

Abstract
Background
Sudden unexplained nocturnal death syndrome (SUNDS) remains an autopsy negative entity with unclear etiology. Arrhythmia has been implicated in SUNDS. Mutations/deficiencies in intercalated disc components have been shown to cause arrhythmias. Human cardiomyopathy‐associated 1 (XIRP1) and 3 (XIRP2) are intercalated disc–associated, Xin repeats‐containing proteins. Mouse Xirp1 is necessary for the integrity of intercalated disc and for the surface expression of transient outward and delayed rectifier K+ channels, whereas mouse Xirp2 is required for Xirp1 intercalated disc localization. Thus, XIRP1 and XIRP2 may be potentially causal genes for SUNDS.
Methods and Results
We genetically screened XIRP genes in 134 sporadic SUNDS victims and 22 Brugada syndrome (BrS) cases in a Chinese Han population. We identified 16 rare variants (6 were in silico predicted as deleterious) in SUNDS victims, including a novel variant, XIRP2‐E215K. There were also four rare variants (2 were in silico predicted as deleterious) detected in BrS cases, including a novel variant, XIRP2‐L2718P. Interestingly, among these 20 variants, we detected 2 likely pathogenic variants: a nonsense variant (XIRP2‐Q2875*) and a frameshift variant (XIRP2‐T2238QfsX7). Analyzing available Xirp2 knockout mice, we further found that mouse hearts without Xirp2 exhibited prolonged PR and QT intervals, slow conduction velocity, atrioventricular conduction block, and an abnormal infranodal ventricular conduction system. Whole‐cell patch‐clamp detected altered ionic currents in Xirp2 −/− cardiomyocytes, consistent with the observed association between Xirp2 and Nav1.5/Kv1.5 in co‐immunoprecipitation.
Conclusions
This is the first report identifying likely pathogenic XIRP rare variants in arrhythmogenic disorders such as SUNDS and Brugada syndrome, and showing critical roles of Xirp2 in cardiac conduction.
Keywords: Xirp proteins, cardiac conduction, rare variants, sudden cardiac death, sudden unexplained nocturnal death syndrome, Brugada syndrome
Subject Categories: Sudden Cardiac Death
Clinical Perspective
What Is New?
Xirp2‐null hearts exhibited prolonged PR and QT intervals, slow conduction velocity, atrioventricular conduction block, and an abnormal infranodal ventricular conduction system.
Xirp2‐null cardiomyocytes showed altered action potential waveform, partly explained by ionic current defects and protein interaction.
Sixteen XIRP rare variants (6 were in silico predicted as deleterious) were identified in sudden unexplained nocturnal death syndrome victims, and 4 XIRP rare variants (2 were in silico predicted as deleterious) were detected in Brugada syndrome patients.
Among these 20 variants, 2 likely pathogenic variants—a nonsense variant and a frameshift variant—were detected.
What Are the Clinical Implications?
Genetic screening of XIRP genes should be encouraged for clinical cardiac conduction defect patients and sudden cardiac death victims.
The present study raises a possibility that the altered electrophysiology characteristics of XIRP‐null hearts may represent a potential target spot for cardiac conduction defect patients caused by XIRP mutation.
For 100 years, sudden unexplained nocturnal death syndrome (SUNDS) has remained an autopsy negative enigma with uncertain etiology to both forensic pathologists and clinicians. The definition of SUNDS described an entity with a special clinic phenotype, which was reported in Southeast Asia1, 2, 3, 4, 5, 6, 7, 8 and the United States9, 10, 11: (1) prevails preponderantly in males (>90%); (2) >80% of victims are aged between 20 and 40 years; (3) predominantly occurs in Southeast Asia or immigrants from Southeast Asia (such as Hmong in the United States) without a significant disease history; (4) occurs during nocturnal sleep with typical symptoms such as moaning and tachypnea; and (5) there are no pathological changes to identify the cause of death.
Previous evidence has shown that SUNDS might be a cardiac arrhythmia disease sharing overlapping genetic predisposition with Brugada syndrome (BrS) and long QT syndrome.3, 4, 12, 13 However, our postmortem genetic tests on the known lethal cardiac arrhythmia‐associated 39 genes could only possibly account for a small part of the genetic cause of SUNDS victims,14, 15, 16, 17, 18, 19 leaving unclear the genetic etiology of more than 80% of SUNDS cases. Thus, there is a pressing need to identify new susceptibility genes for SUNDS. Because SUNDS shares overlapping genetic causes with BrS, establishing new candidate genes may also expand the list of susceptible genes for this more‐prevalent arrhythmic disease.
The Xin actin‐binding repeat containing family of genes (XIRPs) are promising candidates for SUNDS. The XIRPs are localized to the intercalate discs (ICDs) of cardiomyocytes and have been shown to play important roles for postnatal ICD development and function.20 ICDs are responsible for mechanical and electrical coupling and transducing signals among cardiomyocytes.20 Genetic variants in ICD‐associated genes lead to cardiomyopathy, arrhythmias, and heart failure in both human patients and various genetically engineered animal models.21, 22, 23, 24, 25 Diverse vertebrates, including mammals, possess 2 paralogous genes encoding the XIRP proteins, XIRP1 (Xinα or CMYA1) and XIRP2 (Xinβ or CMYA3).26 Deletion of mouse Xirp1 (mXinα) leads to late‐onset cardiomyopathy with conduction and structural defects in ICDs,27, 28, 29, 30 whereas complete loss of mouse Xirp2 (mXinβ) results in the failure in ICD maturation, severe growth retardation, and early postnatal lethality.31
We have shown that Xirp1 plays important roles in maintaining the electrophysiological functions in the heart. Loss of Xirp1 in ventricular cardiomyocytes leads to significantly reduced transient outward K+ current (ITO) and delayed rectifier K+ current (IK).27 Consistently, Xirp1 −/− ventricular and left atrial‐pulmonary vein cardiomyocytes have prolonged action potential duration (APD), less‐negative maximal diastolic potential, and increased incidence of early afterdepolorization, which may underlie the observed cardiac conduction defects in Xirp1 −/− mice.27, 28, 29, 30 Biochemical experiments demonstrated that Xirp1 is required for the surface expression of ITO and IK channel complexes. Xirp1 directly interacts with the KChIP2 subunit of ITO channel complex27 and the actin‐binding protein, filamin.27, 32, 33 Both KChIP2 and filamin are essential to maintain ITO current density.34, 35 In addition, Xirp1 was shown to directly interact with the actin‐binding protein, cortactin, through which Xirp1 may regulate the level of ICD‐localized cortactin and then the surface expression of IK channels.36, 37
No direct evidence has been provided to support a role for Xirp2 in cardiac electrophysiology. During the first 2 postnatal weeks in the mouse, Xirp2 is rapidly upregulated and localized to the forming ICDs where it recruits other ICD proteins, including Xirp1.38 Furthermore, similar to XIRP1, XIRP2 is an actin‐binding and ‐bundling protein that interacts with actin filaments through highly conserved Xin repeats.32, 39 Because actin cytoskeleton is important for channel trafficking and surface localization,27, 35, 37 both XIRP1 and XIRP2 may also regulate cardiac ion channel function by modulating the actin cytoskeleton. Intriguingly, XIRP1 and XIRP2 are located in highly conserved syntenies with sodium‐channel gene clusters on chromosome loci 3p22.2 and 2q24.3, respectively.20 It has been implicated that genes within highly conserved syntenies may be functionally related.40 The XIRP1‐containing locus also harbors the SCN5A and SCN10A genes that encode cardiac sodium channels and are known to be associated with BrS, long QT syndrome, and SUNDS.14, 19 On the other hand, XIRP2‐containing locus has not been linked to cardiac arrhythmia, but deletion of chromosome band 2q24 has been associated with congenital heart defects.41
We hypothesized that XIRP variants may contribute to the pathogenesis of human cardiac arrhythmia disorders. In this study, genetic screens for 134 sporadic SUNDS cases and for 22 BrS patients were aimed to determine whether variants of XIRP are present and responsible for a proportion of SUNDS. Among a total of 20 rare variants identified, a nonsense (Q2875*) variant and a frameshift (T2238QfsX7) variant have been found in the XIRP2 gene from SUNDS and BrS cases, respectively. These variants, causing premature termination, might induce the nonsense‐mediated mRNA decay pathway42, 43, 44 and result in reduced amount of XIRP2 in the heart. Using mouse knockout models and biochemical approaches, we provided further evidence that Xirp2 is essential for normal cardiac conduction.
Materials and Methods
The data, analytical methods, and study materials will be made available to other researchers upon request for purposes of reproducing the results or replicating the procedure.
Study Population
In this study, 134 sporadic SUNDS cases from 2004 to 2015 were diagnosed and collected by the Department of Forensic Pathology, Zhongshan School of Medicine, Sun Yat‐sen University (China National Forensic Autopsy Center). Inclusion criteria for SUNDS were as follows6, 7: (1) a Chinese Han aged ≥15 years; (2) apparently healthy without any previously significant disease; (3) before experiencing a sudden unexpected death at night during sleep with symptoms of moaning, apnea, and abrupt tic of limbs; and (4) negative postmortem findings of the standard forensic autopsy, histopathology examination, toxicological analysis, and death‐scene investigation.
Twenty‐two BrS cases from 2007 to 2015 were collected from the First Affiliated Hospital of Sun Yat‐sen University to screen XIRP variants. Inclusion criteria for BrS patients were7: (1) a basal ECG showing a BrS type I pattern; (2) at least 1 clinical criterion (documented family history of BrS or sudden cardiac death, and/or symptoms secondary to arrhythmia); and (3) no structural heart disease.
All of these SUNDS and BrS cases were previously screened for deleterious rare variants in 39 primary arrhythmia‐associated genes,7 including SCN5A, MOG1, GPD1L, SCN10A, CACNB2, SCN1B‐4B, KCNE3, KCND3, KCNJ8, KCNQ1, KCNH2, ANK2, KCNE1, KCNE2, KCNJ2, CACNA1C, CAV3, AKAP9, SNTA1, GJA1, KCNJ5, RYR2, CASQ2, ANK2, TRDN, PRKAG2, HCN4, KCNA5, NPPA, NUP155, GJA5, ABCC9, GATA4, GATA5, GATA6, and KCNE1L. A control population of 649 age‐ and ethnic‐matched unrelated healthy Chinese Han (1298 alleles) were provided by the China National GeneBank, BGI‐Shenzhen. None of the control subjects had a history of syncope or cardiovascular disease.
Informed consent was obtained from BrS patients and legal representatives of SUNDS victims. Principles outlined in the Declaration of Helsinki were followed. The project was approved for human research by the ethics committee of Sun Yat‐sen University.
Genetic Variants Analysis
Genomic DNA was extracted from blood samples as reported previously.6, 7, 14, 15, 16, 17, 18, 19 All coding regions and exon‐intron boundaries for XIRP1 and XIRP2 (GenBank NM_194293.2 and NM_152381.5, respectively) were polymerase chain reaction amplified using designed primers (Table S1). Polymerase chain reaction products were directly sequenced on an ABI 3730XL Automated DNA Analyzer (Applied Biosystems, Foster City, CA). Sequencing data were compared with the corresponding reference sequence using SeqManTMII expert sequence analysis software (DNASTAR, Inc, Madison, WI). All suspicious variants were confirmed using the opposite primer. A standard nomenclature for the description of sequence variants has been agreed upon on http://varnomen.hgvs.org/.
