

Abstract
Background
Epidemiological evidence supports an important association between air pollution exposure and hypertension. However, the mechanisms are not clear.
Methods and Results
Our present study found that long‐term exposure to fine particulate matter (PM2.5) causes hypertension and impairs renal sodium excretion, which might be ascribed to lower D1 receptor expression and higher D1 receptor phosphorylation, accompanied with a higher G‐protein–coupled receptor kinase type 4 (GRK4) expression. The in vivo results were confirmed in in vitro studies (ie, PM 2.5 increased basal and decreased D1 receptor mediated inhibitory effect on Na+‐K+ ATPase activity, decreased D1 receptor expression, and increased D1 receptor phosphorylation in renal proximal tubule cells). The downregulation of D1 receptor expression and function might be attributable to a higher GRK4 expression after the exposure of renal proximal tubule cells to PM 2.5, because downregulation of GRK4 by small‐interfering RNA reversed the D1 receptor expression and function. Because of the role of reactive oxygen species on D1 receptor dysfunction and its relationship with air pollution exposure, we determined plasma reactive oxygen species and found the levels higher in PM 2.5‐treated Sprague‐Dawley rats. Inhibition of reactive oxygen species by tempol (4‐hydroxy‐2,2,6,6‐tetramethylpiperidin‐1‐oxyl) reduced blood pressure and increased sodium excretion in PM 2.5‐treated Sprague‐Dawley rats, accompanied by an increase in the low D1 receptor expression, and decreased the hyperphosphorylated D1 receptor and GRK4 expression.
Conclusions
Our present study indicated that long‐term exposure of PM 2.5 increases blood pressure by decreasing D1 receptor expression and function; reactive oxygen species, via regulation of GRK4 expression, plays an important role in the pathogenesis of PM 2.5‐induced hypertension.
Keywords: dopamine receptor, GRK4, hypertension, kidney, PM2.5, sodium excretion
Subject Categories: Hypertension, High Blood Pressure, Risk Factors
Clinical Perspective
What Is New?
Fine particulate matter exposure causes hypertension because of the dysfunction of the renal dopamine D1 receptor.
Increased oxidative stress, associated with increased G‐protein–coupled receptor kinase type 4 levels, impairs renal D1 receptor function and leads to hypertension.
What Are the Clinical Implications?
Particulate matter air pollution might play an important role in the pathogenesis of hypertension.
Restoration of dopamine D1 receptor function by antioxidants may be a therapeutic target for fine particulate matter–caused hypertension.
Epidemiological evidence supports an important association between air pollution exposure and cardiovascular risks.1 The World Health Organization reported that >800 000 premature deaths worldwide per year can be directly attributed to particulate matter air pollution.2 The ESCAPE (European Study of Cohorts for Air Pollution Effects) showed that a 5 μg/m3 increase in estimated annual mean fine particulate matter (PM2.5) was associated with a 13% increased risk of coronary events.3 Recently, the relationship between particulate matter and hypertension is getting more and more attention.4 Dai‐Hua Tsai et al found that inhalable particulate matter short time exposure, a 10 μg/m3 increase in inhalable particulate matter level, is associated with higher nighttime blood pressure and is preceded by associations with the reduced ability of the kidney to excrete sodium during the daytime.5 Numerous investigations have shown that exposure to PM2.5 elucidated potential biological consequences, such as modulation of blood pressure, including systemic proinflammatory oxidative responses, DNA damage, and autonomic nervous system imbalance.5, 6, 7, 8, 9, 10, 11, 12 However, the underlying pathophysiological mechanisms of airborne PM2.5‐mediated hypertension are complex and remain to a large extent unexplored. Studies have shown that hypertension is caused by prohypertensive factors, which include the increased activity of the sympathetic nervous system and the decreased activity of antihypertensive mechanisms, such as sodium excretion.13 The kidney is endowed with local hormonal systems that play major roles in the regulation of sodium transport across the renal proximal tubule (RPT).14 Among those systems, dopamine is taken as an important one, which is synthesized in and secreted from RPT cells and decreases RPT sodium transport,13, 14, 15, 16, 17 via the D1 and D3 receptors, by inhibition of NHE3 at the brush border membrane and Na+‐K+‐ATPase at the basolateral membrane.13 In hypertensive states, the renal D1 receptor is hyperphosphorylated, which leads to its dysfunction, and is ascribed to the higher G‐protein–coupled receptor kinase type 4 (GRK4) expression13, 18 and activity in spontaneously hypertensive rats or hypertensive patients.19, 20, 21 This can be linked to increased oxidative stress.22, 23, 24, 25 Many studies have shown that the impaired intrarenal D1 receptor signaling in hypertension can cause or be caused by oxidative stress.24, 25, 26, 27 Antioxidant supplementation with tempol (4‐hydroxy‐2,2,6,6‐tetramethylpiperidin‐1‐oxyl), a superoxide dismutase (SOD) mimetic, decreases oxidative stress, restores D1 receptor signaling, and lowers blood pressure.24, 25
There are reports that PM2.5 induces hypertension through impaired sodium excretion and increased reactive oxygen species (ROS) and oxidative stress28, 29, 30, 31; ROS has been reported to decrease dopamine receptors in animals.24, 25, 26, 27 Therefore, we hypothesize that PM2.5, via increased ROS levels, increases GRK4 expression and consequently impairs renal D1 receptor function, and leads to hypertension. To test this hypothesis, we exposed Sprague‐Dawley (SD) rats to PM2.5 in vivo. Concurrently, we also exposed immortalized RPT cells in vitro, because they behave similarly to freshly obtained RPT cells, at least with regard to dopamine receptors and its responses to G‐protein stimulation, in this present study.
