


Abstract
Thymine DNA glycosylase (TDG) initiates base excision repair by cleaving the N-glycosidic bond between the sugar and target base. After catalysis, the release of excised base is a requisite step to terminate the catalytic cycle and liberate the TDG for the following enzymatic reactions. However, an atomistic-level understanding of the dynamics of the product release process in TDG remains unknown. Here, by employing molecular dynamics simulations combined with the Markov State Model, we reveal the dynamics of the thymine release after the excision at microseconds timescale and all-atom resolution. We identify several key metastable states of the thymine and its dominant releasing pathway. Notably, after replacing the TDG residue Gly142 with tyrosine, the thymine release is delayed compared to the wild-type (wt) TDG, as supported by our potential of mean force (PMF) calculations. These findings warrant further experimental tests to potentially trap the excised base in the active site of TDG after the catalysis, which had been unsuccessful by previous attempts. Finally, we extended our studies to other TDG products, including the uracil, 5hmU, 5fC and 5caC bases in order to compare the product release for different targeting bases in the TDG–DNA complex.
INTRODUCTION
Thymine DNA glycosylase (TDG) plays an essential role in correcting mismatched/damaged DNA base pair (bp) by cleaving the N-glycosidic bond between the sugar and target base through the base excision repair pathways (1–3). TDG specifically recognizes the double-stranded DNA containing U·G or T·G mismatches caused by the deamination of the cytosine or 5-methylcytosine (5mC), thus preventing the C→T mutation. More recently, TDG has also been found to be involved in an active DNA demethylation process by removing the oxidized products of the 5mC and 5-hydroxymethylcytosine (5hmC), namely the 5-formylcytosine (5fC) and 5-carboxycytosine (5caC), respectively, which are generated by ten-eleven-translocation (Tet) proteins (4–8). This epigenetic modification conducted by TDG has profound influences on maintaining the genome integrity and correctly programming the embryogenic development (9–13).
Despite of recognizing a broad range of substrates, TDG adopts a universal base-flipping mechanism to carry out the catalytic reaction, thereby the target bases can be flipped out of the DNA duplex to reach into the active site of TDG, as observed in several TDG–DNA complexes (1,14–17). For example, in a recent determined TDG–DNA complex (PDB ID: 5hf7) (16), a flipped dU analog is inserted into a binding pocket formed by several TDG residues, including Tyr152, Ala145, Pro153 and Asn191 that can form direct contacts with the uracil, and His151 and Asn140 that indirectly interact with the uracil through water molecules. Notably, the void room left by the flipped base is occupied by a key TDG residue Arg275 from an intercalation loop region (residues 270–281), which prevents the return of the flipped base back to the DNA duplex (see Figure 1A). Finally, after catalysis, the cleaved base is released from the active site, creating an abasic (AP) site within the DNA. Therefore, the product release is a requisite step to terminate the catalytic cycle and liberate the enzyme for subsequent reactions. Recently, several studies have been devoted to capture the ternary product complex for the TDG using crystallographic methods. Hashimoto et al. have recently reported the crystal structures of the 5-hydroxymethyluracil (5hmU) and 5caC bound TDG–DNA complexes (18,19). Nonetheless, Malik et al., using an improved crystallization condition, failed to trap any type of cleaved bases in TDG, suggesting that the product is prone to release from the active site right after the reaction and exhibits low binding affinity with the TDG–DNA complex (20). Based on the high-resolution structures, Malik et al. also proposed a possible product releasing pathway along a solvent-filled channel (20).
Figure 1.

(A) Schematic illustration of the DNA repair process in TDG. (B) The modeled ternary product complex. TDG is shown in gray and DNA chain is shown in orange cartoon. A zoomed-in view of the active site is provided on the right panel, with the thymine represented in green sticks, the key residues that form direct/indirect contacts with the thymine shown in gray sticks, and the AP site in orange sticks. The Pro-rich loop (residues Pro153 to Asn157) and the gating helix (residues Leu143 to Lys148) regions are shown in wheat and cyan, respectively. The intercalated residue Arg275 is highlighted in blue sticks. The references to color are only for the online version.
Although the crystallographic studies have provided atomistic-level understandings of the structural features of the TDG–DNA complexes (21–23), the detailed dynamics of the product release process remains unknown, e.g. is it possible for the product to release through the proposed solvent-filled channel? How does the release process actually happen? In addition, since the ternary product complexes for the TDG system have not been determined yet, we wonder whether a TDG variant exists that can potentially trap the cleaved base in the active site after the catalysis. However, due to the transient dynamics, it is still challenging for the current experimental techniques to directly capture the product release process in TDG. Molecular dynamics (MD) simulation has become a powerful theoretical and computational tool to reveal the dynamic properties of the bio-molecules at an atomistic resolution (24). Moreover, a Markov state model (MSM) can be constructed based on a large number of MD simulations to extract both thermodynamic and kinetic properties for certain conformational changes taken place at a relatively long timescale (i.e. microsecond or longer) that is inaccessible by performing a long unbiased MD simulation (25–30). By discretizing the conformational phase space into many sub-states based on certain metrics, i.e. the root mean square deviation (RMSD), one can construct an MSM using a coarse-grained lag time Δt and then build a transition probability matrix T where each entry Tij represents the transition probability from state i to state j after a certain lag time Δt; the transition only depends on the current state i but not the preceding states (i.e. the markovian process). In this manner, one can investigate the comparatively long timescale dynamics by propagating the transition probability matrix to a given time of interest according to the following equation:
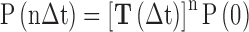 |
By diagonalizing the transition probability matrix T, the stationary distribution can be found as the right eigenvector, corresponding to the eigenvalue of 1. The entries of the vector correspond to the probabilities of each state in the stationary distribution. Other eigenvalues <1 can be used to derive the kinetic properties by calculating the implied timescales (see ‘Materials and Methods’ section 6 for more details). In recent years, the MSM method has been used to study many critical bio-molecular events, including protein folding, protein–ligand recognition and protein conformational dynamics in general (31–46).
In this work, we investigated the dynamics of the product release process in TDG at an atomistic resolution by constructing an MSM based on hundreds of MD simulations with an aggregated simulation time of ∼13 μs. We explored the thymine release through a solvent-filled channel, which is flanked by a Pro-rich loop and a gating helix domain (residues Gly138 to His158, see Figure 1B). Our MSM reveals four metastable states (S1–S4) during the thymine release and predicts that the overall product release process takes place at a timescale of ∼10 μs. Notably, our MD simulations captured a key intermediate state, which can be stabilized by a pre-existing TDG conformation, indicating a conformational selection mode of the binding between TDG and the product. Moreover, for comparative studies, we performed potential of mean force (PMF) calculations to reveal the relative releasing kinetic rates for several alternative bases (e.g. uracil, 5hmU, 5fC and 5caC) by employing the thymine as a reference system. Finally, we explored potential strategies to delay the product release from the active site of TDG, by replacing a glycine located in the releasing channel with a bulkier residue. Our work thus aims at revealing the structural basis of the product release in TDG and providing rational experimental design strategies.
MATERIALS AND METHODS
Modeling of the TDG–DNA complexes bearing one G·T mismatched bp before and after the catalysis
We first built the TDG–DNA complex containing one G·T mispair before the cleavage based on one recently resolved crystal structure (PDB ID: 5hf7) (16). We replaced the Fluorine atom of the flipped 2′-fluoroarabino analogue of dU with H atom, and kept all the DNA chains containing 24 bps. Then, the dU nucleotide was modified to dT by adding one methyl-group on the 5-site of the pyrimidine ring. The missing side chains of several TDG residues were added. This final model of the TDG–DNA complex was subject to energy minimization using Gromacs-4.6 package to relieve the steric clashes (see Supplementary Figure S1A). We then used this minimized TDG–DNA structure to construct the ternary product complex by directly cleaving the N-glycosidic bond of the flipped dT nucleotide, followed by energy minimization (see Supplementary Figure S1B). The sugar conformation of the AP site after the nucleophilic attack by one water molecule was modeled as an α-form rather than a β-form as suggested by previous studies (47). Although the β-form can be an alternative conformation after the product release (16), an α-form is used here since we are focused on the overall product release process starting right after the catalytic reaction. The comparison of these above two minimized TDG–DNA complexes, before and after the catalysis, indicates subtle differences in the active site regions (see Supplementary Figure S1). Finally, the minimized structure of the ternary product complex was used for the subsequent MD simulations.
