20/40 in the left eye. Fundus examination revealed subfoveal yellowish deposit in the right eye and foveal scar in the left eye [Fig. 1]. The right eye FFA showed a central hypofluorescence with a halo of hyperfluorescence and the left eye showed a central small area of hyperfluorescence with surrounding hypofluorescence [Fig. 1]. On FAF, the right eye lesion was brightly hyperautofluorescent in the center, surrounded by a border of hypoautofluorescence. The SD-OCT showed a hyperreflective complex in the outer retinal layers that extended into the Henle's layer with aperture of the ellipsoid zone and the outer limiting membrane [Fig. 2a]. The RPE could not be differentiated from this complex. She received two monthly injections of bevacizumab in the left eye for an erroneous diagnosis of occult CNVM. Two years later, the patient complained of increased metamorphopsia in the right eye. There was a disappearance of the yellowish deposits in the right eye with atrophic changes in the fovea, and on FAF, the lesion now appeared hypoautofluorescent. The SD-OCT scan showed disappearance of the previously accumulated hyperreflective material, baring of the Bruch's membrane, and discontinuity of photoreceptor (PR)-RPE complex [Fig. 2b]. Three years later, the right eye fundoscopy showed the reappearance of the yellowish material (adjacent to the previous atrophic lesion), which was hyperautofluorescent and corresponded to new hyperreflective deposits beneath the ellipsoid zone, at the border of previously identified region of missing PR-RPE complex [Fig. 2c]. The left eye too underwent similar changes with the RPE aperture noted 5 years after presentation [Fig. 2d]. The BCVA was 20/40 in both eyes at 5-year follow-up.
Figure 1.

Fundus photograph of Case 1 showing a subfoveal deposit in the right eye and foveal scar in the left eye (top). Fluorescein angiography showed a central hypofluorescence with a halo of hyperfluorescence in the right eye and a central tiny hyperfluorescence with surrounding hypofluorescence in the left eye (bottom)
Figure 2.
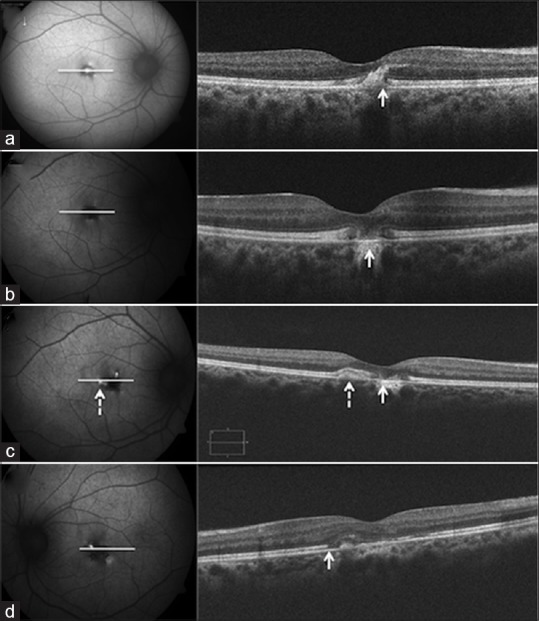
Fundus autofluorescence and corresponding spectral domain-optical coherence tomography scans of right eye (Case 1) showing a brightly hyperautofluorescent lesion in the center, surrounded by a border of hypoautofluorescence, with spectral domain-optical coherence tomography showing a hyperreflective complex in the outer retinal layers, anterior to the retinal pigment epithelium (arrow) (a). Two years later, the lesion now appeared completely hypoautofluorescent with disappearance of the yellowish deposits, and the spectral domain-optical coherence tomography scan showed disappearance of the previously accumulated hyperreflective material, baring of Bruch's membrane, and discontinuity of photoreceptor-retinal pigment epithelium complex (arrow) (b). Three years later, a new hyperautofluorescent material (dashed arrow) appeared adjacent and temporal to previous lesion, appearing as new hyperreflective deposits beneath the photoreceptor-retinal pigment epithelium complex (dashed arrow), at the border of previously identified region of missing photoreceptor-retinal pigment epithelium complex (solid arrow) (c). The left eye too underwent similar changes and aperture of retinal pigment epithelium was noted 5 years after presentation (d)
Case example 2
A 70-year-old male with metamorphopsia (diagnosed as occult CNVM elsewhere) presented with BCVA of 20/30 in both eyes. The right eye had a hypopigmented, yellowish subfoveal lesion that was hyperautofluorescent on FAF, suggestive of AFVD [Fig. 3]. The SD-OCT line scan passing through the lesion showed a pigment epithelium detachment (PED) with hyperreflective material in the outer retinal layers (anterior to PED) that was still persisting 2 years later [Fig. 4a] and developed a small RPE aperture 6 months later [Fig. 4b]. Three years further on, the yellow material disappeared and the lesion became hypoautofluorescent on FAF corresponding to RPE aperture. The PED had collapsed with baring of the Bruch's membrane [Fig. 4c]. Still, 3 years later, the yellowish material, brightly hyperautofluorescent reaccumulated next to the region of RPE aperture and corresponded to the newly appeared hyperreflective material under the ellipsoid zone [Fig. 4d]. The BCVA in the right eye was 20/80. The left eye too had a similar course.
