

Abstract
Introduction
Patients with Parkinson's disease (PD) frequently experience disrupted sleep and several sleep abnormalities are associated with an increased risk of incident PD. However, there are few data concerning the relationship between objectively quantified sleep disruption and the cardinal histopathological features of PD, especially in individuals without clinical PD.
Methods
We studied 269 older adults without Parkinson's disease who had participated in the Rush Memory and Aging Project, and undergone uniform structured neuropathologic evaluations upon death. Sleep fragmentation was measured using actigraphy. Logistic regression models examined the associations of sleep fragmentation proximate to death with the burden of Lewy body pathology and substantia nigra neuron loss.
Results
Greater sleep fragmentation was associated with the presence of Lewy body pathology (OR 1.40; 95% CI 1.05-1.86; p=0.02) and substantia nigra neuron loss (OR 1.43; 95% CI 1.10-1.88; p=0.008) and a higher odds of a pathological diagnosis of PD (OR 2.04; 95% CI 1.34-3.16; p=0.0009). These associations were independent of motor features of parkinsonism, demographic characteristics, and a wide range of medical co-morbidities.
Conclusions
Sleep fragmentation is associated with PD pathology in older adults without PD. These results suggest that sleep fragmentation may be a marker of or risk factor for PD pathology in older adults without PD.
Keywords: sleep, actigraphy, Parkinson's disease, histopathology, Lewy bodies
Introduction
Patients with Parkinson's disease (PD) experience a number of sleep-related problems including insomnia1, sleep fragmentation1, REM sleep behaviour disorder (RBD)2, 3, restless leg syndrome4, 5, excessive daytime sleepiness6, and circadian rhythm disruption7. These symptoms may be present even in the earliest stages of PD, before the initiation of drug therapy, suggesting that they are not solely due to medication effects8-10.
Several sleep abnormalities have also been associated with an increased risk of incident PD. Up to 80% of individuals with REM sleep behavior disorder eventually develop PD or a related synucleinopathy11-13. Excessive daytime sleepiness14and daytime napping15are also associated with an elevated risk of incident PD.
The association between these sleep abnormalities and the risk of subsequent PD has led to the hypothesis that some sleep abnormalities may be markers of pre-clinical PD pathology. REM sleep behaviour disorder in adults without PD is associated with a nigrostriatal dopaminergic deficit as measured by SPECT16 and with the burden of synuclein pathology at death17 and synuclein pathology is commonly found in patients with REM sleep behaviour disorder and a concomitant neurodegenerative syndrome18. However, REM sleep behaviour disorder is relatively uncommon compared to other sleep complaints. Moreover, there are few studies directly linking common objectively quantified sleep abnormalities to PD pathology in older adults without clinical PD.
To address this gap, we analyzed data from 269 older adults without clinical PD who were enrolled in the Rush Memory and Aging Project. Sleep fragmentation was objectively quantified by actigraphy while Lewy body pathology and substantia nigra neuron loss were assessed by structured autopsy. We tested the hypothesis that sleep fragmentation is associated with PD pathology in older adults without PD.
Methods
Participants
We studied participants from the Rush Memory and Aging Project (MAP) – a community based cohort study of older adults that began in 199719. Participants have no dementia at enrolment, undergo annual clinical evaluation, and sign an Anatomical Gift Act for organ donation at death. Actigraphy was added to the study in 2005. For these analyses, we examined data from participants who had at least one actigraphic recording of at least 7 days' duration before death, had undergone a structured assessment for parkinsonism the same year as the last available actigraphic recording, did not have a clinical diagnosis of PD at any study visit, and had undergone structured autopsy. At the time of this study, 1540 participants had been enrolled in the MAP and were alive and eligible for actigraphy, of which 1164 had actigraphy with 1131 having at least 1 recording of at least 7 days' duration, 693 of which had undergone structured assessment of parkinsonism the same year as the last available actigraphic recording. Of these, 276 had died with structured autopsy data available, 269 of which did not have a clinical diagnosis of PD at any time during the study. Primary analyses were performed on these participants.
This study was conducted in accordance with the declaration of Helsinki and approved by the Institutional Review Board of Rush University Medical Center. Written informed consent was obtained from all participants.
Quantification of sleep fragmentation
Sleep fragmentation was assessed by actigraphy (Actical, Phillips Respironics, Bend, OR). Actigraphs were set to record in 15-sec epochs and placed on participants' non-dominant wrist by study staff who returned 10 days later to remove them. Records were visually examined for periods of suspected device removal. In addition, any period 4 hours or greater with no activity counts at all was considered suspicious for device removal. We analyzed recordings that contained at least 7 consecutive days without periods of suspected device removal. Where more than 7 days of data were available, we analyzed only the first 7 days. For each individual, we analyzed the last available actigraphic recording meeting the above criteria, which was done a median [IQR] of 1.4 [0.7,2.7] years prior to death.
Sleep fragmentation, the extent to which sleep is interrupted by awakenings, was quantified with the metric kRA as described and validated elsewhere20, 21. The metric kRA roughly represents the probability per 15-second period of having an arousal (as indicated by a non-zero activity count) following a period of sustained rest (>= ∼5 minutes of inactivity). A greater degree of sleep fragmentation translates to a higher kRA. kRA correlates strongly with polysomnogprahic measures of sleep fragmentation21.
Clinical Diagnosis of Parkinson's Disease and Quantification of Parkinsonism
Participants were considered to have a clinical diagnosis of Parkinson disease if they reported a diagnosis of Parkinson disease and had a history of L-dopa use at any time prior to death.
