

Abstract
Background
Although hydrochlorothiazide (HCTZ) is a well‐established first‐line antihypertensive in the United States, <50% of HCTZ treated patients achieve blood pressure (BP) control. Thus, identifying biomarkers that could predict the BP response to HCTZ is critically important. In this study, we utilized metabolomics, genomics, and lipidomics to identify novel pathways and biomarkers associated with HCTZ BP response.
Methods and Results
First, we conducted a pathway analysis for 13 metabolites we recently identified to be significantly associated with HCTZ BP response. From this analysis, we found the sphingolipid metabolic pathway as the most significant pathway (P=5.8E‐05). Testing 78 variants, within 14 genes involved in the sphingolipid metabolic canonical pathway, with the BP response to HCTZ identified variant rs6078905, within the SPTLC3 gene, as a novel biomarker significantly associated with the BP response to HCTZ in whites (n=228). We found that rs6078905 C‐allele carriers had a better BP response to HCTZ versus noncarriers (∆SBP/∆DBP: −11.4/−6.9 versus −6.8/−3.5 mm Hg; ∆SBP P=6.7E‐04; ∆DBP P=4.8E‐04). Additionally, in blacks (n=148), we found genetic signals in the SPTLC3 genomic region significantly associated with the BP response to HCTZ (P<0.05). Last, we observed that rs6078905 significantly affects the baseline level of 4 sphingomyelins (N24:2, N24:3, N16:1, and N22:1; false discovery rate <0.05), from which N24:2 sphingomyelin has a significant correlation with both HCTZ DBP‐response (r=−0.42; P=7E‐03) and SBP‐response (r=−0.36; P=2E‐02).
Conclusions
This study provides insight into potential pharmacometabolomic and genetic mechanisms underlying HCTZ BP response and suggests that SPTLC3 is a potential determinant of the BP response to HCTZ.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT00246519.
Keywords: blood pressure, lipid metabolites, metabolomics, pharmacogenetics, thiazide diuretics
Subject Categories: Genetic, Association Studies; Hypertension; Cardiovascular Disease
Clinical Perspective
What Is New?
Despite the widespread use of hydrochlorothiazide (HCTZ), the blood pressure (BP) control rates of HCTZ‐treated patients are far from optimal (<50% of patients achieve BP control on monotherapy).
Herein, we integrated pharmacometabolomics and pharmacogenomics data to uncover a novel genetic polymorphism, rs6078905, within the SPTLC3 gene with clinically relevant effects on the BP response to HCTZ.
We found that rs6078905 variant allele carriers (C‐allele) had a significantly better BP response to HCTZ compared with noncarriers.
Leveraging the analysis with lipidomics data further confirmed the influence of SPTLC3 rs6078905 single‐nucleotide polymorphism on the BP response to HCTZ and highlighted sphingolipids as potential mediators regulating the BP response to HCTZ.
What Are the Clinical Implications?
The results of this study suggest that SPTLC3 is a potential determinant of the BP response to HCTZ.
Replicating the findings from this study in other large, well‐designed, independent studies may help advance a personalized medicine approach to antihypertensive therapy.
Introduction
Cardiovascular diseases—including heart disease and stroke—are the leading cause of death globally.1 According to the 2011 US death rate data, more than 2150 Americans die of cardiovascular diseases each day.2 Hypertension has long been recognized as 1 of the leading causes of cardiovascular diseases worldwide,3 and reduction of high blood pressure (BP) has been associated with significant improvement in cardiovascular outcomes.4 Despite the availability of multiple drug classes for treating hypertension, data across the globe suggest that BP control rates, to any given antihypertensive medication, are far from optimal (with <50% control rate).5 This fact is likely influenced, in part, by the empirical “trial and error” approach currently used for selecting antihypertensive medications. Thus, research to identify new therapeutic approaches and biomarkers that can be utilized to better predict the best antihypertensive therapy for each patient is necessary to improve overall heart health globally.
Thiazide diuretics, including hydrochlorothiazide (HCTZ), are 1 of the most commonly prescribed antihypertensive classes and have long been used as first‐line therapy for most patients with uncomplicated essential hypertension.6 Despite the widespread use of this drug class, there remains a gap in knowledge regarding the mechanism underlying thiazide‐induced long‐term BP‐lowering effects.7 Additionally, global data show that only around half of thiazide diuretic–treated patients achieve BP control.8, 9 Thus, additional research is needed to better understand the BP‐lowering mechanism underlying thiazide diuretics and identify predictors that can be used to select patients likely to achieve optimal BP‐lowering response.
Over the past decade, pharmacogenomics studies have identified several promising genetic signatures associated with variability in response to thiazide diuretics.7, 10, 11 Additionally, pharmacometabolomics has been successful in identifying novel pathways and biomarkers influencing patients’ variability in response to different drugs.12, 13, 14 Recently, using an untargeted metabolomics approach, we were able to identify several novel metabolites significantly associated with the BP response to HCTZ.15 Also, pharmacolipidomics represents a promising approach to identify novel biomarkers of drug response and yield mechanistic insights for several drugs.16, 17 Moreover, integrating different omics data is a powerful approach to identify novel signatures and pathways associated with complex traits, including drug response.18, 19 Therefore, in this study, we conducted a pathway analysis of the metabolites (Table S1) we previously identified to be associated with HCTZ BP response,15 to further pursue identification of novel pathways and biomarkers associated with BP response to HCTZ. Additionally, we performed integrated analyses of data from pharmacogenomics and pharmacolipidomics platforms to provide insights into the BP‐lowering mechanism underlying HCTZ and to validate our findings, respectively.
Methods
Pharmacogenomic Evaluation of Antihypertensive Response Study
Biological samples and clinical data used for metabolomics, genomics, and lipidomics analyses were collected as part of the PEAR (Pharmacogenomic Evaluation of Antihypertensive Response) trial (clinicaltrials.gov #NCT00246519). The PEAR genomics and phenotype data used in this study have been made publicly available at the database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap). The design and objectives of the PEAR study have been previously described.20 In brief, PEAR was a prospective study that recruited white and black participants, aged 17 to 65 years, with mild‐to‐moderate hypertension at the University of Florida (Gainesville, FL), Emory University (Atlanta, GA), and the Mayo Clinic (Rochester, MN). All participants had approximately a 4‐week washout period of any antihypertensive therapies and then were randomized to receive 12.5 mg/daily of HCTZ or 50 mg/daily of atenolol (β‐1‐selective blocker) monotherapy for 3 weeks. The HCTZ dose was then increased to 25 mg/daily and atenolol to 100 mg/daily for 6 additional weeks if the BP was greater than 120/70 mm Hg.
Genetic Epidemiology of Responses to Antihypertensives Study
White participants treated with HCTZ, from the GERA (Genetic Epidemiology of Responses to Antihypertensives) study (clinicaltrials.gov #NCT00005520), were used to replicate our pharmacogenetic finding from the PEAR primary analysis. The study design and objectives of the GERA study have been previously described.21 In brief, GERA was a prospective study that recruited hypertensive participants, aged 30 to 59 years, at Emory University (Atlanta, GA), and the Mayo Clinic (Rochester, MN). After enrollment, all participants had an average 4‐week washout period of anti‐HTN therapies followed by a BP assessment. Participants then started taking 25 mg of HCTZ daily for 4 weeks followed by another BP assessment.
