

Abstract
Background
Alzheimer’s disease (AD) is a most common dementia in elderly people. Since AD symptoms resemble those of other neurodegenerative diseases, including idiopathic normal pressure hydrocephalus (iNPH), it is difficult to distinguish AD from iNPH for a precise and early diagnosis. iNPH is caused by the accumulation of cerebrospinal fluid (CSF) and involves gait disturbance, urinary incontinence, and dementia. iNPH is treatable with shunt operation which removes accumulated CSF from the brain ventricles.
Methods
We performed metabolomic analysis in the CSF of patients with AD and iNPH with capillary electrophoresis-mass spectrometry. We assessed metabolites to discriminate between AD and iNPH with Welch’s t-test, receiver operating characteristic (ROC) curve analysis, and multiple logistic regression analysis.
Results
We found significant increased levels of glycerate and N-acetylneuraminate and significant decreased levels of serine and 2-hydroxybutyrate in the CSF of patients with AD compared to the CSF of patients with iNPH. The ROC curve analysis with these four metabolites showed that the area under the ROC curve was 0.90, indicating good discrimination between AD and iNPH.
Conclusions
This study identified four metabolites that could possibly discriminate between AD and iNPH, which previous research has shown are closely related to the risk factors, pathogenesis, and symptoms of AD. Analyzing pathway-specific metabolites in the CSF of patients with AD may further elucidate the mechanism and pathogenesis of AD.
Electronic supplementary material
The online version of this article (10.1186/s40364-018-0119-x) contains supplementary material, which is available to authorized users.
Keywords: Alzheimer’s disease, Idiopathic normal pressure hydrocephalus, Diagnostic marker, Cerebrospinal fluid, Serine, Glycerate, N-acetylneuraminate, 2-hydroxybutyrate
Background
Alzheimer’s disease (AD) is the most common type of dementia in the world, which concerns approximately 60–70% cases of dementia, and it is becoming a significant social issue because of the growing aging population.
In general, AD is diagnosed based on the presence of cognitive impairment and by neuropsychological testing according to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association criteria [1]. Currently, there is no curative or radical treatment for AD, although some medicines have been developed to reduce the symptoms [2]. However, some symptoms of AD are similar to those of other neurodegenerative diseases, such as idiopathic normal pressure hydrocephalus (iNPH). iNPH is caused by the accumulation of cerebrospinal fluid (CSF) in the brain and causes gait disturbance, urinary incontinence, and dementia. In contrast to other dementias, iNPH is treatable by shunt operation that removes the accumulated CSF [3]. Therefore, a precise diagnosis that discriminates patients with AD from patients with iNPH is essential for proper treatment at the early stages of these diseases. Presently, increased phosphate tau (p-tau) and decreased amyloid-beta 1-42 (Aβ42) in the CSF are used as established AD diagnostic markers [4]. However, p-tau accumulates after synaptic degeneration and Aβ42 is difficult to accurately quantify. Therefore, we tried to find additional biomarkers, capable of precisely detecting AD before neurodegeneration progresses.
Recently, accumulated evidence has indicated that AD is a type of metabolic disease in the brain [5, 6]. The brain is the most energy-consuming organ and glucose is an essential and dominant energy source for the brain [7]. Progressive regional cerebral glucose metabolism reduction, correlated with the symptom severity, has been found in the brain of patients with AD [8, 9]. Mills et al. (2013) performed RNA-Seq analysis in the parietal cortex of patients with AD and reported that two enzymes, ACOT1 and ACOT2, which are involved in lipid metabolism, were upregulated. They also found a downregulation of TERC, which is involved in the synthesis of very long chain fatty acids [10]. In addition, impairment of the insulin response, as is seen in diabetes mellitus and metabolic syndrome, was revealed as a risk factor for AD [11–13]. These reports suggested that the pathomechanism of AD is strongly related to a disturbance in brain energy metabolism and homeostasis. The disturbance could induce metabolite alterations in the body fluids of patients with AD, such as in the plasma, serum, and CSF. Therefore, metabolome analysis of body fluids in AD has been actively performed to identify new diagnostic markers. Especially, the CSF is thought to be a superior analyte than other body fluids because it is in direct contact with the extracellular space of the brain and thus directly reflects the biological changes in the pathological brain processes in AD.
So far, several pertinent reports have been published and several diagnostic markers for AD have been suggested. For example, D’ Aniello et al. (2005) conducted high performance liquid chromatography (HPLC) analysis and found L-glutamine was increased and L-asparate was decreased in the CSF of patients with AD than in the CSF of controls [14]. Czech et al. (2012) reported that increased cysteine with decreased uridine was the optimal combination to identify mild AD, and increased cortisol levels were associated with the progression of AD in a European cohort [15]. Ibanez et al. (2012) performed capillary electrophoresis-mass spectrometry (CE-MS) to investigate metabolome changes in the CSF of patients at different AD stages and found that choline, dimethylarginine, arginine, valine, proline, serine, histidine, creatine, carnitine, and suberylglycine could be possible disease progression markers [16]. Furthermore, they conducted ultra-high-performance liquid chromatography-time-of-flight mass spectrometry (TOFMS) and found uracil and uridine were good candidate biomarkers for AD, as has been previously described [15, 17].
However, different metabolomic platforms and methods detect different metabolites, thus utilizing multiple types of metabolomic platforms would provide a wider perspective of metabolomics information under study. In addition, differences in ethnicity, culture, and education may influence decision making regarding the diagnosis, symptoms, and severity of AD [18]. Accordingly, accumulating information related to AD pathology from differential ethnoracial cohorts and with differential methods is essential.
From these points of view, we performed metabolomic analysis of the CSF of patients with AD or iNPH in Japanese cohorts with capillary electrophoresis-TOFMS (CE-TOFMS). Our present findings would support the utility of metabolomics analysis for discriminate between AD and iNPH.
Methods
Subjects
The characteristics of the study patients with AD and iNPH are summarized in Table 1. AD and iNPH were diagnosed according to previously published criteria [1, 19]. Bioresources were largely obtained from the biobank of the National Center for Geriatrics and Gerontology (NCGG, Aichi, Japan) and partly donated by the National Center of Neurology and Psychiatry (NCNP, Tokyo, Japan) and the Department of Biological Regulation, School of Medicine, Tottori University (Tottori, Japan). All samples were obtained with the written informed consent of the patients before sampling between 2011 and 2015. This study was reviewed and approved by the ethics committees of all participating institutes and biobanks.
Table 1.