Methods
The data, analytic methods, and study materials will be made available on request to other researchers for purposes of reproducing the results or replicating the procedure.
PM2.5 Sampling
The sample site was the Daping Hospital, which is ≈1 km from the city center. The nearest main road is in the northeast at a distance of 100 m. Within an ≈200 m radius of the monitoring site is almost completely surrounded by residential areas.
We used a medium volume sampler (model TH‐150; Tianhong Co, Wuhan, China) equipped with a PM2.5 filtration system to collect PM2.5 samples on filters (diameter, 150 mm), according to the reported method.32, 33, 34 The flow rate of medium volume sampler was adjusted to 30 m3/h. After sampling, filters were shredded into small pieces and sonicated using an ultrasonator (KQ‐250DE; Shumei, Kunshan, Jiangsu, China) for 3× 40 minutes in double‐distilled water. The extracted material was frozen, concentrated using lyophilization, and weighed to determine the extraction efficiency. The farinose solid was stored at −80°C until further use. The sampling period started on April 8, 2013, and ended on July 28, 2013. A total of 25 filters were used to collect the PM2.5 samples. To make sure the PM2.5 sample diameter was correct, we performed a scanning electron microscope analysis of the samples to prove the purity of the samples. The analysis confirmed that the particulate matter collected had a diameter of = or <2.5 μm (Figure S1).
Animal Treatment
Male SD rats, 8 to 10 weeks old and initially weighing 198±6 g, were given a standard laboratory chow and ad libitum access to clean drinking water. They were randomly divided into 3 groups (n=56 for control, n=32 for lower dosage PM2.5 treatment, and n=56 for higher dosage PM2.5 treatment). The details of each group are shown in Figure S2. The rats were individually housed in metabolic cages for urine collection and maintained at a temperature of 25±2°C, a relative humidity of 50% to 60%, and a 12‐hour dark‐light cycle. An acclimatization period of 2 days was allowed for the rats before the start of the experiments. The rats were weighed at the beginning of the experiment and just before urine collection and euthanasia. This study was approved by the Third Military Medical University Animal Use and Care Committee. All experiments conformed to the guidelines of the ethical use of animals, and all efforts were made to minimize animal suffering.
PM2.5 was suspended in PBS. To minimize aggregation, particle suspensions were always sonicated for 15 minutes, 3 times, and vortexed before their dilution and before intratracheal administration. Control animals received PBS treatments. The animals were anesthetized with isoflurane and placed supine with extended necks on an angled board.33, 35, 36 An oropharynx instillation of the PM2.5 suspension (3 or 30 μg PM2.5 in 25 μL) or PBS only was instilled (25 μL) via a sterile syringe. SD rats were treated with the PM2.5 solution every 3 days, for 8 weeks. In the present study, 2 dosages of PM2.5 treatment of either lower or higher dose were used. The lower dosage given (3 μg PM2.5) represented 35 μg/m3 per 24 hours PM2.5 in the air and was marked as PM2.5‐L. The higher dosage given (30 μg PM2.5) represented 350 μg/m3 per 24 hours PM2.5 in the air and marked as PM2.5‐H. To confirm the success of the exposure model, we performed a hematoxylin‐eosin stain of the lung after the rats were euthanized (Figure S3). Light microscope detection found the PM2.5 in the lung tissue.
