

Abstract
Objective
To study the influence of the Abelson helper integration site 1 (AHI1) locus associated with MS susceptibility on CD4+ T cell function.
Methods
We characterized the chromatin state of T cells in the MS-associated AHI1 linkage disequilibrium (LD) block. The expression and the role of the AHI1 variant were examined in T cells from genotyped healthy subjects who were recruited from the PhenoGenetic Project, and the function of AHI1 was explored using T cells from Ahi1 knockout mice.
Results
Chromatin state analysis reveals that the LD block containing rs4896153, which is robustly associated with MS susceptibility (odds ratio 1.15, p = 1.65 × 10−13), overlaps with strong enhancer regions that are present in human naive and memory CD4+ T cells. Relative to the rs4896153A protective allele, the rs4896153T susceptibility allele is associated with decreased AHI1 mRNA expression, specifically in naive CD4+ T cells (p = 1.73 × 10−74, n = 213), and we replicate this effect in an independent set of subjects (p = 2.5 × 10−9, n = 32). Functional studies then showed that the rs4896153T risk variant and the subsequent decreased AHI1 expression were associated with reduced CD4+ T cell proliferation and a specific differentiation into interferon gamma (IFNγ)–positive T cells when compared with the protective rs4896153A allele. This T cell phenotype was also observed in murine CD4+ T cells with genetic deletion of Ahi1.
Conclusions
Our findings suggest that the effect of the AHI1 genetic risk for MS is mediated, in part, by enhancing the development of proinflammatory IFNγ+ T cells that have previously been implicated in MS and its mouse models.
MS is a chronic inflammatory disease of the CNS characterized by episodes of neuronal demyelination in genetically susceptible individuals on exposure to environmental triggers. Genome-wide association studies (GWASs) have identified and validated over 110 non–major histocompatibility complex genetic variants associated with MS, and many of these are implicated in the function of CD4+ and CD8+ T cells that are present in MS lesions.1,2 Considerable work over the years in studies of patients with MS and in animal models of MS has implicated proinflammatory CD4+ T helper (Th)1 cells (which produce interferon gamma [IFNγ]) and Th17 cells (interleukin [IL]-17-producers) as important mediators of new relapsing disease activity.3,4 These 2 types of pathogenic cells are implicated in MS and could account for some of the immunologic and clinical heterogeneity of the disease.5
Recent analysis of Th17 cell development and pathogenicity revealed a critical role of IFNγ for the severity of the disease in mouse models of MS. Indeed, in a reporter mouse model, it has been found that Th17 cells could play a pathogenic role in MS by converting their phenotype into a proinflammatory Th1 profile in which committed Th17 cells give rise to a progeny that shifts toward enhanced IFNγ expression.6 Similarly, in humans, there is evidence indicating that, in the presence of IL-12, Th17 cells also produce IFNγ. These cells, which produce both IL-17 and IFNγ, are called Th1/17 cells,7 and, together with “non-classical Th1 cells,” they might contribute to disease pathogenesis through properties shared by both the Th1 and Th17 subsets.8 Moreover, Th17 cells producing IFN-γ are enriched in myelin oligodendrocyte glycoprotein-specific T cells.9 IFNγ produced by these cells could strongly activate macrophages whose infiltration in the CNS correlates with experimental autoimmune encephalomyelitis (EAE) severity.10
It is necessary to understand the functional consequences of disease-associated genetic variations as we strive to unravel the causal chain of events linking genetic risk factors to clinical syndromes. Separate from GWASs, expression quantitative trait locus (eQTL) studies have been used to map a genetic variants contribution to variation in gene expression.11 However, moving from a list of eQTL effects to phenotypic changes, regulation of disease activity remains challenging and is often unfruitful.
In this study, we integrated results from large-scale, genome-wide disease gene discovery studies with eQTL studies to identify the MS susceptibility variant with a transcriptional effect that influences T cell phenotypes. We examined such a variant in the MS susceptibility gene Abelson helper integration site 1 (AHI1) by first characterizing and validating its cis-eQTL effect, and we go on to demonstrate that the AHI1 genetic variant is involved in the regulation of human CD4+ T cell proliferation and IFNγ production. Furthermore, we leveraged a mouse model with genetic deletion of Ahi1 to analyze the effects of Ahi1 deficiency on murine CD4+ T cell function. Consistent with the human results, the mouse results suggest that the mechanism of the AHI1 genetic risk involves the differentiation of naive CD4+ T cells into proinflammatory IFNγ+ T cells, which could contribute to MS onset.
METHODS
PhenoGenetic Project.
The PhenoGenetic Project (PGP) is a biobank of healthy individuals, older than 18 years, who are free from chronic inflammatory infections and metabolic diseases.11 Genotyped individuals (n = 32) of European descent bearing risk (TT), protective (AA), or heterozygous (AT) genotypes for rs4896153 were selected from this cohort and used for this study.
PBMC isolation and CD4+ T cell culture.