Figure 3.

Right eye fundus photograph (left) of Case 2 showing a hypopigmented, subfoveal lesion with hyperautofluorescence on fundus autofluorescence (right)
Figure 4.
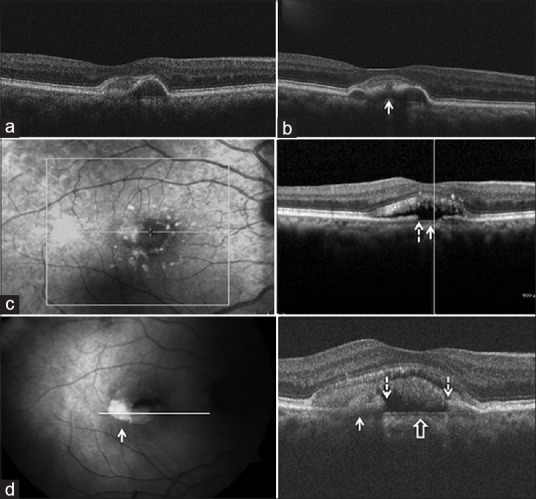
Line scan (case 2) through the lesion shows hyperreflective material anterior to pigment epithelium detachment (a). Two years later, a retinal pigment epithelium aperture developed (arrow) (b). Three years later, it disappeared and became hypoautofluorescent, corresponding to retinal pigment epithelium aperture (dashed arrow) with collapse of pigment epithelium detachment and baring of Bruch's membrane (solid arrow) (c). Three years later, hyperautofluorescence (solid arrow, left panel) is seen corresponding to the newly appeared subretinal hyperreflective material (solid arrow, right panel) (d). The aperture (dashed arrows) had widened, with baring of Bruch's membrane (block arrow)
Case example 3
A 68-year-old male presented with metamorphopsia and BCVA of 20/30 in the right eye and 20/100 in the left eye. The left eye yellowish subfoveal lesion was hyperfluorescent on FFA and corresponded to the RPE aperture on SD-OCT with hyporeflectivity anterior, as well as posterior, to RPE aperture, suggesting a completely reabsorbed vitelliform material [Fig. 5a]. Three years later, the RPE aperture and PED increased, with appearance of new hyperreflective material under the disrupted RPE [Fig. 5b]. Over the next 2 years, fundus examination and SD-OCT showed complete disappearance of the yellowish material both above and below RPE [Fig. 5c]. Two years later, whereas the SD-OCT findings remained the same through the center of the hypoautofluorescent atrophic lesion [Fig. 5d], a new hyperautofluorescent halo appeared around this lesion, with a corresponding hyperreflective material both above and below the RPE [Fig. 6]. The BCVA at last follow-up was 20/30 in the right eye and 20/100 in the left eye.
Figure 5.