Subject characteristics.
| Metabolomics | Validation | |||
|---|---|---|---|---|
| AD | iNPH | AD | iNPH | |
| Subject No. | 39 | 19 | 42 | 38 |
| Mean age (SD) | 73.5 (9.8) | 77.8 (4.5) | 74.1 (9.6) | 77.9 (5.6) |
| Age range | 47-86 | 70-86 | 47-86 | 57-87 |
| P-value | 0.03 | 0.03 | ||
| Male/female | 14/25 | 9/10 | 15/27 | 22/16 |
| P-value | 0.24 | 0.09 | ||
| Mean MMSE (SD) | 21.4 (5.1) | 23.5 (2.9) | 21.2 (5.0) | 22.1 (3.1) |
| P-value | 0.08 | 0.46 | ||
| Missing values | 6 | 0 | 6 | 0 |
Collection of CSF
CSF was collected by lumbar puncture and centrifuged to remove debris. The supernatant was aliquoted into low protein-binding tubes and immediately frozen in liquid nitrogen and stored at −80 °C until use.
Measurement of p-tau and Aβ42
To detect the concentration of p-tau and Aβ42 in the CSF, the enzyme-linked immunosorbent assay systems of INNOTEST β-AMYLOID(1-42) and INNOTEST PHOSPHO-TAU(181P) were used according to the manufacturer’s instructions (Fujibireo Inc., Tokyo, Japan).
Metabolites extraction from CSF
Frozen CSF samples were thawed and 40 μL aliquots were placed into 360 μL of methanol containing internal standards (20 μmol/L each of methionine sulfone and D-camphor-10-sulfonic acid). The solutions were thoroughly mixed, and then both 400 μL of chloroform and 160 μL of Milli-Q water were added, followed by centrifugation at 10,000 × g for 3 min at 4 °C. The aqueous layer was transferred to a 5-kDa-cutoff filter (Human Metabolome Technologies, Tsuruoka, Japan) to remove proteins. The filtrate was dried using a centrifuge concentrator and reconstituted with 50 μL of Milli-Q water containing reference compounds (200 μmol/L each of 3-aminopyrrolidine and trimesic acid) prior to CE-TOFMS analysis.
Metabolome analysis by CE-TOFMS
All CE-TOFMS experiments were performed using an Agilent 1600 Capillary Electrophoresis system (Agilent technologies, Santa Clara, CA), an Agilent 6220 TOF LC/MS system, an Agilent 1200 series isocratic HPLC pump, a G1603A Agilent CE-MS adapter kit, and a G1607A Agilent CE-electrospray ionization (ESI)-MS sprayer kit. In the anionic metabolites analysis, ESI sprayer was replaced with a platinum needle instead of the initial stainless steel needle [20]. The other conditions relating to the CE-ESI-MS sprayer were identical as received. For CE-MS system control and data acquisition, we used the Agilent MassHunter software.
Cationic metabolome analysis
For cationic metabolome analysis, a fused-silica capillary (50 μm i.d. × 100 cm) filled with 1 mol/L formic acid as the electrolyte was used [21]. A new capillary was flushed with the electrolyte for 20 min, and the capillary was equilibrated for 4 min by flushing with the electrolyte before each run. Sample solution was injected at 5 kPa for 3 s and a positive voltage of 30 kV was applied. The temperatures of the capillary and sample trays were maintained at 20 °C and 4 °C, respectively. Methanol/water (50% v/v) containing 0.1 μmol/L hexakis(2,2-difluoroethoxy)phosphazene was delivered as sheath liquid at 10 L/min. ESI-TOFMS was operated in the positive ion mode, and the capillary voltage was set at 4 kV. The flow rate of heated nitrogen gas (heater temperature, 300 °C) was maintained at 10 psig. In TOFMS, the fragmentor, skimmer, and Oct RF voltages were set at 75, 50, and 125 V, respectively. Automatic recalibration of each acquired spectrum was performed using the masses of reference standards ([13C isotopic ion of protonated methanol dimer (2CH3OH + H)]+, m/z 66.06306) and ([hexakis(2,2-difluoroethoxy)phosphazene + H]+, m/z 622.02896). Exact mass data were acquired at the rate of 1.5 cycles/s over a 50 to 1000 m/z range.
Anionic metabolome analysis
For the anionic metabolome analysis, a COSMO(+) capillary (50 μm i.d. × 105 cm, Nacalai Tesque, Kyoto, Japan) filled with 50 mmol/L ammonium acetate (pH 8.5) as the electrolyte was used [20]. Before the first use, a new capillary was successively flushed with the electrolyte, 50 mmol/L acetic acid (pH 3.4), and then the electrolyte again for 10 min each. Before each run, the capillary was equilibrated by flushing with 50 mmol/L acetic acid (pH 3.4) for 2 min and then with the electrolyte for 5 min. Sample was injected at 5 kPa for 30 s and a negative voltage of 30 kV was applied. The temperatures of the capillary and sample trays were maintained at 20 °C and 4 °C, respectively. Ammonium acetate (5 mmol/L) in 50% (v/v) methanol/water solution that contained 0.1 μmol/L hexakis(2,2-difluoroethoxy)phosphazene was delivered as sheath liquid at 10 μL/min. ESI-TOFMS was operated in the negative ion mode, and the capillary voltage was set at 3.5 kV. The flow rate of heated nitrogen gas (heater temperature, 300 °C) was maintained at 10 psig. In TOFMS, the fragmentor, skimmer, and Oct RF voltages were set at 100, 50, and 200 V, respectively. Automatic recalibration of each acquired spectrum was performed using the masses of reference standards ([13C isotopic ion of deprotonated acetate dimer (2CH3COOH − H)]−, m/z 120.03841) and ([hexakis(2,2-difluoroethoxy)phosphazene + deprotonated acetate(CH3COOH − H)]−, m/z 680.03554). Exact mass data were acquired at the rate of 1.5 cycles/s over a 50 to 1000 m/z range.
Statistical analyses
Comprehensive metabolic data were processed using our proprietary software (MasterHands) [22–24]. The peaks were identified by matching m/z values and normalized migration times of corresponding authentic standard compounds. Statistical analysis was performed using Welch’s t-test, receiver operating characteristic (ROC) curve analysis, and Pearson’s correlation analysis in R version 3.3.2 (2016-10-31) [25]. Multiple logistic regression analysis was performed with Statflex ver. 6 (Artech Co., Ltd., Osaka, Japan). A P-value <0.05 was considered to be significant.