Both PEAR and GERA studies were approved by the institutional review board at each study site. All participants provided written informed consent before participation in the study.
Hydrochlorothiazide Blood Pressure Response Measurement
PEAR BP was measured pre‐HCTZ (at baseline) and 9 weeks after HCTZ monotherapy treatment. BP data were obtained from home, office, and ambulatory daytime and nighttime BP measurements,20 as explained in Data S1. Of note, the BP used from the PEAR participants is a composite weighted average of the home, office, and ambulatory daytime and nighttime data, which has been shown to be a more‐accurate measurement of BP response with a better signal‐to‐noise ratio and more power to identify genetic predictors of BP response.22
GERA white participants had their BP measured in triplicate by a trained assistant using a random zero sphygmomanometer (Hawksley and Sons, Ltd; West Sussex, UK).21 HCTZ BP response was measured by calculating the difference between post‐HCTZ BP and pre‐HCTZ BP readings.
Genomics
A total of 228 white participants treated with HCTZ monotherapy in the PEAR study were included in the primary genetic analysis. Additionally, we used data from 148 black participants treated with HCTZ monotherapy in PEAR and 196 white participants treated with HCTZ monotherapy in GERA as 2 independent cohorts used for the replication efforts in this study. Details of the genotyping, quality control, and imputation performed in the PEAR and GERA studies are described in Data S1.
Lipidomics
PEAR white participants (n=40) were selected from the upper and lower quartiles of BP response to HCTZ for lipidomics analysis. Given the fact that sex hormones have been shown to exhibit sex‐associated differences in sphingolipids levels as sphingomyelins,23, 24 we selected only samples from female participants treated with HCTZ for the primary analysis of the lipidomics data. Lipidomics profiling was conducted on fasting baseline plasma samples using multidimensional mass spectrometry‐based shotgun lipidomics, as described in Data S1.
Experimental Approach
We utilized a 4‐step analytic approach, which is outlined in Figure 1.
Figure 1.

Overall framework analyses. BP indicates blood pressure; GERA, Genetic Epidemiology of Responses to Antihypertensives; HCTZ, hydrochlorothiazide; PEAR, Pharmacogenomic Evaluation of Antihypertensive Responses; SNPs, single‐nucleotide polymorphisms.
Metabolomics pathway analysis (step 1)
First, we conducted a pathway analysis for the 13 metabolites we recently identified to be significantly associated with changes in both systolic and diastolic BP response to HCTZ (false discovery rate, <0.05).15 Pathway analysis was conducted using MPINet R‐based tool (http://cran.r-project.org/web/packages/MPINet/), which has previously been used to identify novel pathways associated with complex traits, including drug response.25 A false discovery rate–adjusted P value of <0.05 was used to account for multiple comparisons. From the pathway analysis, the pathway with the lowest P value was selected to move forward to step 2.
Genomics association analysis (step 2)
We selected single‐nucleotide polymorphisms (SNPs) within genes involved in the selected pathway from step 1 (sphingolipid metabolism pathway; Table S2; Figure S1). Gene regions were defined as the full transcript ±2 kb. A total of 78 SNPs were extracted after excluding SNPs with minor allele frequency <5% and after linkage disequilibrium (LD) pruning. LD pruning was conducted using the PLINK software option (–indep‐pairwise 50 5 0.3), which is based on removing SNPs within a 50‐SNP sliding window that shifts 5 SNPs along with each move and considering an r 2 threshold greater than 0.3, as previously suggested.26, 27 Genetic analyses were then conducted to test the association between these SNPs and BP response in PEAR whites treated with HCTZ monotherapy. Association analysis was conducted using PLINK software,28 based on an additive genetic model with age, sex, baseline BP, and principal components (principal components 1 and 2) as adjustment variables. From this analysis, SNPs with an false discovery rate‐adjusted P value of <0.05 were considered significant. Hardy–Weinberg equilibrium for the selected SNPs was evaluated using exact test, and SNPs with a Hardy–Weinberg equilibrium P value <1E‐03 were excluded from the analyses.
Replication (step 3)
SNPs that were significantly associated with HCTZ BP response in step 2 were then tested for replication in 2 independent cohorts of participants treated with HCTZ. The first group included 148 blacks treated with HCTZ in PEAR study. The other group included 196 white participants treated with HCTZ in the GERA study. In blacks, because of the differences in recombination rates and linkage disequilibrium patterns between blacks and whites, SNPs identified in whites may not be significant in blacks; however, other SNPs in the region may show significant association with the phenotype. Thus, replication was considered significant in blacks if the genomic region, in which the index SNP is detected in whites, has a significant association of P<0.05 with the BP response to HCTZ. Additionally, to confirm the specificity of the replicated genetic signals to HCTZ, we also tested their association in whites treated with atenolol monotherapy in the PEAR study (n=214).
Validation (step 4)
SNPs that were identified in step 2 and replicated in step 3 are located in genes within the sphingolipid metabolic pathway and are also significantly associated with HCTZ BP response. Therefore, we hypothesized that the association between these significant SNPs and HCTZ BP response might be mediated by their effect on either sphingomyelins or ceramides, which are involved in the sphingolipid metabolic pathway (most significant pathway identified in step 1). Thus, to test our hypothesis and to confirm the association of the sphingolipid metabolic pathway to HCTZ BP response, we tested the effect of the replicated SNPs, in step 3, on sphingomyelins and ceramides, as discussed below.
First, the normality of each sphingolipid was tested using Shapiro–Wilk and Kolmogorov–Smirnov tests. Samples with levels >4 SDs from the mean were considered outliers and were removed from the analysis. Association between each sphingolipid (sphingomyelins or ceramides) and rs6078905 SNP was then performed. For normally distributed sphingolipids, the ANOVA test was used to test the association between each sphingolipid and rs6078905 SNP; Kruskal–Wallis was used for non‐normally distributed sphingolipids. Multiple linear regression was also used to test the association between each lipid and rs6078905 SNP, assuming an additive genetic model, with adjustment for age. Pearson's correlation was used to assess the correlation between significant sphingolipids, identified from this analysis, with HCTZ BP responses. A partial correlation analysis was also conducted to test the correlation between sphingolipids and HCTZ BP response, with adjustment for age and baseline BP.
Statistical Analyses
Characteristics of study participants were analyzed using descriptive statistics. Numerical variables are represented as mean±SD, and categorical variables are presented as percentages. All statistical analyses were carried out with SAS (version 9.3; SAS Institute Inc, Cary, NC) and SPSS software (version 17.0 for Windows; SPSS Inc, Chicago, IL).
Results
Baseline characteristics and HCTZ BP responses of PEAR and GERA participants included in the genomics analyses, and PEAR participants included in the metabolomics analyses, are described in Table 1. In Table S3, we described the baseline characteristics and HCTZ BP response of the subset of PEAR participants who were included in the lipidomics analyses.