Fundus photograph (left) and fluorescein angiography (middle) of left eye (Case 3) showing a hyperfluorescent subfoveal lesion, and retinal pigment epithelium aperture on spectral domain-optical coherence tomography (right) with hyporeflectivity anterior (dashed arrow), as well as posterior (solid arrow), to retinal pigment epithelium aperture, suggesting a completely reabsorbed vitelliform material (a). Three years later, the pigment epithelium detachment increased, with appearance of new hyperreflective material under the disrupted retinal pigment epithelium (arrow) (b). Two years later, spectral domain-optical coherence tomography showed the complete disappearance of the hyperreflective material both above and below retinal pigment epithelium, with focal choroidal excavation, due to increase in the pigment epithelium detachment (c). Two years later, the spectral domain-optical coherence tomography findings remained the same through the center of the hypoautofluorescent lesion (d)
Figure 6.
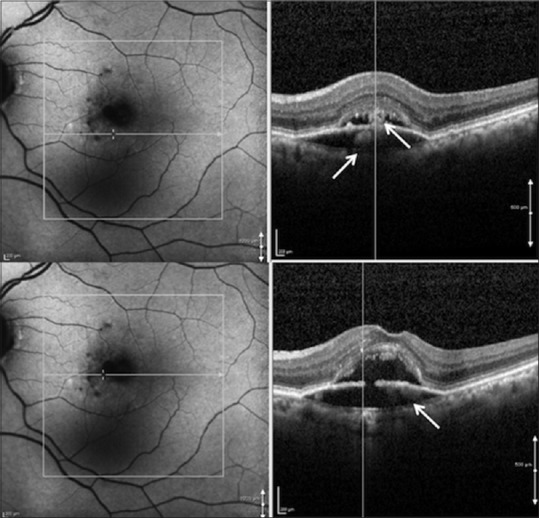
Same visit as in Figure 5 showing a new hyperautofluorescent halo appearing around the hypoautofluorescent lesion, with a corresponding hyperreflective material both above and below the retinal pigment epithelium (top right and bottom right) on spectral domain-optical coherence tomography scans at different regions
Case example 4
A 82-year-old male with metamorphopsia had BCVA 20/30 in the right eye and 20/100 in the left eye. An ill-defined staining of subfoveal lesion on FA led to a diagnosis of occult CNVM in the left eye, for which he received two intravitreal bevacizumab injections. The FAF imaging showed a mixed pattern of the FAF [Fig. 7a]. Two years later, the lesion showed an increasing hypoautofluorescence [Fig. 7b]. The SD-OCT scan revealed three different stages of the disease as the lesion showed varying levels of AF, i.e., vitelliform stage through the hyperautofluorescent part of the lesion [Fig. 8a], vitelliruptive stage through the junction of hyper- and hypoautofluorescent parts [Fig. 8b], and RPE aperture through the completely hypoautofluorescent part of the lesion [Fig. 8c].
Figure 7.
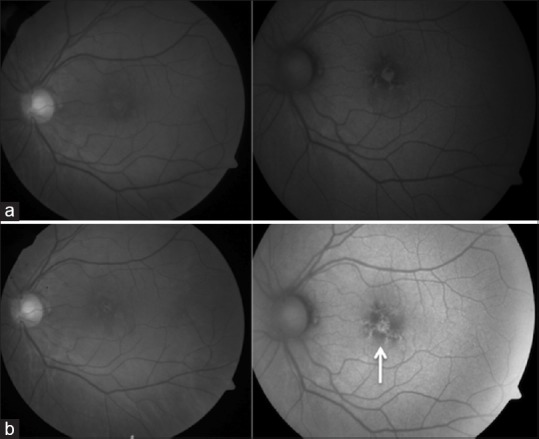
Fundus photograph (left) and autofluorescence (right) of left eye of Case 4 showing a subfoveal lesion with a mixed pattern of autofluorescence (a). Two years later, the lesion showed an increasing hypoautofluorescence (arrow) (b)
Figure 8.