Results
First, we performed comprehensive metabolic analysis of the CSF to identify characteristic metabolites in the patients with AD and iNPH with the screening subjects indicated in Table 1 (column: Metabolomics). Eighty-three anionic and 60 cationic metabolites were detected in this analysis (Additional file 1). Among these, 18 metabolites showed a P-value <0.05 and an area under the ROC curve (AUC) > 0.7 between AD and iNPH with several missing values (Table 2). Therefore, to validate the concentration of these metabolites in the CSF of patients with AD and iNPH, we repeated CE-TOFMS with additional samples (Table 1, column: Validation). Undecanoate (PubChem ID: 8180) and N-acetylhistidine (PubChem ID: 273,260) were excluded from further analysis as false positives because they are not metabolized in the human brain [26]. Nine metabolites indicated P-value <0.05 and an AUC > 0.7 between AD and iNPH without missing values (Table 3). Of these metabolites, glycerate (PubChem ID: 752) and N-acetylneuraminate (Neu5Ac, PubChem ID: 3568) were increased in the CSF of patients with AD than of patients with iNPH, while the other seven metabolites were decreased.
Table 2.
Statistically significant metabolites between AD and iNPH in the metabolomics
| Mean concentration, μmol/L (SD) | Welch’s t-test | ROC | AD/iNPH | Valid value | Missing value | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PubChem ID | Metabolite | AD | iNPH | P-value | AUC | Fold change | AD | iNPH | AD | iNPH | ||
| 752 | Glycerate | 60.61 | (30.94) | 26.23 | (6.22) | 6.E-07 | 0.88 | 2.31 | 32 | 16 | 7 | 3 |
| 1060 | Pyruvate | 45.22 | (13.45) | 68.34 | (12.95) | 1.E-04 | 0.89 | 0.66 | 12 | 16 | 27 | 3 |
| 3441 | 2-Oxoisopentanoate | 4.29 | (1.19) | 5.64 | (1.29) | 0.001 | 0.78 | 0.76 | 30 | 19 | 9 | 0 |
| 3568 | N-Acetylneuraminate | 16.75 | (3.89) | 13.35 | (3.2) | 0.001 | 0.76 | 1.25 | 39 | 19 | 0 | 0 |
| 617 | Serine | 24.63 | (3.9) | 30.12 | (6.15) | 0.002 | 0.77 | 0.82 | 39 | 19 | 0 | 0 |
| 273,260 | N-Acetylhistidinea | 1.19 | (0.88) | 0.88 | (0.92) | 0.002 | 0.72 | 1.36 | 39 | 19 | 0 | 0 |
| 8180 | Undecanoatea | 0.54 | (0.17) | 0.71 | (0.09) | 0.003 | 0.81 | 0.76 | 17 | 7 | 22 | 12 |
| 3,527,278 | 4-Methyl-2-oxopentanoate | 2.64 | (0.85) | 3.51 | (0.98) | 0.003 | 0.74 | 0.75 | 39 | 19 | 0 | 0 |
| 11,266 | 2-Hydroxybutyrate | 18.08 | (5.98) | 23.14 | (6.06) | 0.005 | 0.74 | 0.78 | 39 | 19 | 0 | 0 |
| 525 | Malate | 1.17 | (0.33) | 1.54 | (0.46) | 0.005 | 0.73 | 0.76 | 39 | 19 | 0 | 0 |
| 602 | Alanine | 32.03 | (8.53) | 42.82 | (14.42) | 0.006 | 0.77 | 0.75 | 39 | 19 | 0 | 0 |
| 876 | Methionine | 2.79 | (1.08) | 3.67 | (1.07) | 0.006 | 0.76 | 0.76 | 39 | 19 | 0 | 0 |
| 205 | Threonine | 27.56 | (6.43) | 34.81 | (9.53) | 0.006 | 0.73 | 0.79 | 39 | 19 | 0 | 0 |
| 232 | Arginine | 23.08 | (4.5) | 26.79 | (4.6) | 0.006 | 0.71 | 0.86 | 39 | 19 | 0 | 0 |
| 64,969 | 3-Methylhistidine | 1.12 | (0.88) | 1.81 | (0.92) | 0.010 | 0.79 | 0.62 | 39 | 19 | 0 | 0 |
| 1081 | Citramalate | 1.20 | (0.96) | 0.57 | (0.12) | 0.010 | 0.74 | 2.12 | 19 | 8 | 20 | 11 |
| 1175 | Urate | 20.84 | (11.36) | 30.61 | (15.74) | 0.023 | 0.73 | 0.68 | 39 | 19 | 0 | 0 |
| 866 | Lysine | 30.00 | (5.84) | 36.35 | (10.66) | 0.023 | 0.72 | 0.82 | 39 | 19 | 0 | 0 |
aUndecanoate and N-Acetylhistidine were excluded from further analysis
AD Alzheimer’s disease, iNPH idiopathic normal pressure hydrocephalus, AUC area under the curve, ROC receiver operator characteristic
Table 3.
Statistically significant metabolites between AD and iNPH in the validation
| Mean concentration, μmol/L (SD) | Welch’s t-test | ROC | AD/iNPH | ||||
|---|---|---|---|---|---|---|---|
| Metabolite | AD | iNPH | P-valuea | AUC | Fold change | ||
| Serine | 29.1 | (5.3) | 34.5 | (4.9) | 1.6E-04 | 0.78 | 0.84 |
| Glycerate | 54.6 | (22.2) | 37.3 | (12.9) | 2.0E-04 | 0.71 | 1.46 |
| 3-Methylhistidine | 1.1 | (0.9) | 2.1 | (1.2) | 6.4E-04 | 0.82 | 0.52 |
| Threonine | 29.0 | (7.0) | 39.4 | (12.9) | 7.4E-04 | 0.77 | 0.74 |
| Methionine | 2.6 | (1.0) | 3.6 | (1.2) | 9.4E-04 | 0.77 | 0.71 |
| Urate | 19.9 | (8.6) | 28.7 | (9.9) | 1.3E-03 | 0.78 | 0.70 |
| N-Acetylneuraminate | 16.1 | (3.9) | 12.9 | (3.2) | 1.9E-03 | 0.75 | 1.25 |
| Alanine | 33.0 | (8.4) | 42.4 | (12.6) | 3.9E-03 | 0.73 | 0.78 |
| 2-Hydroxybutyrate | 17.5 | (4.6) | 22.2 | (6.7) | 0.009 | 0.72 | 0.79 |
aBonferroni correction was applied
AD Alzheimer’s disease, iNPH idiopathic normal pressure hydrocephalus, AUC area under the curve, ROC receiver operator characteristic
Next, we performed multiple logistic regression analysis with these nine metabolites, setting age as a covariate. Using stepwise regression, we found statistical significance for four metabolites, serine (PubChem ID: 617), glycerate, Neu5Ac, and 2-hydroxybutylate (2-HB, PubChem ID: 11,266) (Fig. 1 and Table 4). Formulation of the regression coefficient of these four metabolites was as follows; (−0.1198) × age + (−0.2508) × serine + (0.05715) × glycerate + (0.37226) × Neu5Ac + (−0.1705) × 2-HB + 12.3001. When the cutoff value was set to 9.73, sensitivity and specificity were highest (Fig. 2, AUC = 0.90, sensitivity = 0.86, specificity = 0.84, and the odds ratio was 32.0).