Genomic data of East Asian (EAS) from 1000 Genomes Project Phase 3 (http://browser.1000genomes.org/, all 5008 alleles, EAS 1008 alleles, Southern Han Chinese 210 alleles, Han Chinese in Beijing 206 alleles), Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/, all 121 412 alleles, EAS 8654 alleles), and Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/, all 277 264 alleles, EAS 18 870 alleles) were accessed to compare allele frequency. Variants absent from the local database, ExAC, gnomAD, and database of single‐nucleotide polymorphism (http://www.ncbi.nlm.nih.gov/snp) were defined as novel. Variants identified with a minor allele frequency (MAF) less than 0.01 were termed as rare. If the MAF was higher than 0.01, variants were regarded as common. Online in silico prediction tools SIFT (http://sift.jcvi.org/), Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2/), and CONDEL (http://bg.upf.edu/fannsdb/) were used to evaluate deleteriousness of missense variants. Candidate variants were filtered based on the following: MAF, type of variant, in silico prediction (SIFT, Polyphen‐2, and CONDEL), and related published literature. The “radical” variants, including nonsense and frameshift, were treated as candidate variants unless identified as common variants. Filtered variants were predicted to be deleterious if all the 3 software made concordant predictions. All the filtered variants were then classified to 5 categories based on their clinical significance (pathogenic, likely pathogenic, variant of uncertain significance, likely benign, and benign) according the American College of Medical Genetics Guidelines Revisions.45
Mouse Models
All animal procedures were approved and conducted in accord with institutional guidelines. Xirp2 −/− mice were reported on previously.31 Mice expressing β‐galactosidase under the control of endogenous Xirp2 regulatory elements were generated by crossing mice bearing Xirp2 tm1a(KOMP)Wtsi allele (obtained from UC Davis KOMP Repository) with mice expressing Cre recombinase in the germline (EK‐Cre, a kind gift from Dr Baoli Yang, University of Iowa), which resulted in Xirp2 +/lacZ‐tm2 mice expressing LacZ under the control of the endogenous Xirp2 promoter.
Optical Mapping
Optical mapping was carried out as described previously.29 The cannulated, perfused heart was further stained with di‐4‐ANEPPS as a voltage‐sensitive dye. The stained heart was paced at the basal portion of the right ventricle from a stimulator as described.29 Fluorescence emission from the paced heart was collected and processed to form an image detecting a 5×5 mm area of the epicardium. Spatial and temporal filtering were utilized in data postprocessing. Isochronal maps of activation spread (activation map) and conduction velocity measurements were derived using Cardioplex software (RedShirtImaging).
ECG Recordings
ECG recordings of age‐matched Xirp2 +/+ and Xirp2 −/− littermates were performed under anesthesia with sodium pentobarbital (50 mg/kg, intraperitoneally) as described previously.46 ECG signals were recorded with an ECG amplifier and recorder (RS3400; Gould Instruments, Basingstoke, UK). ECG parameters, such as P wave, QRS, QT, and R‐R intervals, were calculated from ECG tracings as described previously.28 Bazett's formula was used to calculate corrected QT interval.
Electrophysiological Studies of Isolated Ventricular Myocytes
Ventricular myocytes were enzymatically isolated from age‐matched Xirp2 +/+ and Xirp2 −/− mouse hearts as described previously.27 The whole‐cell patch‐clamp was performed in ventricular myocytes to acquire action potential (AP) characteristics and ionic current properties, as described in detail.27, 29 The ionic currents studied included the inward Na+ current (INa), the transient outward K+ current (ITO), the delayed rectifier outward K+ current (IK), and the Ba2+‐sensitive inward rectifier K+ current (IK1).
Histology and Immunofluorescence Microscopy
Whole‐heart X‐gal and immunofluorescence stainings were carried out as described previously.31, 47 Rabbit anti‐hyperpolarization‐activated cyclic nucleotide‐gated K+ channel 4 (HCN4; AB5808; Millipore, Burlington, MA) or rabbit anti‐connexin 40 (Cx40; AB1726; Millipore) was used as the primary antibody to facilitate the identification of atrioventricular node and His‐bundle/bundle branches.48 After staining with rhodamine‐conjugated secondary antibody, the section was stained with Alexa Fluor 488–conjugated wheat germ agglutinin (WGA488). Sections were imaged with a Leica TCS SPE confocal microscope (Leica Microsystems, Wetzlar, Germany). Under the same conditions, fluorescence intensity and particle size were estimated and compared from 5 confocal images each of Xirp2 +/+ and Xirp2 −/− heart sections with ImageJ (NIH, Bethesda, MD), as described previously.38 Histological staining for acetylcholine esterase (AChE) activity was performed as described previously49 on the adjacent section to that used in immunofluorescence for identifying the cardiac conduction system.
Co‐Immunoprecipitation and Western Blot
Hearts harvested from 2 adult wild‐type mice were pulverized in liquid nitrogen and incubated in extraction buffer containing 20 mmol/L of HEPES pH7.2, 120 mmol/L of NaCl, 10 mmol/L of MgCl2, 1 mmol/L of DTT, 1× protease inhibitor cocktail without EDTA (Sigma‐Aldrich, St. Louis, MO), 5% glycerol, and 0.5% Triton X‐100 at 4°C for 15 minutes. Total lysate was clarified by centrifugation at 10 000g for 10 minutes. Co‐immunoprecipitation (Co‐IP) was carried out by incubating Dynabeads protein G cross‐linked rabbit anti‐Kv1.5 (AB9786; Chemicon International, Billerica, MA), rabbit anti‐Nav1.5 (ASC005; Alomone Labs, Jerusalem, Israel), mouse anti‐Kv4.2 (NeuroMab, Davis, CA), rabbit anti‐GAPDH (Cell Signaling Technology, Danvers, MA), U1697 (rabbit antimouse Xirp1 peptide, aa# 564–583 of mXinα),28 U1040 (rabbit antimouse Xirp2 C‐terminus, aa# 3255–3278 of mXinβ),31 or U1013 (rabbit antimouse Xirp1, aa# 1–532 of mXinα)28, 31 with clarified total lysate overnight. After washing, immunoprecipitates were boiled in gel sample buffer and analyzed by western blot as previously described.31
Statistical Analysis
Genotype frequencies were calculated by the genotype counting method. Data from electrophysiological studies are presented as median±interquartile range or mean±SEM. Statistical analysis, including ANOVA, Student t test, or Mann–Whitney U rank‐sum test, was used to test the difference between 2 groups. A P value <0.05 was considered statistically significant.
Results
Demographics of Study Population
Death age of 134 SUNDS cases (3 females) was 30.8±7.6 years (range, 15–55). All these apparently healthy SUNDS victims without family history of sudden death were sporadic and lacked clinical records. There were no significant pathological alterations identified to elucidate these sudden deaths after comprehensive forensic autopsy examination.
All but 4 (father and son from 1 family, 2 brothers from another family) of the 22 BrS patients (2 females) were unrelated. Age at the time of diagnosis was 42.2±10.8 years (range, 20–60). A total of 18 of 22 BrS cases had suffered previous syncope and seizures. A total of 9 of 22 BrS had a family history of sudden unexpected death or BrS. A total of 8 of 22 BrS cases had suffered an episode of spontaneous ventricular fibrillation and had a cardioverter defibrillator implanted (Figure 1 shows a representative BrS ECG).
Figure 1.
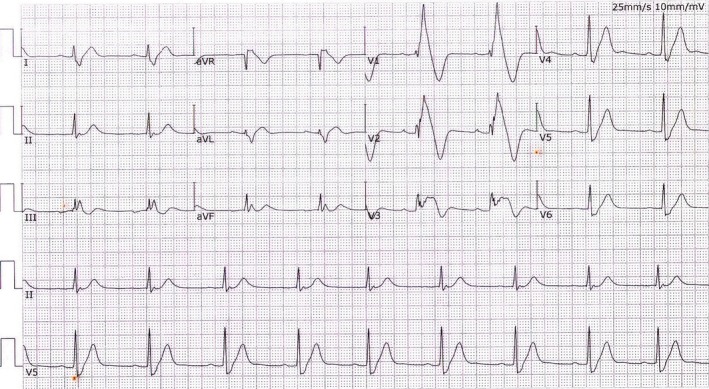
A representative Brugada syndrome (BrS) ECG. This typical BrS ECG (ST‐segment elevation among the right precordial leads V1 and V2) was recorded from a 56‐year‐old male who had recurrent episodes of syncope and ventricular fibrillation with implantable cardioverter defibrillator treatment. This patient tested negative for structural heart disease by magnetic resonance imaging and echocardiograph examination.
Rare Variants in Chinese Sporadic SUNDS Victims and BrS Patients
Sixteen rare variants in XIRP genes were identified in 134 (11.2%) sporadic SUNDS cases (Table 1). Five of the variants (T4A, E75K, Q856R, A930V, and T1830M) were located in the XIRP1 coding region, and the other 11 variants (V51A, S52L, E215K, A288S, E737K, I1899N, V2121I, D2177N, N2233S, Q2875*, and R3223H) were found in the XIRP2 coding region. One variant was a nonsense variant (Q2875*) located in exon 9 of the XIRP2 gene with substitution c.8623C>T whereas the remaining 15 were missense variants (Table 1). Among these 16 rare variants, 1 was a novel variant (E215K) located in exon 4 of the XIRP2 gene in SUNDS case E89. Of these 16 rare variants, 6 were in silico predicted as deleterious (A930V, E215K, E737K, I1899N, Q2875*, and R3223H). Interestingly, rare variant A930V of XIRP1 and deleterious common variant V1743I of XIRP2 (Table S2, found in SUNDS with MAF=0.005 in 105 Southern Han Chinese from 1000 Genomes Project Phase 3 and with MAF more than 0.01 in EAS from ExAC, 1000 Genomes Project Phase 3, and gnomAD) are located very close to each other on the consensus sequence (Table S3) of the highly conserved regions immediately after the Xin repeats of XIRP1 and XIRP2,26 which possess 2 previously identified α‐actinin‐binding regions50 (Table S4). The variant, R3223H, is located within the consensus sequences for nuclear localization signal (NLS) of XIRP2, whereas nonsense variant Q2875* should result in a lack of this NLS (Figure 2). The majority of the remaining missense variants are located within the highly conserved XIRP isoform‐specific regions at the N‐terminus and at the sequences immediately downstream of the Xin repeats,26 where limited functional information is known (Table S4).
Table 1.