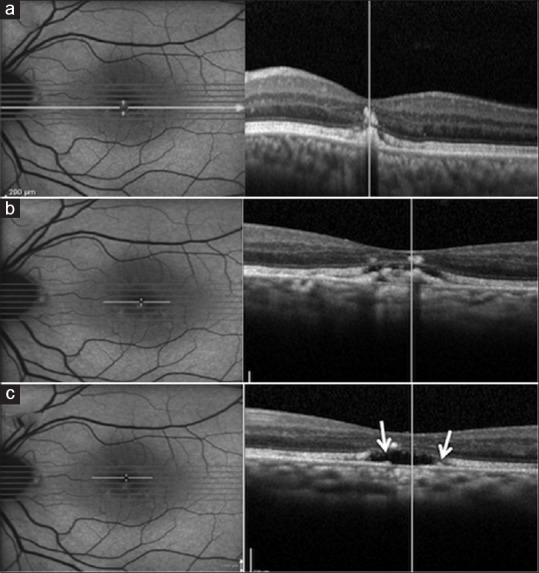
The spectral domain-optical coherence tomography scan revealed three different stages of the disease at the same time point within the lesion showing varying levels of autofluorescence, i.e., vitelliform stage through the hyperautofluorescent part of the lesion (a), vitelliruptive stage through the junction of hyper- and hypoautofluorescent parts (b), and retinal pigment epithelium aperture (arrows) through the completely hypoautofluorescent part of the lesion (c)
Discussion
An RPE aperture has been recently described as a new finding, previously unreported, in the evolution of avascular PED.[6] We observed this finding in four of our patients of AFVD during a long-term follow up, two of which had a coexisting avascular PED. The multimodal imaging analysis revealed that this aperture in the RPE corresponded to the clinical disappearance of the yellow vitelliform material and conversion of the lesions showing hyperautofluorescence to hypoautofluorescence on the FAF. This finding is consistent with that described by Querques et al. where the lesion representing drusenoid material revealed focal hyperautofluorescence preceding the development of RPE aperture and turned hypoautofluorescent with its atrophic progression.[6] Besides showing the RPE aperture, the SD-OCT in our patients also revealed the disappearance of the hyperreflective vitelliform deposits till it appeared again years later. The yellowish appearing vitelliform material in AFVD represents the PR outer segments debris and lipofuscin accumulated between the neurosensory retina and the RPE.[2,4] While the FFA findings may often be inconclusive to differentiate AFVD from other conditions mimicking it, multimodal imaging including FAF and SD-OCT are extremely valuable in confirming its diagnosis, staging the disease, and studying its natural course.[5] Pierro et al. described a common finding of a well-defined subretinal thickening of the RPE on SD-OCT in all of 72 eyes they studied, with varying features of FFA and indocyanine green angiography.[7] They believed that metamorphopsia in these patients resulted from compression of the neurosensory retina overlying the subretinal vitelliform deposit, and visual loss occurred due to thinning of the neurosensory retina.[7] The OCT helps in preventing misdiagnosing AFVD as CNVM by showing a subretinal lesion with medium-high reflectivity with a neurosensory wedge-shaped elevation.[8]
While the PR layer gets elevated and separated from RPE, various OCT-based studies have shown RPE layer to be linear and not elevated.[4,9] Puche et al. described different morphological features in eyes with AFVD using high-resolution SD-OCT.[10] The subretinal vitelliform material was mostly heterogeneous, highly reflective, and dome shaped. Some eyes showed hyperreflective clumps within the outer plexiform and outer nuclear layers. The PR inner segment/outer segment (IS/OS) interface, now labeled as the ellipsoid zone showed high reflectivity, and appeared irregular and discontinuous in few eyes. While some eyes showed discrete RPE detachments, few showed RPE irregularities with hyperreflective mottling on its inner aspect. The subretinal accumulation of vitelliform material was followed by alterations in PR IS/OS interface, pigment migration toward inner retinal layers, and fluid accumulation. Changes in the RPE included atrophy/hypertrophy or rarely sub-RPE deposits. Chen et al. have described intraretinal hyperreflective foci in patients with the acquired vitelliform lesions, which represent cells of the same origin as those found in vitelliform lesions.[11] Aperture of the ellipsoid zone facilitates inward migration of the vitelliform material, followed by its lateral dissipation within the inner retinal layers. None have reported RPE aperture as seen in our study.
As described by Querques et al.,[6] our series has the common findings of the absence of any rippling or retraction at the edges of the RPE aperture (differentiating it from RPE rip or tear) and hypoautofluorescence on FAF corresponding to the aperture on SD-OCT. However, the subretinal fluid is present in only two of our eyes with coexisting PED, which differentiates it from the previous series that had subretinal fluid in all eyes. Furthermore, though we did not quantitatively assess the size of the RPE aperture, it did not show any apparent increase in size over a follow-up of as long as 2 years as seen in case example 3 [Fig. 5b and c].