Fig. 1.
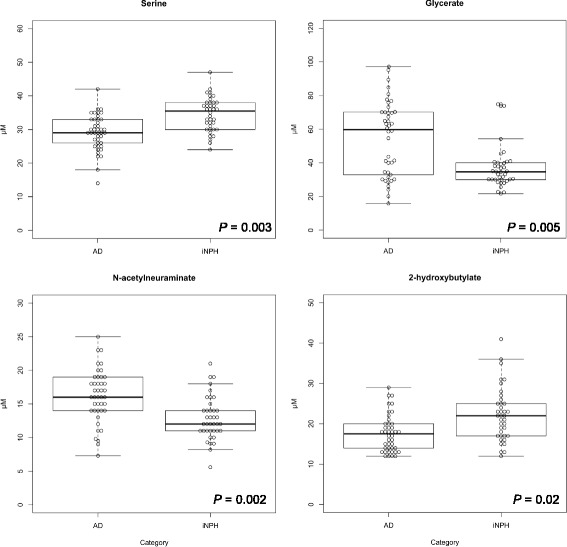
Statistically significant metabolites in the cerebrospinal fluid between Alzheimer’s disease and idiopathic normal pressure hydrocephalus. Four metabolites were statistically significant in the multiple logistic regression analysis
Table 4.
Statistically significant metabolites in multiple logistic regression analysis
| Metabolite | P-value | Odds ratio | CI 95% |
|---|---|---|---|
| Serine | 0.002 | 0.78 | 0.66-0.91 |
| Glycerate | 0.009 | 1.06 | 1.01-1.10 |
| N-Acetylneuraminate | 0.003 | 1.45 | 1.14-1.85 |
| 2-Hydroxybutyrate | 0.034 | 0.84 | 0.72-0.99 |
Age was used as a covariate
Fig. 2.
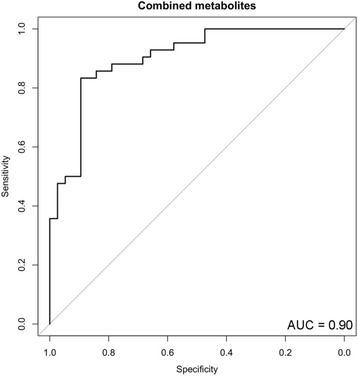
Receiver operator characteristic (ROC) curve analysis of statistically significant metabolites in the cerebrospinal fluid between Alzheimer’s disease (AD) and idiopathic normal pressure hydrocephalus (iNPH). ROC curve analysis was performed to compare the predictive power of AD and iNPH with the combined metabolites
Further, we examined the correlation between p-tau, Aβ42, and these four metabolites. Correlation coefficient values were −0.33, 0.35, 0.55, and −0.27 between p-tau and serine, glycerate, Neu5Ac, and 2-HB, respectively, showing a weak to moderate correlation between p-tau and the four metabolites (Fig. 3). On the other hand, the correlation coefficients between Aβ42 and the four metabolites were 0.10, −0.35, 0.18, and 0.01 for serine, glycerate, Neu5Ac, and 2-HB, respectively, showing weak or absent correlations for the first three metabolites and a negative correlation for glycerate (Fig. 4). When ROC curve analysis was performed between AD and iNPH with p-tau and Aβ42, the AUC values were 0.94 and 0.71 for p-tau and Aβ42, respectively (Fig. 5). These results indicate that these metabolites combined may have a discriminatory power equal to that of p-tau.
Fig. 3.
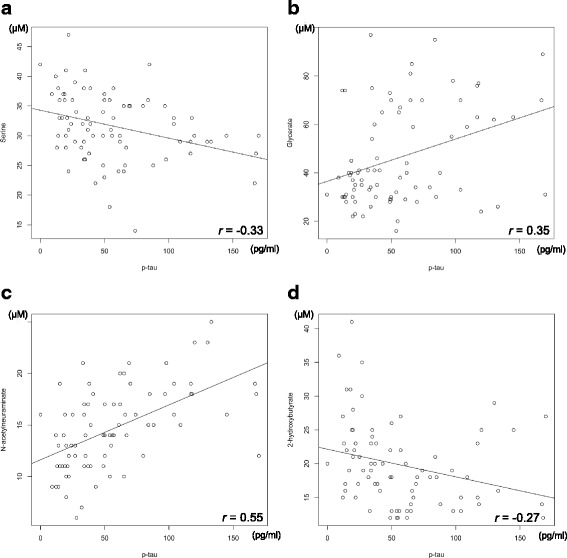
Correlation diagram of p-tau versus the four metabolites. (a) serine, (b) glycerate, (c) Neu5Ac, and (d) 2-HB with regression lines are indicated
Fig. 4.
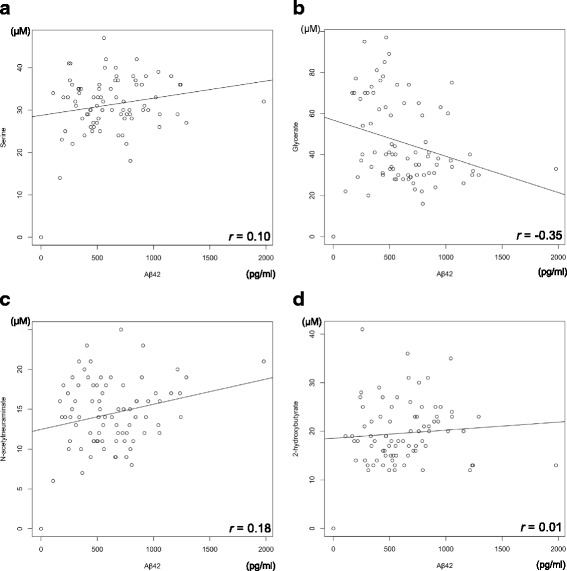
Correlation diagram of Aβ42 versus the four metabolites. (a) serine, (b) glycerate, (c) Neu5Ac, and (d) 2-HB with regression lines are indicated
Fig. 5.