Setup of the MD simulations and equilibration of the ternary product complex
All the MD simulations were performed using GROMACS 4.6 package (48–50). The ternary product complex was centered in a triclinic box with a minimal system and box wall distance set to 7.0 Å. The whole system was solvated in a simple point-charge (SPC) water box (51), and in order to neutralize the system and ensure an ionic concentration of 0.15M, we added 93 Na+ and 44 Cl− ions. The final system contains 51 935 atoms. We employed the AMBER99sb force field with PARMBSC0 corrections for the nucleic acid to describe the whole system (52–55). The amber force fields of the thymine and other TDG products, including the uracil, 5fC, 5hmU and 5caC, were generated using the Antechamber module implemented in the AmberTools package (56,57), and the RESP charges were calculated after a full optimization of each base using the Hartree-Fork methods under the basis set of 6–31G* (see Supplementary Figure S2 for the complete charge parameters of all the above bases). A switched Van der Waals type was used with a cutoff of 12 Å. We set a cutoff for the short-range electrostatic interactions to 12 Å and treated the long-range electrostatic interactions using the Particle-Mesh Ewald summation method (58). LINCS algorithm was used to constrain all the chemical bonds (59).
Starting from the above modeled ternary product complex, we first performed energy minimization using the steepest decent method, followed by a 500-ps restrained MD simulation by constraining all the heavy atoms of the system (protein, nucleic acid and the thymine). We then performed temperature annealing by increasing the temperature from 50 K to 310 K within 200 ps. Finally, we equilibrated the whole system by performing one unbiased 100-ns NVT MD simulation with a time step of 2 fs at 310 K using the velocity rescaling thermostat (60). The final snapshot from the above MD trajectory was used for the subsequent pulling simulations.
Performing Steered MD (SMD) simulations to obtain the initial product release pathways
Next, in order to obtain an initial product releasing pathway, we employed Steered MD (SMD) to pull the thymine out of the TDG active site using the last snapshot of the above 100-ns MD trajectory. A close examination on the TDG structure reveals no alternative channel or pocket from the TDG surface for the product to release except for the solvent-filled channel as identified by former studies (20) (see Supplementary Figure S3C). To determine the dominant product releasing pathway, we conducted two sets of SMD simulations: with and without pre-defined pulling directions. For the first setup, we pulled the center of mass (COM) of the thymine along the abovementioned solvent-filled channel toward two opposite directions using two DNA P atoms as the reference points, respectively, (denoted as ref a and ref b in Supplementary Figure S3A) and using a pulling force constant of 0.3 kJ/mol/Å2. Notably, all three parallel SMD simulations show that the product fails to release along the pulling direction toward the ref b (see Supplementary Figure S3D). In contrast, the product can be readily released from the solvent-filled channel in all three parallel SMD simulations toward the ref a (see Supplementary Figure S3D). In addition, we designed a second SMD setup by only increasing the distance between the COM of the thymine and the Cα atoms of two TDG residues Asn191 and Ile134, respectively, without defining any pulling direction in order to automatically determine which releasing path the product would use (denoted as ref c in Supplementary Figure S3A). Consistent with the results from the first SMD setup, the product is unambiguously released through the solvent-filled channel in all above SMD simulations (see Supplementary Figure S3E and F). Taken together, our results confirm that the TDG product is determined to release along the solvent-filled channel, as also suggested by former experimental studies (20).
Moreover, former quantum-mechanical/molecular-mechanical studies have indicated that the protonated form of the His151 is critical for the catalytic reaction in TDG (61). To evaluate the role of the His151 in product release process, we designed three different TDG systems with varied His151 forms, namely the ε-histidine form (HIE), the protonated form (HIP) and the δ–histidine form (HID) (see Supplementary Figures S1, S4A and B). For each system, using the same pulling parameters, we then performed one set of SMD simulations to exam how different His151 forms might affect the product release process. As shown in Supplementary Figure S4C, the results show that for all three systems, the product can be readily released from the active site within 20 ns, suggesting that different His151 forms have no substantial influences on the product release process. We thus used the HIE form of the His151 throughout this work. Next, we performed two additional SMD simulations along the ref a using two different pulling force constants, 0.1 and 0.2 kJ/mol/Å2. We finally performed geometric clustering for all the conformations derived from the SMD simulations along the solvent-filled channel and chose representative conformations as the starting structures to shoot more unbiased MD simulations.
Geometric clustering of the SMD conformations
Based on the initial thymine release pathways, we then performed geometric clustering by at first projecting all the high-dimensional SMD conformations onto a low-dimensional space using time-structure independent component analysis (tICA) (62–65). tICA can efficiently capture the slowest dynamics for a certain conformational change in a system by constructing a time-lagged correlation matrix. This information, however, is hard to be extracted using other related dimension-reduction techniques (e.g. the principle component analysis, or PCA). In recent years, tICA has been successively applied to study many dynamic processes when constructing the MSM for bio-molecules (25,28,34,46). Different metrics, i.e. distance, contact number etc, can be used as the order parameters for the tICA. In current work, we selected a total of 513 distance pairs between the heavy atoms of the thymine and several other TDG/DNA motifs (see Supplementary Figure S5A), specified as following:
heavy atoms of thymine – Cα atoms of TDG residues K107 to L124.
heavy atoms of thymine – Cα atoms of TDG residues G138 to H158.
heavy atoms of thymine – 3 sidechain heavy atoms of TDG residue N191.
heavy atoms of thymine – 7 sidechain heavy atoms of TDG residue Y152.
heavy atoms of thymine – 8 P atoms of DNA nucleotides out of the active site.
We then projected each SMD conformation onto the top four slowest tICs, followed by clustering the projected conformations into 100 classes using the K-centers algorithm implemented in the MSMbuilder package (25,66). Finally, from each cluster, the center conformation was selected as the representative structure for the subsequent unbiased MD simulations.
Seeding unbiased MD simulations
In addition to the above 100 representative conformations, we selected 25 additional conformations in which the thymine located near to the exit of the release channel in order to ensure enough transitions could be observed. Then, the above chosen 125 conformations were directly subject to the unbiased 100-ns NVT MD simulation using the same MD setup as used to equilibrate the initial ternary product complex. Finally, we collected a total of 125 MD trajectories (100 ns each) with an aggregated simulation time of 12.5 μs and used this final simulation dataset to construct the MSM.
Construction and validation of the MSM
We employed a splitting and lumping procedure to construct the MSM. We first did geometric clustering for all the conformations from the above 125 100-ns MD simulations (in total of 1 250 000 MD snapshots) into different number of microstates by performing tICA projection followed by K-centers clustering using the same distance metric as described before (see Supplementary Figure S5A). Then, to dissect the detailed thymine release mechanism in TDG, we further lumped a 500-state MSM into four macrostates using the PCCA+ method (67), detailed as below:
Clustering MD conformations into different number of microstates
To evaluate the effects of microstate number and the correlation lag-time on the stability of the MSM, we performed tICA projections by constructing the time-lagged correlation matrix using three different correlation lag-times: 20, 30 and 40 ns, and for each case, we clustered the MD conformations into 500, 600, 700 and 800 states, respectively. Then, for each clustering setup (in total of 3 × 4 different clustering), we constructed the transition count matrix C in which each entry Cij was obtained by counting the transition numbers for each microstate pair (i and j) from the unbiased MD simulations. This raw count matrix was then symmetrized and converted to a transition probability matrix T by normalizing each entry by the sum of the row.
Next, we calculated the eigen-function of the TPM from which the kinetic properties were obtained by calculating the implied timescales according to the following equation:
 |
where, μk is the k-th eigenvalue of the transition probability matrix T with lag time τ, each implied timescale curve indicates the average transition time between two sets of conformations. Therefore, we can readily estimate at what timescale the slowest transition, in most cases also the principal conformational change of interest, might take place. For each clustering, the slowest implied timescale curves are all converged at about tens of microseconds (see Supplementary Figure S6), suggesting our MSM is robust to the different choices of either the microstate number or correlation lag-times.
To reveal the metastable states involved in the thymine release process, we chose the 500-state MSM at the lag-time of 20 ns as a representative model and further lumped the 500 microstates into four macrostates using the Robust Perron Cluster Cluster Analysis (PCCA+) method implemented in MSMbuilder 2.7 package (67).