Rare Variants of XIRP Genes in SUNDS Cases
| Case | Gene | Age, y | Nucleotide Change | Amino Acid Change | Type of Variant | dbSNP | Chinese Han | EAS | Prediction In Silicoa | ACMG Classificationb | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Local Database | (CHS+CHB) in 1000G | ExAC | 1000G | gnomAD | |||||||||||
| Allele Count | MAF | Allele Count | MAF | MAF | MAF | MAF | |||||||||
| E145 | XIRP1 | 33 | c.10A>G | p.T4A | missense | rs780225117 | 1298 | 0 | 416 | 0 | 0.00012 | ··· | 0.000273 | neutral | VUS |
| E144 | XIRP1 | 20 | c.223G>A | p.E75K | missense | rs749373055 | 1298 | 0 | 416 | 0 | 0.00127 | ··· | 0.00159 | neutral | VUS |
| E7 | XIRP1 | 36 | c.2567A>G | p.Q856R | missense | rs751104961 | 1298 | 0 | 416 | 0 | 0.00047 | ··· | 0.000371 | neutral | VUS |
| E128 | XIRP1 | 24 | c.2789C>T | p.A930V | missense | ··· | 1298 | 0 | 416 | 0 | ··· | ··· | 0.000116 | deleterious | VUS |
| E97 | XIRP1 | 30 | c.5489C>T | p.T1830M | missense | rs142746395 | 1298 | 0.005393 | 416 | 0.01442308 | 0.00451 | 0.007 | 0.00482 | neutral | VUS |
| E101 | XIRP2 | 31 | c.152T>C | p.V51A | missense | rs775164919 | 1298 | 0.00077 | 416 | 0 | 0 | ··· | 0.000266 | neutral | VUS |
| AKAP9 | c.11135G>A | p.R3712Q | missense | rs186148498 | 1298 | 0 | 416 | 0.00480769 | 0.00832 | 0.006 | 0.00885 | deleterious | VUS | ||
| E113 | XIRP2 | 19 | c.155C>T | p.S52L | missense | rs567637067 | 1298 | 0 | 416 | 0 | 0.00152 | 0.003 | 0.00123 | neutral | VUS |
| E89 | XIRP2 | 37 | c.643G>A | p.E215K | missense | ··· | 1298 | 0 | 416 | 0 | ··· | ··· | ··· | deleterious | VUS |
| E136 | XIRP2 | 40 | c.862G>T | p.A288S | missense | rs552549925 | 1298 | 0.00077 | 416 | 0 | 0.00349 | 0.001 | 0.00377 | neutral | VUS |
| KCNH2 | c.3110A>T | p.D1037V | missense | ··· | 1298 | 0 | 416 | 0 | ··· | ··· | ··· | deleterious | VUS | ||
| ZS032 | XIRP2 | 35 | c.2209G>A | p.E737K | missense | rs201995517 | 1298 | 0 | 416 | 0 | 0.00035 | ··· | 0.000106 | deleterious | VUS |
| CACNA1C | c.4393T>C | p.F1465L | missense | ··· | 1298 | 0 | 416 | 0 | ··· | ··· | ··· | deleterious | VUS | ||
| ZS120 | XIRP2 | 42 | c.5696T>A | p.I1899N | missense | rs748428961 | 1298 | 0 | 416 | 0 | 0.00082 | ··· | 0.000582 | deleterious | VUS |
| ZS146 | XIRP2 | 20 | c.6361G>A | p.V2121I | missense | rs181373166 | 1298 | 0.00154 | 416 | 0.00721 | 0.003385 | 0.006 | 0.002875 | neutral | VUS |
| E122 | XIRP2 | 30 | c.6529G>A | p.D2177N | missense | rs113038974 | 1298 | 0 | 416 | 0 | 0.0007 | ··· | 0.000748 | neutral | VUS |
| E135 | XIRP2 | 33 | c.6698A>G | p.N2233S | missense | rs778173531 | 1298 | 0.00077 | 416 | 0 | 0 | ··· | 0 | neutral | VUS |
| E78 | XIRP2 | 38 | c.8623C>T | p.Q2875* | nonsense | rs201881932 | 1298 | 0.00077 | 416 | 0.00240385 | 0.00093 | 0.001 | 0.00101 | deleterious | Likely pathogenic |
| E148 | XIRP2 | 27 | c.9668G>A | p.R3223H | missense | rs778569774 | 1298 | 0 | 416 | 0 | 0 | ··· | 0 | deleterious | VUS |
ACMG indicates American College of Medical Genetics and Genomics; CHB, Han Chinese in Beijing; CHS, Southern Han Chinese; EAS, East Asian; MAF, minor allele frequency; SUNDS, sudden unexplained nocturnal death syndrome; VUS, variant of uncertain significance.
Evaluated by 3 in silico tools (SIFT, Polyphen‐2, and CONDEL) and harboring “radical” variants or not.
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Figure 2.
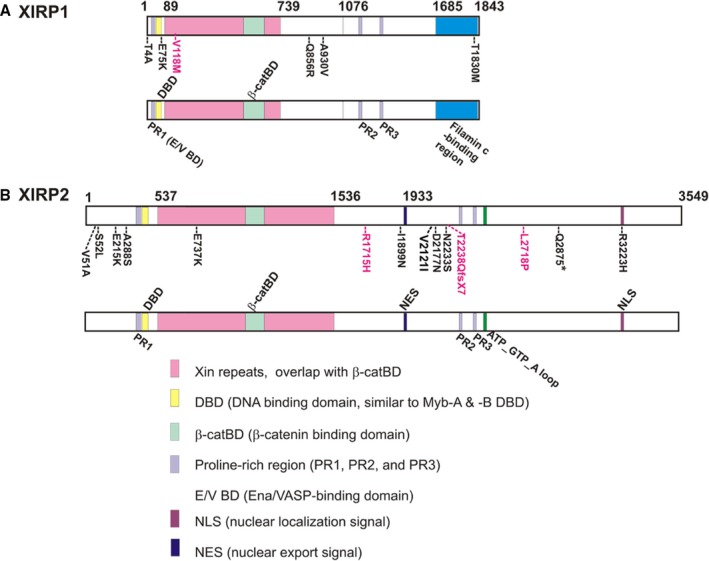
Schematic diagrams depict relative positions of XIRP rare variants to their respective domains/regions. The variant positions identified from sudden unexplained nocturnal death syndrome (black font) and Brugada syndrome (red font) cases are shown on the top diagram, whereas the known domains and unknown, but conserved, regions are shown in the bottom diagram of each XIRP protein. The Xin‐repeat regions (pink) were identified from human XIRP and mouse XIRP, and experimentally verified as actin‐binding and ‐bundling domains from XIRP 39 and mouse small Xirp1‐S (mXinα).32 Within the Xin repeats, the overlapped β‐catenin‐binding domain (β‐catBD, light green) was experimentally defined in Xirp1‐S.32 The β‐catBD is highly conserved among all Xin‐containing proteins from all animal species.26 Both Ena/VASP‐binding domain (E/V BD, light purple) and filamin c‐binding region (blue) were experimentally identified from human XIRP1.33 However, these similar sequences were not found in XIRP2. Based on sequence similarity, DNA‐binding domain (DBD) and proline‐rich (PR) region, including SH3‐binding motifs of PR‐1, PR‐2, and PR‐3, were found in both XIRP proteins.26 On the other hand, nuclear export signal (NES), nuclear localization signal (NLS), and ATP_GTP_A loop were only found in XIRP2.26 Conserved sequences immediately after the Xin‐repeat region (aa# 739–1076 of XIRP1 and aa# 1536–1933 of XIRP2) were slightly divergent between XIRP1 and XIRP2, but each was highly conserved across all species examined.26
There were also 4 rare variants (Table 2) identified in 22 (18.2%) BrS cases, including 3 missense variants (V118M in XIRP1; R1715H and L2718P in XIRP2), and 1 frameshift variant (c.6712‐6713delAC [p.T2238QfsX7] in XIRP2). Among these rare variants, L2718P was novel and located in exon 9 of the XIRP2 gene. V118M in XIRP1 and T2238QfsX7 in XIRP2 were predicted as deleterious variants. The enrichment of deleterious rare variants in XIRP1 and XIRP2 in both SUNDS and BrS cases suggests that both XIRP proteins may be important determinants in normal cardiac rhythms.
Table 2.
Rare Variants of XIRP Genes in BrS Patients
| Case | Gene | Age, y | Nucleotide Change | Amino Acid Change | Type of Variant | dbSNP | Chinese Han | EAS | Prediction In Silicoa | ACMG Classificationb | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Local Database | (CHS+CHB) in 1000G | ExAC | 1000G | gnomAD | |||||||||||
| Allele Count | MAF | Allele Count | MAF | MAF | MAF | MAF | |||||||||
| ZS106 | XIRP1 | 53 | c.352G>A | p.V118M | missense | rs115823205 | 1298 | 0.002311 | 416 | 0 | 0 | 0 | 0.0000530 | deleterious | VUS |
| SCN5A | c.5676delC | p.T1893PfsX29 | frameshift | ··· | 1298 | 0 | 416 | 0 | ··· | ··· | ··· | deleterious | Pathogenic | ||
| SCN5A | c.5692C>T | p.R1898C | missense | rs373118001 | 1298 | 0 | 416 | 0 | 0.000116 | 0.002 | 0.0000530 | deleterious | VUS | ||
| ZS109 | XIRP2 | 55 | c.5144G>A | p.R1715H | missense | rs552071889 | 1298 | 0.00077 | 416 | 0 | 0.000233 | 0 | 0.000266 | neutral | VUS |
| ZS116 | XIRP2 | 42 | c.6712_6713delAC | p.T2238QfsX7 | frameshift | rs749764225 | 1298 | 0 | 416 | 0 | 0.000117 | ··· | 0.000117 | deleterious | Likely pathogenic |
| IPS | XIRP2 | 57 | c.8153T>C | p.L2718P | missense | ··· | 1298 | 0 | 416 | 0 | ··· | ··· | ··· | neutral | VUS |
BrS, Brugada syndrome; CHB, Han Chinese in Beijing; CHS, Southern Han Chinese; EAS, East Asian; MAF, minor allele frequency; VUS, variant of uncertain significance.
Evaluated by 3 in silico tools (SIFT, Polyphen‐2, and CONDEL) and harboring “radical” variants or not.
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
According to our previous molecular autopsy investigation, all of the SUNDS (except for SUNDS cases E101, E136, and ZS032 [Table 1]) and BrS cases (except for BrS case ZS106 [Table 2]) with XIRP rare variants were negative for deleterious rare variants in 39 genes associated with lethal arrhythmias.7
Common Variants Identified in Chinese Sporadic SUNDS and BrS Cases
Twenty‐nine nonsynonymous common variants were identified in SUNDS cases (Table S2), including 7 in XIRP1 and 22 in XIRP2. All of these 29 variants were identified in EAS and Chinese Han, and 24 of the 29 were identified in BrS cases. The remaining 5 variants (rs75802875, rs181539061, rs77219745, rs3749002, and rs143084183) were not detected in the BrS cases (Table S2). Characters of XIRP common variants in SUNDS and BrS cases are shown in Table S2.
Xirp2 Is Required for Formation of Infranodal Ventricular Conduction System
Supporting a causative role of XIRP1 rare variants in SUNDS and BrS, our previous studies showed that mouse Xirp1 is important for maintenance of ICD integrity.25, 27, 28 Through its interactions with β‐catenin,32 p120‐catenin,36 KChIP2,27 filamin,27, 32 and cortactin,36 Xirp1 influences surface expression of channels responsible for the transient outward K+ current (ITO) and delayed K+ rectifier current (IK).25, 27 Loss of Xirp1 in mice led to abnormal AP and conduction defects.27, 28, 29, 30 Although complete loss of Xirp2 resulted in postnatal lethality,25, 31 a small number of Xirp2 −/− mice survived to postnatal day (P) 20. Studies with these Xirp2 −/− mice also revealed evidence to suggest a causative role of some XIRP2 rare variants, such as nonsense Q2875* and frameshift T2238QfsX7 variants expressing little or no XIRP2, in SUNDS and BrS.
(i) Xirp2 −/− Mouse Hearts Exhibited Slow Conduction Velocity, Prolonged QT Interval, and Atrioventricular Conduction Block
Previous echocardiographic comparisons on wild‐type and Xirp2 −/− mice showed a diastolic dysfunction associated with these Xirp2 −/− mice.31 Using an optical mapping technique, we further detected a 45% reduction in conduction velocity in Xirp2 −/− left ventricles compared with Xirp2 +/+ controls (Figure 3B). As can be seen in Figure 3A, when paced at the base of the right ventricle, activation in Xirp2 +/+ heart spread immediately and smoothly throughout the left ventricle, whereas it took more time in the Xirp2 −/− hearts to reach the same activated area. These results indicate a conduction defect associated with Xirp2 −/− ventricles.
Figure 3.
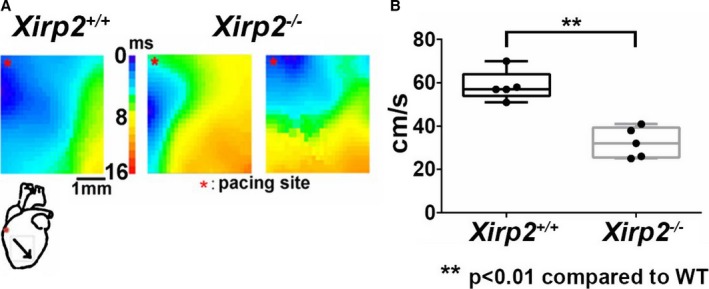
Optical mapping on the front surface of left ventricles from Xirp2 +/+ and Xirp2 −/− hearts. A, Activation maps of ventricles induced by electrical pacing. Left panel is the heatmap from 1 Xirp2 +/+ ventricle, whereas middle and right panels are maps from 2 Xirp2 −/− ventricles. The scale bar of the heatmap represents the time required for action potential (AP) propagating from the site of electric stimulation to a given ventricle position, which ranges from the shortest time (blue color, 0 ms) to the longest time (red color, 16 ms). The heart diagram underneath Xirp2 +/+ map depicts the site of electrical stimulation (*) and the activation direction (oblique arrow). Note that Xirp2 −/− ventricles exhibited slow AP propagation. B, Calculated conduction velocity of Xirp2 +/+ (black boxplots) and Xirp2 −/− (gray boxplots) left ventricles: on each box, the central mark is the median, the edges of the box are the 25th and 75th percentiles, the whiskers extend to the most extreme data points. **P<0.01, significant differences between 2 groups. n, number of mice tested (5 mice each group). WT indicates wild type.