Although this study excluded participants with a clinical diagnosis of PD, some participants may have had unrecognized parkinsonism. Thus, parkinsonism was quantified by structured physical examination using a 26-item modified version of the motor portion of the United Parkinson's Disease Rating Scale as described previously22, 23. Scores in four Parkinsonian domains (gait disturbance, bradykinesia, rigidity, tremor) were derived from the 26 items assessed by adding the number of points for each item in each domain, dividing by the maximal possible number of points in that domain, and scaling to between 0 and 100. An average of these four parkinsonian domain scores was calculated to yield a global parkinsonism metric. This was square root transformed prior to analysis to ensure normality.
Quantification of PD Pathology
The median [IQR] time between death and post-mortem analysis was 6.8 hours [5.7,8.5]. After death, brains were removed, hemisected, cut into 1cm slabs, and fixed in 4% paraformaldehyde. Neuron loss in the substantia nigra was assessed in hematoxylin and eosin stained 6 micron paraffin-embedded sections of the mid to rostral midbrain near the exit of cranial nerve III23, 24using a semi-quantitative 3-point scale where 0=no significant neuron loss, 1= mild and 2=moderate/severe. To quantify Lewy body pathology, 6 micron paraffin-embedded sections from the cingulate, entorhinal, midfrontal, middle temporal, and inferior parietal cortices and the substantia nigra were immunostained with antibodies to alpha synuclein (pSyn-64; 1:20,000; Wako Chemical USA Inc; Richmond, VA). The distribution of Lewy bodies was graded on a semi-quantitative scale (0 = none, 1 = brainstem or limbic predominant, 2 = neocortical) according to modified version of published criteria25. A participant was considered to have a pathological diagnosis of PD if they had any Lewy body pathology, and moderate or greater nigral neuron loss26.
Quantification of Other Pathologies
Macroscopic cerebral infarcts were assessed by gross visual examination of 1cm slabs from both hemispheres of brain tissue. All grossly visualized infarcts were subsequently confirmed by histological review as previously described27. Infarcts were considered present if 1 or more chronic infarcts were visible. The degree of arteriolosclerosis was characterized by assessment of hematoxylin and eosin stained sections of vessels in the anterior basal ganglia using a semi-quantitative scale from 0 (none) to 4 (severe)28. Arteriolosclerosis was considered present if graded mild or greater. To quantify Alzheimer's disease pathology, 6 micron paraffin-embedded sections from the mid-frontal gyrus, middle temporal gyrus, inferior parietal gyrus, entorhinal cortex, and hippocampus were stained with a modified Bielschowsky silver stain. Neurofibrillary tangles, diffuse plaques, and neuritic plaques were counted in the region that appeared to have the maximum density of each pathological index, and a composite measure of global Alzheimer's disease pathology was created as previously described by dividing each raw count by the standard deviation of the mean for the same index for that region across the entire cohort, and averaging the scaled scores to yield a composite measure29.
Assessment of Other Covariates
Covariates included age, sex, and self-reported years of education. Time lag between actigraphy and death was derived from the difference between the start date of actigraphy and date of death.
In addition to quantifying sleep fragmentation, actigraphic data was also used to calculate mean total daily activity, hours of rest per day, and interdaily stability30-32– a measure of circadian rhythmicity.
Participants also reported on their own sleep. Participants were asked about their estimated average daily total sleep time. As well, participants were asked how often they were troubled by waking up in the night, how often they had difficulty falling asleep at night, how often they were so sleepy during the day that they needed to nap, and how often they awoke feeling rested in the morning, with responses on an ordinal scale ranging from 0 (“never”) to 5 (“very often”). A participant was considered to have a sleep complaint if the response was “often” or “very often” (trouble falling asleep, waking up at night, or sleepy during the day) or “never” or “almost never” (waking rested in the morning) and the total number of sleep complaints was summed. Use of sleep medications was determined by interview and examination of medication containers, and coded by the Medi-Span system.
Vascular disease risk was assessed using a self-report questionnaire and calculated by allocating one point for the presence of each risk factor including a history of hypertension, history of diabetes and history of smoking and then subsequently summing the total number of points. A higher score indicates higher vascular risk burden. History of thyroid disease, history of cancer and history of urinary incontinence were also self-reported. The body mass index (BMI) was calculated as weight in kilograms divided by height in meters squared (kg/mˆ2). Depressive symptoms were assessed with a ten-item version33 of the Center of Epidemiologic Studies depression scale34, and a score representing the number of symptoms experienced in the past week was tabulated. Neuroticism, an indicator of proneness to psychological distress, was assessed using responses to a self-report questionnaire35 containing six items from the neuroticism scale of the Revised NEO Personality Inventory36.
To assess cognition, participants underwent a battery of 19 cognitive tests spanning 5 domains, and a composite measure computed as described previously37. This measure is scaled such that 0 represents the mean score of all participants at baseline, positive scores indicate better performance, and 1 unit represents approximately 1 standard unit of performance.
Statistical Analyses
Prior to analysis, the distribution of all variables was visually inspected and variables were transformed to assure normality and centered and normalized for computational efficiency, as appropriate.
In a set of preliminary analyses, we used unadjusted logistic regression models to examine the relationship between sleep fragmentation and each of three outcomes: presence of any Lewy body pathology at death, presence of mild or greater nigral cell loss at death, and presence of a pathological diagnosis of Parkinson disease. In our primary analyses, we then repeated these analyses using models adjusted for age at death, sex, education, and time lag between actigraphy and death. In secondary analyses we used ordinal logistic regression models to examine the relationship between sleep fragmentation and severity of Lewy body pathology or nigral cell loss at death, measured on ordinal semi-quantitative scales as described above.