Peripheral venous blood was obtained from healthy control volunteers in compliance with protocols approved by the Institutional Review Board of Partners Healthcare as part of the PGP. Peripheral blood mononuclear cells (PBMCs) were separated by Ficoll-Paque PLUS (GE Healthcare, Piscataway, NJ) gradient centrifugation. PBMCs were frozen at a concentration of 1–3 × 107 cells/mL in 10% DMSO and stored at −80°C. CD4+CD45RO−CD45RA+ naive T cells were isolated from cryopreserved PBMCs by negative selection (Miltenyi Biotec, Auburn, CA). Naive CD4+ T cells were cultured in 96-well round bottom plates (Costar, Cambridge, MA) at 2 × 104 cells/well in serum-free X-Vivo 15 medium (BioWhittaker, Walkersville, MD) and stimulated with plate-bound anti-CD3 OKT3 (BioXCell, 10 μg/mL) and anti-CD28 (BioXCell, 1 μg/mL) antibodies for 6–8 days.
Mouse CD4+ T cell isolation and activation.
Ahi1−/− mice were generated as described previously12 and backcrossed >10 generations onto a Friend Virus B NIH Jackson background. CD4+ T cells were isolated from the spleens of Ahi1−/− and littermate control mice by negative selection (Miltenyi Biotec, Auburn, CA) and cultured in 96-well round bottom plates (Costar, Cambridge, MA) at 1 × 105 cells/well in serum-free X-Vivo 15 medium (BioWhittaker, Walkersville, MD) supplemented with 1:1000 BME and stimulated with plate-bound anti-CD3 (BioXCell, 0.25–2 μg/mL) and anti-CD28 (BioXCell, 0.25–2 μg/mL) antibodies for 4 days. For Th1 cell polarization, recombinant IL-12 was used at 10 ng/mL. For Th17 cell polarization, recombinant IL-6 (10 ng/mL) and transforming growth factor–β1 (3 ng/mL) were used. All recombinant proteins were from R&D Systems (Minneapolis, MN).
Cytokine measurement by flow cytometry.
CD4+ T cells were stimulated for 4 hours with PMA (50 ng/mL) and Ionomycin (250 ng/mL, both from Sigma-Aldrich, St. Louis, MO) in the presence of GolgiStop (BD Biosciences), then washed and fixed/permeabilized with Cytofix/Cytoperm (BD Biosciences, San Jose, CA) according to manufacturer's instructions. Cells were stained with 7-AAD (BD Biosciences), or Aqua LIVE/DEAD Fixable stain (Life Technologies, Carlsbad, CA) and FITC-IFNγ (clone 25723; R&D Systems). Data were acquired on a FACSCalibur or LSRII (BD Biosciences) and analyzed with FlowJo software (TreeStar, Ashland, OR).
RNA isolation and qPCR.
RNA was isolated using the Qiagen Plus Micro Kit (Qiagen, Venlo, the Netherlands) and converted to cDNA via reverse transcriptase by random hexamers and MuLV transcriptase (Applied Biosystems, Foster City, CA). Samples were subjected to real-time PCR analysis on PRISM 7000 Sequencer Detection System (Applied Biosystems) under standard conditions. The primers used for the human AHI1 were purchased from Applied Biosystems: isoform A forward primer 5′- CCAGCTAATCATGTGGCTAGTGAAACACTG-3′; reverse primer 5′ CCTCAGGGCTTAAAGGAGGGGATGC-3′; isoform B forward primer 5′- TCAGACCGACAGTCACTTTGCTGAA-3′; and reverse primer 5′ TGGTGGGATCCCAGGTCGGCTCAGT-3′. Values are represented as the difference in Ct values normalized to β2-microglobulin for each sample as per the following formula: relative RNA expression = (2−dCt) × 103. Murine Ahi1 was measured using commercially available assays (Applied Biosystems). Relative mRNA abundance was normalized against Gapdh.
Proliferation assay.
Cells were cultured for 48 hours before the addition of 1 μCi of [3H]-thymidine for 16 hours. The cells were harvested and quantified using an automated sample harvester (Perkin-Elmer, Waltham, MA). [3H]-thymidine uptake is expressed in radiation counts per minute.
Statistical analysis.
Statistical analysis was performed using Prism version 7.0 and R version 3.2.1. Cytokine and gene expression data were analyzed using unpaired t tests. Results from figure 1A are from Raj et al.11 and represent meta-analysis of cis-eQTLs at false discovery rate 0.05 in CD4+ T cells from 407 individuals.
Figure 1. SNAP plot of the chromatin state overlapping the SNPs in LD with the AHI1 cis-eQTL and MS susceptibility variant, rs4896153 in CD4+ T cells.

(A) Naive (CD4+CD25−CDRA+) and memory (CD4+CD25−CDRA−) CD4+ T cell–specific chromatin state mapping was generated using the chromHMM algorithm in 1-Mb and 100-kb views surrounding the AHI1 locus. ChIP-seq data generated by the ENCODE/ROADMAP project reveal various chromatin marks including enhancers, heterochromatin, and polycomb, and various transcription marks are color coded. All the discovered SNPs from dbGAP137 are listed including their p value of association to MS, the recombination rate. The LD structure is represented in the r-square value calculated from the Broad SNAP server and is assigned in red. The strongest MS-associated variant rs4896153 (green) and the strongest eQTL variant rs6908428 are labeled. (B) Zoom in of the region overlapping the rs13197384 showing the chromatin state in naive and memory T cells. eQTL = expression quantitative trait locus; LD = linkage disequilibrium; TSS = transcription start site.
RESULTS
AHI1 locus is associated with MS susceptibility and has a cis-eQTL effect.