Table 1.
Characteristics of White PEAR Participants Involved in the Genomics and Metabolomics Analyses
| Characteristics | PEAR HCTZ Monotherapy (Genomics, N=228 Whites) | PEAR HCTZ Monotherapy (Metabolomics, N=123 Whites) | PEAR HCTZ Monotherapy (Genomics, N=148 Blacks) | GERA HCTZ Monotherapy (Genomics, N=196 Whites) |
|---|---|---|---|---|
| Age, mean (SD), y | 50±9.5 | 50.7±8.9 | 47.4±8.8 | 48.5±7.3 |
| Women, N (%) | 91 (40) | 57 (46.7) | 92 (62.2) | 84 (43) |
| BMI, mean (SD) kg×m−2 | 30.30±4.90 | 33±4.90 | 31.53±5.41 | 31.30±5.57 |
| Pretreatment office SBP, mean (SD) mm Hg | 151.80±12.40 | 153.46±12.24 | 151.37±13.44 | 142.70±12.60 |
| Pretreatment office DBP, mean (SD) mm Hg | 98.10±5.80 | 98.12±6.30 | 99.23±6.16 | 95.60±5.70 |
| Office SBP response, mean (SD) mm Hg | −11.00±12.80 | −10.80±12.94 | −15.6±14.37 | −10.90±13.00 |
| Office DBP response, mean (SD) mm Hg | −5.01±7.17 | −4.39±6.97 | −9.27±8.67 | −6.26±8.83 |
| Composite SBP response, mean (SD) mm Hg | −8.50±7.02 | −9.3±6.90 | −12.61±7.81 | NA |
| Composite DBP response, mean (SD) mm Hg | −4.68±4.79 | −5.11±4.87 | −7.56±5.32 | NA |
Continuous variables are presented as mean±SD, and categorical variables are presented as n (%). A composite blood pressure represents a composite of office, home, and ambulatory blood pressure measurements. BMI indicates body mass index; DBP, diastolic blood pressure; GERA, Genetic Epidemiology of Responses to Antihypertensives; HCTZ, hydrochlorothiazide; PEAR, Pharmacogenomic Evaluation of Antihypertensive Responses; SBP, systolic blood pressure.
Metabolomics Pathway Analysis
The pathway analysis revealed the sphingolipid metabolism pathway as the most significant pathway (P=5.8E‐05; Table 2). We then tested the association between the 78 SNPs, within the 14 genes directly involved in the sphingolipid metabolism canonical pathway, and HCTZ BP response. From this analysis, we identified a significant signal, rs6078905 SNP, associated with the BP response to HCTZ (diastolic BP [DBP], P=4.7E‐04; q=3E‐02; and systolic BP [SBP], P=6.8E‐04; q=4E‐02; Figure 2; Table S4). This SNP is located within the serine palmitoyltransferase, long chain base subunit 3 (SPTLC3) gene, which is involved in the rate‐limiting step of sphingolipid synthesis. We found that patients carrying the CC genotype of rs6078905 SNP had better responses (SBP/DBP=−11.4/−6.8 mm Hg) compared with those carrying the CT (SBP/DBP=−9.0/−4.9 mm Hg) and TT genotypes (SBP/DBP=−6.7/−3.5 mm Hg; Figure 2). Additionally, in silico analysis using transformed fibroblast cells from the Genotype‐Tissue Expression (GTEx) project http://www.gtexportal.org/home/ revealed that CC genotype carriers (who had a better HCTZ BP response in PEAR) had higher SPTLC3 expression levels than CT and TT genotypes (P=6E‐11).
Table 2.
Significant Pathways (FDR <0.05) From the Metabolomics Pathway Analysis
| Pathway Name | Metabolitesa | Genesb | P Valuec | Q Valuec |
|---|---|---|---|---|
| Sphingolipid metabolism | 2 | 14 | 5.8E‐05 | 1.2E‐03 |
| Visual phototransduction | 2 | 26 | 1.4E‐04 | 1.2E‐03 |
| Phospholipases | 2 | 27 | 1.7E‐04 | 1.2E‐03 |
| Fatty acid alpha oxidation | 2 | 28 | 2.1E‐04 | 1.2E‐03 |
| Ceramide degradation | 2 | 28 | 3.3E‐04 | 1.5E‐03 |
| Triacylglycerol degradation | 2 | 29 | 9.2E‐04 | 3.5E‐03 |
| Sphingosine and sphingosine‐1‐phosphate metabolism | 2 | 10 | 1.6E‐03 | 5.5E‐03 |
FDR indicates false discovery rate.
Number of metabolites mapped to pathways, of the 13 significant metabolites associated with hydrochlorothiazide blood pressure response.
Number of genes identified within each pathway.
P values and q values were generated using the MPINet R‐based tool (http://cran.r-project.org/web/packages/MPINet/) based on Humancyc (https://humancyc.org/) as the pathways database source.
Figure 2.
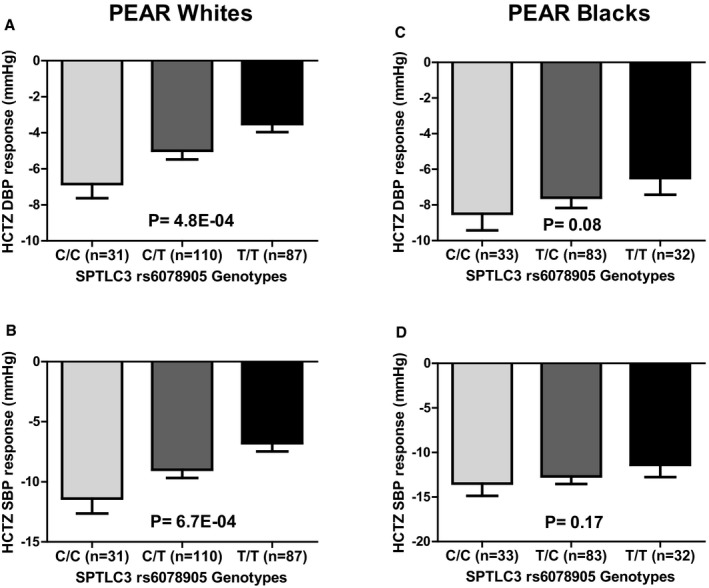
The effect of rs6078905 polymorphism on the blood pressure response of whites and blacks treated with hydrochlorothiazide in the PEAR (Pharmacogenomic Evaluation of Antihypertensive Responses) study. Blood pressure responses were adjusted for baseline blood pressure, age, sex, and population substructure, and P values represented are for contrast of adjusted means between different genotype groups. Error bars represent SEM. DBP indicates diastolic blood pressure; HCTZ, hydrochlorothiazide; SBP, systolic blood pressure; SPTLC3, serine palmitoyltransferase, long chain base subunit 3 (A) Diastolic blood pressure response in PEAR white participants, (B) Systolic blood pressure response in PEAR white participants, (C) Diastolic blood pressure response in PEAR black participants, (D) Systolic blood pressure response in PEAR black participants.