Validation of the MSM
To exam if our MD simulations are sufficient to build a reliable MSM, we sub-sampled the original MD simulations by choosing subsets of the conformations from each MD trajectory, with each subset data containing an aggregated simulations time of (125 × 50 ns), (125 × 60 ns), (125 × 70 ns), (125 × 80 ns), (125 × 90 ns) and (125 × 100 ns), respectively. Then for each subset data, we used a correlation lag-time of 20 ns for the tICA projection and clustered the corresponding MD conformations into 500 states. We then projected each set of conformations onto the same top two tIC vectors as that for the complete dataset (see Supplementary Figure S7). The results show a clear convergence of the MD samplings along the two slowest tICs, suggesting that our MD simulations are fairly sufficient to build a reliable MSM. Next, to exam if the kinetics (i.e. the timescale at which the slowest event takes place) would be changed or not, we constructed the MSM for each subset data and calculated the implied timescales with the errors estimated by bootstrapping the trajectory-list 100 times. The results clearly show that, for each subset data, the slowest transition times are consistently converged to about tens of microseconds (see Supplementary Figure S8), indicating that the MSM is reliable to predict reasonable kinetic properties. To further validate our MSM, we performed the Chapman–Kolmogorov test for several microstates that are the most populated from our MD simulations. The results show that the evolution of the residence probability of the microstates observed from the raw MD simulations data is in good agreement with the values predicted by the MSM (see Supplementary Figure S9).
Calculations of the mean first massage time (MFPT) and stationary distributions
We finally calculated the mean first massage time (MFPT) for each transition and the equilibrium population for each state. To estimate the corresponding errors, we generated 100 trajectory lists with each containing 125 randomly chosen trajectories from the original MD simulations. Then for each trajectory list, we constructed the corresponding TPM from which a 10-ms long Monte Carlo (MC) simulation was then generated. This MC trajectory is so long that the stationary distribution for each state and the MFPT value for each transition can be readily obtained. Finally, the mean and standard deviation were calculated by averaging over the results from the above 100 bootstrapping.
Setup of the pulling simulations for the PMF calculations
To evaluate the relative kinetic rates of the product release between the varied systems (including the uracil, 5fC, 5caC, 5hmU in wild-type (wt) TDG, and the thymine in G142Y mutant TDG) and the thymine release in wt TDG that is served as a reference system, we carried out the PMF calculations for each of the product release processes along the dominant releasing path. The PMF shows the free energy changes along a certain reaction coordinate (RC). Comparisons of the free energy landscapes between different systems can thus provide the relative kinetic rates for certain transitions. Here, we chose the distance between the COM of each product and one backbone P atom of the DNA chain as the RC for the PMF calculations (defined as drc), which is the same reference point we used for the initial SMD simulations (see Supplementary Figure S10). We then performed a number of constant velocity SMD (cv-SMD) simulations for each of the above six systems to calculate the PMF for the product release along the drc using the Jarzynski’s equality that evaluates the free energy difference between two states from the work done through many realizations of the process connecting the two states (68):
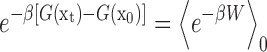 |
where, β = 1/kBT (kB is the Boltzmann constant, and T is the temperature in kelvin), x denotes a time-dependent RC evolving from an initial value of x0 at time t = 0 to xt = x(t) at a certain time t, and the angular bracket averages over an ensemble of trajectories starting from an equilibrated distribution of the initial state and arriving to the final state corresponding to xt. Employing the same protocol as described by previous simulation studies using the Jarzynski method (69,70), we divided the product releasing channel into two sections, with the drcvalue, serving as the RC x here, decreased from ∼20 Å to 15 Å (section I) and then from 15 Å to 9 Å (section II). Notably, these two sections encompass the S1→S2 and S2→S3 transitions for the thymine release in the wt TDG, respectively, as seen from the calculated drcvalue based on our MSM (the mean drcvalue is ∼16 Å for S2 and ∼9 Å for S3, see Supplementary Figure S10 and the main text for the results of the MSM).
We, at first constructed the ternary product complexes for each product based on the thymine system. For each case, we then performed one 100-ns MD simulations to fully relax the structure and the final MD snapshots were used as the input structures for the cv-SMD simulations. Then, for each of the six system, namely the thymine (in both wt and G142Y mutant TDG), uracil, 5fC, 5caC and 5hmU systems, we performed eight 5-ns cv-SMD simulations for each section (8 × 2 × 6 = 96 simulations in total, which amounts to ∼0.5 μs simulation time in aggregation for the PMF calculations), with a pulling rate v = ∼1 Å/ns and a force constant k = 30 kcal/mol/Å. We applied the force on the COM of the product and calculated the external work using the following equation:
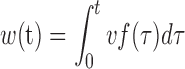 |
where, the f(τ) is the applied external force at time τ. We finally resorted to the second order cumulant expansion of the external work to calculate the free energy changes (69,70):
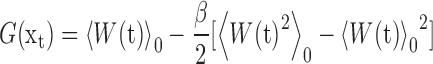 |
where, we define 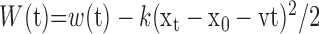 .
.
Generating a base-flipping pathway for the G142Y mutant TDG–DNA complex using Targeted MD (TMD) simulations
To exam whether the G142Y substitution might potentially affect the base-flipping process, we performed the TMD simulation to obtain a base-flipping pathway for the dT nucleotide. To achieve this, we at first built one interrogating complex and one recognition complex for the TDG–DNA system in which the mispaired dT was in an intrahelical and an extrahelical form, respectively. The above modeled TDG–DNA complex prior to the catalysis serves as the model of recognition complex (see Supplementary Figure S1A). To model the interrogating complex, we employed the double-stranded DNA chains from the above recognition complex and constructed an intrahelical T·G-mispair containing DNA duplex (see the Supplementary Figure S11A for the energy minimized DNA structure). We then relaxed the DNA backbones by performed one 7 ns MD simulations and the last snapshot was used for modeling the final TDG–DNA binding complex (see the Supplementary Figure S11A). Notably, we adopted the TDG conformation that forms non-specific complex with the DNA duplex where the intercalated residue Arg275 is lying along the minor groove (PDB ID: 2rba) (21). This model was subjected to the energy minimization and served as the final model of interrogating complex. Finally, we introduced the G142Y mutation to both the interrogating complex and recognition complex (see the Supplementary Figure S12A and B), and performed TMD simulations to obtain the base-flipping pathway by targeting the interrogating complex to recognition complex and constraining the backbone P atoms of two DNA ends using a force constant of 500 kcal/mol/Å2 (see Supplementary Figure S11B). Several discontinuous regions were selected as the targeting regions using a pulling force constant of 5 kcal/mol/Å2, including 16 P atoms of several DNA nucleotides locating at the middle part of the DNA chain; the heavy atoms of the mispaired dT nt and one of its adjacent nucleotide; the heavy atoms of the intercalated residue Arg275; the Cα atoms of the TDG residue from Cys276 to Glu303 (see Supplementary Figure S11B). The amber force field 99SB with the parambsr0 correction was used to describe the TDG–DNA system.
RESULTS
Structural features of the ternary product complex
We built the ternary product complex based on the crystal structure of the TDG–DNA complex prior to the catalysis (PDB ID: 5hf7) (16), which was at first subject to an energy minimization (see Supplementary Figure S1A). Then, by directly cleaving the N-glycosidic bond between the sugar and the base, we obtained a locally stabilized ternary product complex (see Figure 1B, Supplementary Figure S1B and ‘Materials and Methods’ section 1 for more details of the model construction). In this structure, the thymine can form direct packing interactions with the residue Tyr152, while the same interaction can also be found before the catalysis (see Supplementary Figure S1A). The 5-methyl group of the thymine locates in a hydrophobic pocket formed by residues Pro153 and Ala145 (see Figure 1B). In addition, the thymine O2 atom forms a hydrogen bond (HB) with the Ile139 backbone N-H; the N3 and O4 atoms can form two HBs with the side chain of residue Asn191 by expelling one crystal water molecule as observed in the crystal structure (PDB ID: 5hf7); the O4 atom can also indirectly interact with the His151 through one water molecule. Moreover, we treated the sugar group at the AP site, after a nucleophilic attack by a water molecule, as an α-form rather than a β-from, as implicated by previous studies (47,71,72). Taken together, in general the thymine can retain all the major interactions with TDG before and after the catalysis (see Supplementary Figure S1). This modeled ternary product complex was finally used for investigating the product release process.