Although our primary analyses excluded participants with a clinical diagnosis of Parkinson's disease, some of the participants in these analyses may have unrecognized motor parkinsonism or PD. To account for this, we augmented the above models with a term for global parkinsonism as determined above.
We further augmented these models with terms for actigraphically quantified daily hours of rest, total daily activity, and circadian rhythmicity; self report sleep measures and medications; medical co-morbidities including depression, vascular disease risk factors (hypertension, diabetes and smoking), and thyroid disease; physical symptoms including urinary incontinence; personality traits such as neuroticism; body habitus as measured by BMI; cognitive impairment measured using a set of 19 cognitive tests; and other neuropathological measures including the presence/absence of cerebral gross infarct pathology and arteriolosclerosis and the severity of Alzheimer's disease pathology.
All analyses were performed using R programming language38. For each predictor in each ordinal logistic regression model, we assessed the proportional odds assumption by using a likelihood ratio test to compare a full model incorporating the proportional odds assumption to a model where the proportional odds assumption was relaxed for that predictor. The proportional odds assumption was considered to be met if the two model fits did not differ at p<0.05.
Results
Characteristics of the Study Participants
The clinical characteristics of the 269 participants included in this study are summarized in Table 1. The median [IQR] time between last actigraphy and death was 1.4 years [0.7-2.7]. 72% of the participants were female and the mean age at death was 90.9 years. Median [IQR] sleep fragmentation, as measured by the actigraphic metric kRA, was 0.026 [0.022-0.031]. Lewy Body pathology was found in 23% (n=62) of participants at death and mild or greater nigral neuronal loss was observed in 46% (n=125) of individuals. 19 (7%) participants were considered to have a pathologic diagnosis of PD (moderate or greater nigral neuronal loss and presence of Lewy bodies). The median sleep fragmentation was almost 20% higher in participants with a pathological diagnosis of PD compared those without (median [IQR] 0.030 [0.020, 0.040] in participants with a pathological diagnosis of PD vs. 0.026 [0.022, 0.031] in those without; p=0.013)
Table 1. Characteristics of the Study Population.
N=269.
| Clinical Characteristics | Without pathological diagnosis of PD n=250 (Median [IQR] or Number [%]) | With pathological diagnosis of PD n=19 (Median [IQR] or Number [%]) | P-value |
|---|---|---|---|
| Sleep Fragmentation (kRA) | 0.026 [0.022, 0.031] | 0.03 [0.02,0.04] | 0.01 |
| Age at Death (Years) | 91 [86.8,94.7] | 91 [88.6, 93.6] | 0.81 |
| Female Sex | 182 [73%] | 12 [63%] | 0.52 |
| Education (Years) | 15 [12,16] | 15 [14, 16] | 0.32 |
| Circadian Rhythmicity (Interdaily Stability) | 0.50 [0.37, 0.60] | 0.43 [0.33, 0.55] | 0.26 |
| Total Daily Activity (Counts) | 123732[68320, 207644] | 100417 [67218, 141604] | 0.14 |
| Hours of Rest per Day | 16.7 [14.7,18.6] | 17.5 [15.9, 18.4] | 0.32 |
| Reported Sleep Hours per Day | 8.0 [7.0, 8.0] | 8.0 [7.0, 9.0] | 0.28 |
| Number of Sleep Complaints | 1.0 [0.0, 2.0] | 1.0 [0.0, 1.0] | 0.49 |
| Use of Sleep Medications | 24 [9.6%] | 2 [10%] | >0.99 |
| Number of Depressive Symptoms | 1.0 [0.0, 2.25] | 1.0 [0.0, 2.25] | 0.56 |
| Severity of Neuroticism | 14.0 [12.0, 18.0] | 17.0 [12.5, 20.0] | 0.09 |
| Vascular Disease Risk | 1.0 [1.0, 2.0] | 1.0 [0.5, 1.0] | 0.25 |
| Thyroid Disease | 65 [26%] | 6 [32%] | 0.74 |
| Urinary Incontinence | 122 [49%] | 8 [42%] | 0.75 |
| BMI | 25.0 [22.2, 28.1] | 24.4 [22.0, 27.1] | 0.69 |
| Composite Global Cognition | -0.41 [-1.16, 0.12] | -1.20 [-1.64, -0.28] | 0.10 |
| Severity of Parkinsonism at Last Examination | 13.2 [7.6, 19.4] | 16.8 [13.2, 23.0] | 0.08 |
| Severity of Lewy Body Pathology at Death | <0.01 | ||
| None | 207 [83%] | 0 [0%] | |
| Subcortical | 20 [8%] | 6 [32%] | |
| Cortical | 33 [9%] | 13 [68%] | |
| Substantia Nigra Cell Loss at Death | <0.01 | ||
| None | 144 [58%] | 0 [0%] | |
| Mild | 100 [40%] | 0 [0%] | |
| Moderate to Severe | 6 [2%] | 19 [100%] | |
| Time Lag Between Actigraphy and Death (Days) | 517 [248,968] | 525 [266.5, 972.5] | 0.85 |
Sleep Fragmentation and Lewy Body Pathology
We then examined the relation between sleep fragmentation and Lewy body pathology. In an unadjusted logistic regression model, each 1 SD greater sleep fragmentation was associated with an over 40% higher odds of having Lewy Body pathology, an effect that remained significant after controlling for age, sex, education, and time lag between actigraphy and death (Table 2, Models A-B). Furthermore, sleep fragmentation was also associated with a greater distribution of Lewy Body pathology; each 1 SD higher sleep fragmentation was associated with a nearly 40% higher odds of having a more advanced stage of Lewy body pathology (Figure 1; Table S1, Models A-B).