Replication
We tested the association between the rs6078905 SNP and HCTZ BP response in PEAR blacks and we found a trend toward significance with DBP response (P=0.08) and SBP response (P=0.17; Figure 2). We also tested the association between all the SNPs extracted from the 14 genes and HCTZ BP response in blacks and performed a meta‐analysis, using an inverse variance‐weighted fixed‐effect model as implemented in METAL,29 with the association results from whites. The results from this meta‐analysis showed the SNP rs6078905 as the most significant signal associated with both SBP (meta‐analysis, P=0.00061) and DBP (meta‐analysis, P=0.00014; Tables S5 and S6).
Given the differences in the recombination rates and the LD patterns between blacks and whites, we also checked the SPTLC3 region of PEAR blacks treated with HCTZ where we found rs6105039 SNP significantly associated with HCTZ BP response (DBP response, P=1E‐02; SBP response, P=3E‐02; Figure S2). According to the PEAR genomics data, the SNP rs6078905 has a minor allele frequency of 37% in whites and 50% in blacks. However, the minor allele frequency of the SNP rs6105039 was 15% in blacks, but it was absent in whites. Consistent with these frequencies, data from the 1000 Genome, phase 3, Project have also shown that the rs6105039 SNP has a minor allele frequency of 15% African ancestry in Southwest USA, but it was absent in the Northern Europeans from Utah population, which might be the reason that rs6105039 was not observed in our analysis in PEAR whites.
Testing the association between rs6078905 SNP and atenolol BP response in PEAR white participants revealed no significant association (SBP response, P=0.6; DBP response, P=0.8), which suggests that the SPTLC3 rs6078905 SNP and the sphingomyelin pathway might be specific for the BP response to thiazides. We performed a post hoc power calculation to make sure that we are not getting a false‐negative result because of lack of power. The post hoc power analysis revealed that we had 91% power to detect an effect size f2 of 0.052 (which we observed with HCTZ BP response) in the 214 white atenolol‐treated participants, using a linear multiple regression model, assuming an alpha‐level=0.05 and 2‐sided tails hypothesis.
Validation
Of note, using a multidimensional mass spectrometry–based shotgun lipidomics approach, we identified 9 lipid classes, as discussed in Data S1. However, we focused only on the sphingomyelins and ceramides measured (26 sphingomyelins and 23 ceramides), because they are the sphingolipids involved in the sphingolipid metabolic pathway. Testing the association between the SPTLC3 rs6078905 SNP and the baseline levels of each sphingolipid revealed a significant association, defined as false discovery rate <0.05, between SPTLC3 rs6078905 and the baseline levels of sphingomyelins N24:2, N24:3, N16:1, and N22:1 after adjustment for age (Table 3). Of note, sphingomyelins N24:2, N24:3, N16:1, and N22:1 are highly correlated, with a correlation coefficient (r) ranging from 0.73 to 0.88. We found that participants carrying the CC genotypes (who had a better response to HCTZ) have higher baseline sphingomyelins (N24:2, N24:3, N16:1, and N22:1) compared with CT and TT carriers (Figure 3).
Table 3.
Top Significant Sphingolipids Associated With the SPTLC3 rs6078905 SNP (With P Value Less Than 0.05)
| Lipids | P Valuea | Q Value |
|---|---|---|
| SM N24:2 | 0.001 | 0.0163b |
| SM N24:3 | 0.001 | 0.0163b |
| SM N16:1 | 0.001 | 0.0163b |
| SM N22:1 | 0.004 | 0.049b |
| SM N22:2 | 0.006 | 0.0588 |
| SM N23:1 | 0.012 | 0.084 |
| SM N24:1 | 0.011 | 0.084 |
| SM N20:1 | 0.016 | 0.098 |
| CER OH_N24:2 | 0.04 | 0.2178 |
CER indicates ceramide; SM, sphingomyelin.
P values were generated by testing the association between rs6078905 SNP and each 1 of the 49 measured sphingolipids, with adjustment for age.
q values represent sphingolipids with q<0.05.
Figure 3.
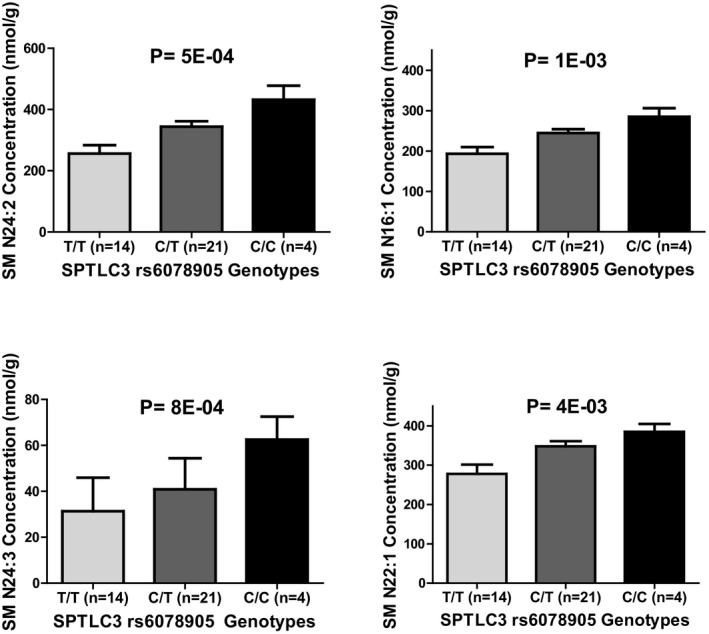
The effect of rs6078905 polymorphism on sphingomyelin concentrations of SM N24:2, SM N24:3, SM N16:1, and SM N22:1 in whites treated with hydrochlorothiazide in the PEAR (Pharmacogenomic Evaluation of Antihypertensive Responses) study. P values were generated using a linear regression model adjusted for age. SM indicates sphingomyelin; SPTLC3, serine palmitoyltransferase, long chain base subunit 3.
To further confirm whether sphingomyelins N24:2 and N24:3 are associated with HCTZ BP response; we tested their association with HCTZ BP response. This analysis revealed a significant association between sphingomyelin N24:2 baseline levels and HCTZ BP response (DBP, r=−0.42; P=0.007; SBP, r=−0.36; P=0.026; Figure 4), and a trend toward significance between sphingomyelin N16:1 and N24:3 baseline levels and HCTZ BP response (Table S7).
Figure 4.

The correlation between Sphingomyelin N24:2 and hydrochlorothiazide blood pressure response (A) Systolic blood pressure response, (B) Diastolic blood pressure response. P values and r values were generated using partial correlation with adjustment for age and baseline blood pressure. DBP indicates diastolic blood pressure; HCTZ, hydrochlorothiazide; SBP, systolic blood pressure; SM, sphingomyelin.