To further show a conduction defect in the Xirp2 −/− hearts, we performed ECG recordings on age‐matched Xirp2 +/+ and Xirp2 −/− mice. Each of a representative ECG trace is shown in Figure 4A and 4B for a Xirp2 +/+ and a Xirp2 −/− mouse younger than P18. Table 3 summarizes the ECG parameters calculated from tracings of 12 Xirp2 +/+ and 8 Xirp2 −/− mice at P14 to P17. Significantly prolonged PR interval (representing the period of atrioventricular (A‐V) conduction) and prolonged QT and QTc intervals (representing the period between depolarization and repolarization of ventricle), but no changes in P wave and QRS interval, were detected in Xirp2 −/− mice. These results further suggest a defect in the infranodal ventricular conduction system in Xirp2 −/− mice. Interestingly, all of the P18 Xirp2 −/− mice examined had a severe A‐V conduction block. Figure 4C shows 2 examples of such an A‐V conduction block with many P waves (indicated by the arrows) followed by no QRS wave.
Figure 4.
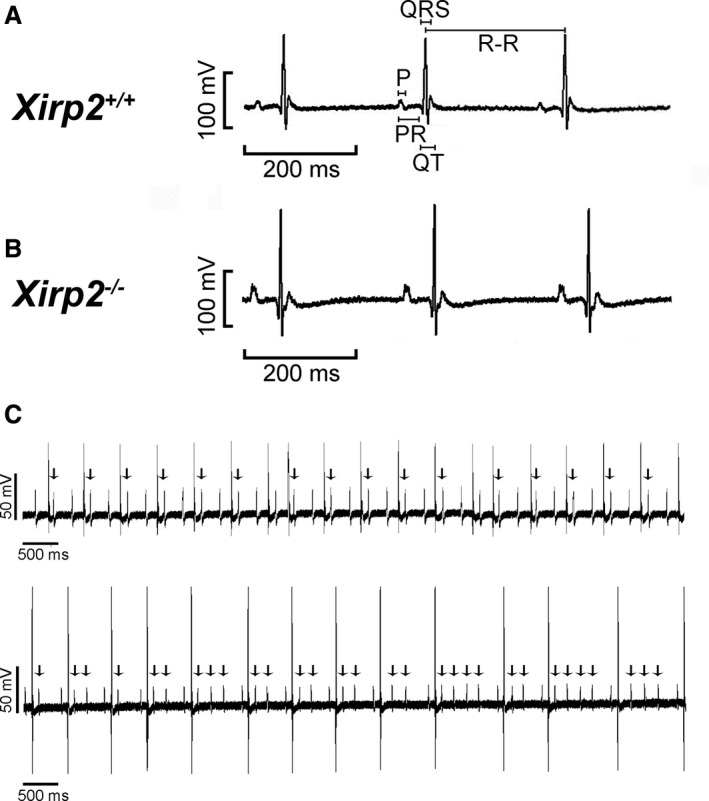
ECG tracings from age‐matched Xirp2 +/+ and Xirp2 −/− mice. A, ECG trace of a representative Xirp2 +/+ mouse at P14 to P17. The durations for P wave as well as PR, QRS, QT, and R‐R intervals are defined in this trace. B, ECG trace of a representative Xirp2 −/− mice at P14 to P17. C, Atrioventricular conduction (A‐V) blocks detected in all tested Xirp2 −/− mice at P18. Two representative ECG traces showed such A‐V block with many P waves followed by missing QRS (indicated by arrows).
Table 3.
Conduction Parameters (14‐ to 17‐day‐old) and Incidence of A‐V block (18‐day‐old) Determined by Surface ECG Recording From Xirp2 +/+ and Xirp2 −/− Mice
| Xirp2 +/+ | Xirp2 −/− | |
|---|---|---|
| No. of 14‐ to 17‐day‐old mice tested | 12 | 8 |
| P wave, ms | 11.8±0.3 | 12.6±0.9 |
| PR interval, ms | 39.2±0.3 | 45.9±3.5a |
| QRS interval, ms | 13.5±0.5 | 13.5±1.2 |
| QT interval, ms | 24.8±0.9 | 31.1±1.5a |
| QTc interval, ms | 17.4±0.6 | 21.2±0.7b |
| R‐R interval, ms | 217.5±10.0 | 223.0±24.9 |
| Number of 18‐day‐old mice tested | 7 | 7 |
| Incidence of A‐V conduction block | 0% | 100%b |
Values are presented as mean±SEM. A‐V indicates atrioventricular conduction; QTc, corrected QT interval.
P<0.05.
P<0.01 significant difference between Xirp2 +/+ and Xirp2 −/− mice.
(ii) Xirp2 Expressed in Atria and Ventricular Conduction System and Complete Loss of Xirp2 Resulted in Underdevelopment of His‐Bundle and Purkinje Fibers
The A‐V block phenotype suggested an involvement of Xirp2 in the ventricular conduction system. Therefore, we first sought to better define the expression profile of Xirp2 in the mouse heart. We generated mice expressing β‐galactosidase under the control of endogenous Xirp2 regulatory elements from an LacZ gene inserted into the Xirp2 locus (Xirp2 lacZ−tm2; Figure 5A). In adult heterozygous Xirp2 lacZ−tm2/+ hearts, the β‐galactosidase signal was diffused and homogeneous in the atria, but heterogeneous in the ventricles (Figure 5B, panel b), whereas no signal was detected in Xirp2 +/+ hearts (Figure 5B, panel a). En face view of the right ventricular endocardial surface showed that LacZ was widely expressed, but the strongest signal was associated with Purkinje fiber‐like structures, which are similar to that in β‐galactosidase‐stained hearts from CCS‐lacZ transgenic mice51 (Figure 5B, panel c).
Figure 5.
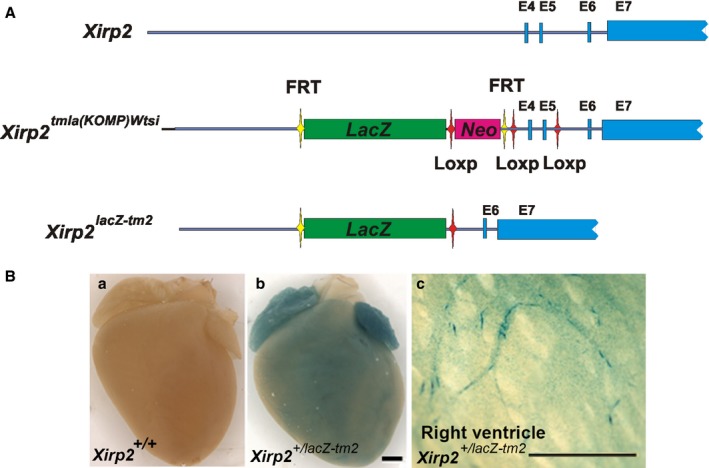
β‐galactosidase (β‐Gal) expression under the endogenous Xirp2 promoter in Xirp2 +/lacZ‐tm2 mice. A, Schematic representations of endogenous Xirp2 locus, targeted locus (Xirp2 tm1a(KOMP)Wtsi) and Xirp2 lacZ‐tm2 locus. After verification of homologous recombination and germline transmission, the resulting mice carrying targeted locus with the replacement of endogenous exon 4 (E4) and E5 by the DNA fragment containing FRT‐LacZ‐Loxp‐Neo‐FRT‐LoxP‐E4‐E5‐LoxP from the KOPM targeting vector were bred with EK‐Cre transgenic mice to generate Xirp2 lacZ‐tm2 mice (with a knock‐in LacZ gene under the control of Xirp2 promoter/enhancer and a disruption of Xirp2 expression called lacZ‐tm2 distinguished from our original Xirp2 knockout mice). B, β‐gal stain of P20 wild‐type (Xirp2 +/+) and Xirp2 +/lacZ‐tm2 mouse hearts. a, Wild‐type heart serves as a negative stain control; b, heterozygous Xirp2 +/lacZ‐tm2 heart shows strong β‐gal in atria and weak/diffuse stain in ventricle; and c, dissected Xirp2 +/lacZ‐tm2 right ventricle at the endocardium view shows positive stain on what appears to be ventricular conduction system including Purkinje fibers. Bar in b=1 mm for a and b; bar in c=1 mm.
The above Xirp2 expression pattern, together with conduction defects observed in Xirp2‐null hearts, led us to hypothesize that Xirp2 may play an important role in the development of the ventricular conduction system. We thus performed histochemical stain for AChE activity (a marker for the ventricular conduction system49) on serial sections of entire hearts prepared from Xirp2 +/+ and Xirp2 −/− mice. Examples of such stains at Purkinje fiber regions are shown in Figure 6 for comparisons. AChE activities, as detected by the brown color stains, were clearly diminished from the endocardial surface of the right ventricle of Xirp2 −/− hearts (Figure 6), suggesting that the Purkinje fibers might not be properly developed.
Figure 6.
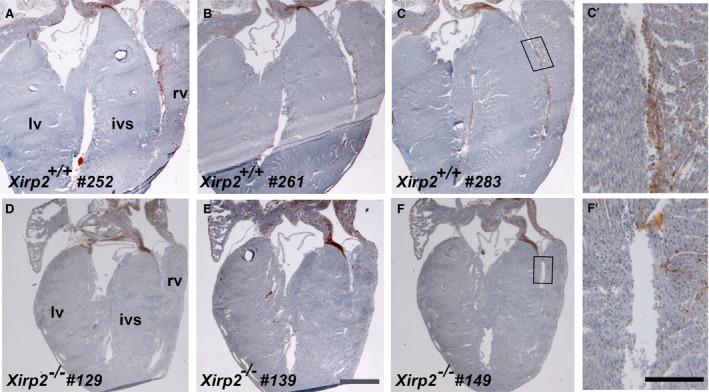
Acetylcholine esterase (AChE) stain revealing a reduction in Purkinje fibers of P18 Xirp2 −/− heart. Serial 10‐μm sections (sectioning from frontal/ventral to dorsal) of entire hearts from Xirp2 +/+ and Xirp2 −/− mice were histochemically stained for AChE activity (brown) to reveal the ventricular conduction system. Purkinje fibers were positively stained in representative sections of Xirp2 +/+ heart (a, section #252; b, #261; and c, #283), whereas comparable sections of Xirp2 −/− heart showed a drastically reduced in AChE activity (d, section #129; e, #139; and f, #149). Bar in e=1 mm for a through f. rv, right ventricle; lv, left ventricle; ivs, interventricular septum. (c′ and f′) higher magnification of the box regions in c and f, respectively. Bar in f’=0.2 mm for c′ and f′.
To further investigate the expression of HCN4 and Cx40 in the atrioventricular node and His‐bundle and bundle branches, we immunofluorescently labeled an adjacent section to that stained for AChE activity. As can be seen in Figure 7 (panels a and e), AChE staining was not different in the atrioventricular node as identified by anti‐HCN4 staining between Xirp2 +/+ and Xirp2 −/− hearts. Similarly, immunofluorescent intensity of HCN4 within the atrioventricular node was undistinguished (Figure 7). Cx40 is a major gap junction protein of the His‐bundle and bundle branches, and Cx40‐null mice are characterized by bundle branch block.52 Reduced expression of Cx40 in Xirp2 −/− hearts might be a reason for the observed A‐V block. Supporting this hypothesis, we found that immunofluorescent intensity and size of Cx40 puncta were significantly reduced in the His‐bundle of Xirp2 −/− hearts (Figure 8). Our results suggest that XIRP2 plays a role in the infranodal conduction system, and mutation of XIRP2 could contribute to conduction defects.