Figure 1. Sleep Fragmentation and Severity of Lewy Body Pathology In Adults Without Parkinson's Disease.
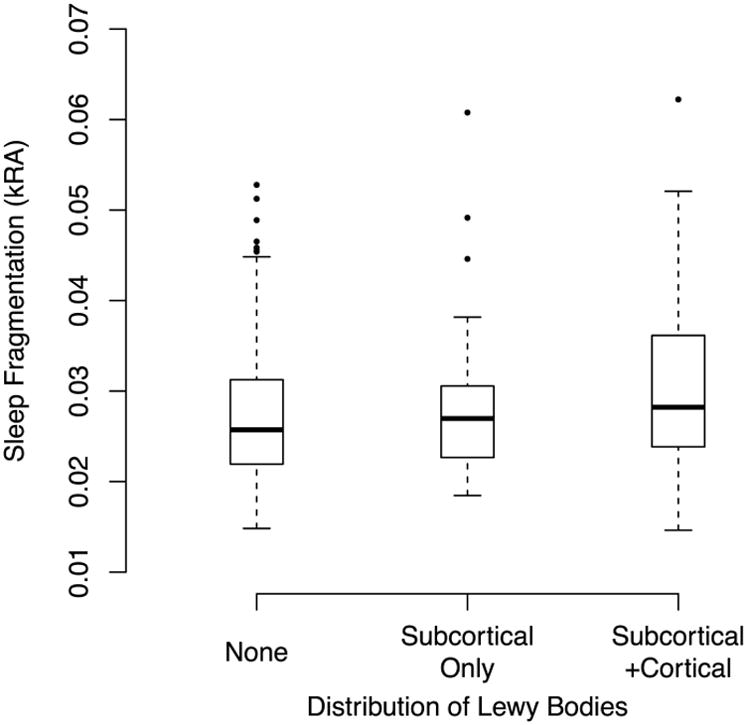
Solid bars indicate medians. Boxes indicate interquartile ranges.
Sleep Fragmentation and Substantia Nigra Cell Loss
Next, we examined the association between sleep fragmentation and nigral neuron loss. In an unadjusted logistic regression model, each 1SD higher sleep fragmentation was associated with 43% higher odds of having at least mild nigral cell loss at death, an effect that remained significant after controlling for age, sex, education, and time lag between actigraphy and death (Table 3, Models A-B). Moreover, the degree of nigral cell loss was associated with sleep fragmentation; each 1 SD greater sleep fragmentation was associated with 50% higher odds of having more severe nigral cell loss (Figure 2; Table S1, Models A-B).
Figure 2. Sleep Fragmentation and Severity of Substantia Nigra Cell Loss in Adults Without Parkinson's Disease.
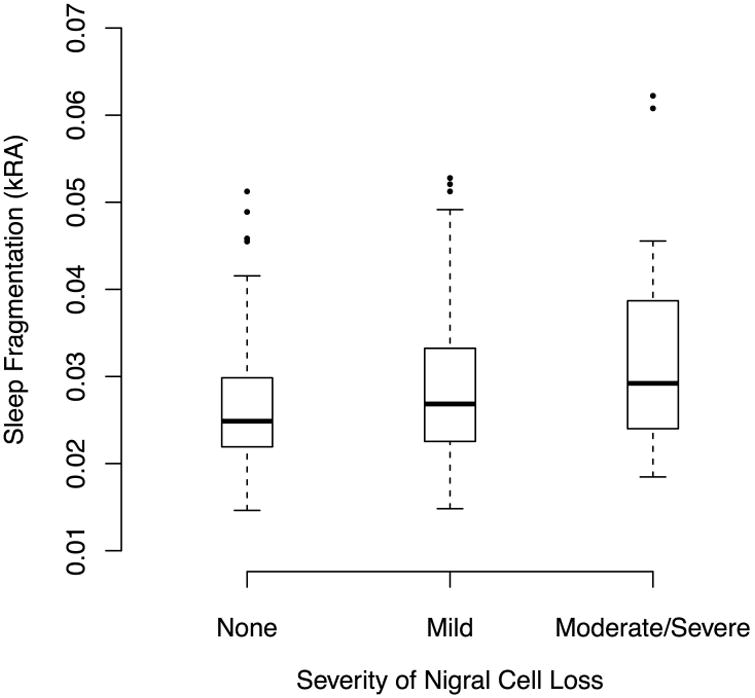
Solid bars indicate medians. Boxes indicate interquartile ranges.
Sleep Fragmentation and Pathological Diagnosis of Parkinson's Disease
In an unadjusted logistic regression model, each 1 SD higher sleep fragmentation was associated with a nearly 2-fold higher odds of having a pathological diagnosis of PD in older adults without clinical Parkinson disease, an effect that remained significant after controlling for age, sex, education, and time lag between actigraphy and death (Figure 3; Table 2, Models A-B).
Figure 3. Sleep Fragmentation and Pathological Diagnosis of PD in Adults Without Parkinson's Disease. Solid bars indicate medians.