Of note, SPTLC3 rs6078905 SNP did not replicate in whites treated with HCTZ in GERA. Post hoc power analysis showed that using a multiple regression model, we had 89% power to identify a similar effect size, as observed between SNP rs6078905 and HCTZ BP response, in the 196 participants included from GERA, with the assumption of an alpha‐level=0.05 and 2‐sided tails hypothesis. Although we were not able to replicate the rs6078905 SNP in GERA, this lack of replication does not negate the importance of the sphingolipid metabolism pathway and SPTLC3 rs6078905 association with HCTZ BP response. We believe that the lack of replication observed in GERA might be attributed to different reasons, including the differences in the BP response phenotypes used in PEAR (composite BP) compared to GERA (office BP).
Discussion
Thiazide diuretics, including HCTZ, have been a cornerstone in treating hypertensive patients for more than 5 decades, and, currently, they are ranked among the most commonly prescribed first‐line antihypertensives globally. However, the mechanism underlying the long‐term BP‐lowering effect of thiazide diuretics is not well understood, and data reveal that <50% of patients treated with thiazide diuretics achieve optimal BP goals. In this study, we conducted a metabolomics pathway analysis that highlights the sphingolipid metabolism as a pathway that might be involved in the long‐term mechanism underlying HCTZ BP response. Additionally, using genomics data, we were able to identify rs6078905 SNP in the SPTLC3 gene as a potential determinant of the BP response to HCTZ in whites. We were also able to replicate the SPTLC3 genomic region and its association with the BP response to HCTZ in blacks. Moreover, leveraging our analyses with lipidomics data further confirmed the influence of the SPTLC3 rs6078905 SNP on the BP response to HCTZ, and sheds light on the association between sphingomyelins and the interindividual variability observed in the BP response to HCTZ.
The results from the lipidomics analysis revealed a significant association between SPTLC3 rs6078905 SNP and sphingomyelins levels. Consistent with these results, Demirkan et al30 have previously reported an association between rs680379 SNP, located 20 kb away from the 5‐prime of the SPTLC3 gene, and sphingolipids levels. We tested the LD between the rs6078905 SNP and the rs680379 SNP reported by Demirkan et al and they were not in LD (D′=0.13; R 2=0.01). Additionally, we did not find an association between the rs680379 SNP and HCTZ BP response (SBP, P=0.82; DBP, P=0.61). These results reveal the novelty of the genetic signal identified in this study and its association with sphingomyelins and HCTZ BP response.
Sphingomyelin and its metabolites have an influential effect on vascular tone31, 32 and have been reported to be involved in the mechanism underlying BP regulation.33, 34, 35 Animal and human data show that disruption in membrane lipids, including sphingomyelins, is closely linked with impaired ion transport and cytosolic calcium concentrations in various forms of HTN.36, 37, 38 Moreover, in the vasculature, biologically active sphingomyelin metabolites, such as sphingosine‐1‐phosphate (S1P), have been reported as acute vasoconstrictors in most vessels.39, 40, 41 S1P is a lipid mediator formed by the metabolism of sphingomyelins.42 In the kidney, the target organ of thiazides, intravenous and intrarenal arterial administration of S1P caused renal vasoconstriction.39, 41 Additionally, studies have shown that S1P, acting through S1P1 receptors, regulates sodium excretion by affecting transport mechanisms in the renal medulla, possibly by modulating the activity of the epithelial sodium channel.43
Studies have also shown that S1P can regulate the activity of various ion channels, including potassium channels,44, 45, 46 which have previously been proposed to be of importance in the mechanism underlying thiazide diuretics BP response.47, 48 S1P has also been shown to be involved in the mobilization of calcium from intracellular stores, influx of extracellular calcium through L‐type calcium channels,49, 50 and activation of Rho‐kinase.32, 51 Interestingly, Rho‐kinase was previously shown to be involved in the pathogenesis of hypertension52 and cardiovascular diseases.53, 54 Additionally, Zhu et al have shown that thiazide diuretics induced vasodilation by reducing the expression of Rho‐kinase significantly in the vascular smooth muscle.55 Therefore, we suggest that sphingomyelins and their active biological metabolites (ie, S1P) might be involved in the long‐term mechanism underlying thiazide diuretics BP response through the Rho‐kinase pathway—the latter is involved in the contraction of the vascular smooth muscle. Thus, further work on sphingomyelins, S1P, and their metabolic signaling pathways might identify novel biomarkers associated with thiazide diuretics and provide more insights into the mechanism underlying BP regulation.
Our study has several limitations. First, our sample size was relatively small, which limited our power to identify additional novel signals associated with HCTZ BP response. However, using genomics, metabolomics, and lipidomics data from participants treated with HCTZ added to the breadth and the depth of our analyses and helped us to identify and confirm the importance of the sphingomyelins metabolic pathway as a potential pathway associated with HCTZ BP response. Second, we found a significant association between SPTLC3 rs6078905 and HCTZ BP response in PEAR whites and found a trend toward significance in PEAR blacks; however, this finding did not replicate in whites treated with HCTZ in GERA. This lack of replication in GERA might have several explanations. It is, of course, possible that the finding in PEAR is a false positive, thus explaining the failure to replicate. However, other possibilities also exist. First, the discrepancy in measuring BP in PEAR compared with GERA might be 1 of the reasons that contributed to the failure of replicating the rs6078905 signal in GERA, especially with the small sample size used. In PEAR, the BP response was based on a composite of office, home, and ambulatory BP measurements, which we have shown previously to be a more‐accurate measurement of BP response with a better signal‐to‐noise ratio,22 whereas in GERA the BP response in GERA participants was based on office measurements, which might have more signal‐to‐noise ratio compared with the composite BP used in PEAR. Another explanation is that the rs6078905 might not be the causal signal; presumably, it might be in LD with a rare causal signal that might be driving its effect on HCTZ BP response. Therefore, more work is still needed to test whether this SNP or another SNP in LD with this SNP can be used as a predictor for HCTZ BP response. Another limitation of this study is that it was conducted only in females. Several factors led us to do this. First was that resources were limited for running a larger sample set, and given the fact sex hormones have been shown to exhibit sex‐associated differences in sphingolipid levels, we considered it most scientifically robust to perform lipidomics analysis in a single sex group. We chose to focus on females because we have previously shown that the adverse metabolic effects of thiazides are more common in females than males.56 Future studies are still needed to test the effect of sphingolipids on HCTZ BP response in males. Last, the results presented here were identified in mild‐to‐moderate hypertensive participants recruited in the PEAR study. Testing the findings from this study in severe hypertensive patients is still needed, which might provide novel insights in the mechanism underlying BP regulation.
Our study also has several strengths. Using metabolomics, genomics, and lipidomics data to identify pathways and markers associated with drug response is an innovative and powerful approach. We believe that using multiple “omics” approaches, similar to the one presented here, can help in uncovering novel pathways and biomarkers that were not identified using genome‐wide association studies data alone. These pathways and biomarkers hold the promise to provide more insight in drug‐response mechanisms and facilitate the development of new drugs based on a deeper understanding of determinants of drug‐response phenotypes.