The AHI1 locus has long been associated with MS. In 2011, a GWAS identified rs11154801A with an odds ratio (OR) of 1.13 (p = 1 × 10−13) in the AHI1 locus, and the association was initially attributed to the MYB gene based on its proximity to the most associated variant.13 In a subsequent study using the ImmunoChip array, the association with rs11154801A was replicated (OR 1.11; p = 2.3 × 10−9) and assigned to the AHI1 gene.2 Neither of these studies densely genotyped this region; thus, we leveraged results of a large-scale imputation based meta-analysis of the IMSGC to identify the most strongly associated single nucleotide polymorphisms (SNPs) within this locus. In this analysis,2 the original lead SNP, rs11154801A was genome-wide significant (p = 1.0 × 10−12). However, another SNP in the region had a more statistically significant p value: rs4896153T (OR 1.15, p = 1.65 × 10−13). These 2 SNPs are in moderate linkage disequilibrium (LD), with an r2 of 0.55 and D′ of 0.96 in the European panel data of the 1000 Genome Project. Of note, rs4896153 was not present in either of the previously published studies that reported rs11154801A as the most associated SNP. We therefore used rs4896153 as the tag SNP for this MS susceptibility locus.
Next, we tested whether rs4896153 exhibited a cis-eQTL effect in any of the nearby genes using a large-scale data set of naive CD4+ T cells from healthy genotyped individuals. We found that the rs4896153T risk allele is associated with lower RNA expression of AHI1 (p = 2.14 × 10−24); however, an SNP nearby, rs6908428, had a stronger effect (p = 8.77 × 10−31) after correction for batch, age, and/or gender effects (table 1 and figure e-1, http://links.lww.com/NXI/A5). These 2 SNPs are in moderate LD, with an r2 of 0.58 and D′ of 0.98. Of interest, rs6908428 and rs4896153 did not alter the expression of the neighboring genes in naive T cells, suggesting that AHI1 is the main target of these 2 SNPs (table 1). When rs6908428 and rs4896153 are modeled jointly with the expression of AHI1 as the outcome in CD4+ T cells, the p values are 0.026 and 0.537, respectively. This result indicates that rs6908428 has a marginally independent association with the expression of AHI1, but that rs4896153 does not, and that rs6908428 may be more likely to be the functional variant. However, 1 technical explanation for the superior eQTL effect of rs6908428 (MAF 0.3173, 1000 Genomes) is that it has a better imputation INFO score of 0.96 vs 0.55 for rs4896153 (MAF 0.3007, 1000 Genomes) in our sample of 211 subjects. For instance, the MAF of rs6908428 is much closer to the 1000 Genomes MAF of 0.31, than the MAF of rs4896153 (0.467). The lower imputation metric of rs4896153 implies a less accurate dosage measurement, thereby reducing power and likely artificially diminishing the level of statistical significance. In addition, the rs6908428 SNP is also a cis-eQTL for AHI1 in PBMCs of MS subjects (p = 2 × 10−22).14
Table 1.
rs4896153 and rs6908428 SNPs influence specifically AHI1 expression but not other neighboring genes in naive CD4+ T cells
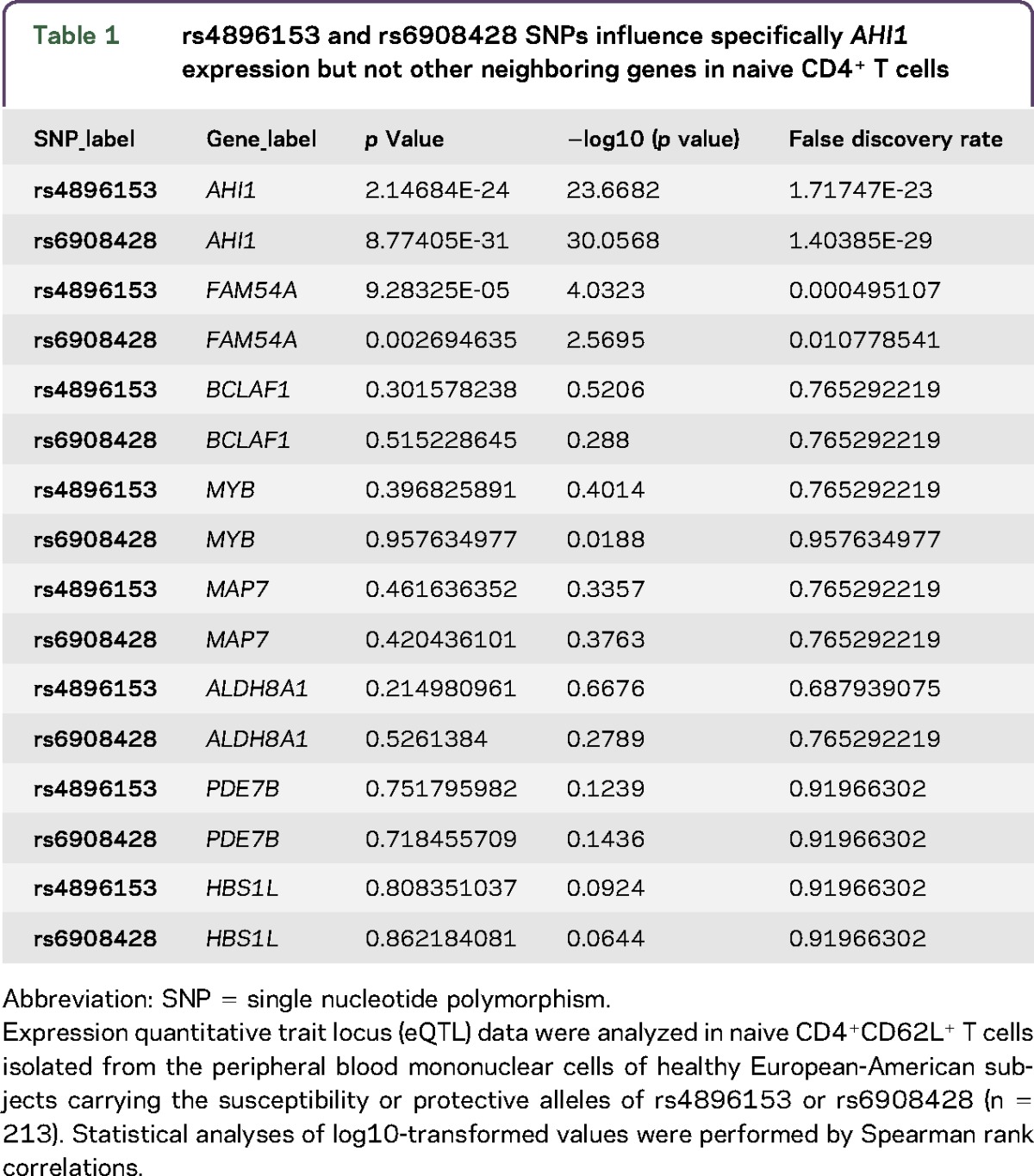
To visualize the MS susceptibility LD region associated with a cis-eQTL effect on AHI1, an SNAP plot was generated (figure 1, A and B). Eleven SNPs (red and purple) are in an LD block with rs4896153 (green) as the lead MS SNP. Ten SNPs are in strong LD with this lead SNP and are considered to be the “critical set” of SNPs that are likely to include the causal variant. To prioritize variants among this set of ten SNPs, we first looked at reference epigenomic data from CD4+ T cells. The transcription start site (TSS) of AHI1 is flanked by enhancer marks in both naive and memory CD4+ T cells, consistent with this gene's expression in this cell type (figure 1, A and B). The SNP rs13197384 (purple) is 4 bp downstream of the +1 TSS of AHI1 and