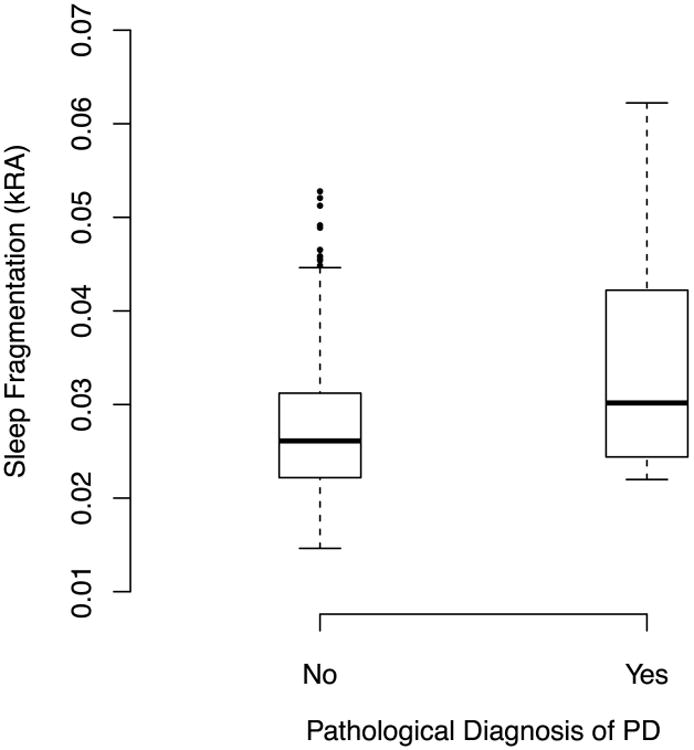
Boxes indicate interquartile ranges.
Consideration of Potential Confounders
In models further adjusted for the degree of parkinsonism as assessed by a modified UPDRS, greater sleep fragmentation remained associated with Lewy body pathology, nigral neuron loss, and a pathological diagnosis of PD (Tables 2 and S1; Model C).
Physical activity and circadian rhythmicity may plausibly influence sleep fragmentation and have been associated with clinical Parkinson disease. Moreover, several self-report sleep measures have been associated with the risk of Parkinson's disease. However, in models further adjusted for actigraphically quantified total daily rest, total daily activity, and circadian rhythmicity; and self-reported sleep duration, quality, and medications, greater sleep fragmentation remained associated with Lewy body pathology, nigral neuron loss, and a pathological diagnosis of PD (Tables 2 and S1; Models D-E).
A number of medical co-morbidities, personality traits, and health behaviours are associated with risk for Parkinson's disease. However, in models adjusted for depressive symptoms, neuroticism, vascular disease risk factors (diabetes, hypertension, and smoking), thyroid disease, urinary incontinence, body mass index, and cognitive function, greater sleep fragmentation remained significantly associated with Lewy body pathology and nigral neuron loss (Tables 2 and S1; Model F), although the statistical significance of the association with a pathological diagnosis of PD was somewhat attenuated, reflecting in part the smaller number of participants included in this analysis.
We have previously reported that sleep fragmentation is associated with AD pathology39, and with cerebrovascular pathology including infarcts and arteriolosclerosis40. However, in models adjusted for the presence of macroscopic cerebral infarcts and arteriolosclerosis, and the burden of Alzheimer's disease pathology, greater sleep fragmentation remained significantly associated with Lewy body pathology, nigral neuron loss and a pathological diagnosis of PD (Tables 2 and S2; Model G).
Discussion
In this study of 269 community dwelling older adults without a clinical diagnosis of Parkinson's disease, greater sleep fragmentation, as objectively measured by actigraphy, was associated with a greater burden of Lewy body pathology and substantia nigra neuron loss as measured by histology, and with a higher odds of having a pathological diagnosis of Parkinson's disease. These associations were independent of motoric signs of parkinsonism; actigraphic measures of physical activity, circadian rhythmicity, and rest; self-report sleep measures; health behaviours; medical and psychiatric co-morbidities including cognitive impairment; and the burden of cerebrovascular and Alzheimer's disease pathology. Taken together, these results suggest that sleep fragmentation may be an independent marker of or contributor to PD pathology in older community dwelling adults without a clinical diagnosis of PD. They raise the possibility that non-invasive quantitative measures of sleep fragmentation such as actigraphy may be useful in the identification of patients likely to have PD pathology for the purpose of clinical trials or early intervention. Moreover, if sleep fragmentation is a contributor to PD pathology, then interventions aimed at treating sleep fragmentation or its specific causes may be useful in preventing the development of PD pathology.
Several studies have reported associations between self-reported sleep measures and imaging41 or histological17 measures of PD pathology in patients with a clinical diagnosis of PD. However, these studies examined only patients with clinical PD, and relied on self-report sleep measures, which correlate only partially with objective measures. One study reported an association between polysomnographically confirmed idiopathic REM sleep behaviour disorder and dopamine transporter binding as assessed by SPECT16; however there was no histopathological assessment of the cardinal neuropathological features of PD, including Lewy bodies and nigral neuron loss. Meanwhile, several studies have reported associations between sleep measures and incident clinical PD11-15. However, these studies also lacked histopathological assessment of PD pathology. This study extends this previous work by showing that a non-invasive objective measure of sleep quality is associated with both Lewy body pathology and nigral neuron loss – the classic pathological features of PD – even in older adults without clinical PD, and independent of motor features of parkinsonism.
One potential interpretation of our findings is that Parkinson disease pathology may contribute to sleep fragmentation, either through direct disruption of sleep regulatory circuits or by leading to other symptoms such as depression, motor dysfunction, or nocturia which may disrupt sleep. Arguing against the latter possibility is our observation that the association between sleep fragmentation and PD pathology is independent of motor features of parkinsonism, depression, urinary incontinence, and a host of other symptoms and medical co-morbidities. In support of the possibility that PD pathology may directly affect sleep regulatory circuits, individuals with established clinical PD can have substantial loss of neurons in sleep regulatory regions such as the orexinergic lateral hypothalamus 42. Moreover, one contemporary model of the spread of alpha synuclein pathology suggests that alpha synuclein pathology may be found in sleep regulatory regions such as the sublaterodoral nucleus, peri-locus ceruleus, and magnocellular reticular formation even before the substantia nigra26, 43.