Sub-RPE changes in AFVD have been rarely mentioned in clinical or clinicopathologic studies.[10,12,13,14] Our short case series demonstrated a sub-RPE hyperreflective lesion following development of RPE aperture indicating a sub-RPE migration of the vitelliform material and absorption from thereof. Previously, a well-circumscribed elevation of RPE has been reported by Saito et al., with an underlying lesion beneath with a moderate-high reflectivity.[15] Clinicopathologic studies on AFVD have revealed atrophy of PRs and RPE,[12,13,14] collection of eosinophilic material,[12] pigmented cells containing periodic acid–Schiff (PAS)-positive material and pigmented cells containing lipofuscin in the subretinal space.[13,15] Dense eosinophilic material and PAS-positive material have also been shown in the space between Bruch's membrane and RPE.[12,13] Based on our observations in these four patients, we suggest that this migration of the vitelliform material may have been facilitated by an aperture in the RPE.
As AFVD progresses, a change in the composition of the vitelliform lesions changes the clinical and morphological appearance of these lesions. The acquired vitelliform material that collects as a result of impaired phagocytosis of shed PR outer segments due to dysfunctional RPE is believed to show a gradual spontaneous resolution due to the ongoing PR loss.[2] While the RPE tears associated with PED/wet AMD occur secondary to hydrostatic forces by the sub-RPE fluid/membrane,[16,17] it may be likely that this phenomenon in AFVD may represent the loss of integrity of RPE cells that have undergone atrophy following a chronic detachment. It is difficult to say whether the regression of vitelliform material leads to progressive atrophy of the RPE as hypothesized in context to drusenoid PEDs,[6] or the aperture of the RPE is an additional mechanism for the disappearance of the vitelliform material. Histological demonstration of intraretinal RPE cells and vitelliform lesions in a large drusenoid PED[18] and the coexistence of serous PEDs in two of our patients with AFVD may suggest a common underlying mechanism in the pathogenesis of RPE aperture.
Our series has a few limitations of being a retrospective study. The follow-up scans obtained by the SD-OCT (Cirrus HD-OCT [Carl Zeiss Meditec, Dublin, CA, USA) lack the exact registration of the lesions. Two of our patients showed PED, a finding more consistent with AMD than AFVD. However, these patients lacked the typical findings of AMD. Nevertheless, AMD has been reported as one of the non-AFVD entities associated with acquired vitelliform lesions.[2] A small number of eyes, variable follow-up, lack of quantitative assessment of the RPE aperture, and lack of histologic correlation are other drawbacks of our study.
Conclusion
Complications such as CNVM, macular hole, and retinal detachment have been occasionally reported in AFVD. Our study suggests RPE aperture as another possible complication in these eyes. We also demonstrate for the first time that there is progressive accumulation of the vitelliform material even after the lesions have reached the vitelliruptive and atrophic stage in the natural course of the AFVD. We believe that the RPE aperture beginning simultaneous or subsequent to the onset of vitelliruptive stage may occur during the natural course of AFVD.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Gass JD. A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc. 1974;72:139–56. [PMC free article] [PubMed] [Google Scholar]
- 2.Freund KB, Laud K, Lima LH, Spaide RF, Zweifel S, Yannuzzi LA. Acquired Vitelliform Lesions: Correlation of clinical findings and multiple imaging analyses. Retina. 2011;31:13–25. doi: 10.1097/IAE.0b013e3181ea48ba. [DOI] [PubMed] [Google Scholar]
- 3.Querques G, Forte R, Querques L, Massamba N, Souied EH. Natural course of adult-onset foveomacular vitelliform dystrophy: A spectral-domain optical coherence tomography analysis. Am J Ophthalmol. 2011;152:304–13. doi: 10.1016/j.ajo.2011.01.047. [DOI] [PubMed] [Google Scholar]