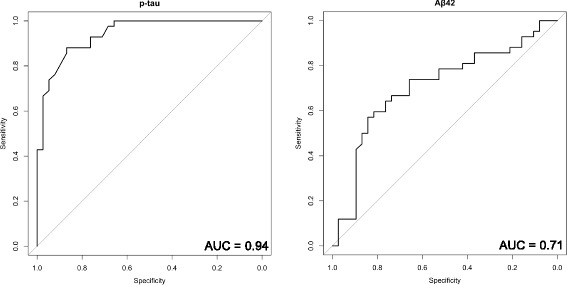
Receiver operator characteristic (ROC) curve analysis of p-tau and Aβ42 in the cerebrospinal fluid between Alzheimer’s disease and idiopathic normal pressure hydrocephalus. ROC curve analysis was performed to compare the predictive power of p-tau (left) and Aβ42 (right)
Discussion
We found the combination of four metabolites, serine, glycerate, Neu5Ac, and 2-HB could contribute to distinguishing AD from iNPH (Table 4, Fig. 2). We searched the KEGG Pathway Database [27] (http://www.genome.jp/kegg/pathway.html) and found that glycerate is the intermediate metabolite in the pentose phosphate pathway (PPP, KEGG ID: map00030), amino acid metabolism (KEGG ID: map00260), and glycerolipid metabolism (KEGG ID: map00561) in humans.
PPP is one of the glucose metabolic pathways generating pentose and nicotinamide adenine dinucleotide phosphate (NADPH). NADPH has an antioxidant reducing activity and an important role in opposing oxidative stress [28]. In the brain of patients with AD, oxidative stress signatures are observed at the very early stage of the disease [29–31] and PPP is activated to provide NAPDH to counteract oxidative stress [32, 33]. In addition, in the state of hypoxia, which is reportedly a risk factor for AD [34], the PPP preferentially metabolizes glucose instead of the common glycolysis pathway [35], also suggesting the activation of PPP in the brain of patients with AD. Together, increased glycerate in the CSF of patients with AD may reflect the activation of PPP to compensate for the failure in brain functions that accompany AD progression.
Glycerate is also generated in amino acid metabolism, glycine, serine, and threonine. Some reports have indicated alterations in amino acid metabolism in AD [36–38]. Especially, Madeira et al. reported increased levels of D-serine and total serine concentrations in the CSF of patients with AD [39]. They indicated that the amyloid beta oligomer activated serine racemase, an enzyme which converts L-serine to D-serine. In our study, although we did not detect D-serine sole concentration, total serine concentration was decreased in the patients with AD (Fig. 1). However, as we included patients with iNPH as controls, it is difficult to directly compare our results to those of the other studies, which included healthy individuals as controls. However, there is a report indicating phosphatidylserine synthase (PSS) was activated in the aging rat brain [40]. PSS incorporates serine into phosphatidylethanolamine or phosphatidylcholine and generates phosphatidylserine. As aging is a risk factor for AD [40] and phosphatidylserine administration improved several cognitive measures in AD [41], serine reduction in the CSF of patients with AD may reflect the neuroprotective role of PSS. Moreover, serine is converted to hydroxypyruvate by transamination, followed by conversion to glycerate by glycerate dehydrogenase [42]. Additionally, glycerate is generated in the serine degradation pathway [43, 44]. Therefore, increased glycerate and decreased serine in the CSF of patients with AD seem to be justifiable.
Another group indicated that there was no statistically significant difference in serine concentration between the CSF of patients with AD and that of controls [14, 45]. However, these studies were performed with relatively small sample sizes and amino acids concentration in the CSF seemingly influenced by the content of daily diet [46], further studies with larger sample sizes are needed to elucidate the relations between serine concentration in the CSF and AD.
Glycerate is also produced during glycerolipid metabolism. Malaisse et al. indicated that triglyceride species were increased in the brain tissue of Type II diabetic rats [47]. Type II diabetes is known as a risk factor for AD [48, 49] and AD is known as the Type III diabetes [50], suggesting glycerolipid metabolism accelerated in the AD brain and may result in the accumulation of glycerate in the CSF.
Other than glycerate, Neu5Ac was increased in the CSF of AD. Neu5Ac is the most abundant sialic acid in nature and a component of gangliosides [51]. Gangliosides are abundant in neural cell membranes and have important roles in the organization of lipid rafts [52]. Lipid rafts are the subdomains of the plasma membrane that integrate numerous types of lipid proteins having important roles in cell signaling, cell-cell adhesion, and intracellular vesicular trafficking. There are some reports indicating that lipid rafts contain many types of AD associated proteins [53–56] and aberrations in the structure of the lipid rafts are considered to lead to AD [57]. Kracun et al. indicated that there was significant decrease of gangliosides in the brain of patients with AD and in the aging population, suggesting accelerated degradation of gangliosides accompanied by neuronal cell death [58]. Hence, the increased level of Neu5Ac in the CSF of patients with AD may reflect that neuronal and lipid raft destruction may accompany AD progression.
In this study, 2-HB was decreased in the CSF of patients with AD. 2-HB is derived from 2-ketobutyrate (2-KB) dehydration by lactate dehydrogenase (LDH) [59]. 2-KB is an important intermediate metabolite of amino acid metabolism, which is reportedly altered in AD [36–38]. In addition, in the brain of AD model mice, LDH expression was decreased [60], which may result in decreased 2-HB production. Hence, the four metabolites detected in this study are likely related to AD risk factors, pathogenesis, and/or symptoms (Fig. 6). Also, all these metabolites are found at the first time as the AD related ones in the CSF.
Fig. 6.
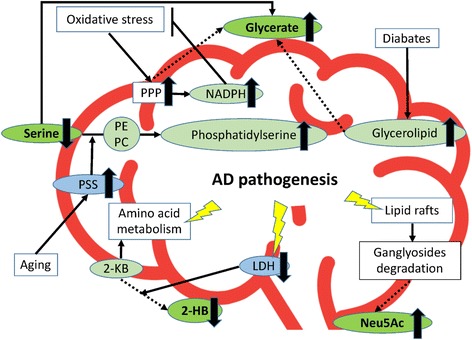
A model of the relationship of Alzheimer’s disease-specific metabolites indicated in this study. Serine, glycerate, Neu5Ac, and 2-HB are depicted with the AD-related metabolites, phenomenon, structures and metabolic pathways. Metabolites detected in this study are indicated in green oval with bold letters. Other related metabolites are indicated in light green oval. Enzymes are indicated in light blue oval. The phenomena, structure and pathways known to participate with AD pathogenesis are indicated in square. Valid allows indicate the interactions previously reported. Dotted allows indicate predicted interactions. Red bold line indicates the brain and CSF boundary. 2-KB: 2-ketobutyrate; 2-HB: 2-hydroxybutyrate; AD: Alzheimer’s disease; LDH: lactate dehydrogenase; NADPH: nicotinamide adenine dinucleotide phosphate; Neu5Ac: N-acetylneuraminate; PC: phosphatidylcholine; PE: phosphatidylethanolamine; PPP: pentose phosphate pathway; PSS: phosphatidylserine synthase
The metabolites described here likely participate in several pathways; however, we could not find other statistically significant metabolites included in these pathways. Easily detectable metabolites differ from metabolomic platforms and methods; therefore, other metabolites participating in the pathways indicated here could not be detected in this study. Further analyses with several metabolomic platforms are needed to complement metabolomic information for AD.