overlaps with a region of transcription at the 5′ or 3′, specifically in naive CD4+ T cells (figure 1, A and B). This SNP is therefore highlighted as being in an area of differential transcriptional activity between naive and memory T cells by existing reference epigenomic data.15 Next, we investigated the functional potential of these SNPs using RegulomeDB and HaploReg V4.2: rs13197384 (TSS), rs7750586 (upstream), and rs9399148 (upstream); all have evidence of functionality based on these SNPs altering known target sequences for DNA-binding motifs and being present within DNase hypersensitivity sites that mark chromosomal segments bound to a DNA-binding protein in naive CD4+ T cells (supplementary material, table e-1, http://links.lww.com/NXI/A6). These data prioritize these 3 SNPs for follow-up functional assessment. In an attempt to understand the influence of the SNP position within the enhancer region in CD4+ T cells, we used the R package motifbreakR version 1.4.016 to identify putative transcription factor (TF) binding sites that were likely to be disrupted by the studied polymorphism. TF motifs were obtained from the JASPAR 2014 database (177 motifs for homo sapiens),17 and the motifbreakR algorithm was applied with default settings: the weighted sum method (weighted by relative entropy) was used to obtain scores, and putative bindings sites were reported if the scaled score (between 0 and 1) was greater than or equal to 0.8 for the reference or alternative allele. Binding sites whose unscaled and unweighted scores for the reference and alternative allele did not differ by at least 0.4 were considered as unaffected by the SNP and not reported (supplementary material, table e-2, http://links.lww.com/NXI/A7). We identified candidate TFs whose binding could be affected by the presence of 1 of these 3 SNPs. For instance, the transcription repressor CTCF also known as 11-zinc finger protein or CCCTC-binding factor that is involved in the induction of IFNγ signaling18 overlaps the rs13197384 variant (relative matching score = 0.8). These findings suggest that the rs13197384 variant, which is in LD with the lead MS SNP, may be involved in the regulation of AHI1 transcription in CD4+ T cells.
AHI1 eQTL replication in T cell receptor stimulated T cells.
To study the functional consequences of rs4896153 and its set of linked SNPs, we investigated the correlation between the rs4896153T risk allele and the adaptive immune system of healthy subjects. We used venous blood from healthy subjects, which allows for the examination of the immune system without any confounding effects of systemic inflammation or immune-modulating therapies in patients with autoimmune diseases.19 Healthy genotyped subjects free of autoimmune and chronic infectious diseases were selected from the PGP who were homozygous for either rs4896153T (risk allele) or rs4896153A (protective allele). Using microbeads, naive CD4+ T cells were isolated from PBMCs by negative selection. We first replicated the prior observation that rs4896153 has a cis-eQTL effect in naive CD4+ T cells (figure 2A). Specifically, the rs4896153T risk allele showed significantly lower expression of AHI1 than the rs4896153A protective allele (ρ −0.837, p = 4.29 × 10−9, n = 32), as measured by quantitative PCR. This cis-eQTL effect was present for both of the 2 major AHI1 isoforms: isoform A (figure 2B, mRNA = NM_017651.4, NM_001134830.1, NM_001134831.1; protein = NP_001128303.1) and isoform B (figure 2C, mRNA = NM_001134832.1; protein = NP_001128304.1). AHI1 isoform B lacks the SRC Homology 3 (SH3) domain that is found in isoform A; SH3 is a signaling domain involved in mediating the interaction of proteins involved in signaling pathways.20
Figure 2. The MS risk allele rs4896153T is associated with decreased AHI1 expression in naive CD4+ T cells and T cell receptor–stimulated CD4+ T cells.