An alternative interpretation of our findings is that sleep fragmentation may contribute to the development of PD pathology. Potential mechanisms may include promotion of oxidative stress, or impairment of the clearance of toxic proteins. Both sleep fragmentation44 and sleep deprivation45 can contribute to brain oxidative stress in model organisms and brain oxidative stress in turn has been implicated in the pathogenesis of PD46-48. Sleep may also play an important role in clearing proteins and metabolites from the brain interstitial space49 and while intracellular alpha synuclein in an important component of Lewy bodies, an important role for extracellular alpha synuclein in PD pathogenesis has been proposed50.
We have previously shown that in addition to Lewy body pathology, sleep fragmentation as measured by actigraphy in various subgroups of older adults is also associated with Alzheimer's disease pathology39, cerebrovascular pathology40, cell loss in the intermediate nucleus of the hypothalamus51, and regional cortical gray matter volumes52. In the present study, sleep fragmentation was associated with PD pathology even when controlling for several of these other pathologies. However, it is likely that actigraphically quantified sleep fragmentation, considered in isolation, is not entirely specific for PD pathology. There are several potential reasons for this. First, it is possible that multiple pathologies may lead to sleep fragmentation by disrupting the same sleep circuits, as is the case with other brain functions such as cognition53. Second, sleep fragmentation may lead to pathophysiological mechanisms (e.g. impaired clearance of metabolites from the brain interstitial space49, oxidative stress44) that may simultaneously predispose to multiple brain pathologies. Third, not all sleep fragmentation is identical and it is possible that specific patterns (e.g. NREM vs. REM sleep) or causes of sleep fragmentation may be more specific to PD pathology. Ultimately, studies to identify the causes and patterns of sleep fragmentation most strongly associated with PD pathology, and combining these with other clinical and genetic predictors, will be needed to reliably identify older adults with PD pathology.
Anumber of methodological considerations merit discussion. First, as with all cross-sectional observational studies, the causal direction of the association between sleep fragmentation and PD pathology could not be determined. Longitudinal studies examining the association between sleep fragmentation and in-vivo imaging markers of PD pathology may help clarify this, although ultimately controlled studies to assess the impact of sleep interventions on markers of PD pathology will be needed. Second, although the severity of sleep fragmentation was objectively quantified, as noted above, specific causes or patterns of sleep fragmentation (for instance sleep disorders such as sleep apnea, restless legs, or REM sleep behavior disorder; environmental disturbances such as noise or light; sleep disruption secondary to pathology in sleep regulatory circuits) or patterns (REM sleep vs. NREM sleep)could not be distinguished. Studies incorporating measures of environmental sleep disturbance, specific sleep disorders, and more detailed sleep assessment (e.g. assessment of sleep stages) are needed. Third, PD pathology was assessed in a relatively circumscribed set of brain regions. Future studies assessing PD pathology in other sleep regulatory centers (e.g. the hypothalamus) will be helpful in shedding light on whether PD pathology in specific sleep-regulating brain regions may be a cause of sleep fragmentation in older persons. Finally, the study population consisted predominantly of older women of European descent, which may limit generalizability to other population groups.
The study also had several strengths. First, sleep fragmentation was objectively and non-invasively quantified, avoiding the disadvantages of both self-report and laboratory-based sleep measures such as polysomnography. Moreover, we assessed participants' habitual sleep at home rather than their ability to sleep in a laboratory. Second, motor parkinsonism was carefully assessed by blinded structured physical examination, allowing us to determine the association between sleep fragmentation and PD pathology independent of motor features of PD. Third we directly assessed the cardinal histopathological features of PD, including Lewy bodies and substantia nigra neuron loss, rather than relying on indirect measures. Finally, participants were deeply clinically characterized allowing us to account for a wide range of potential confounders.
Supplementary Material
Acknowledgments
We thank the participants in the Memory and Aging Project and their families, and the staff of the Rush Alzheimer's Disease Center.
This study was funded by National Institutes of Health R01AG052488, R01AG043379, R01AG15819, R01AG17917, and R01NS078009; Canadian Institutes of Health Research MOP125934, MMC112692, and MSH136642; the Illinois Department of Public Health, and the Robert C. Borwell Endowment Fund.
Funding Sources: As above.
Footnotes
Financial Disclosure/ Conflict of Interest Concerning Research Related to the Manuscript: The authors have no conflicts of interest to disclose.
- Research project: A. Conception, B. Organization, C. Execution;
- Statistical Analysis: A. Design, B. Execution, C. Review and Critique;
- Manuscript: A. Writing of the first draft, B. Review and Critique.