Recently, fluorodeoxyglucose (FDG)-positron emission tomography (PET), which detects the cerebral metabolic rates of glucose, has been used to diagnose AD [61], indicating the usefulness of measuring metabolic pathways for diagnosing AD. Other than glucose metabolic rates, several studies have indicated numerous types of metabolic pathways to be influenced relatively early in AD progression [6, 12, 62]. However, it is still difficult to make a precise and early diagnosis for AD at present. To establish the methods of precise and early diagnosis, comprehensive analyses for the pathway specific metabolites altered in AD would be effective.
In this study, the concentration of the four metabolites and established AD biomarker, p-tau, in the CSF were weakly correlated, although another established AD biomarker, Aβ42, did not show similarly significant results (Figs. 3, 4). In addition, according to the AUC value, the four metabolites and p-tau showed the same power in discriminating AD from iNPH (Figs. 2, 5). Since p-tau is expected to be a good surrogate marker for AD progression [63], the four metabolites indicated here could also be additional surrogate markers. Moreover, metabolic alterations are reportedly seen at a relatively early stage of AD [6, 12, 62], and the four metabolites could be capable of detecting AD earlier than p-tau may detect it.
To validate these results and speculations, utilizing differential metabolite detection methods with additional larger sample size including healthy persons, patients with AD at an early stage, and/or mild cognitive impairment will reveal further information for AD pathogenesis and early diagnosis. We believe that the integration and combination of such information could contribute to developing new diagnostic markers for AD and to expanding the understanding of AD.
Conclusions
In this study, we found four metabolites that closely participate in the PPP, glycerolipid metabolism, amino acid metabolism, and lipid raft integration were significantly altered in the CSF of patients with AD compared to the CSF of patients with iNPH. All these biological pathways have been demonstrated to be associated with AD in previous reports. Additionally, the combination of these metabolites could discriminate between AD and iNPH with a power equal to that of p-tau and indicated moderate correlation with p-tau. In future studies, the combination of these and additional metabolites included in the metabolic pathways altered in AD would be useful to classify potential patients with AD earlier and with greater precision.
Acknowledgements
We thank Nobuyoshi Shimoda, Ph.D. for helpful discussions. We thank Editage (www.editage.jp) for English language editing.
Funding
This research was supported by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation of Japan (10-45), The Research Funding for Longevity Sciences (26-20), “Development of Diagnostic Technology for Detection of miRNA in Body Fluids” grant from the Japan Agency for Medical Research and Development and New Energy and Industrial Technology Development Organization.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its Additional file 1.
Abbreviations
- 2-HB
2-hydroxybutyrate
- AD
Alzheimer’s disease
- AUC
Area under the ROC curve
- CE-ESI-MS
Capillary electrophoresis-electrospray ionization-mass spectrometry
- CE-MS
Capillary electrophoresis-mass spectrometry
- CE-TOFMS
Capillary electrophoresis-time-of-flight-mass spectrometry
- CSF
Cerebrospinal fluid
- HPLC
High performance liquid chromatography
- iNPH
Idiopathic normal pressure hydrocephalus
- Neu5Ac
N-acetylneuraminate
- PSS
Phosphatidylserine synthase
Additional file
The list of metabolites detected in the comprehensive metabolic analysis. Cerebrospinal fluid of patients with Alzheimer’s disease and idiopathic normal pressure hydrocephalus was analyzed with capillary electrophoresis-time-of-flight mass spectrometry and detected metabolites were listed. Values are indicated as μM. ND, not detected. (XLSX 135 kb)
Authors’ contributions
YN and SN designed the experiments. AH, SI, AS, FS, MM, and TS performed the metabolome analysis. YN and AH wrote the initial draft, KO reviewed the results and performed critical reading and editing of the manuscript. MK performed the statistical analyses. MB, KH, SY, YG, and KU collected the CSF samples and provided the medical records. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of NCGG, NCNP and Tottori University, with the committee’s reference numbers of 443-6, A-2013-056 and 2188, respectively. Informed consents were obtained from all donors.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40364-018-0119-x) contains supplementary material, which is available to authorized users.
Contributor Information
Yuki Nagata, Email: nagata@ncgg.go.jp.
Akiyoshi Hirayama, Email: hirayama@ttck.keio.ac.jp.
Satsuki Ikeda, Email: satsuki@ttck.keio.ac.jp.
Aoi Shirahata, Email: aoiioa14@ttck.keio.ac.jp.
Futaba Shoji, Email: futa54@ttck.keio.ac.jp.
Midori Maruyama, Email: green12@ttck.keio.ac.jp.
Mitsunori Kayano, Email: kayano@obihiro.ac.jp.
Masahiko Bundo, Email: ibundou@ncgg.go.jp.
Kotaro Hattori, Email: hattori@ncnp.go.jp.
Sumiko Yoshida, Email: syoshida@ncnp.go.jp.
Yu-ichi Goto, Email: goto@ncnp.go.jp.
Katsuya Urakami, Email: kurakami@med.tottori-u.ac.jp.
Tomoyoshi Soga, Email: soga@sfc.keio.ac.jp.
Kouichi Ozaki, Email: ozakikk@ncgg.go.jp.
Shumpei Niida, Email: sniida@ncgg.go.jp.