Figure 7.
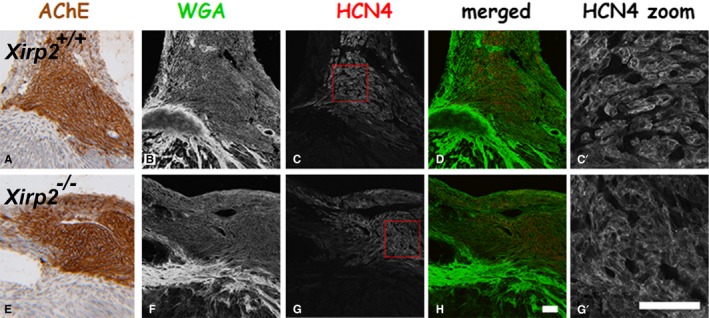
Confocal microscopy for hyperpolarization‐activated cyclic nucleotide‐gated K+ channel 4 (HCN4) localization and histological stains for acetylcholine esterase (AChE) activity and wheat germ agglutinin (WGA). P18 Xirp2 +/+ (top raw) and Xirp2 −/− (bottom raw) hearts were serially cryosectioned. Adjacent serial 10‐μm sections were used for AChE activity stain (brown color) and for double‐label immunofluorescence with anti‐HCN4 (red color) and WGA stain (green color) to identify the atrioventricular (AV) node and to examine HCN4 expression level. a and e, AChE stain identifying the AV node. b and f, WGA stain outlining the tissue. c and g, HCN4‐positive region showing the AV node. d and h, Merged color images of HCN4 and WGA. c’ and g’, Zoom in the boxed areas in c and g, respectively. Bar in h=10 μm for a through h; bar in g′=10 μm for c’ and g′.
Figure 8.
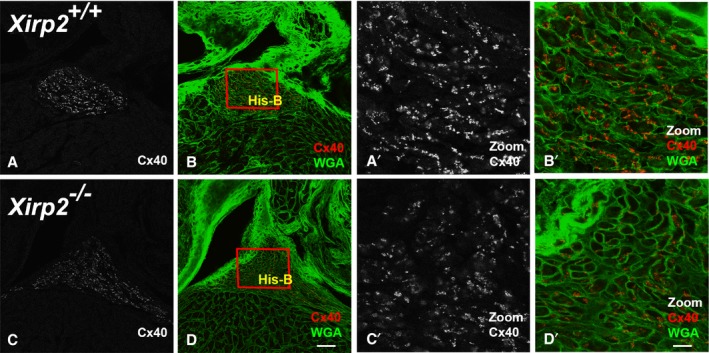
Reduced expression of connexin 40 (Cx40) in the His‐bundle of Xirp2 −/− hearts. Confocal microscopy of P18 Xirp2 +/+ (top raw) and Xirp2 −/− (bottom raw) heart sections double labeling for Cx40 (red) and wheat germ agglutinin (WGA) (green). The adjacent section stained for acetylcholine esterase (AChE) was used to identify His‐bundles (data not shown). Panels b and d show the merged images. Box regions containing His‐bundles (His‐B) were zoomed in to show a significant reduction in Cx40 expression in Xirp2 −/− heart (c’ and d’), compared with Xirp2 +/+ heart (a′ and b′). Bar in d=40 μm for a through d; bar in d′=10 μm for a′ through d′.
Xirp2 −/− Ventricular Myocytes Exhibited Altered AP, Reduced ITO, and Increased IK and IK1
Our previous studies showed that Xirp2 is required for normal ICD localization of Xirp1 and many other ICD components, such as N‐cadherin, desmoplakin, and Cx43,31, 38 and that Xirp1 can influence electrophysiological and conductive properties of ventricular myocytes.27, 28, 29 Taking together with the fact that both Xirp1 and Xirp2 are Xin repeats‐containing and ICD‐associated proteins, we hypothesized that Xirp2 would also play an important role in the electrophysiology of ventricular myocytes. Using the whole‐cell patch‐clamp technique in current‐clamp mode, we detected that the AP of Xirp2 −/− ventricular myocytes had significantly increased AP amplitude, less negative maximal diastolic potential, and shorter APD at 20%, 50%, and 90% repolarization levels (APD20, APD50, and APD90), as compared with those obtained from Xirp2 +/+ cells (Figure 9B). Superimposing 2 traces together (Figure 9A) clearly demonstrated differences in depolarization and repolarization, and, more important, showed a rise in the resting membrane potential of Xirp2 −/− myocytes, which suggests an easy initiation of the next AP wave.
Figure 9.
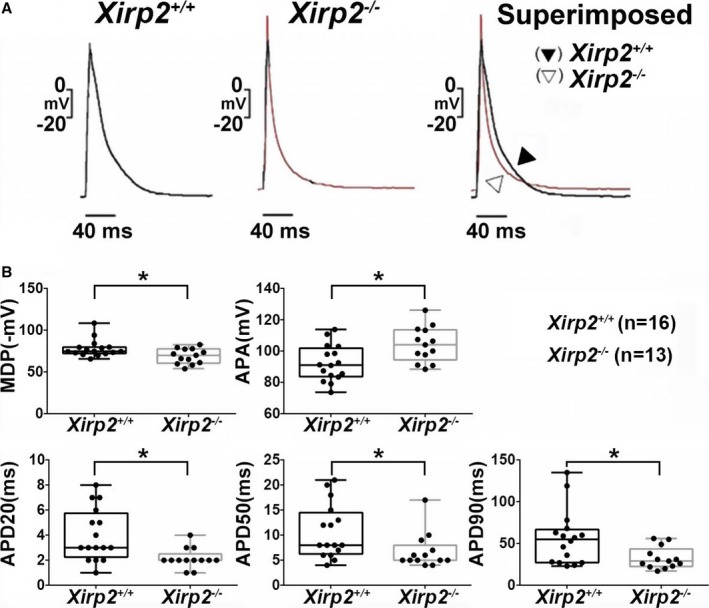
Action potential (AP) configurations of ventricular myocytes prepared from Xirp2 +/+ and Xirp2 −/− mice. A, Representative tracings of AP waveforms in Xirp2 +/+ (left) and Xirp2 −/− (middle) ventricular myocytes driven electrically. Superimposed comparison (right) reveals differences in AP configurations. B, Comparison of AP characteristics between the Xirp2 +/+ (black boxplots) and Xirp2 −/−population (gray box plots): on each box, the central mark is the median, the edges of the box are the 25th and 75th percentiles, and the whiskers extend to the most extreme data points. Significant changes in AP characteristics of Xirp2 −/− ventricular myocytes. APA indicates AP amplitude; MDP, maximal diastolic potential. APD20, APD50, and APD90: APD measured at 20%, 50%, and 90% repolarization, respectively. *P<0.05, significant difference between Xirp2 +/+ and Xirp2 −/− myocytes. n, number of myocytes tested.
To further understand mechanisms accounting for the altered AP waveform in Xirp2 −/− myocytes, the whole‐cell patch‐clamp technique in voltage‐clamp mode was used to measure membrane ionic currents. As shown in Figure 10A (panel c), the INa current densities of Xirp2 −/− myocytes at tested voltages were not significantly different from that of Xirp2 +/+ myocytes; thus, the INa current alone cannot account for the observed increased AP amplitude in Xirp2 −/− myocytes. This finding is in contrast to our previous report29 that Xirp1 −/− myocytes had significantly increased INa current density. On the other hand, depressed ITO currents and current densities were detected in Xirp2 −/− myocytes at pulsed voltages from +20 to +60 mV (Figure 10B). The ITO plays a critical role for the early phase of repolarization in myocytes,53 and the decrease in the ITO may contribute partly to the observed increase in AP amplitude of Xirp2 −/− myocytes. Another outward delayed rectifier, IK, can affect the duration of the repolarization of an AP. As shown in Figure 10C, significant increases in amplitudes and current densities of IK were detected in Xirp2 −/− myocytes compared with control cells. Given that the IK currents contribute to the delayed phase of repolarization of AP,53 the increase in IK current density likely led to the observed decrease in APD50 and APD90 in Xirp2 −/− myocytes. In addition to ITO and IK, inward rectifier IK1 current maintains resting membrane potential and contributes to the terminal phase of repolarization.53 As shown in Figure 10D, significant increases in this IK1 inward currents and current densities at negative voltage from −70 to −120 mV were observed in Xirp2 −/− myocytes compared with control cells.
Figure 10.
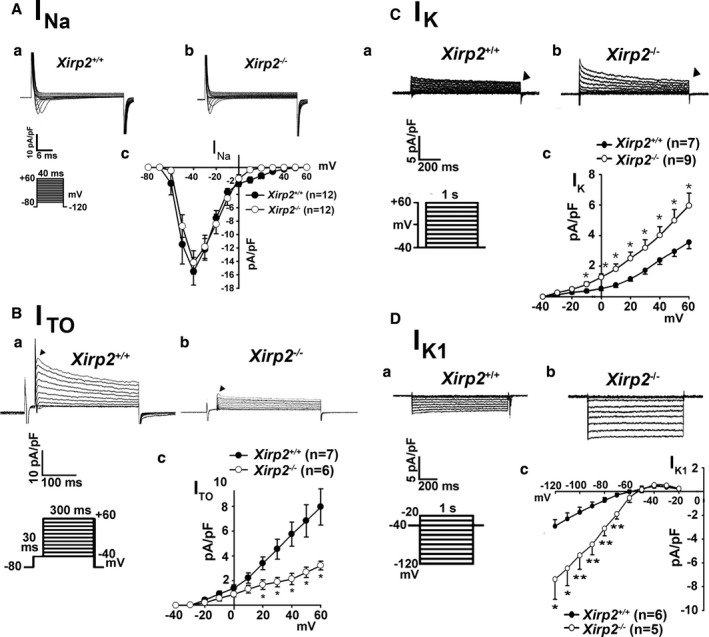
Various voltage‐dependent ionic currents in ventricular myocytes isolated from Xirp2 +/+ and Xirp2 −/− mice. A, Inward Na+ current (IN a) densities. A series of IN a currents were elicited on depolarization from a holding potential of −120 mV to testing potential from −80 to +60 mV for 40 ms in 1 Xirp2 +/+ (a) and 1 Xirp2 −/− (b) ventricular myocyte. Size scales and clamp protocol are shown below Xirp2 +/+ tracings. (c) summarizes current density‐voltage relationships of IN a in 2 groups of myocytes. n, number of myocytes tested. B, Transient outward K+ current (ITO) densities. Membrane currents were elicited on depolarization from a holding potential of −80 to −40 mV for 30 ms and then to test potentials from −40 to +60 mV for 300 ms. Examples of ITO currents (indicated by triangle) recorded from 1 Xirp2 +/+ (a) and 1 Xirp2 −/− (b) ventricular myocyte. Size scales and clamp protocol are shown below Xirp2 +/+ tracings. (c) summarizes current density‐voltage relationships of ITO in 2 groups of myocytes. n, number of myocytes tested. *P<0.05, significant difference. C, Delayed rectifier outward K+ current (IK) densities. A series of IK currents were elicited on depolarization from a holding potential of −40 mV to a test potential from −40 to +60 mV for 1000 ms. Examples of IK currents (indicated by triangle) recorded from 1 Xirp2 +/+ (a) and 1 Xirp2 −/− (b) ventricular myocyte. Size scales and clamp protocol are shown below Xirp2 +/+ tracings. (c) summarizes current density‐voltage relationships of IK in 2 groups of myocytes. n, number of myocytes tested. *P<0.05, significant difference. D, Ba2+‐sensitive inward rectifier K+ current (IK 1) densities. A series of IK 1 currents were elicited on depolarization from a holding potential of −40 mV to a test potential from −20 to −120 mV for 1000 ms in 1 Xirp2 +/+ (a) and 1 Xirp2 −/− (b) ventricular myocyte. Size scales and clamp protocol are shown below Xirp2 +/+ tracings. (c) summarizes current density‐voltage relationships of IK 1 in 2 groups of myocytes. The difference in current densities before and after Ba2+ treatment was plotted against the test potentials to obtain current density‐voltage relationships. n, number of myocytes tested. *P<0.05; **P<0.01; significant difference.