SS: 2A, 2B, 3A
LY: 2C, 3B
JS: 1C, 3B
DAB: 1A, 1B, 1C, 3B
ASB: 1A, 1B, 1C, 3B
ASL: 1A, 2A, 2B, 2C, 3B
Full Financial Disclosures for the Past Year: SS: nothing to disclose
LY: nothing to disclose
JS: NIH P30AG010161, R01AG042210, UH2NS100599
DAB: NIH P30AG010161, RF1AG015819, R01AG017917, RF1AG036042, U01AG046152, Robert C. Borwell Endowment Fund
ASB: NIH R01AG17917, R01AG43379, R01NS078009, R01AG47976, R01AG56352
ASL: National Institute on Aging grant R01AG052488; Canadian Institutes of Health Research grants MOP125934, MOP336806, and MSH136642
References
- 1.Gjerstad MD, Wentzel-Larsen T, Aarsland D, Larsen JP. Insomnia in Parkinson's disease: frequency and progression over time. J Neurol Neurosurg Psychiatry. 2007;78(5):476–479. doi: 10.1136/jnnp.2006.100370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gagnon JF, Bedard MA, Fantini ML, et al. REM sleep behavior disorder and REM sleep without atonia in Parkinson's disease. Neurology. 2002;59(4):585–589. doi: 10.1212/wnl.59.4.585. [DOI] [PubMed] [Google Scholar]
- 3.Sixel-Doring F, Trautmann E, Mollenhauer B, Trenkwalder C. Associated factors for REM sleep behavior disorder in Parkinson disease. Neurology. 2011;77(11):1048–1054. doi: 10.1212/WNL.0b013e31822e560e. [DOI] [PubMed] [Google Scholar]
- 4.Wong JC, Li Y, Schwarzschild MA, Ascherio A, Gao X. Restless legs syndrome: an early clinical feature of Parkinson disease in men. Sleep. 2014;37(2):369–372. doi: 10.5665/sleep.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krishnan PR, Bhatia M, Behari M. Restless legs syndrome in Parkinson's disease: a case-controlled study. Movement disorders: official journal of the Movement Disorder Society. 2003;18(2):181–185. doi: 10.1002/mds.10307. [DOI] [PubMed] [Google Scholar]
- 6.Breen DP, Williams-Gray CH, Mason SL, Foltynie T, Barker RA. Excessive daytime sleepiness and its risk factors in incident Parkinson's disease. J Neurol Neurosurg Psychiatry. 2013;84(2):233–234. doi: 10.1136/jnnp-2012-304097. [DOI] [PubMed] [Google Scholar]
- 7.Videnovic A, Noble C, Reid KJ, et al. Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA neurology. 2014;71(4):463–469. doi: 10.1001/jamaneurol.2013.6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhawan V, Dhoat S, Williams AJ, et al. The range and nature of sleep dysfunction in untreated Parkinson's disease (PD). A comparative controlled clinical study using the Parkinson's disease sleep scale and selective polysomnography. Journal of the neurological sciences. 2006;248(1-2):158–162. doi: 10.1016/j.jns.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Wetter TC, Collado-Seidel V, Pollmacher T, Yassouridis A, Trenkwalder C. Sleep and periodic leg movement patterns in drug-free patients with Parkinson's disease and multiple system atrophy. Sleep. 2000;23(3):361–367. [PubMed] [Google Scholar]
- 10.Arnulf I, Konofal E, Merino-Andreu M, et al. Parkinson's disease and sleepiness: an integral part of PD. Neurology. 2002;58(7):1019–1024. doi: 10.1212/wnl.58.7.1019. [DOI] [PubMed] [Google Scholar]
- 11.Iranzo A, Fernandez-Arcos A, Tolosa E, et al. Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PLoS One. 2014;9(2):e89741. doi: 10.1371/journal.pone.0089741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Postuma RB, Iranzo A, Hogl B, et al. Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: a multicenter study. Ann Neurol. 2015;77(5):830–839. doi: 10.1002/ana.24385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med. 2013;14(8):744–748. doi: 10.1016/j.sleep.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Abbott RD, Ross GW, White LR, et al. Excessive daytime sleepiness and subsequent development of Parkinson disease. Neurology. 2005;65(9):1442–1446. doi: 10.1212/01.wnl.0000183056.89590.0d. [DOI] [PubMed] [Google Scholar]
- 15.Gao J, Huang X, Park Y, et al. Daytime napping, nighttime sleeping, and Parkinson disease. Am J Epidemiol. 2011;173(9):1032–1038. doi: 10.1093/aje/kwq478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisensehr I, Linke R, Noachtar S, Schwarz J, Gildehaus FJ, Tatsch K. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder. Comparison with Parkinson's disease and controls. Brain. 2000;123(Pt 6):1155–1160. doi: 10.1093/brain/123.6.1155. [DOI] [PubMed] [Google Scholar]
- 17.Postuma RB, Adler CH, Dugger BN, et al. REM sleep behavior disorder and neuropathology in Parkinson's disease. Mov Disord. 2015;30(10):1413–1417. doi: 10.1002/mds.26347. [DOI] [PubMed] [Google Scholar]
- 18.Boeve BF, Silber MH, Ferman TJ, et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med. 2013;14(8):754–762. doi: 10.1016/j.sleep.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res. 2012;9(6):646–663. doi: 10.2174/156720512801322663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim AS, Yu L, Costa MD, et al. Quantification of the fragmentation of rest-activity patterns in elderly individuals using a state transition analysis. Sleep. 2011;34(11):1569–1581. doi: 10.5665/sleep.1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA. Sleep Fragmentation and the Risk of Incident Alzheimer's Disease and Cognitive Decline in Older Persons. Sleep. 2013;36(7):1027–1032. doi: 10.5665/sleep.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett DA, Shannon KM, Beckett LA, Wilson RS. Dimensionality of parkinsonian signs in aging and Alzheimer's disease. The journals of gerontology Series A, Biological sciences and medical sciences. 1999;54(4):M191–196. doi: 10.1093/gerona/54.4.m191. [DOI] [PubMed] [Google Scholar]
- 23.Buchman AS, Shulman JM, Nag S, et al. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann Neurol. 2012;71(2):258–266. doi: 10.1002/ana.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider JA, Li JL, Li Y, Wilson RS, Kordower JH, Bennett DA. Substantia nigra tangles are related to gait impairment in older persons. Annals of neurology. 2006;59(1):166–173. doi: 10.1002/ana.20723. [DOI] [PubMed] [Google Scholar]