The blood pressure and 24‐hour urine volume and sodium excretion were measured in conscious rats on the indicated exposure time points. After 8 weeks of being exposed to PM2.5‐L, PM2.5‐H, and PBS, treated rats were randomly assigned into 2 groups: one group that drank tap water served as a control, and the other group drank tap water with 1.0 mmol/L tempol that was changed 2 times per day, for 3 weeks (n=12 in each group).
Blood Pressure and Urine Collection
Systolic blood pressure, diastolic blood pressure, and mean arterial blood pressure were measured every week using a computerized, noninvasive, tail‐cuff plethysmography system in conscious rats (BP‐98A; Softron, Tokyo, Japan). The animals were individually restrained in a clear acrylic restrainer at an ambient temperature of 37°C to 38°C for 15 minutes. Five blood pressure values were recorded per rat, and the average was taken. To ensure the reliability of the measurements, a training period of 1 week was established before the start of the experiment for the rats to adapt to the method. Blood pressure measurements were performed between 3 and 5 pm.
SD rats were acclimated in metabolic cages for at least 2 days before urine was collected for 24 hours every week. After urine collection, sodium and potassium concentrations in urine samples were measured using a flame photometer 480 (Ciba Corning Diagnostics, Norwood, MA).
Cell Culture and Sample Preparation
Immortalized RPT cells from microdissected Sl segments of the RPTs from 4‐ to 8‐week‐old Wistar‐Kyoto rats were cultured at 37°C in 95% air/5% CO2 atmosphere in DMEM/F‐12 with transferrin (5 μg/mL), insulin (5 μg/mL), epidermal growth factor (10 ng/mL), dexamethasone (4 μg/mL), and 5% fetal bovine serum on a 100‐mm Petri dish. The final concentration of PM2.5 (1–200 µg/mL) was mixed evenly into the culture medium, 24 hours prior for the PM2.5 exposure group, and an equal volume of PBS was mixed into the culture medium for the control group. Cells were made quiescent by incubating for 2 hours in the media without fetal bovine serum before adding the drugs. The cells (80% confluence) were extracted in ice‐cold lysis buffer, sonicated, kept on ice for 1 hour, and centrifuged at 16 000g for 30 minutes. All samples were stored at −80°C until use.
Immunoblotting
In the in vivo study, immediately after euthanasia (or after thawing), the renal cortex (upper pole, left kidney) was homogenized in buffer (10 mmol/L Tris‐HCl, 250 mmol/L sucrose, 2 mmol/L phenylmethylsulfonyl fluoride, and protease inhibitor cocktail; pH 7.4) and centrifuged at 24 000g for 25 minutes at 4°C. The upper fluffy layer of the pellet, which is considered as the membrane fraction, was resuspended in the homogenization buffer.37, 38 The samples were quickly frozen and stored at −80°C until use. For the in vitro study, the RPT cells were lysed in a lysis buffer, sonicated, placed on ice for 1 hour, and centrifuged at 16 000g for 30 minutes. The supernatant was stored at −80°C until use.
After boiling the homogenates in sample buffer (35 mmol/L Tris‐HCl, pH 6.8, 4% SDS, 9.3% dithiothreitol, 0.01% bromophenol blue, and 30% glycerol) at 95°C for 5 minutes, 50 μg of protein was separated by SDS‐PAGE (10% polyacrylamide) and then electroblotted onto nitrocellulose membranes (Amersham Life Science, Arlington, TX). The blots representing the nonspecific antibodies were blocked overnight with 5% nonfat dry milk in PBS with Tween 20 (0.05% Tween 20 in 10 mmol/L PBS [isotonic]) at 4°C under constant shaking, then incubated with polyclonal rabbit anti‐rat GRK4 receptor antibodies (1:400; Santa Cruz Biotechnology, Santa Cruz, CA), overnight at 4°C. The other antibodies were anti‐D1 dopamine receptor antibody (1:400; Millipore, Billerica, MA), anti–c‐Myc (phosphorylated T58+S62) antibody (1:400; Abcam, Cambridge, UK), and rabbit polyclonal GAPDH (1:500; Santa Cruz Biotechnology). The membranes were then further incubated using infrared‐labeled secondary antibodies (donkey anti‐rabbit IRDye 800; Li‐Cor Biosciences, Lincoln, NE) to bind to the primary antibody at room temperature for 1 hour. The membranes were washed 3 times in PBS with Tween 20. The bound complex was detected using the Odyssey Infrared Imaging System (Li‐Cor Biosciences). The images were analyzed using the Odyssey Application Software to obtain the integrated intensities.