(A) Naive CD4+ T cells were isolated from the peripheral blood of healthy subjects bearing risk (TT), protective (AA), or heterozygous (AT) genotypes for rs4896153, and AHI1 expression was measured by Taqman qPCR. The 2 major isoforms of AHI1 (B) isoform A (mRNA = NM_017651.4, NM_001134830.1, NM_001134831.1; protein = NP_001128303.1) and (C) isoform B (mRNA = NM_001134832.1; protein = NP_001128304.1) were measured in genotype individuals from (1A) by SYBR green qPCR. (D) Naive CD4+ T cells were stimulated with anti-CD3/CD28 for 0–48 hours, and AHI1 expression was measured by Taqman qPCR. Comparing the AA and TT genotypes, all data points are p < 0.01. Each dot represents an individual. Data are represented as mean ± SEM.
To investigate the temporal expression of AHI1 in CD4+ cells, we measured AHI1 in genotyped samples over time in naive CD4+ T cells after T cell receptor (TCR) stimulation with anti-CD3 and anti-CD28 monoclonal antibodies. We found that AHI1 mRNA expression is rapidly induced within 4 hours of stimulation then decreases over 24 hours, increasing again by 48 hours with the cis-eQTL effect remaining significant during TCR stimulation (p < 0.01 for all time points) (figure 2D).
AHI1 risk variant promotes a CD4+IFNγ+ T cell phenotype.
Previous reports described AHI1 as an oncogene that enhances cancer cell proliferation.21–26 To investigate the effects of the rs4896153 genotype on CD4+ T cell function beyond RNA expression, we measured (1) T cell proliferation by [3H]-thymidine incorporation assays and (2) T cell cytokine profile. Naive CD4+ T cells isolated from PBMCs of healthy genotyped subjects homozygous for either the AHI1 risk (n = 8) or protective (n = 13) alleles were exposed to TCR stimulation followed by analysis of T cell proliferation (day 3) and cytokine expression (day 7). No significant difference was seen in proliferative capacity between the genotype categories (figure 3A). Of interest, T cells bearing the rs4896153TT genotype yielded significantly greater frequency of IFNγ+ T cells than the rs4896153AA genotype in TCR stimulated cells (p = 0.008) (figure 3B). Within the IFNγ-positive cells, more IFNγ was expressed in cells from subjects with the risk genotype as measured by mean fluorescence intensity (MFI) (figure 3C), providing a link between an MS genetic susceptibility and a proinflammatory T cell phenotype. The effect of the AHI1 risk allele on T cell phenotype may be specific to IFNγ because we did not find any significant alteration of IL-17 expression when naive CD4+ T cells carrying the AHI1 risk or protective alleles were polarized under Th17 cell condition (data not shown).
Figure 3. Decreased AHI1 in human CD4+ T cells is associated with decreased proliferation and increased IFNγ production.

(A) Naive CD4+ T cells were isolated from the peripheral blood of healthy subjects bearing risk (TT), protective (AA), or heterozygous (AT) alleles for rs4896153, and cells were activated with anti-CD3/CD28 followed by the analysis of cell proliferation by [3H]-thymidine incorporation 60 hours after stimulation. (B, C) A subset of these cells was further stimulated for 7 days, and IFNγ cytokine expression was measured by flow cytometry. (B) Frequency of CD4+IFNγ+ T cells bearing risk (TT) or protective (AA) alleles for the rs4896153 was analyzed by intracellular staining using flow cytometry. (C) Mean fluorescent intensity (MFI) of IFNγ expression from the activated T cells is shown. p Values were generated using Student's t test. Each dot represents an individual. Data are represented as mean ± SEM. IFNγ = interferon gamma.
Increased IFNγ expression in Ahi1 knockout CD4+ T cells.
To further explore the role of AHI1 in the regulation of IFNγ production by CD4+ T cells, we first measured Ahi1 expression in murine CD4+ T cells. Naive CD4+ T cells were isolated from spleens of wild-type (WT) mice, and cells were TCR stimulated for several time points. RNA lysates were prepared at each time point, and murine Ahi1 mRNA expression was assessed by quantitative PCR. In agreement with AHI1 expression in activated human CD4+ T cells, we found that the mRNA levels of murine Ahi1 are moderately induced on TCR activation (4–6 hours) but rapidly decreased later as shown at 24–72 hours, suggesting a regulatory role of Ahi1 in the differentiation, phenotype, or function of CD4+ T cells (figure 4A and figure e-2A, http://links.lww.com/NXI/A5). Similarly, the rapid but reversible increase in Ahi1 expression was also observed in CD4+T cells polarized under Th1 cells, a T cell subset that is known to express IFNγ+ T cells.