In summary, to our knowledge, this is the first study to highlight the association between the sphingomyelin metabolism pathway and HCTZ BP response. We showed that this association might be mediated by the SPTLC3 gene that influences the production of sphingomyelins, in which we identified and replicated a significant association between the SPTLC3 genomic region and HCTZ BP response. In conclusion, this study illustrates the importance of the sphingolipid metabolic pathway in HCTZ BP response. Additional research on this pathway may open new avenues for new drug development and provide us with more insights into the mechanism underlying the long‐term BP‐lowering effects of thiazide diuretics.
Sources of Funding
PEAR was supported by the National Institute of Health Pharmacogenetics Research Network grant U01‐GM074492 and the National Center for Advancing Translational Sciences under the award number UL1 TR000064 (University of Florida); UL1 TR000454 (Emory University); and UL1 TR000135 (Mayo Clinic). PEAR was also supported by funds from the Mayo Foundation. The metabolomics work was funded by NIGMS (National Institute of General Medical Sciences) grant RC2‐GM092729 “Metabolomics Network for Drug Response Phenotype.” Additional support for this work includes: Shahin is supported by AHA predoctoral fellowship award #14PRE20460115, and Fiehn is funded through NIH DK097154.
Disclosures
None.
Supporting information
Data S1. Supplemental methods.
Table S1. Thirteen Metabolites Significantly Associated With Hydrochlorothiazide Blood Pressure Response of Whites in the PEAR HCTZ Monotherapy Study
Table S2. Genes in the Sphingolipid Metabolism Canonical Pathway
Table S3. Characteristics of White PEAR Participants Included in the Lipidomics Analyses
Table S4. The Effect of the 78 Polymorphisms Selected From the 14 Genes Involved in the Sphingolipid Metabolism Canonical Pathway on Hydrochlorothiazide Blood Pressure Responses
Table S5. Top Signals Associated With Diastolic Blood Pressure Response to Hydrochlorothiazide (With a P Value <0.05) From the Meta‐Analysis Between White and Black Participants in the PEAR Study
Table S6. Top Signals Associated With Systolic Blood Pressure Response to Hydrochlorothiazide (With a P Value <0.05) From the Meta‐Analysis Between White and Black Participants in the PEAR Study
Table S7. Correlation Between Sphingomyelins N24:2, N24:3, N16:1, and N22:1 With Hydrochlorothiazide Blood Pressure Response
Figure S1. The 14 genes involved in the sphingolipid metabolism canonical pathway.
Figure S2. The effect of rs6105039 polymorphism on the blood pressure response of blacks treated with hydrochlorothiazide in the PEAR study.
Acknowledgments
We acknowledge and thank the participants included in the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) and Genetic Epidemiology of Responses to Antihypertensives (GERA) study. We also thank PEAR and GERA support staff and study physicians.
(J Am Heart Assoc. 2018;7:e006656 DOI: 10.1161/JAHA.117.006656.)
References
- 1. Global, regional, and national age‐sex specific all‐cause and cause‐specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–e322. [DOI] [PubMed] [Google Scholar]
- 3. Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–223. [DOI] [PubMed] [Google Scholar]
- 4. Law MR, Morris JK, Wald NJ. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta‐analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ. 2009;338:b1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thoenes M, Neuberger HR, Volpe M, Khan BV, Kirch W, Bohm M. Antihypertensive drug therapy and blood pressure control in men and women: an international perspective. J Hum Hypertens. 2010;24:336–344. [DOI] [PubMed] [Google Scholar]
- 6. Weber MA, Schiffrin EL, White WB, Mann S, Lindholm LH, Kenerson JG, Flack JM, Carter BL, Materson BJ, Ram CV, Cohen DL, Cadet JC, Jean‐Charles RR, Taler S, Kountz D, Townsend R, Chalmers J, Ramirez AJ, Bakris GL, Wang J, Schutte AE, Bisognano JD, Touyz RM, Sica D, Harrap SB. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Hypertens. 2014;32:3–15. [DOI] [PubMed] [Google Scholar]
- 7. Shahin MH, Johnson JA. Mechanisms and pharmacogenetic signals underlying thiazide diuretics blood pressure response. Curr Opin Pharmacol. 2016;27:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Materson BJ. Variability in response to antihypertensive drugs. Am J Med. 2007;120:S10–S20. [DOI] [PubMed] [Google Scholar]
- 9. Materson BJ, Reda DJ, Cushman WC, Massie BM, Freis ED, Kochar MS, Hamburger RJ, Fye C, Lakshman R, Gottdiener J. Single‐drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med. 1993;328:914–921. [DOI] [PubMed] [Google Scholar]
- 10. Turner ST, Boerwinkle E, O'Connell JR, Bailey KR, Gong Y, Chapman AB, McDonough CW, Beitelshees AL, Schwartz GL, Gums JG, Padmanabhan S, Hiltunen TP, Citterio L, Donner KM, Hedner T, Lanzani C, Melander O, Saarela J, Ripatti S, Wahlstrand B, Manunta P, Kontula K, Dominiczak AF, Cooper‐DeHoff RM, Johnson JA. Genomic association analysis of common variants influencing antihypertensive response to hydrochlorothiazide. Hypertension. 2013;62:391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shahin MH, Sa AC, Webb A, Gong Y, Langaee T, McDonough CW, Riva A, Beitleshees AL, Chapman AB, Gums JG, Turner ST, Boerwinkle E, Scherer SE, Sadee W, Cooper‐DeHoff RM, Johnson JA. Genome‐wide prioritization and transcriptomics reveal novel signatures associated with thiazide diuretics blood pressure response. Circ Cardiovasc Genet. 2017;10:e001404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cooper‐Dehoff RM, Hou W, Weng L, Baillie RA, Beitelshees AL, Gong Y, Shahin MH, Turner ST, Chapman A, Gums JG, Boyle SH, Zhu H, Wikoff WR, Boerwinkle E, Fiehn O, Frye RF, Kaddurah‐Daouk R, Johnson JA. Is diabetes mellitus‐linked amino acid signature associated with beta‐blocker‐induced impaired fasting glucose? Circ Cardiovasc Genet. 2014;7:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Oliveira FA, Shahin MH, Gong Y, McDonough CW, Beitelshees AL, Gums JG, Chapman AB, Boerwinkle E, Turner ST, Frye RF, Fiehn O, Kaddurah‐Daouk R, Johnson JA, Cooper‐DeHoff RM. Novel plasma biomarker of atenolol‐induced hyperglycemia identified through a metabolomics‐genomics integrative approach. Metabolomics (Los Angel). 