MSM reveals four metastable states during the thymine release in TDG
To determine the dominant product releasing pathway, we have performed two sets of SMD simulations starting from one well-equilibrated ternary product complex. We are able to confirm that the solvent-filled channel flanked by the Pro-rich loop (residues Pro153 to Asn157) and the gating helix (residues Leu143 to Lys148) region is the major product release pathway (see ‘Materials and Methods’ section 3 for more details). Four metastable states have been clearly identified from the MSM during the thymine release in TDG, namely S1–S4 states (see Figure 2). At a glimpse, the product passes through the releasing channel by at first visiting the S1 and S2 states (see Figure 2A). Then the release pathway is promptly broadened up so that the thymine can be directly released to the bulk water regions. Accordingly, the S3 and S4 states can be viewed as the released states.
Figure 2.

The MSM reveals four metastable states for the thymine release in TDG. (A) For each of the S1 (green), S2 (purple), S3 (blue) and S4 (gray) states, random conformations of the thymine were chosen according to the stationary distributions. For each thymine conformation, only C2 atom is shown in sphere. The active site residue Tyr152 is also shown as a reference point. (B). The four-state kinetic network derived from the MSM. The size of each circle is roughly proportional to the corresponding equilibrium population: 42.2 ± 5.0% (S1); 0.8 ± 0.1% (S2); 28.4 ± 3.7% (S3); 28.6 ± 3.8% (S4). The MFPT for the forward transition is also provided above each arrow. (C–E). Representative conformations for the S1 (C), S2 (D) and S3 (E) states. The structure was randomly selected from the most populated microstate for each macrostate. Key residues and water molecules that interact with the thymine are shown in sticks, and the HBs between the thymine and neighboring molecules are highlighted with dashed lines. Refer to Figure 1 for other representations.
Specifically, in the S1 state, the most populated state among the four, the thymine remains stable in the active site and forms a similar interacting network with the neighboring residues or water molecules as observed in the initial ternary product complex, except that two additional waters enter into the active site and form two extra HBs with the N1 and O2 atoms of the thymine (compare Figures 1B and 2C). Then, the S1 state can transit to S2, the least stable state. In S2, the thymine loses its interactions with the active site residues Tyr152, Ile139 and Asn191, while it is stuck within a region surrounded by the Pro-rich loop, the gating helix, and the DNA backbone. The thymine can actually form two HBs with the Met144 and Ala145 backbone N-H, one HB with the P-O− group at the AP site, and can also maintain hydrophobic contacts with the side chains of residues Pro153, Phe111 and Met144 (see Figure 2D). Notably, several water molecules can be found in the active site, occupying the void region left by the released thymine and forming water chains connecting the thymine and several active site residues, e.g. His151 and Asn191 (see Figure 2D). This water-mediated interacting network greatly compensates the energy cost after the thymine leaves the active site. From S2, the thymine can directly transit to either the S3 or the S4 state, while in the S3 state, the thymine has already reached at the interface between TDG and the bulk region, though in some of the configurations, the thymine can still form direct contacts with either the DNA chains or the N-terminal region of TDG (see Figure 2E). In one representative structure shown in Figure 2E where the thymine locates at the exit of the release channel, the N4 atom of the thymine can form one HB with the backbone oxygen of Pro153, and the other polar atoms are fully occupied by forming HBs with the solvent waters. In S4, however, the thymine is fully exposed to the solvents (see Supplementary Figure S13).
Our MSM predicts that the overall product release process in TDG occurs at a timescale of ∼10 μs, with the S1→S2 and S2→S3/S4 transitions taken place at comparable kinetic rates (see Figure 2B). As noted before, the S1→S2 transition results in impairing several HBs between the thymine and active site residues, i.e. Asn191, Ile139 and Tyr152. On the other hand, the contribution of the solvent waters to the stability of the thymine in the S2 state remains constant because the number of water molecules surrounding the thymine are comparable for the S1 and S2 states (see Figure 3B), which suggests that the thymine in S2 tends to return back to the active site due to its unstable features. Notably, the S2→S3 transition greatly increases the solvent accessible areas of the thymine owning to the sudden widening of the exit channel (see Figures 2A and 3A), reflected from a significant increase of the surrounding water molecules in the S3 and S4 states (see Figure 3B). This solvation process significantly compensates for the energy loss between the thymine and TDG residues, i.e. Pro153, Phe111 and Met144. Finally, we found that as long as the thymine reaches to the S3 state, the subsequent release process becomes relatively fast, taking place at about hundreds of nanoseconds. Interestingly, from some of the unbiased MD simulations, we indeed observed direct transitions of the thymine from S3 to the bulk region (see next section).
Figure 3.

(A) The free energy profile of the MD conformations projected onto the two slowest tICs, with each state from S1 to S4 labeled on the plot. (B) Solvents actively facilitate the thymine release. Four polar groups from the thymine, either serving as a hydrogen donor (HD) or acceptor (HA), were chosen for the analysis. The number of water molecules were then calculated using a distance cutoff of 3 Å between two heavy atoms. Finally, for each state, the probability of different number of water molecules surrounding the thymine is plotted.
Consistently, the free energy landscape of the MD conformations along the two slowest tICs clearly shows that the first tIC corresponds to the S1→S2→S3 or S1→S2→S4 transitions that take place at a timescale of ∼10 μs, according to the MFPT calculations (see Figure 2B). We then calculated the correlation coefficients between the first tIC and each of the order parameters used for constructing the MSM, consisting of 513 distance pairs. We pinpoint the exact distance pair that has the largest anti-correlation coefficients with the first tIC, i.e. the distance between the Cα atom of the TDG residue Gly138 and the O2 atom of the thymine (see Supplementary Figure S5B), which reflects how much the product has been released away from the active site. On the other hand, the second tIC is mainly attributed to the S3→S4 transition, which occurs at about hundreds of nanosecond (see Figure 3A).
It is also worth to note that although the S3 and S4 states can be further lumped into a single state to form a three-state MSM due to their comparatively fast transitions (compare Supplementary Figure S14A and B), they are biologically distinguishable. First, the S3 state is much closer to the interface between the narrow channel and the solvents compared to the S4 state (see Figure 2A), hence, there is a higher probability of the product to form direct contacts with TDG in S3. This idea is supported by the observation that the average distance between the thymine and the active site residue Asn191 is apparently smaller in S3 than that in S4 (see Supplementary Figure S14D). Moreover, the probability to fully solvate the thymine in S3, i.e. with 5, 6 or 7 surrounding waters, is noticeably lower than that in S4 (see Figure 3B), further suggesting that the product experiences different biological environments between S3 and S4. Accordingly, to illustrate a complete process of the product release in TDG, we adopted the four-state MSM.
Direct observation of the thymine release from the S3 to solvents within 100-ns MD simulations
After arriving at the S3 state (see Figure 2E), the thymine can readily release to the solvents, which has been directly observed from our 41 independent 100-ns MD simulations (see Figure 4). According to the direction of the thymine release, we classify these 41 MD trajectories into two groups, with each representing one thymine release pathway (path A and B). Particularly, in path A, captured in 28 out of 41 MD trajectories, the thymine releases ‘upward’ in the viewpoint represented in Figure 4, either above (red and blue paths in Figure 4A) or even in the perpendicular direction (green, cyan and purple paths in Figure 4A) to the extending direction of the DNA chain. On the other hand, in path B, observed in 13 MD trajectories, the thymine releases ‘downward’, either along the minor groove direction of the DNA chain (green and purple paths in Figure 4B) or by interacting with the N-terminus domain of TDG (cyan, red and blue paths in Figure 4B). Notably, in both pathways, prior to releasing to the solvents, the thymine can visit a region surrounded by several TDG residues, i.e. Arg110, Phe111, Met144 and Pro155, and the minor groove of the DNA chain (see Figure 2A and black dashed circles in Figure 4). After diffusing around the abovementioned region at a nanosecond timescale, the thymine can finally release to the solvents through one of the above pathways. In summary, as long as the product arrives at the exit of the releasing channel, the subsequent releasing process takes place almost immediately.