Figure 4. Ahi1 genetic deficiency decreases T cell proliferation and upregulates IFNγ production.

(A) Ahi1 mRNA expression in activated T cells. Wild-type (WT) naive CD4+ T cells were isolated from the spleens of naive WT mice, and cells were activated with anti-CD3/CD28 for up to 72 hours (h). Ahi1 mRNA expression was measured by Taqman and calculated relatively to the house keeping gene Gapdh. (B, C) Effects of Ahi1 on T cell proliferation and IFNγ expression. Naive CD4+ T cells were isolated from the spleens of Ahi1−/− or WT littermates, and cells were activated with anti-CD3/CD28 (0.25 μg/mL) for (B) 60 hours to measure T cell proliferation by [3H]-thymidine incorporation or for (C) 4 days to measure IFNγ production by intracellular staining using flow cytometry. Data represent the average of 3 independent experiments. Each dot represents a mouse. Data are represented as mean ± SEM. IFNγ = interferon gamma.
Next, we took advantage of total Ahi1 knockout (Ahi1−/−) mice27 to measure the role of Ahi1 in T cells using Ahi1 genetic deletion. Spleens were dissected from Ahi1−/− and WT littermates, and naive CD4+ T cells were sorted and activated with mouse specific anti-CD3/CD28 antibodies. We found that, using a more physiologic stimulation that does not bypass costimulatory pathways (low anti-CD3/CD28 concentrations, each mAb at 0.25 μg/mL), CD4+ T cells from Ahi1−/− mice exhibited impaired cell proliferation compared with their WT counterparts (figure 4B). Strikingly, we detected increased frequency of IFNγ-producing T cells in Ahi1−/− T cells (figure 4C), an effect that is in agreement with the human data. To study whether Ahi1 regulates T cell cytokine production under polarizing conditions, we analyzed IFNγ and IL-17A expressions in CD4+ T cells polarized toward a proinflammatory Th1 or Th17 fate. There was a trend implicating a modest increase in IFNγ+ frequency among Th1 cells (figure e-3A, http://links.lww.com/NXI/A5). Moreover, analysis of IL-17A in Ahi1−/− T cells polarized under Th17 condition did not reveal any significant alteration of IL-17A expression in the absence of Ahi1 (figure e-3B, http://links.lww.com/NXI/A5). Altogether, these findings suggest that Ahi1 regulates specifically IFNγ-producing CD4+ T cells in the absence of exogenous polarizing molecules.
DISCUSSION
Rare mutations in AHI1/Ahi1 have been reported to cause Joubert syndrome, a ciliopathic disorder, characterized by malformations of the cerebellar vermis and brainstem as well as clinical manifestations that include, breathing irregularities, hypotonia, developmental delays, and ocular motor apraxia.28 The AHI1 protein contains an N-terminal coiled-coil domain, 7 WD40 domains, and an SH3 domain; it is the only protein known to contain both a WD40 domain and an SH3 domain, suggesting a novel role in cell signaling for this protein.29 In chronic myeloid leukemia cells, AHI1 knockdown showed reduced TNFα, IL-4, and IL-2 cytokine production.30 On the other hand, overexpression of AHI1 conferred a growth advantage,25 which is consistent with our mouse data in which reduced Ahi1 expression in CD4+ T cells was associated with reduced proliferative capacity. However, the decrease in AHI1 expression did lead to increased cytokine production at least for IFNγ, suggesting that the role of AHI1 in primary human T cells may involve distinct signaling pathways in comparison with tumor cells. In MS, proinflammatory cytokines such as IFNγ, IL-17, IL-22, and GM-CSF are present in elevated amounts in the CSF and CNS lesions of MS patients compared with healthy controls, especially during the active phase of the disease.31–34 The frequency of IFNγ vs IL-17 responses in EAE can influence the localization of inflammation in the CNS.35 Robust Th1 responses producing elevated levels of IFNγ induced CD4+ T cell infiltration in the spinal cord. By contrast, high IL-17 levels caused CD4+ T cell infiltration preferentially into the brain parenchyma and induced atypical EAE.5 In our study, although the AHI1 risk variant was associated with an increase in IFNγ production by CD4+ T cells, we did not detect a significant alteration in IL-17 levels. It should be noted that recent studies in several immunologic diseases including sarcoidosis and Crohn disease identified a subset of pathogenic IFNγ-producing Th17 cells called Th17.1 cells expressing chemokine receptors CCR6 and CXCR3.36,37 These cells are believed to be derived from classically polarized Th17 cells and differentiate into a Th1-like phenotype in which they produce significant IFNγ but little amounts of IL-17.38 Of interest, in patients with MS, circulating Th17 cells that respond to myelin peptides express high levels of IFNγ, suggesting that these autoreactive T cells are plastic and express distinct cytokine profiles from that identified in vitro.39 A more extensive cytokine characterization of CD4+ T cells expressing the AHI1 risk variant may address this issue further.