References
- 1.Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, Delacourte A, Galasko D, Gauthier S, Jicha G, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 2.Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer’s disease. Ther Adv Neurol Disord. 2013;6:19–33. doi: 10.1177/1756285612461679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams RD, Fisher CM, Hakim S, Ojemann RG, Sweet WH. Symptomatic occult hydrocephalus with normal cerebrospinal-fluid pressure. New Engl J Med. 1965;273:117–126. doi: 10.1056/NEJM196507152730301. [DOI] [PubMed] [Google Scholar]
- 4.Kanai M, Matsubara E, Isoe K, Urakami K, Nakashima K, Arai H, Sasaki H, Abe K, Iwatsubo T, Kosaka T, et al. Longitudinal study of cerebrospinal fluid levels of tau, a beta1-40, and a beta1-42(43) in Alzheimer's disease: a study in Japan. Ann Neurol. 1998;44:17–26. doi: 10.1002/ana.410440108. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Zhong C. Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol. 2013;108:21–43. doi: 10.1016/j.pneurobio.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, Castellano A, Pifferi F, Bocti C, Paquet N, et al. Brain fuel metabolism, aging, and Alzheimer's disease. Nutrition. 2011;27:3–20. doi: 10.1016/j.nut.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouzier-Sore AK, Voisin P, Bouchaud V, Bezancon E, Franconi JM, Pellerin L. Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur J Neurosci. 2006;24:1687–1694. doi: 10.1111/j.1460-9568.2006.05056.x. [DOI] [PubMed] [Google Scholar]
- 8.Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer's disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging. 2005;32:486–510. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- 9.Mosconi L, Tsui WH, De Santi S, Li J, Rusinek H, Convit A, Li Y, Boppana M, de Leon MJ. Reduced hippocampal metabolism in MCI and AD: automated FDG-PET image analysis. Neurology. 2005;64:1860–1867. doi: 10.1212/01.WNL.0000163856.13524.08. [DOI] [PubMed] [Google Scholar]
- 10.Mills JD, Nalpathamkalam T, Jacobs HI, Janitz C, Merico D, Hu P, Janitz M. RNA-Seq analysis of the parietal cortex in Alzheimer's disease reveals alternatively spliced isoforms related to lipid metabolism. Neurosci Lett. 2013;536:90–95. doi: 10.1016/j.neulet.2012.12.042. [DOI] [PubMed] [Google Scholar]
- 11.Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- 12.Pasinetti GM, Eberstein JA. Metabolic syndrome and the role of dietary lifestyles in Alzheimer's disease. J Neurochem. 2008;106:1503–1514. doi: 10.1111/j.1471-4159.2008.05454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, Palumbo PJ. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997;145:301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- 14.D'Aniello A, Fisher G, Migliaccio N, Cammisa G, D'Aniello E, Spinelli P. Amino acids and transaminases activity in ventricular CSF and in brain of normal and Alzheimer patients. Neurosci Lett. 2005;388:49–53. doi: 10.1016/j.neulet.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 15.Czech C, Berndt P, Busch K, Schmitz O, Wiemer J, Most V, Hampel H, Kastler J, Senn H. Metabolite profiling of Alzheimer's disease cerebrospinal fluid. PLoS One. 2012;7:e31501. doi: 10.1371/journal.pone.0031501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibanez C, Simo C, Martin-Alvarez PJ, Kivipelto M, Winblad B, Cedazo-Minguez A, Cifuentes A. Toward a predictive model of Alzheimer's disease progression using capillary electrophoresis-mass spectrometry metabolomics. Anal Chem. 2012;84:8532–8540. doi: 10.1021/ac301243k. [DOI] [PubMed] [Google Scholar]
- 17.Ibanez C, Simo C, Barupal DK, Fiehn O, Kivipelto M, Cedazo-Minguez A, Cifuentes A. A new metabolomic workflow for early detection of Alzheimer's disease. J Chromatogr A. 2013;1302:65–71. doi: 10.1016/j.chroma.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Chin AL, Negash S, Hamilton R. Diversity and disparity in dementia: the impact of Ethnoracial differences in Alzheimer’s disease. Alzheimer Dis Assoc Disord. 2011;25:187–195. doi: 10.1097/WAD.0b013e318211c6c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mori E, Ishikawa M, Kato T, Kazui H, Miyake H, Miyajima M, Nakajima M, Hashimoto M, Kuriyama N, Tokuda T, et al. Guidelines for management of idiopathic normal pressure hydrocephalus: second edition. Neurol Med Chir (Tokyo) 2012;52:775–809. doi: 10.2176/nmc.52.775. [DOI] [PubMed] [Google Scholar]
- 20.Soga T, Igarashi K, Ito C, Mizobuchi K, Zimmermann HP, Tomita M. Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal Chem. 2009;81:6165–6174. doi: 10.1021/ac900675k. [DOI] [PubMed] [Google Scholar]
- 21.Soga T, Heiger DN. Amino acid analysis by capillary electrophoresis electrospray ionization mass spectrometry. Anal Chem. 2000;72:1236–1241. doi: 10.1021/ac990976y. [DOI] [PubMed] [Google Scholar]
- 22.Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009;69:4918–4925. doi: 10.1158/0008-5472.CAN-08-4806. [DOI] [PubMed] [Google Scholar]
- 23.Sugimoto M, Kawakami M, Robert M, Soga T, Tomita M. Bioinformatics tools for mass spectroscopy-based Metabolomic data processing and analysis. Curr Bioinforma. 2012;7:96–108. doi: 10.2174/157489312799304431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugimoto M, Wong DT, Hirayama A, Soga T, Tomita M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics. 2010;6:78–95. doi: 10.1007/s11306-009-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ihaka R and Gentleman R. R: a language for data analysis and graphics. J Comp Graph Stat. 1996;5:299-314.
- 26.Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, et al. HMDB: the human Metabolome database. Nucleic Acids Res. 2007;35:D521–D526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–d361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grant CM. Metabolic reconfiguration is a regulated response to oxidative stress. J Biol. 2008;7:1. doi: 10.1186/jbiol63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 31.Zhu X, Lee HG, Casadesus G, Avila J, Drew K, Perry G, Smith MA. Oxidative imbalance in Alzheimer's disease. Mol Neurobiol. 2005;31:205–217. doi: 10.1385/MN:31:1-3:205. [DOI] [PubMed] [Google Scholar]
- 32.Palmer AM. The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer's disease. J Neural Transm (Vienna) 1999;106:317–328. doi: 10.1007/s007020050161. [DOI] [PubMed] [Google Scholar]
- 33.Russell RL, Siedlak SL, Raina AK, Bautista JM, Smith MA, Perry G. Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch Biochem Biophys. 1999;370:236–239. doi: 10.1006/abbi.1999.1404. [DOI] [PubMed] [Google Scholar]
- 34.Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, Song W. Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A. 2006;103:18727–18732. doi: 10.1073/pnas.0606298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hakim AM, Moss G, Gollomp SM. The effect of hypoxia on the pentose phosphate pathway in brain. J Neurochem. 1976;26:683–688. doi: 10.1111/j.1471-4159.1976.tb04437.x. [DOI] [PubMed] [Google Scholar]