Xirp2 Is Associated With Both Nav1.5 and Kv1.5
Ion channels interact with structural proteins for trafficking, stability, and function. A fraction of Nav1.5 (α‐subunit of INa channel) and Kv1.5 (α‐subunit of Ik,slow1, 1 of the delayed rectifier IK channels) are targeted to ICDs.54, 55 Co‐IP from mouse heart lysates showed that both anti‐Kv1.5 and anti‐Nav1.5 co‐pelleted Xirp2, but neither Xirp1‐S (small variant/previously called mXinα)20 nor Xirp1‐L (long variant/previously called mXinα‐a)20 (Figure 11A). On the other hand, anti‐Kv4.2 and control anti‐GAPDH antibodies did not pellet any Xirp proteins. Contrarily, when co‐IP experiments were performed with anti‐Xirp, both Kv1.5 and Nav1.5 were clearly immunoprecipitated with U1040 antibody (specific for Xirp2) against the peptide fragment from aa# 3255 to 3278 of Xirp2,31 but not with U1697 antibody (specific for Xirp1) against the peptide fragment from aa# 564 to 588 of Xirp1,28 U1013 antibody specific for all Xirp,28 or control anti‐GAPDH antibody (Figure 11B). Interestingly, U1013 anti‐Xirp antibody raised against Xirp1 fragment (aa# 1–532) also cross‐reacted with Xirp2 (Figure 11A) in western blot, but it did not co‐pellet Nav1.5 or Kv1.5 (lane 5 of Figure 11B). Although the exact reason for this failure of co‐IP remains unclear, a weak cross‐reactivity of U1013 to Xirp2 or/and a lesser amount of antigen Xirp2 as compared with Xirp1 in total lysate may greatly reduce the sensitivity in this assay. Alternatively, binding of U1013 to the highly conserved region of Xin repeats in Xirp2 may disrupt potential interactions between this region and the Kv1.5/Nav1.5. On the other hand, the epitopes recognized by U1040 are located at the extreme C‐terminus of Xirp2, which may not be required for Xirp2 to interact with Kv1.5 and Nav1.5.
Figure 11.
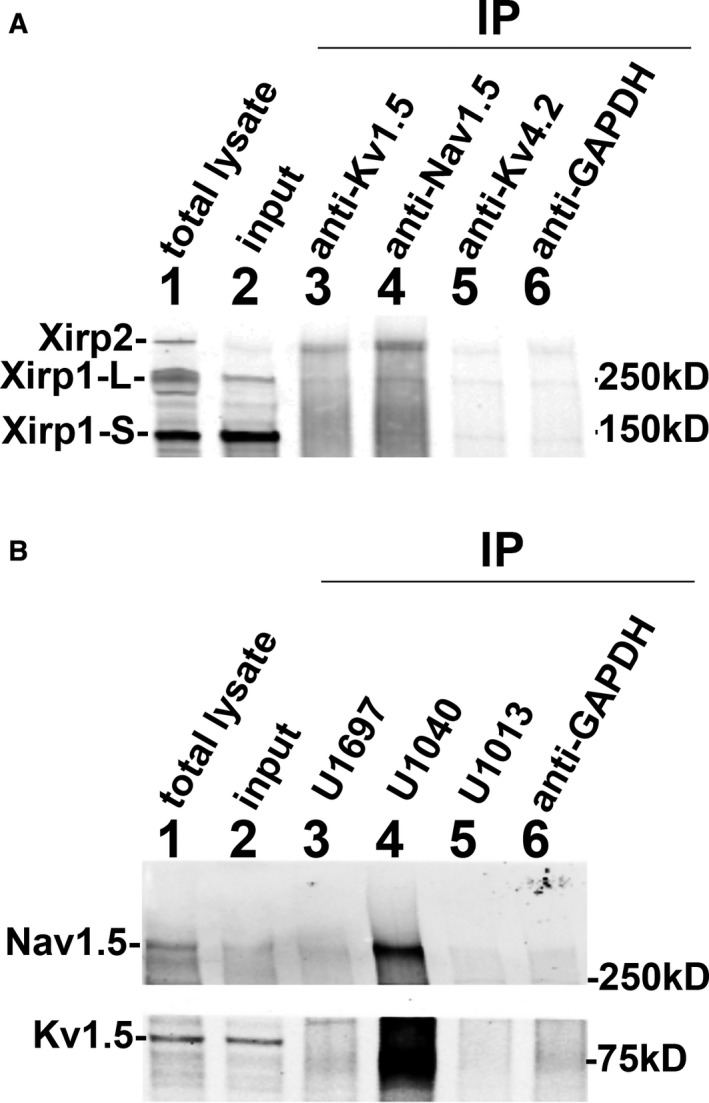
Co‐immunoprecipitation (Co‐IP) experiments. A, Co‐IPs were performed with various antibodies against α‐subunits of different channels to examine the possible associations between XIRP and channel components. Total lysates (lane 1) and clarified supernatants as inputs (lane 2) were prepared from adult wild‐type mouse hearts as described under Methods. The aliquot of input was used for immunoprecipitation (IP) with various primary antibodies cross‐linked to Dynabeads protein G. The primary antibodies included rabbit anti‐Kv1.5 (lane 3), rabbit anti‐Nav1.5 (lane 4), mouse anti‐Kv4.2 (lane 5), and rabbit anti‐GAPDH (lane 6). The resulting immunoprecipitates were fractionated and immunoblotted with rabbit anti‐Xirp1 (U1013) antibody that recognized both Xirp1‐S (small isoform), Xirp‐L (large isoform), and Xirp2. Xirp2 was co‐pelleted in the immunoprecipitates with anti‐Kv1.5 and with anti‐Nav1.5, but neither with anti‐Kv4.2 nor with anti‐GAPDH. B, Reverse Co‐IPs were performed with cross‐linked rabbit antibody U1697 (specific to Xirp1), U1040 (specific to Xirp2), U1013 (cross‐reacted to both Xirp1 and Xirp2), or anti‐GAPDH antibody. The resulting immunoprecipitates were immunoblotted with anti‐Kv1.5 (lower panel), anti‐Nav1.5 (top panel), or anti‐Kv4.2 (data not shown). Both Kv1.5 and Nav1.5 are co‐pelleted in the immunoprecipitates with anti‐Xirp2 U1040 antibody.
Discussion
Mouse Xirp Genes Are Critical for Cardiac Conduction
Our previous studies have clearly demonstrated that mouse hearts without Xirp1 led to disrupted ICD, hypertrophy, and cardiomyopathy with conduction defects.27, 28, 29, 30 In the present study, we have further shown that Xirp2‐null hearts also exhibit cardiac conduction defects (ie, slower conduction velocity, A‐V block, and prolonged QT interval). Xin repeat‐containing protein family, Xirp1 and Xirp2, function as scaffolding proteins to interact with components of actin cytoskeleton, N‐cadherin‐mediated junction, and ionic channel assembly.1 Therefore, Xirp proteins play essential roles in regulating not only the ICD structure and function, but also the surface expression of ion channels, influencing action potential propagation in the heart. Xirp2 initiates the formation of ICD and localizes Xirp1 to ICD, whereas Xirp1 can further stabilize and maintain ICD integrity. Significant proportions of key ion‐channel assemblies required for controlling AP have been shown to localize to ICDs through their scaffolding or anchoring proteins,36, 37, 54, 55, 56 and defects in these localization processes can lead to arrhythmias and cardiac sudden death.56
Cardiac conduction defects observed in Xirp2‐null mice may account for the pathogenesis of the nonsense variant (Q2875*) and frameshift variant (T2238QfsX7) in XIRP2 from SUNDS and BrS, respectively. Both variants contain premature termination codon, which could cause nonsense‐mediated mRNA decay, a universal mRNA degradation pathway in eukaryotes that monitors and eliminates abnormal mRNAs to avoid the generation of harmful proteins.42, 43, 44 Nonsense‐mediated mRNA decay has been reported in long QT syndrome patients attributed to hERG nonsense mutations57 and a BrS patient attributed to a CACNA1C splicing mutation.43 Therefore, the Q2875* nonsense variant and the T2238QfsX7 frameshift variant would likely induce nonsense‐mediated mRNA decay and express a reduced amount of XIRP2, which could contribute to the No. E78 SUNDS case and the No. ZS116 BrS case, respectively.
XIRP1 and XIRP2 Represent Class of Novel Susceptible Genes for Human Cardiac Arrhythmias Disorders With Conduction Defect Such as SUNDS and BrS
In this study, we provided the following experimental evidences to support that both XIRP1 and XIRP2 are novel susceptible genes for SUNDS and BrS. First, we showed that SUNDS and BrS cases unexplained by variants in 39 known arrhythmia‐susceptibility genes are enriched with rare variants in XIRP1 and XIPR2. Some of these variants affect highly conserved regions of the XIRP1 and XIRP2 proteins and many of them are predicted to be deleterious by in silico prediction tools. Next, with a mouse model, we showed that (1) Xirp2 is strongly expressed in the ventricular conduction system; (2) Xirp2 −/− mice exhibit slow conduction velocity, prolonged PR and QT intervals, and A‐V conduction block; (3) loss of Xirp2 leads to reduced activity or expression of markers for the ventricular conduction system, including AChE and Cx40; and (4) Xirp2 −/− ventricular myocytes have altered AP configuration and changes in membrane ionic currents. Finally, we showed that Xirp2 is associated with important cardiac ion channel components, Nav1.5 and Kv1.5. Collectively, this evidence, together with our previous findings that Xirp1 regulates surface expression of ITo and IK channels,27, 29, 36 strongly supports the overall concept that XIRP1 and XIRP2, encoding 2 ICD‐associated proteins, are important for cardiac electrophysiology and that they constitute a novel family of candidate genes for cardiac arrhythmia disorders, including SUNDS and BrS.
SUNDS, a clinical conundrum in both forensic and clinical medicine, has a strong genetic underpinning based on reported molecular pathological studies.3, 14, 15, 16, 17, 18, 19 Accumulated evidence suggests that SUNDS is related primarily to cardiac arrhythmia diseases, such as BrS,58 long QT syndrome,59 sick sinus syndrome,60 and cardiac conduction disease.61 However, we have previously shown that in a Chinese Han population, genetic variants in 39 arrhythmia‐associated genes could only possibly account for a small part of sporadic SUNDS cases,14, 15, 16, 17, 18, 19 suggesting the existence of unknown susceptible genes. In this study, we identified 20 rare variants (8 were in silico predicted as deleterious; Tables 1 and 2) in XIRP1 and XIRP2 genes of SUNDS and BrS cases, and all of these rare variants were found either absent or less frequent in Chinese Han, and East Asian from a local database, 1000 Genomes Project Phase 3, ExAC, and gnomAD data sets.
How Do Identified Rare Variants Contribute to Pathogenesis of SUNDS and BrS?
Variants altering the function of XIRP1 and XIRP2 could cause cardiac electrophysiology defects by several mechanisms. We have reported that Xirp1 regulates the ITO and IK channels by directly interacting with KChIP2, filamin, and cortactin.27, 36 Xirp1‐null cardiomyocytes have prolonged APD, alterations in ionic currents, and increased incidence of early afterdepolorization, and Xirp1‐null hearts show prolonged QT interval and conduction defects.27, 28, 29 In the current study, we further showed that Xirp2 is required for the formation of infranodal ventricular conduction system and normal AP configuration; and that Xirp2 is associated with Nav1.5 and Kv1.5. Therefore, variants in Xirp1 or Xirp2 may disrupt their interaction with the channel complexes and lead to dysregulation of channel functions. Interaction between ion channels and cytoskeletal proteins are important for their trafficking, stability, and function. For example, Nav1.5 E1053K variant causes BrS by blocking the interaction between Nav1.5 and cytoskeletal protein Ankyrin‐G, leading to significant reduction of Nav1.5 at the T‐tubules and ICDs.62 Our co‐IP results suggest that Xirp2 could be important for localizing Nav1.5 and Kv1.5 to ICDs.