In this study, we confirm that rs4896153, an MS susceptibility variant, has a strong cis-eQTL effect on overall AHI1 mRNA expression, affecting both AHI1 isoforms, and that this effect is maintained for at least 48 hours after TCR stimulation. To assess the functional outcome of this SNP on CD4+ T cells, we measured proliferation and cytokine production following TCR stimulation. While we did not see a significant allelic difference in proliferation of human T cells, we consistently saw visibly less cell clonal expansion in the rs4896153TT cultures, an effect noticeable after only 24 hours of differentiation (data not shown). We postulate a role for AHI1 early on in TCR activation, given its peak mRNA expression 4–6 hours after anti-CD3/CD28 stimulation of both mouse and human CD4+ T cells. AHI1 is localized to the mother centriole, and for TCR signaling, the reorientation and movement of the centriole and associated Golgi apparatus toward the contact area between a T cell and an antigen-presenting cell is required after an initial contact.40 It is therefore possible that AHI1, being localized to the mother centriole and involved in actin organization, may play a role in the formation or stabilization of the TCR synapse, which could be a mechanism for its association with MS.
Identification of the causal variant (the SNP/s that affect the trait), and determining their exact mechanism of action, is one of the main challenges and provides the next step in GWAS interpretation.41 Our analysis of the GWAS identified MS variants near the AHI1 gene and prioritized an LD block with several SNPs that are associated with decreased AHI1 expression in CD4+ T cells carrying the MS risk allele. We identified 3 SNPs, rs13197384, rs7750586, and rs9399148, with suggestive functionality based on (1) being present within DNase hypersensitivity sites and (2) the potential alteration of TF binding to their DNA motifs in CD4+ T cells. We identified candidate TFs whose binding could be affected by the presence of 1 of these 3 SNPs including the transcription repressor CTCF that is involved in the induction of IFNγ signaling18 in which the CTCF consensus binding site overlaps with rs13197384. Further functional studies, such as minigene reporter assays, are warranted to identify causal variants that influence a disease trait in MS.
We report that the rs4896153T allele reduces AHI1 RNA expression and is associated with a greater percentage of IFNγ-producing CD4+ T cells. While additional functional studies are required to further delineate the role of AHI1 in MS susceptibility, our results expand the repertoire of dysfunctional cytokine responses that are genetically implicated in MS susceptibility and may help establish a scaffold on which to assemble other susceptibility variants.
ACKNOWLEDGMENT
The authors thank the participants of PGP for their time and specimens that they contributed.
GLOSSARY
- AHI1
Abelson helper integration site 1
- EAE
experimental autoimmune encephalomyelitis
- eQTL
expression quantitative trait locus
- GWAS
genome-wide association study
- IFNγ
interferon gamma
- IL
interleukin
- LD
linkage disequilibrium
- MFI
mean fluorescence intensity
- OR
odds ratio
- PBMC
peripheral blood mononuclear cell
- PGP
PhenoGenetic Project
- SNP
single nucleotide polymorphism
- TCR
T cell receptor
- TF
transcription factor
- Th
T helper
- TSS
transcription start site
- WT
wild type
AUTHOR CONTRIBUTIONS
B.J.K. designed the study, performed experiments, analyzed data, and drafted the manuscript. T.S.B. performed experiments. C.W. performed statistical analyses. H.-U.K. and N.P. analyzed and interpreted data. J.R.B. and R.J.F. provided Ahi1−/− mice and splenocytes. E.M.B. designed the study and interpreted data. P.L.D.J. and W.E. designed the study, interpreted data, supervised the study, and wrote the manuscript.
STUDY FUNDING
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the NIH under award number R01AI130547 (W.E.).
DISCLOSURE
B.J. Kaskow, T.S. Buttrick, H.-U. Klein, C. White, and J.R. Bourgeois report no disclosures. R.J. Ferland received research support from NIH, RPI, AMC, March of Dimes Foundation, and NYSTEM. N. Patsopoulos received research support from Intel Corporation, National MN Society, and TileDb, Inc. E.M. Bradshaw reports no disclosures. P.L. De Jager is on the advisory board of TEVA Neuroscience, Genzyme/Sanofi, and Celgene; received speaker honoraria from Biogen Idec, Healthcare Analytics, Pfizer, and TEVA; is on the editorial board of Journal of Neuroimmunology and Multiple Sclerosis Journal; is an associate editor of Neuroepigenetics; and received research support from Biogen, Eisai, UCB, Pfizer, Sanofi/Genzyme, NIH, and National MS Society. W. Elaman reports no disclosures. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Patsopoulos NA, Esposito F, Reischl J, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol 2011;70:897–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International Multiple Sclerosis Genetics Consortium, Beecham AH, Patsopoulos NA, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 2013;45:1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsushita T, Tateishi T, Isobe N, et al. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS One 2013;8:e61835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darlington PJ, Touil T, Doucet JS, et al. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann Neurol 2013;73:341–354. [DOI] [PubMed] [Google Scholar]
- 5.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med 2008;14:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee YK, Turner H, Maynard CL, et al. Late developmental plasticity in the T helper 17 lineage. Immunity 2009;30:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. J Exp Med 2007;204:1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maggi L, Capone M, Giudici F, et al. CD4+CD161+ T lymphocytes infiltrate Crohn's disease-associated perianal fistulas and are reduced by anti-TNF-alpha local therapy. Int Arch Allergy Immunol 2013;161:81–86. [DOI] [PubMed] [Google Scholar]