Figure 4.

Direct observations of the thymine release from the S3 state to the solvents from multiple 100-ns MD simulations. According to the releasing direction, two groups of release pathways are shown (A and B). For each group, five representative MD trajectories are demonstrated using different colors (blue, red, green, black and purple), in which the starting conformations of the MD trajectories are highlighted within dashed black circles, and the last MD snapshots (released states) are represented with spheres.
The relative movements between TDG and DNA chains facilitate the product release in a conformational selection mode
Our MSM indicates that the S2 state is a key intermediate state before the thymine releases to the solvents where several residues, i.e. Pro153 from the Pro-rich loop, along with Met144 and Ala145 from the gating helix region, can form direct contacts with the thymine (see Figure 2D). To see whether TDG or DNA underwent any structural changes to stabilize the thymine in the S2 state, we measured the relative distance between the Pro-rich loop and the gating helix domain for each macrostate. Notably, in S2, the distance between the COM of the gating helix and that of the Pro-rich loop region (denoted as d1 in Figure 5A) lies predominately around the high-value range (∼12 Å) compared to other three states, in which the d1 largely remains at a low value (∼9 Å) (see Figure 5A, C, D and E). The significant variation of d1 suggests that the above two TDG domains undergo an opening motion when the thymine reaches to the S2 state. Moreover, to examine the relative movements between TDG and the DNA chains, we calculated the distance between the gating helix domain and the P atom at the AP site on the DNA (denoted as d2 in Figure 5A). Again, the gating helix region shows notable movements relative to the DNA chain in the S2 state (d2 ∼ 12 Å), higher than that in the other states (d2 ∼10 Å; see Figure 5A, C, D and F).
Figure 5.

The dynamics of the Pro-rich loop and the gating helix regions play key roles in stabilizing the thymine in the S2 state via a conformational selection mechanism. (A–D) Structure highlights on the Pro-rich loop and the gating helix regions for each metastable state from the S1 to S4 states, respectively. The same representative structures from Figure 2 were used for the analyses. For each state, the starting minimized TDG–DNA complex (in transparent) was superimposed as a reference. (E and F) Projection of the MD conformations onto two RCs: one is the first tIC, and the other one is the distance d1 (denoted in A) between the COMs of the gating helix and the Pro-rich loop regions (E), or the distance d2 (also denoted in A) between the COM of the Pro-rich loop and the P atom at the AP site (F), with the location of each metastable state labeled on the plot.
The above results indicate that the opening motion of the gating helix and the Pro-rich loop in TDG, coupled with the relative movements between the gating helix and the DNA chain, can potentially enlarge the releasing channel, which in turn facilitates the thymine transiting to the S2 state after it leaves the active site. It is also worth to note that in the S1 state, even though the gating helix region is predominately in the closed state, it is also able to explore the open-state configurations, i.e. with both d1 and d2 values reaching to ∼12 Å, comparable to those in the S2 state (see Figure 5E and F). These findings suggest that the above opening motion is an intrinsic property of TDG, thereby transition of the thymine from S1 to S2 can selectively stabilize the transiently accessed open-state of the gating helix, implying a conformational-selection recognition mode. Since the S2 state is the requisite state for the thymine to finally release to the solvents, the above structural re-arrangement of the TDG domains and the DNA chain becomes a key regulator for the product release.
The product release in the G142Y mutant TDG is delayed compared to the wt
Our model suggests that the thymine can release through a narrow, solvent-filled channel (see Figure 2A). It is expected, therefore, that if the release channel is blocked by certain residues with bulky side chains, the product release process would be inhibited, so that one can potentially trap the product in the active site. Following this idea, we attempted to design a mutant TDG with bulkier residues lying within the product release channel. Notably, in the S2 state, Pro153 and Gly142 are relatively close to the thymine compared to other TDG residues (see Figure 6A). Owing to a smaller residue size of glycine, we thus pinpoint Gly142 as a potential candidate for further mutagenesis studies. In addition, in Uracil-DNA glycosylase (UDG), which is structurally related to TDG, the counterpart residue of Gly142 is a tyrosine (1), and people have successively captured one excised uracil in the active site of UDG in the crystal form (73). We therefore constructed a G142Y mutant TDG system for further studies (see Figure 6C).
Figure 6.

The thymine release in the G142Y mutant TDG is delayed in comparison with the wt TDG. (A) Two TDG residues, Gly142 and Pro153, are shown to clamp the thymine in the S2 state. For the S1 and S2 states, the average distance between the COM of the thymine and the Cα atom of each residue from Gly138 to His158 was calculated. The lighter blue color indicates a shorter distance. (B) The PMF curves along the drc for the wt (in blue) and G142Y mutant (in orange) systems. The barrier height changes of the mutant system in reference to the wt TDG system for the S1→S2 and S2→S3 transitions (denoted as Δh12 and Δh23, respectively) are ∼7 kBT and 1 kBT, respectively. In the background, the mean and fluctuation of the drc values calculated based on the MSM are represented by colored boxes for S1 in green, S2 in magenta and S3 in blue (see the right panel in Supplementary Figure S10). The location of the key intermediate state is marked by colored stars. (C) The initial structure of the G142Y mutant TDG system used for the PMF calculations. (D and E) The observed intermediate states for the wt (D) and G142Y systems (E) (marked as colored stars in B), which are equivalent to the S2 state from the MSM.
To evaluate the relative kinetic rates of the thymine release between the wt and the G142Y systems, we carried out the PMF calculations for the thymine release along the solvent-filled channel in both systems (refer to the ‘Materials and Methods’ section 7 for the details of the PMF setup). We chose the distance between the COM of the thymine and one backbone P atom of the DNA chain as the RC for the PMF calculations (defined as drc). As shown in Figure 6B, for the wt system, we observe two transition events, with the drcvalue decreased from ∼20 Å to ∼15 Å and from ∼15 Å to ∼10 Å (see Figure 6B), which correspond to the S1→S2 and S2→S3 transitions, respectively, as observed from our MSM (compare Figure 6B and Supplementary Figure S10). Moreover, the intermediate state identified from the PMF landscape demonstrates very similar structural features to the S2 state (see Figure 6D). That is, the thymine is trapped between the Pro-rich region and the gating helix region by forming two HBs with the backbone N-H of Met144 and Ala145, one HB with the P-O− group at the AP site, and forming nonpolar contacts with Pro153. The consistencies between the MSM and the PMF results suggest that our PMF calculations are capable of providing a reasonable free energy profile for the product release along the solvent-filled channel.
Compared to the wt system, the free energy barrier for the S1→S2 transition in the G142Y system is increased by ∼7.3 kBT, which corresponds to ∼1000-fold (ΔG# ≈ e−7.3) decrease in the transition rate (see Figure 6B), suggesting that the G142Y substitution has profound influences on the product release process. The captured intermediate state shows that the Tyr142 has to rotate away from the releasing channel in order to allow the product release, and forms π–π stacking with the thymine (see Figure 6E). In addition, for the S2→S3 transition, the barrier heights are similar to each other for these two systems, suggesting that after arriving at the S2 state, the subsequent kinetic rates would be similar for both systems. Taken together, the PMF calculations suggest that the G142Y mutation in TDG can delay the product release in comparison with the wt.
Comparisons of product release for different bases in TDG
In addition to the G·T mismatch, TDG also targets on other types of damaged DNA nucleotides, such as G·U, G·5hmU, G·5fC and G·5caC base pairs (8,74,75). To estimate the releasing kinetics for the above four nucleotides, we performed the PMF calculations for each base along the same releasing path (drc). We, at first constructed the ternary product complex for each base system based on the original thymine system. For each complex, we then performed one 100-ns MD simulations to fully relax the structure and the final snapshots were used as the input structures for the pulling simulations. As shown in Figure 7A, for both uracil and 5hmU systems, the product forms very similar interaction networks with the TDG residues, as observed for the thymine, by forming HBs with Asn191 and Ile139. On the other hand, the formyl oxygen of 5fC and carboxyl-group of 5caC can form an additional HB with the Tyr152 backbone N-H, and form nonpolar contacts with the methyl group of Ala145. Moreover, owing to the cytosine ring, several key interactions present for the thymine, uracil, and 5hmU systems, are partially (i.e. Asn191) or completely destroyed (i.e. His151) in the 5fC and 5caC systems (see Figure 7A). Finally, for each of the four ternary product complexes, we employed the same pulling protocol and parameters as used for the thymine system to calculate the PMF.