Furthermore, the rare variants we identified in XIRP1 and XIRP2 may modulate the electrophysiological properties of cardiomyocytes by regulating ICD maturation and maintenance. ICDs are a unique structure to the heart and they contain cell‐cell junctions for mechanical and electrical coupling. ICDs also harbor voltage‐gated sodium channels and potassium channels.63 Accumulated evidence suggests that the cell‐cell junctions at ICDs modulate ionic currents.36, 37, 64
Finally, it is possible that the rare variants we identified in XIRP2 may cause defects in the ventricular conduction system. The Xirp gene family is a downstream target of Nkx2‐5,25, 50, 65, 66 and it is well established that Nkx2‐5 plays an important role in the development of the ventricular conduction system and variants in Nkx2‐5 cause A‐V blocks.67
Five In Silico–Predicted Deleterious Rare Variants Identified in This Study Are Located to Highly Conserved Regions of XIRP Proteins
In addition to a nonsense variant (Q2875*) and a frameshift variant (T2238QfsX7) discussed above, 5 missense rare variants (A930V, E215K, E737K, I1899N, and L2718P) identified in this study intrigued us to speculate on their pathogenic role in patients. Among these in silico–predicted deleterious variants, XIRP2 E215K and XIRP2 L2718P were absent from ExAC, gnomAD, and database of single‐nucleotide polymorphism and were defined as novel. XIRP1 A930V, XIRP2 I1899N, and XIRP2 L2718P variants were located to the highly conserved regions immediately after the Xin repeats of both XIRP proteins.26 In contrast, XIRP2 E215K variant was located at the N‐terminal region of XIRP2 protein. This conserved N‐terminal sequence was only found in the Xirp2 proteins from humans and primates.26
XIRP1 A930 is a highly conserved residue among multiple mammalian species26 and its surrounding sequence is homologous to the XIRP2 region26 harboring the V1743I polymorphism (Table S3). In silico software predicted that A930V might alter actin cytoskeleton dynamics. Supporting this prediction, the V1743I polymorphism and A930V variant are located within the regions homologous to an α‐actinin‐binding site in mouse Xirp2.50 We proposed that the A930V variant is most probably the genetic cause of the No. E128 SUNDS case, but the biophysical mechanisms should be elucidated by future electrophysiological, biochemical, and functional studies. Within this highly conserved region, we also identified a XIRP1 Q856R variant from a SUNDS case and a XIRP2 R1715H variant from a BrS case (Figure 2).
The novel XIRP2 E215K variant led to the replacement of glutamic (E) by lysine (K) at the N‐terminus of XIRP2 and was in silico predicted as a deleterious variant. The N‐terminal sequence from aa#1 to #222 of human (also chimpanzee and rhesus monkey) XIRP2 is not found in mouse and chicken Xirp2 proteins, and therefore this region is primate specific.26 It should be noted that within this primate‐specific region, we found a total of 3 rare variants (V51A, S52L, and E215K) associated with SUNDS cases (Figure 2).
XIPR2 E737K is located in 1 of the highly conserved Xin repeats of XIPR2, and therefore this variant may interfere with the interaction between Xirp2 and actin filaments.
The novel XIRP2 L2718P variant is located very close to XIRP2 Q2875* variant within the conserved sequence region at C‐terminus of all Xirp2 examined, which is, however, not found in all Xirp1 examined.26 In the current study, we have shown that Xirp2, but not Xirp1, is capable of associating with Nav1.5 and Kv1.5 (Figure 11). Although the interacting sites for Nav1.5 and Kv1.5 have not been mapped on XIRP2, this conserved C‐terminal region may possess such sites. If it is proven to be true, this novel variant could interfere with the interactions and then affect INa and IK channel activities.
Within the conserved C‐terminus of XIRP2, there exists a sequence (aa#3201–3229) highly homologous to consensus NLS. The importance of this NLS in XIRP2 was suggested by the identification of a missense variant R3223H within the NLS, a nonsense variant Q2875*, and a frameshift variant T2238QfsX7. These variants might be able to either disrupt or delete this NLS signal and then affect normal XIRP2's function. However, the exact functions of R3223 in XIRP2 will need to be experimentally studied.
The predicted functional defects in all 20 rare variants of XIRP were summarized in Table S5. Together with all findings above, we proposed that all these identified XIRP rare variants in both SUNDS and BrS may contribute to the corresponding SUNDS or BrS cases (Tables 1 and 2).
Study Limitations
The relatively small sample size for genetic testing as well as the absence of clinic information (for SUNDS) and genetic investigation of family members (for both SUNDS and BrS) limited a precise assessment of linkage between clinic and genetic phenotypes.
Although our data from mouse Xirp knockout studies support that Xirp proteins are important in conduction system and ion channel functions, direct characterization of mice carrying these XIRP variants to be established by CRISPR technology is still needed to define their causal roles in SUNDS and BrS cases.
Conclusions
Despite several study limitations, this first report identifying likely pathogenic XIRP rare variants in BrS cases and in the largest SUNDS cohort reported provided molecular and pathological evidences that XIRP rare variants may contribute to the genetic cause of arrhythmogenic disorders such as SUNDS and BrS.
Sources of Funding
This work was supported by the Key Program (81430046) from National Natural Science Foundation of China (Cheng), by the grants (NSC98‐2923‐B‐016‐001‐MY3) from National Science Council Foundation of Taiwan (Loh), and by the grant (HL107383) from NIH (Lin).
Disclosures
None.
Supporting information
Table S1. Sequences and Annealing Temperatures of Primers
Table S2. Characters of XIRP Common Variants in Sudden Unexplained Nocturnal Death Syndrome (SUNDS) and Brugada Syndrome (BrS)
Table S3. Nearby Sequence Comparison Between A930V Rare Variant of XIRP1 and V1743I Common Variant of XIRP2
Table S4. Amino Acid Residues Covering Known Functional Domains and Highly Conserved Regions of XIRP1 and XIRP2 Proteins
Table S5. Summary of Predicted Functional Defects in Rare Variants of XIRP
Acknowledgments
We thank the legal representatives of the victims and BrS patients for their cooperation. We thank Robert F. Corliss (Department of Pathology and Laboratory Medicine and Waisman Center, University of Wisconsin, Madison, WI) for his assistance in pathological analysis.
(J Am Heart Assoc. 2018;7:e006320 DOI: 10.1161/JAHA.117.006320.)29306897
This article was handled independently by N.A. Mark Estes, III, as a guest editor.
Note
Contributor Information
Qinchuan Wang, Email: Qinchuan.Wang@jhmi.edu.
Shih‐Hurng Loh, Email: shloh@mail.ndmctsgh.edu.tw.
Jianding Cheng, Email: chengjd@mail.sysu.edu.cn.
References
- 1. Gaw AC, Lee B, Gervacio‐Domingo G, Antzelevitch C, Divinagracia R, Jocano FJ. Unraveling the enigma of Bangungut: is sudden unexplained nocturnal death syndrome (SUNDS) in the Philippines a disease allelic to the Brugada syndrome? Philipp J Intern Med. 2011;49:165–176. [PMC free article] [PubMed] [Google Scholar]
- 2. Nimmannit S, Malasit P, Chaovakul V, Susaengrat W, Vasuvattakul S, Nilwarangkur S. Pathogenesis of sudden unexplained nocturnal death (lai tai) and endemic distal renal tubular acidosis. Lancet. 1991;338:930–932. [DOI] [PubMed] [Google Scholar]
- 3. Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, Tunsanga K, Kuasirikul S, Malasit P, Tansupasawadikul S, Tatsanavivat P. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96:2595–2600. [DOI] [PubMed] [Google Scholar]
- 4. Vatta M, Dumaine R, Varghese G, Richard TA, Shimizu W, Aihara N, Nademanee K, Brugada R, Brugada J, Veerakul G, Li H, Bowles NE, Brugada P, Antzelevitch C, Towbin JA. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11:337–345. [DOI] [PubMed] [Google Scholar]
- 5. Nakajima K, Takeichi S, Nakajima Y, Fujita MQ. Pokkuri Death Syndrome; sudden cardiac death cases without coronary atherosclerosis in South Asian young males. Forensic Sci Int. 2011;207:6–13. [DOI] [PubMed] [Google Scholar]
- 6. Cheng J, Makielski JC, Yuan P, Shi N, Zhou F, Ye B, Tan BH, Kroboth S. Sudden unexplained nocturnal death syndrome in Southern China: an epidemiological survey and SCN5A gene screening. Am J Forensic Med Pathol. 2011;32:359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang L, Tester DJ, Lang D, Chen Y, Zheng J, Gao R, Corliss RF, Tang S, Kyle JW, Liu C, Ackerman MJ, Makielski JC, Cheng J. Does sudden unexplained nocturnal death syndrome remain the autopsy‐negative disorder: a gross, microscopic, and molecular autopsy investigation in Southern China. Mayo Clin Proc. 2016;91:1503–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Z, Mu J, Chen X, Dong H. Sudden unexplained nocturnal death syndrome in central china (Hubei): a 16‐year retrospective study of autopsy cases. Medicine (Baltimore). 2016;95:e2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baron RC, Thacker SB, Gorelkin L, Vernon AA, Taylor WR, Choi K. Sudden death among Southeast Asian refugees. An unexplained nocturnal phenomenon. JAMA. 1983;250:2947–2951. [PubMed] [Google Scholar]
- 10. Kirschner RH, Eckner FA, Baron RC. The cardiac pathology of sudden, unexplained nocturnal death in Southeast Asian refugees. JAMA. 1986;256:2700–2705. [PubMed] [Google Scholar]
- 11. Young E, Xiong S, Finn L, Young T. Unique sleep disorders profile of a population‐based sample of 747 Hmong immigrants in Wisconsin. Soc Sci Med. 2013;79:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sangwatanaroj S, Prechawat S, Sunsaneewitayakul B, Sitthisook S, Tosukhowong P, Tungsanga K. New electrocardiographic leads and the procainamide test for the detection of the Brugada sign in sudden unexplained death syndrome survivors and their relatives. Eur Heart J. 2001;22:2290–2296. [DOI] [PubMed] [Google Scholar]
- 13. Sutphin BS, Boczek NJ, Barajas‐Martínez H, Hu D, Ye D, Tester DJ, Antzelevitch C, Ackerman MJ. Molecular and functional characterization of rare CACNA1C variants in sudden unexplained death in the young. Congenit Heart Dis. 2016;11:683–692. [DOI] [PubMed] [Google Scholar]
- 14. Liu C, Tester DJ, Hou Y, Wang W, Lv G, Ackerman MJ, Makielski JC, Cheng J. Is sudden unexplained nocturnal death syndrome in Southern China a cardiac sodium channel dysfunction disorder? Forensic Sci Int. 2014;236:38–45. [DOI] [PubMed] [Google Scholar]
- 15. Zhao Q, Chen Y, Peng L, Gao R, Liu N, Jiang P, Liu C, Tang S, Quan L, Makielski JC, Cheng J. Identification of rare variants of DSP gene in sudden unexplained nocturnal death syndrome in the southern Chinese Han population. Int J Legal Med. 2016;130:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu C, Zhao Q, Su T, Tang S, Lv G, Liu H, Quan L, Cheng J. Postmortem molecular analysis of KCNQ1, KCNH2, KCNE1 and KCNE2 genes in sudden unexplained nocturnal death syndrome in the Chinese Han population. Forensic Sci Int. 2013;231:82–87. [DOI] [PubMed] [Google Scholar]
- 17. Huang L, Liu C, Tang S, Su T, Cheng J. Postmortem genetic screening of SNPs in RyR2 gene in sudden unexplained nocturnal death syndrome in the southern Chinese Han population. Forensic Sci Int. 2014;235:14–18. [DOI] [PubMed] [Google Scholar]