- 25.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 26.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 27.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62(7):1148–1155. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 28.Buchman AS, Leurgans SE, Nag S, Bennett DA, Schneider JA. Cerebrovascular disease pathology and parkinsonian signs in old age. Stroke. 2011;42(11):3183–3189. doi: 10.1161/STROKEAHA.111.623462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett DA, Wilson RS, Schneider JA, et al. Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer's disease. Neurology. 2003;60(2):246–252. doi: 10.1212/01.wnl.0000042478.08543.f7. [DOI] [PubMed] [Google Scholar]
- 30.Sokolove PG, Bushell WN. The chi square periodogram: its utility for analysis of circadian rhythms. J Theor Biol. 1978;72(1):131–160. doi: 10.1016/0022-5193(78)90022-x. [DOI] [PubMed] [Google Scholar]
- 31.van Someren EJ, Hagebeuk EE, Lijzenga C, et al. Circadian rest-activity rhythm disturbances in Alzheimer's disease. Biol Psychiatry. 1996;40(4):259–270. doi: 10.1016/0006-3223(95)00370-3. [DOI] [PubMed] [Google Scholar]
- 32.Witting W, Kwa IH, Eikelenboom P, Mirmiran M, Swaab DF. Alterations in the circadian rest-activity rhythm in aging and Alzheimer's disease. Biol Psychiatry. 1990;27(6):563–572. doi: 10.1016/0006-3223(90)90523-5. [DOI] [PubMed] [Google Scholar]
- 33.Bennett DA, Wilson RS, Schneider JA, Bienias JL, Arnold SE. Cerebral infarctions and the relationship of depression symptoms to level of cognitive functioning in older persons. Am J Geriatr Psychiatry. 2004;12(2):211–219. [PubMed] [Google Scholar]
- 34.Roberts RE, Vernon SW. The Center for Epidemiologic Studies Depression Scale: its use in a community sample. The American journal of psychiatry. 1983;140(1):41–46. doi: 10.1176/ajp.140.1.41. [DOI] [PubMed] [Google Scholar]
- 35.Buchman AS, Boyle PA, Wilson RS, Leurgans SE, Arnold SE, Bennett DA. Neuroticism, extraversion, and motor function in community-dwelling older persons. Am J Geriatr Psychiatry. 2013;21(2):145–154. doi: 10.1016/j.jagp.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costa PT, McCrae RR. Normal personality assessment in clinical practice: The NEO Personality Inventory. Psychological Assessment. 1992;4(1):5–13. [Google Scholar]
- 37.Wilson RS, Beck TL, Bienias JL, Bennett DA. Terminal cognitive decline: accelerated loss of cognition in the last years of life. Psychosomatic medicine. 2007;69(2):131–137. doi: 10.1097/PSY.0b013e31803130ae. [DOI] [PubMed] [Google Scholar]
- 38.Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing; 2013. [Google Scholar]
- 39.Lim AS, Yu L, Kowgier M, Schneider JA, Buchman AS, Bennett DA. Modification of the Relationship of the Apolipoprotein E ε4 Allele to the Risk of Alzheimer Disease and Neurofibrillary Tangle Density by Sleep. JAMA neurology. 2013;70(12):1544–1551. doi: 10.1001/jamaneurol.2013.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim AS, Yu L, Schneider JA, Bennett DA, Buchman AS. Sleep Fragmentation, Cerebral Arteriolosclerosis, and Brain Infarct Pathology in Community-Dwelling Older People. Stroke. 2016;47(2):516–518. doi: 10.1161/STROKEAHA.115.011608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kotagal V, Albin RL, Muller ML, et al. Symptoms of rapid eye movement sleep behavior disorder are associated with cholinergic denervation in Parkinson disease. Ann Neurol. 2012;71(4):560–568. doi: 10.1002/ana.22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thannickal TC, Lai YY, Siegel JM. Hypocretin (orexin) cell loss in Parkinson's disease. Brain. 2007;130(Pt 6):1586–1595. doi: 10.1093/brain/awm097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell and tissue research. 2004;318(1):121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 44.Nair D, Zhang SX, Ramesh V, et al. Sleep fragmentation induces cognitive deficits via nicotinamide adenine dinucleotide phosphate oxidase-dependent pathways in mouse. Am J Respir Crit Care Med. 2011;184(11):1305–1312. doi: 10.1164/rccm.201107-1173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villafuerte G, Miguel-Puga A, Rodriguez EM, Machado S, Manjarrez E, Arias-Carrion O. Sleep deprivation and oxidative stress in animal models: a systematic review. Oxid Med Cell Longev. 2015;2015:234952. doi: 10.1155/2015/234952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD. Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson's Disease Research Group. Ann Neurol. 1992;(32 Suppl):S82–87. doi: 10.1002/ana.410320714. [DOI] [PubMed] [Google Scholar]
- 47.Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology. 1996;47(6 Suppl 3):S161–170. doi: 10.1212/wnl.47.6_suppl_3.161s. [DOI] [PubMed] [Google Scholar]
- 48.Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol. 2003;53(Suppl 3):S26–36. doi: 10.1002/ana.10483. discussion S36-28. [DOI] [PubMed] [Google Scholar]
- 49.Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee HJ, Bae EJ, Lee SJ. Extracellular alpha--synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10(2):92–98. doi: 10.1038/nrneurol.2013.275. [DOI] [PubMed] [Google Scholar]
- 51.Lim AS, Ellison BA, Wang JL, et al. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer's disease. Brain. 2014;137(Pt 10):2847–2861. doi: 10.1093/brain/awu222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim AS, Fleischman DA, Dawe RJ, et al. Regional Neocortical Gray Matter Structure and Sleep Fragmentation in Older Adults. Sleep. 2016;39(1):227–235. doi: 10.5665/sleep.5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69(24):2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.