Figure 7.

(A) Initial structures of four ternary product complexes for the uracil, 5hmU, 5fC and 5caC. These conformations are the input structures for the PMF calculations. The excised base is shown in green stick; key HBs are highlighted with dashed lines. (B) Comparisons of the PMF landscapes between the thymine (in blue) and each of the four altered base systems (in orange) along drc. The relative barrier heights for the S1→S2 transition in the uracil, 5hmU, 5fC and 5caC systems, comparing to the thymine system (Δh12), are ∼−5 kBT, −2 kBT, −1 kBT and −3 kBT, respectively. The relative barrier heights for the S2→S3 transition (Δh23) in the uracil, 5fC and 5caC systems are around ∼-5 kBT, −1 kBT and −1 kBT, respectively. The mean and fluctuation of the drc values calculated based on the MSM are represented with colored boxes for S1 in green, S2 in magenta and S3 in blue. The location of the key intermediate state is marked by colored stars. (C) The key intermediate states for each system (marked as colored stars in B). The products are shown in purple sticks for uracil, 5fC and 5caC but green sticks for 5hmU. The key HBs are indicated with dashed lines.
The PMF landscapes reveal two major transition events for the uracil, 5fC, and 5caC as observed for the thymine, however, only one dominant transition is observed for the 5hmU (see Figure 7B). In specific, during the S1→S2 transition, both the uracil and 5caC demonstrate lower transition barriers than that for the thymine (Δh12 < 0), suggesting faster releasing rates for these two products (see Figure 7B). The 5fC, however, exhibits a similar barrier height to the thymine (see Figure 7B). On the other hand, for the S2→S3 transition, the 5fC and 5caC show comparable barrier heights with the thymine, but the uracil demonstrate an even lower transition barrier. Therefore, in summary, comparing to the thymine, both the uracil and 5caC have faster releasing rates while the 5fC exhibits a similar releasing rate. Furthermore, from the structural perspectives, the observed intermediate state that is equivalent to the S2 state of the thymine shows that the uracil can form two HBs with the G142 backbone N-H and the P-O− at the AP site, demonstrating distinct structural features from the S2 state of the thymine (see Figure 7C). In contrast, for the 5fC and 5caC, the intermediate state can form one HB with the Ala145 backbone N-H via the formyl group in 5fC and the carboxyl group in 5caC. In addition, owning to the negative charge of the 5caC, two water molecules can form HBs with the carboxyl group of the base and the 5fC can form one HB with the P-O− at the AP site (see Figure 7C).
Interestingly, the 5hmU demonstrates quite different dynamic properties compared to all other bases. Remarkably, we identified an alternative binding site of the 5hmU in TDG at drc = ∼18 Å comparing to its initial location at drc = ∼20 Å (marked as blue star in Figure 7B). In this structure, the HBs between Asn191 and 5hmU that are present in the initial ternary model are switched to the backbone atoms of the residues Asn140 and Gly142 (compare Figure 7A and C), and the 5-hydroxylmethly group forms one HB with the P-O− at the AP site. Furthermore, different from other four bases, only one transition event was captured for the 5hmU as drcdecreased from ∼18 Å to ∼10 Å, with a transition barrier of similar height to that of the S1→S2 transition for the thymine.
In conclusion, the four different bases exhibit either a faster (e.g. uracil, 5caC and 5hmU) or a similar (e.g. 5fC) product releasing rate comparing to the thymine. Moreover, different bases share a common releasing mechanism by which the base can interact with both the Pro-rich loop and the gating helix regions. However, owing to their unique chemical structures, the specific contacting points vary for different bases.
DISCUSSION
Close examination of the TDG–DNA complex reveals only one most likely product release pathway filled with solvent waters as observed in the recent crystal structure (16). Previous experimental studies have suggested that the cleaved bases exhibit very weak binding affinities with TDG, and the following releasing process takes place very fast. Here, we employed extensive MD simulations (with an aggregated simulation time of ∼13 μs) combined with the MSM construction to reveal the dynamics of the product release in TDG at an atomistic resolution. We identify four metastable states during the thymine release in TDG (S1–S4 states), with S1 termed as the bound state, S3 and S4 as the released states. Essentially, the S2 state is a key intermediate state that can be transiently stabilized by the Pro-rich loop and the gating helix regions of TDG and several water molecules. As long as the thymine reaches to the S3 state, the subsequent release process takes place very fast and has been directly captured from our unbiased 100-ns MD simulations. Our MSM suggests that the overall release process of the thymine in TDG occurs at ∼10 μs. Given that TDG belongs to the family II of the UDG superfamily, similar product release pathways can be readily deduced for other UNG members that are structurally related to TDG.
Energetics and role of solvent waters during the thymine release
As shown in Figures 2A and 3B, the releasing pathway of the thymine is a very narrow, solvent-filled channel. The transition of the thymine from the S1 to S2 state is an enthalpy unfavorable process because it needs to break several interactions with the TDG residues in the active site, i.e. Asn191, Ile139, His151 and Tyr152. On the other hand, the number of the water molecules surrounding the thymine is comparable in the S1 and S2 states (see Figure 3B). Therefore, the S2 state is energetically less stable than the S1 state. Further release of the thymine from S2 to S3 greatly increases the solvent accessible area of the thymine and breaks most of its interactions with the TDG residues. Notably, the prompt increase of the entropy plays an important role in promoting the product release from the active site to the solvents, as reflected from the second tIC shown in Figure 3A. After arriving at the S3 state, the solvation become the main driving force to control the dynamics of the thymine by forming up to six HBs with the polar groups of the thymine (see Figure 3B). Accordingly, the solvents play an important role in facilitating the product release in TDG during the whole release process: first by mediating the interactions between the thymine and the active site residues during the S1→S2 transition; then by solvating the thymine once it leaves the narrow channel.
Intrinsic motions of TDG promotes the product release
Our findings suggest that the intrinsic dynamics of TDG and its coupled motions with the DNA chains are important for the product release. The Pro-rich loop and the gating helix regions of TDG maintain an intrinsic opening motion even when the thymine is still trapped in the S1 state (see Figure 5E). This opening motion can significantly enlarge the release channel, therefore, to accommodate the dissociating thymine. Interestingly, the transition of the thymine from S1 to S2 can take advantage of the above open-state TDG by interacting with both the gating helix and the AP site of the DNA duplex, suggesting a conformational selection mechanism of recognition. Although the S2 state is less stable comparing to other metastable states, it is the key intermediate state connecting the bound state and the released states (see Figure 2B). In this regard, the conformational selection from S1 to S2 for the TDG and thymine interaction is an efficient mechanism to promote the product release from the active site.
The G142Y mutation slows down the product release in TDG
Former experimental efforts have been devoted to trapping the product in the active site of the TDG–DNA complex (18–20). However, due to the transient product releasing event, the structure basis for the product-bound TDG–DNA complex remains unclear. This work designed a mutant G142Y TDG in which, comparing to the wt TDG, the thymine release process is delayed by the side chain of the replaced Tyr142 that locates within the release channel (see Figure 6B). The PMF calculations show that the thymine release in the G142Y mutant TDG exhibits ∼1000-fold reduction in the releasing rate comparing to the wt system, reaching a releasing timescale of ∼10 ms. The findings shed light on further experimental trials along this route to potentially capture the excised base in the active site of the TDG–DNA complex.
Notably, our control simulations suggest that the G142Y substitution has no substantial effects on either the TDG structure or the dynamics of the base-flipping process. We compared the 100-ns MD simulations for the wt and G142Y TDG–DNA complexes. For both systems, the TDG structure reaches to a well-equilibrated conformation, as reflected from the converged RMSD values for the TDG backbones (see Supplementary Figure S15A). In comparison, the G142Y system is shown to exhibit ∼1 Å higher RMSD fluctuations compared to the wt system. Further comparisons of the MD snapshots at 100 ns for these two systems demonstrate that the above structural difference mainly lies in the gating helix region where the G142Y substitution is introduced (see Supplementary Figure S15B). Notably, the above minor structural perturbation has no effects on either the secondary structures of the gating helix domain or the location of the residue Tyr142 within the product release channel. Moreover, we performed the TMD simulation to exam whether the G142Y substitution might potentially affect the base-flipping process (see Supplementary Figure S12A and B and the ‘Materials and Methods’ section for more details of the model construction and the TMD setup). By targeting the interrogating complex to the recognition complex, it is shown that the residue Tyr142 is not lying on the base-flipping pathway; it is thus unlikely to make impacts on the dynamics of the base-flipping process (see Supplementary Figure S12C and D and Supplementary Movie for the complete base-flipping process).