- 18. Huang L, Tangs S, Peng L, Chen Y, Cheng J. Molecular autopsy of desmosomal protein plakophilin‐2 in sudden unexplained nocturnal death syndrome. J Forensic Sci. 2016;61:687–691. [DOI] [PubMed] [Google Scholar]
- 19. Zhang L, Zhou F, Huang L, Wu Q, Zheng J, Wu Y, Yin K, Cheng J. Association of common and rare variants of SCN10A gene with sudden unexplained nocturnal death syndrome in Chinese Han population. Int J Legal Med. 2017;131:53–60. [DOI] [PubMed] [Google Scholar]
- 20. Wang Q, Lin JL, Erives AJ, Lin CI, Lin JJ. New insights into the roles of Xin repeat‐containing proteins in cardiac development, function, and disease. Int Rev Cell Mol Biol. 2014;310:89–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–714. [DOI] [PubMed] [Google Scholar]
- 22. Li J, Swope D, Raess N, Cheng L, Muller EJ, Radice GL. Cardiac tissue‐restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of β‐catenin signaling. Mol Cell Biol. 2011;31:1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lombardi R, Marian AJ. Molecular genetics and pathogenesis of arrhythmogenic right ventricular cardiomyopathy: a disease of cardiac stem cells. Pediatr Cardiol. 2011;32:360–365. [DOI] [PubMed] [Google Scholar]
- 24. Swope D, Cheng L, Gao E, Li J, Radice GL. Loss of cadherin‐binding proteins beta‐catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol Cell Biol. 2012;32:1056–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Q, Lin JL, Wu KH, Wang DZ, Reiter RS, Sinn HW, Lin CI, Lin CJ. Xin proteins and intercalated disc maturation, signaling and diseases. Front Biosci (Landmark Ed). 2012;17:2566–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grosskurth SE, Bhattacharya D, Wang Q, Lin JJ. Emergence of Xin demarcates a key innovation in heart evolution. PLoS One. 2008;3:e2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan FC, Cheng CP, Wu KH, Chen YC, Hsu CH, Gustafson‐Wagner EA, Lin JL, Wang Q, Lin JJ, Lin CI. Intercalated disc‐associated protein, mXinα, influences surface expression of ITO currents in ventricular myocytes. Front Biosci (Elite Ed). 2011;3:1425–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gustafson‐Wagner EA, Sinn HW, Chen YL, Wang DZ, Reiter RS, Lin JL, Yang B, Williamson RA, Chen J, Lin CI, Lin JJ. Loss of mXinα, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am J Physiol Heart Circ Physiol. 2007;293:H2680–H2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lai YJ, Chen YY, Cheng CP, Lin JJ, Chudorodova SL, Roshchevskaya IM, Roshchevsky MP, Chen YC, Lin CI. Changes in ionic currents and reduced conduction velocity in hypertrophied ventricular myocardium of Xinα‐deficient mice. Anatol J Cardiol. 2007;7:90–92. [PMC free article] [PubMed] [Google Scholar]
- 30. Lai YJ, Huang EYK, Yeh HI, Chen YL, Lin JJ, Lin CI. On the mechanisms of arrhythmias in the myocardium of mXinα‐deficient murine left atrial‐pulmonary veins. Life Sci. 2008;83:272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Q, Lin JL, Reinking BE, Feng HZ, Chan FC, Lin CI, Jin JP, Gustafson‐Wagner EA, Scholz TD, Yang B, Lin JJ. Essential roles of an intercalated disc protein, mXinβ, in postnatal heart growth and survival. Circ Res. 2010;106:1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Choi S, Gustafson‐Wagner EA, Wang Q, Harlan SM, Sinn HW, Lin JL, Lin JJ. The intercalated disk protein, mXinα, is capable of interacting with β‐catenin and bundling actin filaments. J Biol Chem. 2007;282:36024–36036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van der Ven PF, Ehler E, Vakeel P, Eulitz S, Schenk JA, Milting H, Micheel B, Fürst DO. Unusual splicing events result in distinct Xin isoforms that associate differentially with filamin c and Mena/VASP. Exp Cell Res. 2006;312:2154–2167. [DOI] [PubMed] [Google Scholar]
- 34. Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, Nguyêñ‐Trân VT, Gu Y, Ikeda Y, Chu PH, Ross J, Giles WR, Chien KR. A defect in the Kv channel‐interacting protein 1 (KChIP2) gene leads to a complete loss of I to and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801–813. [DOI] [PubMed] [Google Scholar]
- 35. Petrecca K, Miller DM, Shrier A. Localization and enhanced current density of the Kv4.2 potassium channel by interaction with the actin‐binding protein filamin. J Neurosci. 2000;20:8736–8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Q, Lu TL, Adams E, Lin JL, Lin JJ. Intercalated disc protein, mXinα, suppresses p120‐catenin‐induced branching phenotype via its interactions with p120‐catenin and cortactin. Arch Biochem Biophys. 2013;535:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng L, Yung A, Covarrubias M, Radice GL. Cortactin is required for N‐cadherin regulation of Kv1.5 channel function. J Biol Chem. 2011;286:20478–20489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Q, Lin JL, Chan SY, Lin JJ. The Xin repeat‐containing protein, mXinβ, initiates the mutation of the intercalated discs during postnatal heart development. Dev Biol. 2013;374:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pacholsky D, Vakeel P, Himmel M, Löwe T, Stradal T, Rottner K, Fürst DO, van der Ven PF. Xin repeats define a novel actin‐binding motif. J Cell Sci. 2004;117:5257–5268. [DOI] [PubMed] [Google Scholar]
- 40. Moreno‐Hagelsieb G, Trevino V, Perez‐Rueda E, Smith TF, Collado‐Vides J. Transcription unit conservation in the three domains of life: a perspective from Escherichia coli. Trends Genet. 2001;17:175–177. [DOI] [PubMed] [Google Scholar]
- 41. Nimmakayalu M, Noble N, Horton VK, Willing M, Copeland S, Sheffield V, Nagy PL, Wassink T, Patil S, Shchelochkov OA. 2q24 deletions: further characterization of clinical findings and their relation to the SCN cluster. Am J Med Genet. 2012;158A:2767–2774. [DOI] [PubMed] [Google Scholar]
- 42. Holbrook JA, Neu‐Yilik G, Hentze MW, Kulozik AE. Nonsense‐mediated decay approaches the clinic. Nat Genet. 2004;36:801–808. [DOI] [PubMed] [Google Scholar]
- 43. Fukuyama M, Ohno S, Wang Q, Shirayama T, Itoh H, Horie M. Nonsense‐mediated mRNA decay due to a CACNA1C splicing mutation in a patient with Brugada syndrome. Heart Rhythm. 2014;11:629–634. [DOI] [PubMed] [Google Scholar]
- 44. Dai Y, Li W, An L. NMD mechanism and the function of Upf proteins in plant. Plant Cell Rep. 2016;35:5–15. [DOI] [PubMed] [Google Scholar]
- 45. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ovsepyan AA, Panchenkov DN, Prokhortchouk EB, Telegin GB, Zhigalova NA, Golubev EP, Sviridova TE, Matskeplishvili ST, Skryabin KG, Buziashvili UI. Modeling myocardial infarction in mice: methodology, monitoring, pathomorphology. Acta Naturae. 2011;3:107–115. [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Q, Reiter RS, Huang QQ, Jin JP, Lin JJ. Comparative studies on the expression patterns of three troponin T genes during mouse development. Anat Rec. 2001;263:72–84. [DOI] [PubMed] [Google Scholar]
- 48. Liang X, Wang G, Lin L, Lowe J, Zhang Q, Bu L, Chen Y, Chen J, Sun Y, Evans SM. HCN4 dynamically marks the first heart field and conduction system precursors. Circ Res. 2013;113:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carbonell LM. Esterases of the conductive system of the heart. J Histochem Cytochem. 1956;4:87–95. [DOI] [PubMed] [Google Scholar]
- 50. Huang HT, Brand OM, Mathew M, Ignatiou C, Ewen EP, McCalmon SA, Naya FJ. Myomaxin is a novel transcriptional target of MEF2A that encodes a Xin‐related α‐actinin‐interacting protein. J Biol Chem. 2006;281:39370–39379. [DOI] [PubMed] [Google Scholar]
- 51. Rentschler S, Vaidya DM, Tamaddon H, Degenhardt K, Sassoon D, Morley GE, Jalife J, Fishman GI. Visualization and functional characterization of the developing murine cardiac conduction system. Development. 2001;128:1785–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simon AM, Goodenough DA, Paul DL. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr Biol. 1998;8:295–298. [DOI] [PubMed] [Google Scholar]
- 53. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. [DOI] [PubMed] [Google Scholar]
- 54. Mays DJ, Foose JM, Philipson LH, Tamkun MM. Localization of the Kv1.5 K+ channel protein in explanted cardiac tissue. J Clin Invest. 1995;96:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shy D, Gillet L, Abriel H. Cardiac sodium channel Nav1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta. 2013;1833:886–894. [DOI] [PubMed] [Google Scholar]
- 56. Wang Q, Lin JJ. Xin scaffolding proteins and arrhythmias. J Cardiol Clin Res. 2013;1:1011. [PMC free article] [PubMed] [Google Scholar]
- 57. Gong Q, Zhang L, Vincent GM, Horne BD, Zhou Z. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense‐mediated mRNA decay in human long‐QT syndrome. Circulation. 2007;116:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brugada P. Brugada syndrome: more than 20 years of scientific excitement. J Cardiol. 2016;67:215–220. [DOI] [PubMed] [Google Scholar]
- 59. Kanters JK, Yuan L, Hedley PL, Stoevring B, Jons C, Bloch Thomsen PE, Grunnet M, Christiansen M, Jespersen T. Flecainide provocation reveals concealed Brugada syndrome in a long QT syndrome family with a novel L1786Q mutation in SCN5A. Circ J. 2014;78:1136–1143. [DOI] [PubMed] [Google Scholar]
- 60. Chakrabarti S, Wu X, Yang Z, Wu L, Yong SL, Zhang C, Hu K, Wang QK, Chen Q. MOG1 rescues defective trafficking of Na(v)1.5 mutations in Brugada syndrome and sick sinus syndrome. Circ Arrhythm Electrophysiol. 2013;6:392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hothi SS, Ara F, Timperley J. p.Y1449C SCN5A mutation associated with overlap disorder comprising conduction disease, Brugada syndrome, and atrial flutter. J Cardiovasc Electrophysiol. 2015;26:93–97. [DOI] [PubMed] [Google Scholar]
- 62. Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin‐G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101:17533–17538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR, Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A. 2012;109:E2134–E2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Agullo‐Pascual E, Cerrone M, Delmar M. Arrhythmogenic cardiomyopathy and Brugada syndrome: diseases of the connexome. FEBS Lett. 2014;588:1322–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang DZ, Reiter RS, Lin JL, Wang Q, Williams HS, Krob SL, Schultheiss TM, Evans S, Lin JJ. Requirement of a novel gene, Xin, in cardiac morphogenesis. Development. 1999;126:1281–1294. [DOI] [PubMed] [Google Scholar]
- 66. Lin JJ, Gustafson‐Wagner EA, Sinn HW, Choi S, Jaacks SM, Wang DZ, Evans S, Lin JL. Structure, expression, and function of a novel intercalated disc protein, Xin. J Med Sci. 2005;25:215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jay PY, Harris BS, Maguire CT, Buerger A, Wakimoto H, Tanaka M, Kupershmidt S, Roden DM, Schultheiss TM, O'Brien TX, Gourdie RG, Berul CI, Izumo S. Nkx2‐5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest. 2004;113:1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences and Annealing Temperatures of Primers
Table S2. Characters of XIRP Common Variants in Sudden Unexplained Nocturnal Death Syndrome (SUNDS) and Brugada Syndrome (BrS)
Table S3. Nearby Sequence Comparison Between A930V Rare Variant of XIRP1 and V1743I Common Variant of XIRP2
Table S4. Amino Acid Residues Covering Known Functional Domains and Highly Conserved Regions of XIRP1 and XIRP2 Proteins
Table S5. Summary of Predicted Functional Defects in Rare Variants of XIRP