2016;12:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaddurah‐Daouk R, Weinshilboum R. Metabolomic signatures for drug response phenotypes: pharmacometabolomics enables precision medicine. Clin Pharmacol Ther. 2015;98:71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shahin MH, Gong Y, McDonough CW, Rotroff DM, Beitelshees AL, Garrett TJ, Gums JG, Motsinger‐Reif A, Chapman AB, Turner ST, Boerwinkle E, Frye RF, Fiehn O, Cooper‐DeHoff RM, Kaddurah‐Daouk R, Johnson JA. A genetic response score for hydrochlorothiazide use: insights from genomics and metabolomics integration. Hypertension. 2016;68:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaddurah‐Daouk R, Baillie RA, Zhu H, Zeng ZB, Wiest MM, Nguyen UT, Watkins SM, Krauss RM. Lipidomic analysis of variation in response to simvastatin in the cholesterol and pharmacogenetics study. Metabolomics. 2010;6:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu C, Kong H, Qu F, Li Y, Yu Z, Gao P, Peng S, Xu G. Application of plasma lipidomics in studying the response of patients with essential hypertension to antihypertensive drug therapy. Mol Biosyst. 2011;7:3271–3279. [DOI] [PubMed] [Google Scholar]
- 18. Ji Y, Hebbring S, Zhu H, Jenkins GD, Biernacka J, Snyder K, Drews M, Fiehn O, Zeng Z, Schaid D, Mrazek DA, Kaddurah‐Daouk R, Weinshilboum RM. Glycine and a glycine dehydrogenase (GLDC) SNP as citalopram/escitalopram response biomarkers in depression: pharmacometabolomics‐informed pharmacogenomics. Clin Pharmacol Ther. 2011;89:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song IS, Lee do Y, Shin MH, Kim H, Ahn YG, Park I, Kim KH, Kind T, Shin JG, Fiehn O, Liu KH. Pharmacogenetics meets metabolomics: discovery of tryptophan as a new endogenous OCT2 substrate related to metformin disposition. PLoS One. 2012;7:e36637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson JA, Boerwinkle E, Zineh I, Chapman AB, Bailey K, Cooper‐DeHoff RM, Gums J, Curry RW, Gong Y, Beitelshees AL, Schwartz G, Turner ST. Pharmacogenomics of antihypertensive drugs: rationale and design of the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) study. Am Heart J. 2009;157:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chapman AB, Schwartz GL, Boerwinkle E, Turner ST. Predictors of antihypertensive response to a standard dose of hydrochlorothiazide for essential hypertension. Kidney Int. 2002;61:1047–1055. [DOI] [PubMed] [Google Scholar]
- 22. Turner ST, Schwartz GL, Chapman AB, Beitelshees AL, Gums JG, Cooper‐Dehoff RM, Boerwinkle E, Johnson JA, Bailey KR. Power to identify a genetic predictor of antihypertensive drug response using different methods to measure blood pressure response. J Transl Med. 2012;10:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Merrill AH Jr, Wang E, Innis WS, Mullins R. Increases in serum sphingomyelin by 17 beta‐estradiol. Lipids. 1985;20:252–254. [DOI] [PubMed] [Google Scholar]
- 24. Nikkila J, Sysi‐Aho M, Ermolov A, Seppanen‐Laakso T, Simell O, Kaski S, Oresic M. Gender‐dependent progression of systemic metabolic states in early childhood. Mol Syst Biol. 2008;4:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li F, Xu Y, Shang D, Yang H, Liu W, Han J, Sun Z, Yao Q, Zhang C, Ma J, Su F, Feng L, Shi X, Zhang Y, Li J, Gu Q, Li X, Li C. MPINet: metabolite pathway identification via coupling of global metabolite network structure and metabolomic profile. Biomed Res Int. 2014;2014:325697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sobota RS, Shriner D, Kodaman N, Goodloe R, Zheng W, Gao YT, Edwards TL, Amos CI, Williams SM. Addressing population‐specific multiple testing burdens in genetic association studies. Ann Hum Genet. 2015;79:136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burkhardt R, Kirsten H, Beutner F, Holdt LM, Gross A, Teren A, Tonjes A, Becker S, Krohn K, Kovacs P, Stumvoll M, Teupser D, Thiery J, Ceglarek U, Scholz M. Integration of genome‐wide SNP data and gene‐expression profiles reveals six novel loci and regulatory mechanisms for amino acids and acylcarnitines in whole blood. PLoS Genet. 2015;11:e1005510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Demirkan A, van Duijn CM, Ugocsai P, Isaacs A, Pramstaller PP, Liebisch G, Wilson JF, Johansson A, Rudan I, Aulchenko YS, Kirichenko AV, Janssens AC, Jansen RC, Gnewuch C, Domingues FS, Pattaro C, Wild SH, Jonasson I, Polasek O, Zorkoltseva IV, Hofman A, Karssen LC, Struchalin M, Floyd J, Igl W, Biloglav Z, Broer L, Pfeufer A, Pichler I, Campbell S, Zaboli G, Kolcic I, Rivadeneira F, Huffman J, Hastie ND, Uitterlinden A, Franke L, Franklin CS, Vitart V, Nelson CP, Preuss M, Bis JC, O'Donnell CJ, Franceschini N, Witteman JC, Axenovich T, Oostra BA, Meitinger T, Hicks AA, Hayward C, Wright AF, Gyllensten U, Campbell H, Schmitz G. Genome‐wide association study identifies novel loci associated with circulating phospho‐ and sphingolipid concentrations. PLoS Genet. 2012;8:e1002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li H, Junk P, Huwiler A, Burkhardt C, Wallerath T, Pfeilschifter J, Forstermann U. Dual effect of ceramide on human endothelial cells: induction of oxidative stress and transcriptional upregulation of endothelial nitric oxide synthase. Circulation. 2002;106:2250–2256. [DOI] [PubMed] [Google Scholar]
- 32. Ohmori T, Yatomi Y, Osada M, Kazama F, Takafuta T, Ikeda H, Ozaki Y. Sphingosine 1‐phosphate induces contraction of coronary artery smooth muscle cells via S1P2. Cardiovasc Res. 2003;58:170–177. [DOI] [PubMed] [Google Scholar]
- 33. Fenger M, Linneberg A, Jorgensen T, Madsbad S, Sobye K, Eugen‐Olsen J, Jeppesen J. Genetics of the ceramide/sphingosine‐1‐phosphate rheostat in blood pressure regulation and hypertension. BMC Genet. 2011;12:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spijkers LJ, van den Akker RF, Janssen BJ, Debets JJ, De Mey JG, Stroes ES, van den Born BJ, Wijesinghe DS, Chalfant CE, MacAleese L, Eijkel GB, Heeren RM, Alewijnse AE, Peters SL. Hypertension is associated with marked alterations in sphingolipid biology: a potential role for ceramide. PLoS One. 2011;6:e21817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fenger M, Linneberg A, Jeppesen J. Network‐based analysis of the sphingolipid metabolism in hypertension. Front Genet. 2015;6:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rutherford PA, Thomas TH, Laker MF, Wilkinson R. Plasma lipids affect maximum velocity not sodium affinity of human sodium‐lithium countertransport: distinction from essential hypertension. Eur J Clin Invest. 1992;22:719–724. [DOI] [PubMed] [Google Scholar]