Relative product releasing rates for different bases in TDG–DNA complex
By constructing the MSM, we obtained the product releasing timescales for the thymine in the TDG–DNA complex. To estimate the kinetic rates for other bases, we employed the thymine as a reference and performed PMF calculations to evaluate the relative product releasing rates for different bases along the same releasing channel, from which we get to know the releasing timescales for other products. The PMF landscape for the thymine successively captures the main transition events (e.g. S1→S2 and S2→S3) and key intermediate state that have been revealed by the MSM. The consistencies suggest that the PMF setup is appropriate to derive the free energy landscapes for different bases. Our results show that due to the lack of one methyl group, the uracil has a faster releasing rate than the thymine. Similarly, the 5caC also releases faster than the thymine, which is likely caused by the stabilizing effects from the water molecules owning to the carboxyl group of the 5caC. In comparison, the 5fC exhibits a similar releasing rate to the thymine, and the corresponding intermediate state also demonstrates a similar interaction network with the TDG–DNA complex to that for the thymine system, i.e. by forming two HBs with the Ala145 N-H and AP P-O− groups (see Figure 7C). In contrast, the 5hmU can form one HB with the AP P-O− group due to the presence of the 5-hydroxymethyl group right after the catalysis, which is not observed in other base species. This specific interaction leads to a rather different binding site of the 5hmU in TDG, which is captured by our pulling simulations.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the computational support from the Beijing Computational Science Research Center (CSRC) and the Center for HPC, Shanghai Jiao Tong University.
Footnotes
Present address: Lin-Tai Da, Key Laboratory of Systems Biomedicine (Ministry of Education), Shanghai Center for Systems Biomedicine, Shanghai JiaoTong University, 800 Dongchuan Road, Shanghai 200240, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Pujiang Talent Project of Shanghai [17PJ1403600]; Shanghai Center for Systems Biomedicine, Shanghai Jiao Tong University, Startup Fund [WF220441503]; NSFC [11635002 to J.Y., 81502423 to Y.S.]; NSAF [U1530401 to J.Y.]; SJTU Chen Xing Type B Project [16X100080032]. Funding for open access charge: Pujiang Talent Project of Shanghai [17PJ1403600].
Conflict of interest statement. None declared.
REFERENCES
- 1. Schormann N., Ricciardi R., Chattopadhyay D.. Uracil-DNA glycosylases-structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci. 2014; 23:1667–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cortázar D., Kunz C., Saito Y., Steinacher R., Schär P.. The enigmatic thymine DNA glycosylase. DNA Repair. 2007; 6:489–504. [DOI] [PubMed] [Google Scholar]
- 3. McCullough A.K., Dodson M.L., Lloyd R.S.. Initiation of base excision repair: glycosylase mechanism and structure. Annu. Rev. Biochem. 1999; 68:255–285. [DOI] [PubMed] [Google Scholar]
- 4. He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L. et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011; 333:1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y.. Tet proteins can convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science. 2011; 333:1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shen L., Wu H., Diep D., Yamaguchi S., D’Alessio A.C., Fung H.L., Zhang K., Zhang Y.. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013; 153:692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu X., Zhang Y.. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 2017; 18:517–534. [DOI] [PubMed] [Google Scholar]
- 8. Maiti A., Drohat A.C.. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011; 286:35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bellacosa A., Drohat A.C.. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair. 2015; 32:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drohat A.C., Coey C.T.. Role of base excision “Repair” enzymes in erasing epigenetic marks from DNA. Chem. Rev. 2016; 116:12711–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A., Coz M.L., Devarajan K., Wessels A., Soprano D.. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011; 146:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cortázar D., Kunz C., Selfridge J., Lettieri T., Saito Y., Macdougall E., Wirz A., Schuermann D., Jacobs A.L., Siegrist F.. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011; 470:419–423. [DOI] [PubMed] [Google Scholar]
- 13. Song C.X., Szulwach K.E., Dai Q., Fu Y., Mao S.Q., Lin L., Street C., Li Y., Poidevin M., Wu H.. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013; 153:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pidugu L.S., Flowers J.W., Coey C.T., Pozharski E., Greenberg M.M., Drohat A.C.. Structural basis for excision of 5-formylcytosine by thymine DNA glycosylase. Biochemistry. 2016; 55:6205–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang L., Lu X., Lu J., Liang H., Dai Q., Xu G.L., Luo C., Jiang H., He C.. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012; 8:328–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coey C.T., Malik S.S., Pidugu L.S., Varney K.M., Pozharski E., Drohat A.C.. Structural basis of damage recognition by thymine DNA glycosylase: key roles for N-terminal residues. Nucleic Acids Res. 2016; 44:10248–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maiti A., Noon M.S., M.K. A. Jr, Pozharski E., Drohat A.C.. Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:8091–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hashimoto H., Hong S., Bhagwat A.S., Zhang X., Cheng X.. Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012; 40:10203–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hashimoto H., Zhang X., Cheng X.. Activity and crystal structure of human thymine DNA glycosylase mutant N140A with 5-carboxylcytosine DNA at low pH. DNA Repair. 2013; 12:535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Malik S.S., Coey C.T., Varney K.M., Pozharski E., Drohat A.C.. Thymine DNA glycosylase exhibits negligible affinity for nucleobases that it removes from DNA. Nucleic Acids Res. 2015; 43:9541–9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maiti A., Morgan M.T., Pozharski E., Drohat A.C.. Crystal structure of human thymine DNA glycosylase bound to DNA elucidates sequence-specific mismatch recognition. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8890–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barrett T.E., Savva R., Panayotou G., Barlow T., Brown T., Jiricny J., Pearl L.H.. Crystal structure of a G:T/U mismatch-specific DNA glycosylase: mismatch recognition by complementary-strand interactions. Cell. 1998; 92:117–129. [DOI] [PubMed] [Google Scholar]
- 23. Baba D., Maita N., Jee J.-G., Uchimura Y., Saitoh H., Sugasawa K., Hanaoka F., Tochio H., Hiroaki H., Shirakawa M.. Crystal structure of thymine DNA glycosylase conjugated to SUMO-1. Nature. 2005; 435:979–982. [DOI] [PubMed] [Google Scholar]
- 24. Karplus M., Mccammon J.A.. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002; 9:646–652. [DOI] [PubMed] [Google Scholar]
- 25. Harrigan M.P., Sultan M.M., Hernández C.X., Husic B.E., Eastman P., Schwantes C.R., Beauchamp K.A., Mcgibbon R.T., Pande V.S.. MSMBuilder: statistical models for biomolecular dynamics. Biophys. J. 2017; 112:10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chodera J.D., Noé F.. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014; 25:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prinz J.H., Wu H., Sarich M., Keller B., Senne M., Held M., Chodera J.D., Schütte C., Noé F.. Markov models of molecular kinetics: generation and validation. J. Chem. Phys. 2011; 134:174105. [DOI] [PubMed] [Google Scholar]
- 28. Shukla D., Hernández C.X., Weber J.K., Pande V.S.. Markov state models provide insights into dynamic modulation of protein function. Acc. Chem. Res. 2015; 48:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Noé F., Fischer S.. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr. Opin. Struct. Biol. 2008; 18:154–162. [DOI] [PubMed] [Google Scholar]
- 30. And W.C.S., Pitera J.W., Suits F.. Describing protein folding kinetics by molecular dynamics simulations. 1. Theory†. J. Phys. Chem. B. 2004; 108:2084–2089. [Google Scholar]
- 31. Ryckbosch S.M., Wender P.A., Pande V.S.. Molecular dynamics simulations reveal ligand-controlled positioning of a peripheral protein complex in membranes. Nat. Commun. 2017; 8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu L., Fu K.S., Zeng X., Huang X.. Elucidating conformational dynamics of multi-body systems by constructing Markov State Models. PCCP. 2016; 18:30228–30235. [DOI] [PubMed] [Google Scholar]