- 4.Benhamou N, Souied EH, Zolf R, Coscas F, Coscas G, Soubrane G. Adult-onset foveomacular vitelliform dystrophy: A study by optical coherence tomography. Am J Ophthalmol. 2003;135:362–7. doi: 10.1016/s0002-9394(02)01946-3. [DOI] [PubMed] [Google Scholar]
- 5.Grob S, Yonekawa Y, Eliott D. Multimodal imaging of adult-onset foveomacular vitelliform dystrophy. Saudi J Ophthalmol. 2014;28:104–10. doi: 10.1016/j.sjopt.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Querques G, Capuano V, Costanzo E, Corvi F, Querques L, Introini U, et al. Retinal pigment epithelium aperture: A previously unreported finding in the evolution of avascular pigment epithelium detachment. Retina. 2016;36(Suppl 1):S65–72. doi: 10.1097/IAE.0000000000001231. [DOI] [PubMed] [Google Scholar]
- 7.Pierro L, Tremolada G, Introini U, Calori G, Brancato R. Optical coherence tomography findings in adult-onset foveomacular vitelliform dystrophy. Am J Ophthalmol. 2002;134:675–80. doi: 10.1016/s0002-9394(02)01685-9. [DOI] [PubMed] [Google Scholar]
- 8.Abengoechea-Hernández S, Elizalde-Montagut J, Fideliz de la Paz-Dalisay M. Photodynamic therapy in adult-onset foveomacular vitelliform dystrophy. Arch Soc Esp Oftalmol. 2007;82:117–20. doi: 10.4321/s0365-66912007000200011. [DOI] [PubMed] [Google Scholar]
- 9.Benhamou N, Messas-Kaplan A, Cohen Y, Gaudric A, Souied EH, Soubrane G, et al. Adult-onset foveomacular vitelliform dystrophy with OCT 3. Am J Ophthalmol. 2004;138:294–6. doi: 10.1016/j.ajo.2004.02.076. [DOI] [PubMed] [Google Scholar]
- 10.Puche N, Querques G, Benhamou N, Tick S, Mimoun G, Martinelli D, et al. High-resolution spectral domain optical coherence tomography features in adult onset foveomacular vitelliform dystrophy. Br J Ophthalmol. 2010;94:1190–6. doi: 10.1136/bjo.2009.175075. [DOI] [PubMed] [Google Scholar]
- 11.Chen KC, Jung JJ, Curcio CA, Balaratnasingam C, Gallego-Pinazo R, Dolz-Marco R, et al. Intraretinal hyperreflective foci in acquired vitelliform lesions of the macula: Clinical and histologic study. Am J Ophthalmol. 2016;164:89–98. doi: 10.1016/j.ajo.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Patrinely JR, Lewis RA, Font RL. Foveomacular vitelliform dystrophy, adult type. A clinicopathologic study including electron microscopic observations. Ophthalmology. 1985;92:1712–8. doi: 10.1016/s0161-6420(85)34097-6. [DOI] [PubMed] [Google Scholar]
- 13.Jaffe GJ, Schatz H. Histopathologic features of adult-onset foveomacular pigment epithelial dystrophy. Arch Ophthalmol. 1988;106:958–60. doi: 10.1001/archopht.1988.01060140104034. [DOI] [PubMed] [Google Scholar]
- 14.Dubovy SR, Hairston RJ, Schatz H, Schachat AP, Bressler NM, Finkelstein D, et al. Adult-onset foveomacular pigment epithelial dystrophy: Clinicopathologic correlation of three cases. Retina. 2000;20:638–49. doi: 10.1097/00006982-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Saito W, Yamamoto S, Hayashi M, Ogata K. Morphological and functional analyses of adult onset vitelliform macular dystrophy. Br J Ophthalmol. 2003;87:758–62. doi: 10.1136/bjo.87.6.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gass JD. Pathogenesis of tears of the retinal pigment epithelium. Br J Ophthalmol. 1984;68:513–9. doi: 10.1136/bjo.68.8.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chowers I, Tiosano L, Audo I, Grunin M, Boon CJ. Adult-onset foveomacular vitelliform dystrophy: A fresh perspective. Prog Retin Eye Res. 2015;47:64–85. doi: 10.1016/j.preteyeres.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Balaratnasingam C, Messinger JD, Sloan KR, Yannuzzi LA, Freund KB, Curcio CA. Histologic and optical coherence tomographic correlates in drusenoid pigment epithelium detachment in age-related macular degeneration. Ophthalmology. 2017;124:644–56. doi: 10.1016/j.ophtha.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]