- 36.Ghauri FY, Nicholson JK, Sweatman BC, Wood J, Beddell CR, Lindon JC, Cairns NJ. NMR spectroscopy of human post mortem cerebrospinal fluid: distinction of Alzheimer's disease from control using pattern recognition and statistics. NMR Biomed. 1993;6:163–167. doi: 10.1002/nbm.1940060210. [DOI] [PubMed] [Google Scholar]
- 37.Kaddurah-Daouk R, Rozen S, Matson W, Han X, Hulette CM, Burke JR, Doraiswamy PM, Welsh-Bohmer KA. Metabolomic changes in autopsy-confirmed Alzheimer's disease. Alzheimers Dement. 2011;7:309–317. doi: 10.1016/j.jalz.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicoli F, Vion-Dury J, Confort-Gouny S, Maillet S, Gastaut JL, Cozzone PJ. Cerebrospinal fluid metabolic profiles in multiple sclerosis and degenerative dementias obtained by high resolution proton magnetic resonance spectroscopy. C R Acad Sci III. 1996;319:623–631. [PubMed] [Google Scholar]
- 39.Madeira C, Lourenco MV, Vargas-Lopes C, Suemoto CK, Brandao CO, Reis T, Leite RE, Laks J, Jacob-Filho W, Pasqualucci CA, et al. D-serine levels in Alzheimer's disease: implications for novel biomarker development. Transl. Psychiatry. 2015;5:e561. doi: 10.1038/tp.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giusto NM, Salvador GA, Castagnet PI, Pasquare SJ, Ilincheta de Boschero MG. Age-associated changes in central nervous system glycerolipid composition and metabolism. Neurochem Res. 2002;27:1513–1523. doi: 10.1023/A:1021604623208. [DOI] [PubMed] [Google Scholar]
- 41.Crook T, Petrie W, Wells C, Massari DC. Effects of phosphatidylserine in Alzheimer's disease. Psychopharmacol Bull. 1992;28:61–66. [PubMed] [Google Scholar]
- 42.Van Schaftingen E. D-glycerate kinase deficiency as a cause of D-glyceric aciduria. FEBS Lett. 1989;243:127–131. doi: 10.1016/0014-5793(89)80113-9. [DOI] [PubMed] [Google Scholar]
- 43.Snell K. The duality of pathways for serine biosynthesis is a fallacy. Trends Biochem Sci. 1986;11:241–243. doi: 10.1016/0968-0004(86)90184-2. [DOI] [Google Scholar]
- 44.Hart CE, Race V, Achouri Y, Wiame E, Sharrard M, Olpin SE, Watkinson J, Bonham JR, Jaeken J, Matthijs G, Van Schaftingen E. Phosphoserine aminotransferase deficiency: a novel disorder of the serine biosynthesis pathway. Am J Hum Genet. 2007;80:931–937. doi: 10.1086/517888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biemans EA, Verhoeven-Duif NM, Gerrits J, Claassen JA, Kuiperij HB, Verbeek MM. CSF d-serine concentrations are similar in Alzheimer's disease, other dementias, and elderly controls. Neurobiol Aging. 2016;42:213–216. doi: 10.1016/j.neurobiolaging.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 46.Grimes MA, Cameron JL, Fernstrom JD. Cerebrospinal fluid concentrations of large neutral and basic amino acids in Macaca Mulatta: diurnal variations and responses to chronic changes in dietary protein intake. Metabolism. 2009;58:129–140. doi: 10.1016/j.metabol.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 47.Malaisse WJ, Zhang Y, Louchami K, Sener A, Portois L, Carpentier YA. Brain phospholipid and triglyceride fatty acid content and pattern in type 1 and type 2 diabetic rats. Neurosci Lett. 2006;409:75–79. doi: 10.1016/j.neulet.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 48.Janson J, Laedtke T, Parisi JE, O'Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–481. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 49.Biessels GJ, Kappelle LJ. Increased risk of Alzheimer's disease in type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem Soc Trans. 2005;33:1041–1044. doi: 10.1042/BST0331041. [DOI] [PubMed] [Google Scholar]
- 50.de la Monte SM, Wands JR. Alzheimer's disease is type 3 diabetes–evidence reviewed. J Diabetes Sci Technol (Online) 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sonnino S, Mauri L, Chigorno V, Prinetti A. Gangliosides as components of lipid membrane domains. Glycobiology. 2007;17:1r–13r. doi: 10.1093/glycob/cwl052. [DOI] [PubMed] [Google Scholar]
- 52.Cantu L, Del Favero E, Sonnino S, Prinetti A. Gangliosides and the multiscale modulation of membrane structure. Chem Phys Lipids. 2011;164:796–810. doi: 10.1016/j.chemphyslip.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 53.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harris B, Pereira I, Parkin E. Targeting ADAM10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res. 2009;1296:203–215. doi: 10.1016/j.brainres.2009.07.105. [DOI] [PubMed] [Google Scholar]
- 55.Hur JY, Welander H, Behbahani H, Aoki M, Franberg J, Winblad B, Frykman S, Tjernberg LO. Active gamma-secretase is localized to detergent-resistant membranes in human brain. FEBS J. 2008;275:1174–1187. doi: 10.1111/j.1742-4658.2008.06278.x. [DOI] [PubMed] [Google Scholar]
- 56.Parkin ET, Turner AJ, Hooper NM. Amyloid precursor protein, although partially detergent-insoluble in mouse cerebral cortex, behaves as an atypical lipid raft protein. Biochem J. 1999;344(Pt 1):23–30. doi: 10.1042/bj3440023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Furukawa K, Ohmi Y, Ohkawa Y, Tokuda N, Kondo Y, Tajima O, Furukawa K. Regulatory mechanisms of nervous systems with glycosphingolipids. Neurochem Res. 2011;36:1578–1586. doi: 10.1007/s11064-011-0494-2. [DOI] [PubMed] [Google Scholar]
- 58.Kracun I, Rosner H, Drnovsek V, Heffer-Lauc M, Cosovic C, Lauc G. Human brain gangliosides in development, aging and disease. Int J Dev Biol. 1991;35:289–295. [PubMed] [Google Scholar]
- 59.Rosalki SB, Wilkinson JH. Reduction of alpha-ketobutyrate by human serum. Nature. 1960;188:1110–1111. doi: 10.1038/1881110a0. [DOI] [PubMed] [Google Scholar]
- 60.Newington JT, Rappon T, Albers S, Wong DY, Rylett RJ, Cumming RC. Overexpression of Pyruvate Dehydrogenase Kinase 1 and lactate Dehydrogenase a in nerve cells confers resistance to Amyloid β and other toxins by decreasing mitochondrial respiration and reactive oxygen species production. J Biol Chem. 2012;287:37245–37258. doi: 10.1074/jbc.M112.366195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mosconi L, Berti V, Glodzik L, Pupi A, De Santi S, de Leon MJ. Pre-clinical detection of Alzheimer’s disease using FDG-PET, with or without Amyloid imaging. J Alzheimers Dis. 2010;20:843–854. doi: 10.3233/JAD-2010-091504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cruz T, Gleizes M, Balayssac S, Mornet E, Marsal G, Millan JL, Malet-Martino M, Nowak LG, Gilard V, Fonta C. Identification of altered brain metabolites associated with TNAP activity in a mouse model of hypophosphatasia using untargeted NMR-based metabolomics analysis. J Neurochem. 2017;140:919–940. doi: 10.1111/jnc.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blennow K, Zetterberg H, Minthon L, Lannfelt L, Strid S, Annas P, Basun H, Andreasen N. Longitudinal stability of CSF biomarkers in Alzheimer's disease. Neurosci Lett. 2007;419:18–22. doi: 10.1016/j.neulet.2007.03.064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its Additional file 1.