- 33. Wu H., Paul F., Wehmeyer C., Noé F.. Multiensemble Markov models of molecular thermodynamics and kinetics. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E3221–E3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Da L.T., Pardoavila F., Liang X., Silva D.A., Lu Z., Xin G., Dong W., Huang X.. Bridge helix bending promotes RNA polymerase II backtracking through a critical and conserved threonine residue. Nat. Commun. 2016; 7:11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Plattner N., Noé F.. Protein conformational plasticity and complex ligand-binding kinetics explored by atomistic simulations and Markov models. Nat. Commun. 2015; 6:7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Malmstrom R.D., Kornev A.P., Taylor S.S., Amaro R.E.. Allostery through the computational microscope: cAMP activation of a canonical signalling domain. Nat. Commun. 2015; 6:7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Da L.T., Chao E., Duan B., Zhang C., Zhou X., Yu J.. A jump-from-cavity pyrophosphate ion release assisted by a key lysine residue in T7 RNA polymerase transcription elongation. PLoS Comp. Biol. 2015; 11:e1004624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Silva D.A., Weiss D.R., Pardo A.F., Da L.T., Levitt M., Wang D., Huang X.. Millisecond dynamics of RNA polymerase II translocation at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choudhary O.P., Paz A., Adelman J.L., Colletier J.P., Abramson J., Grabe M.. Structure-guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat. Struct. Mol. Biol. 2014; 21:626–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bowman G.R., Voelz V.A., Pande V.S.. Atomistic folding simulations of the five-helix bundle protein λ(6−85). J. Am. Chem. Soc. 2014; 133:664–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Qiao Q., Bowman G.R., Huang X.. Dynamics of an intrinsically disordered protein reveal metastable conformations that potentially seed aggregation. J. Am. Chem. Soc. 2013; 135:16092–16101. [DOI] [PubMed] [Google Scholar]
- 42. Kai J. Kohlhoff, D. S., Morgan Lawrenz, Gregory R. Bowman, David E. Konerding, Dan Belov, Russ B. Altman, Vijay S. Pande. Cloud-based simulations on Google Exacycle reveal ligand-modulation of GPCR activation pathways. Nat. Chem. 2013; 6:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Da L.T., Wang D., Huang X.. Dynamics of pyrophosphate ion release and its coupled trigger loop motion from closed to open state in RNA Polymerase II. J. Am. Chem. Soc. 2012; 134:2399–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang X., Bowman G.R., Bacallado S., Pande V.S.. Rapid equilibrium sampling initiated from nonequilibrium data. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:19765–19769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dodani S.C., Kiss G., Cahn J.K., Su Y., Pande V.S., Arnold F.H.. Discovery of a regioselectivity switch in nitrating P450s guided by molecular dynamics simulations and Markov models. Nat. Chem. 2016; 8:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Da L.T., C. E., Shuai Y., Wu S., Su X.D., Yu J.. T7 RNA polymerase translocation is facilitated by a helix opening on the fingers domain that may also prevent backtracking. Nucleic Acids Res. 2017; 45:7909–7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stivers J.T., Jiang Y.L.. A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev. 2003; 103:2729–2759. [DOI] [PubMed] [Google Scholar]
- 48. Hess B., Kutzner C., van der Spoel D., Lindahl E.. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008; 4:435–447. [DOI] [PubMed] [Google Scholar]
- 49. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J.. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005; 26:1701–1718. [DOI] [PubMed] [Google Scholar]
- 50. Berendsen H.J., van der Spoel D., van Drunen R.. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995; 91:43–56. [Google Scholar]
- 51. Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Hermans J.. Pullman B. Interaction models for water in relation to protein hydration. Intermolecular Forces. 1981; Dordrecht: Reidel Publishing Company; 331–342. [Google Scholar]
- 52. Guy A.T., Piggot T.J., Khalid S.. Single-stranded DNA within nanopores: conformational dynamics and implications for sequencing; a molecular dynamics simulation study. Biophys. J. 2012; 103:1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C.. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006; 65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Joung I.S., Cheatham T.E. III. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008; 112:9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Joung I.S., Cheatham T.E.. Molecular dynamics simulations of the dynamic and energetic properties of alkali and halide ions using water-model-specific ion parameters. J. Phys. Chem. B. 2009; 113:13279–13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A.. Development and testing of a general amber force field. J. Comput. Chem. 2004; 25:1157–1174. [DOI] [PubMed] [Google Scholar]
- 57. Case D.A., Cerutti D.S., Cheatham T.E. III, Darden T.A., Duke R.E., Giese T.J., Gohlke H., Goetz A.W., Greene D., Homeyer N. et al. AMBER17. 2017; San Francisco: University of California; http://ambermd.org/. [Google Scholar]
- 58. Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G.. A smooth particle mesh Ewald method. J. Chem. Phys. 1995; 103:8577–8593. [Google Scholar]
- 59. Hess B., B. H., Berendsen H.J.C., Fraaije J.G.E.M.. LINCS: a linear constraint solver for molecular simulations. J. Comp. Chem. 1997; 18:1463–1472. [Google Scholar]
- 60. Bussi G., Donadio D., Parrinello M.. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007; 126:014101. [DOI] [PubMed] [Google Scholar]
- 61. Kanaan N., Crehuet R., Imhof P.. Mechanism of the glycosidic bond cleavage of mismatched thymine in human thymine DNA glycosylase revealed by classical molecular dynamics and quantum mechanical/molecular mechanical calculations. J. Phys. Chem. B. 2015; 119:12365–12380. [DOI] [PubMed] [Google Scholar]
- 62. Pérezhernández G., Paul F., Giorgino T., De F.G., Noé F.. Identification of slow molecular order parameters for Markov model construction. J. Chem. Phys. 2013; 139:015102. [DOI] [PubMed] [Google Scholar]
- 63. Pande C.R.S., Vijay S.. Improvements in Markov State Model construction reveal many non-native interactions in the folding of NTL9. J. Chem. Theory Comput. 2013; 9:2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Naritomi Y., Fuchigami S.. Slow dynamics of a protein backbone in molecular dynamics simulation revealed by time-structure based independent component analysis. J. Chem. Phys. 2013; 139:215102. [DOI] [PubMed] [Google Scholar]
- 65. Naritomi Yusuke, Fuchigami Sotaro. Slow dynamics in protein fluctuations revealed by time-structure based independent component analysis. J. Chem. Phys. 2011; 134:065101. [DOI] [PubMed] [Google Scholar]
- 66. Bowman G.R., Huang X., Pande V.S.. Using generalized ensemble simulations and Markov state models to identify conformational states. Methods. 2009; 49:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Deuflhard P., Weber M.. Robust Perron cluster analysis in conformation dynamics. Linear Algebra Appl. 2005; 398:161–184. [Google Scholar]
- 68. Jarzynski C. A nonequilibrium equality for free energy differences. Phys. Rev. Lett. 1997; 78:2690–2693. [Google Scholar]
- 69. Jensen M.Ø., Park S., Tajkhorshid E., Schulten K.. Energetics of glycerol conduction through aquaglyceroporin GlpF. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yu J., Yool A.J., Schulten K., Tajkhorshid E.. Mechanism of gating and ion conductivity of a possible tetrameric pore in aquaporin-1. Structure. 2006; 14:1411–1423. [DOI] [PubMed] [Google Scholar]
- 71. Drohat A.C., Maiti A.. Mechanisms for enzymatic cleavage of the N-glycosidic bond in DNA. Org. Biomol. Chem. 2014; 12:8367–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schermerhorn K.M., Delaney S.. A chemical and kinetic perspective on base excision repair of DNA. Acc. Chem. Res. 2014; 47:1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Parikh S.S., Walcher G., Jones G.D., Slupphaug G., Krokan H.E., Blackburn G.M., Tainer J.A.. Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:5083–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Maiti A., Michelson A.Z., Armwood C.J., Lee J.K., Drohat A.C.. Divergent mechanisms for enzymatic excision of 5-formylcytosine and 5-carboxylcytosine from DNA. J. Am. Chem. Soc. 2013; 135:15813–15822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Morgan M.T., Bennett M.T., Drohat A.C.. Excision of 5-halogenated uracils by human thymine DNA glycosylase. Robust activity for DNA contexts other than CpG. J. Biol. Chem. 2007; 282:27578–27586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.