- 9.Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E. Cutting edge: the pathogenicity of IFN-gamma-producing Th17 cells is independent of T-bet. J Immunol 2013;190:4478–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 2011;14:1142–1149. [DOI] [PubMed] [Google Scholar]
- 11.Raj T, Rothamel K, Mostafavi S, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 2014;344:519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsiao YC, Tong ZJ, Westfall JE, Ault JG, Page-McCaw PS, Ferland RJ. Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum Mol Genet 2009;18:3926–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ottoboni L, Keenan BT, Tamayo P, et al. An RNA profile identifies two subsets of multiple sclerosis patients differing in disease activity. Sci Transl Med 2012;4:153ra131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein BE, Stamatoyannopoulos JA, Costello JF, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 2010;28:1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coetzee SG, Coetzee GA, Hazelett DJ. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 2015;31:3847–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathelier A, Zhao X, Zhang AW, et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 2014;42:D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deligianni C, Spilianakis CG. Long-range genomic interactions epigenetically regulate the expression of a cytokine receptor. EMBO Rep 2012;13:819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orent W, McHenry AR, Rao DA, et al. Rheumatoid arthritis-associated RBPJ polymorphism alters memory CD4+ T cells. Hum Mol Genet 2016;25:404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koch CA, Anderson D, Moran MF, Ellis C, Pawson T. SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science 1991;252:668–674. [DOI] [PubMed] [Google Scholar]
- 21.Chen M, Gallipoli P, DeGeer D, et al. Targeting primitive chronic myeloid leukemia cells by effective inhibition of a new AHI-1-BCR-ABL-JAK2 complex. J Natl Cancer Inst 2013;105:405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esmailzadeh S, Jiang X. AHI-1: a novel signaling protein and potential therapeutic target in human leukemia and brain disorders. Oncotarget 2011;2:918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kennah E, Ringrose A, Zhou LL, et al. Identification of tyrosine kinase, HCK, and tumor suppressor, BIN1, as potential mediators of AHI-1 oncogene in primary and transformed CTCL cells. Blood 2009;113:4646–4655. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Chen M, Lobo P, et al. Molecular and structural characterization of the SH3 domain of AHI-1 in regulation of cellular resistance of BCR-ABL(+) chronic myeloid leukemia cells to tyrosine kinase inhibitors. Proteomics 2012;12:2094–2106. [DOI] [PubMed] [Google Scholar]
- 25.Zhou LL, Zhao Y, Ringrose A, et al. AHI-1 interacts with BCR-ABL and modulates BCR-ABL transforming activity and imatinib response of CML stem/progenitor cells. J Exp Med 2008;205:2657–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang X, Zhao Y, Chan WY, et al. Deregulated expression in Ph+ human leukemias of AHI-1, a gene activated by insertional mutagenesis in mouse models of leukemia. Blood 2004;103:3897–3904. [DOI] [PubMed] [Google Scholar]
- 27.Westfall JE, Hoyt C, Liu Q, et al. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J Neurosci 2010;30:8759–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the “molar tooth”. Lancet Neurol 2013;12:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang X, Hanna Z, Kaouass M, Girard L, Jolicoeur P. Ahi-1, a novel gene encoding a modular protein with WD40-repeat and SH3 domains, is targeted by the Ahi-1 and Mis-2 provirus integrations. J Virol 2002;76:9046–9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ringrose A, Zhou Y, Pang E, et al. Evidence for an oncogenic role of AHI-1 in Sezary syndrome, a leukemic variant of human cutaneous T-cell lymphomas. Leukemia 2006;20:1593–1601. [DOI] [PubMed] [Google Scholar]
- 31.Kebir H, Ifergan I, Alvarez JI, et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol 2009;66:390–402. [DOI] [PubMed] [Google Scholar]
- 32.Mellergard J, Edstrom M, Vrethem M, Ernerudh J, Dahle C. Natalizumab treatment in multiple sclerosis: marked decline of chemokines and cytokines in cerebrospinal fluid. Mult Scler 2010;16:208–217. [DOI] [PubMed] [Google Scholar]
- 33.Monteyne P, Van Antwerpen MP, Sindic CJ. Expression of costimulatory molecules and cytokines in CSF and peripheral blood mononuclear cells from multiple sclerosis patients. Acta Neurol Belg 1999;99:11–20. [PubMed] [Google Scholar]
- 34.Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol 1998;20:373–382. [DOI] [PubMed] [Google Scholar]
- 35.Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev 2012;248:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramstein J, Broos CE, Simpson LJ, et al. IFN-gamma-producing T-helper 17.1 cells are increased in sarcoidosis and are more prevalent than T-helper type 1 cells. Am J Respir Crit Care Med 2016;193:1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramesh R, Kozhaya L, McKevitt K, et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med 2014;211:89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sundrud MS, Trivigno C. Identity crisis of Th17 cells: many forms, many functions, many questions. Semin Immunol 2013;25:263–272. [DOI] [PubMed] [Google Scholar]
- 39.Cao Y, Goods BA, Raddassi K, et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med 2015;7:287ra274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kupfer A, Swain SL, Janeway CA, Singer SJ. The specific direct interaction of helper T cells and antigen-presenting B cells. Proc Natl Acad Sci USA 1986;83:6080–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCarthy MI, Hirschhorn JN. Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet 2008;17:R156–R165. [DOI] [PMC free article] [PubMed] [Google Scholar]