- 37. Chi Y, Mota de Freitas D, Sikora M, Bansal VK. Correlations of Na+‐Li+ exchange activity with Na+ and Li+ binding and phospholipid composition in erythrocyte membranes of white hypertensive and normotensive individuals: a nuclear magnetic resonance investigation. Hypertension. 1996;27:456–464. [DOI] [PubMed] [Google Scholar]
- 38. Zicha J, Kunes J, Devynck MA. Abnormalities of membrane function and lipid metabolism in hypertension: a review. Am J Hypertens. 1999;12:315–331. [DOI] [PubMed] [Google Scholar]
- 39. Bischoff A, Czyborra P, Meyer Zu Heringdorf D, Jakobs KH, Michel MC. Sphingosine‐1‐phosphate reduces rat renal and mesenteric blood flow in vivo in a pertussis toxin‐sensitive manner. Br J Pharmacol. 2000;130:1878–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coussin F, Scott RH, Nixon GF. Sphingosine 1‐phosphate induces CREB activation in rat cerebral artery via a protein kinase C‐mediated inhibition of voltage‐gated K+ channels. Biochem Pharmacol. 2003;66:1861–1870. [DOI] [PubMed] [Google Scholar]
- 41. Guan Z, Singletary ST, Cook AK, Hobbs JL, Pollock JS, Inscho EW. Sphingosine‐1‐phosphate evokes unique segment‐specific vasoconstriction of the renal microvasculature. J Am Soc Nephrol. 2014;25:1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spiegel S, Milstien S. Sphingosine‐1‐phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. [DOI] [PubMed] [Google Scholar]
- 43. Zhu Q, Xia M, Wang Z, Li PL, Li N. A novel lipid natriuretic factor in the renal medulla: sphingosine‐1‐phosphate. Am J Physiol Renal Physiol. 2011;301:F35–F41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Himmel HM, Meyer Zu Heringdorf D, Graf E, Dobrev D, Kortner A, Schuler S, Jakobs KH, Ravens U. Evidence for Edg‐3 receptor‐mediated activation of I(K.ACh) by sphingosine‐1‐phosphate in human atrial cardiomyocytes. Mol Pharmacol. 2000;58:449–454. [DOI] [PubMed] [Google Scholar]
- 45. Xu SZ, Muraki K, Zeng F, Li J, Sukumar P, Shah S, Dedman AM, Flemming PK, McHugh D, Naylor J, Cheong A, Bateson AN, Munsch CM, Porter KE, Beech DJ. A sphingosine‐1‐phosphate‐activated calcium channel controlling vascular smooth muscle cell motility. Circ Res. 2006;98:1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim MY, Liang GH, Kim JA, Kim YJ, Oh S, Suh SH. Sphingosine‐1‐phosphate activates BKCa channels independently of G protein‐coupled receptor in human endothelial cells. Am J Physiol Cell Physiol. 2006;290:C1000–C1008. [DOI] [PubMed] [Google Scholar]
- 47. Calder JA, Schachter M, Sever PS. Potassium channel opening properties of thiazide diuretics in isolated guinea pig resistance arteries. J Cardiovasc Pharmacol. 1994;24:158–164. [DOI] [PubMed] [Google Scholar]
- 48. Pickkers P, Hughes AD, Russel FG, Thien T, Smits P. Thiazide‐induced vasodilation in humans is mediated by potassium channel activation. Hypertension. 1998;32:1071–1076. [DOI] [PubMed] [Google Scholar]
- 49. Birchwood CJ, Saba JD, Dickson RC, Cunningham KW. Calcium influx and signaling in yeast stimulated by intracellular sphingosine 1‐phosphate accumulation. J Biol Chem. 2001;276:11712–11718. [DOI] [PubMed] [Google Scholar]
- 50. Mattie M, Brooker G, Spiegel S. Sphingosine‐1‐phosphate, a putative second messenger, mobilizes calcium from internal stores via an inositol trisphosphate‐independent pathway. J Biol Chem. 1994;269:3181–3188. [PubMed] [Google Scholar]
- 51. Ishizawa S, Takahashi‐Fujigasaki J, Kanazawa Y, Matoba K, Kawanami D, Yokota T, Iwamoto T, Tajima N, Manome Y, Utsunomiya K. Sphingosine‐1‐phosphate induces differentiation of cultured renal tubular epithelial cells under Rho kinase activation via the S1P2 receptor. Clin Exp Nephrol. 2014;18:844–852. [DOI] [PubMed] [Google Scholar]
- 52. Wirth A. Rho kinase and hypertension. Biochim Biophys Acta. 2010;1802:1276–1284. [DOI] [PubMed] [Google Scholar]
- 53. Do e Z, Fukumoto Y, Sugimura K, Miura Y, Tatebe S, Yamamoto S, Aoki T, Nochioka K, Nergui S, Yaoita N, Satoh K, Kondo M, Nakano M, Wakayama Y, Fukuda K, Nihei T, Kikuchi Y, Takahashi J, Shimokawa H. Rho‐kinase activation in patients with heart failure. Circ J. 2013;77:2542–2550. [DOI] [PubMed] [Google Scholar]
- 54. Satoh K, Fukumoto Y, Shimokawa H. Rho‐kinase: important new therapeutic target in cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2011;301:H287–H296. [DOI] [PubMed] [Google Scholar]
- 55. Zhu Z, Zhu S, Liu D, Cao T, Wang L, Tepel M. Thiazide‐like diuretics attenuate agonist‐induced vasoconstriction by calcium desensitization linked to Rho kinase. Hypertension. 2005;45:233–239. [DOI] [PubMed] [Google Scholar]
- 56. Cooper‐DeHoff RM, Wen S, Beitelshees AL, Zineh I, Gums JG, Turner ST, Gong Y, Hall K, Parekh V, Chapman AB, Boerwinkle E, Johnson JA. Impact of abdominal obesity on incidence of adverse metabolic effects associated with antihypertensive medications. Hypertension. 2010;55:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Table S1. Thirteen Metabolites Significantly Associated With Hydrochlorothiazide Blood Pressure Response of Whites in the PEAR HCTZ Monotherapy Study
Table S2. Genes in the Sphingolipid Metabolism Canonical Pathway
Table S3. Characteristics of White PEAR Participants Included in the Lipidomics Analyses
Table S4. The Effect of the 78 Polymorphisms Selected From the 14 Genes Involved in the Sphingolipid Metabolism Canonical Pathway on Hydrochlorothiazide Blood Pressure Responses
Table S5. Top Signals Associated With Diastolic Blood Pressure Response to Hydrochlorothiazide (With a P Value <0.05) From the Meta‐Analysis Between White and Black Participants in the PEAR Study
Table S6. Top Signals Associated With Systolic Blood Pressure Response to Hydrochlorothiazide (With a P Value <0.05) From the Meta‐Analysis Between White and Black Participants in the PEAR Study
Table S7. Correlation Between Sphingomyelins N24:2, N24:3, N16:1, and N22:1 With Hydrochlorothiazide Blood Pressure Response
Figure S1. The 14 genes involved in the sphingolipid metabolism canonical pathway.
Figure S2. The effect of rs6105039 polymorphism on the blood pressure response of blacks treated with hydrochlorothiazide in the PEAR study.