Determination of D1 Receptor Phosphorylation by Coimmunoprecipitation
Equal amounts of lysates (1.0 μg protein/mL supernatant from RPT cells or renal cortex membranes) were incubated with dopamine D1 receptor (D1R) antibody (2 μg; Millipore) for 2 hours, followed by protein A/G agarose overnight, with constant rocking at 4°C for 12 hours. The immunoprecipitates were pelleted and washed 3 times with PBS. Then, the pellets were suspended in sample buffer, boiled for 10 minutes, and subjected to immunoblotting with phosphoserine antibody (1:400; Immunechem, Burnaby, BC, Canada). To determine the specificity of the bands, normal rabbit IgG (negative control) and D1 receptor antibody (positive control) were used as the immunoprecipitants. The bound complexes were detected using the Odyssey Infrared Imaging System (Li‐Cor Biosciences). Band density of the serine‐phosphorylated D1R was normalized by band density of D1R. All the bands were quantified by densitometry.
Small‐Interfering RNA
Small‐interfering RNA (siRNA) against GRK4 mRNA and its control, scrambled RNA, was synthesized and purified with reverse‐phased high‐performance liquid chromatography as 25‐mer phosphorothioate‐modified oligodeoxynucleotides (GRK4 siRNA sequence, 5′‐AUCUAAAGAGGUGCAUUGAAUUCUUdTdT‐3′; scrambled RNA sequence, 5′‐TGACGATAAGAACAATAACdTdT‐3′), from nucleotides 412 to 436 and 1752 to 1776, respectively, of the rat GRK4 cDNA. The effects of 50 nmol/L siRNA was compared with scrambled RNA (control). Briefly, cells were grown in 6‐well plates until 60% confluence, and 50 nmol/L siRNA or control RNA was mixed with 6 μL of Oligofectamine in Optimem medium (Invitrogen Life Technologies) and incubated for 24 hours, then switched to growth medium and incubated for another 24 hours. The cells were collected and processed for reverse transcription–polymerase chain reaction for GRK4 to determine the efficiency of siRNA‐induced GRK4 gene silencing.
Cell Counting Kit‐8 Assay
As previously described,39 the number of viable cells was assayed using the Cell Counting Kit‐8 (Dojindo Laboratories, Kumamoto, Japan), per the manufacturer's protocol. Briefly, RPT cells were plated into a 96‐well plate and incubated with Cell Counting Kit‐8 for 2 hours at 37°C. The optical density (OD) values were subsequently measured on a spectrophotometer (PerkinElmer) at 490 nm. Three independent experiments were performed.
Real‐Time Quantitative Polymerase Chain Reaction Detecting System
A total of 2 to 3 μg of total RNA extracted from RPT cells or renal cortex membranes was used to synthesize cDNA, which served as template for the amplification of receptor and GAPDH (as the reference housekeeping gene). For GAPDH, the forward primer was 5′‐GACATGCCGCCTGGAGAAAC‐3′ and the reverse primer was 5′‐AGCCCAGGATGCCCTTTAGT‐3′ (GeneBank Accession No. NM_022928.1). For GRK4, the forward primer was 5′‐ACTTCAGCAGACTGGAAGCA‐3′ and the reverse primer was 5′‐GGTGTCCAGGTTGACTCCTT‐3′ (GeneBank Accession No. NM_017008.4). For c‐Myc, the forward primer was 5′‐ ACTTCAGCAGACTGGAAGCA‐3′ and the reverse primer was 5′‐GGTGTCCAGGTTGACTCCTT‐3′ (GeneBank Accession No. AY679730.1). The amplification was performed with the following conditions: denaturation at 95°C for 3 minutes, followed by 35 cycles at 95°C for 10 seconds and 60°C for 30 seconds. At the end of each run, a melting curve analysis was performed from 65°C to 95°C to monitor primer dimers or nonspecific product formation. The reactions were performed in triplicate. The GRK4 and c‐Myc mRNA expression was normalized for GAPDH mRNA.
Na+‐K+‐ATPase Activity Assay
Na+‐K+‐ATPase activity was determined as the rate of inorganic phosphate released in the presence or absence of ouabain.40, 41 Rat RPT cells were treated with vehicle (distilled water) and D1 receptor agonist,fenoldopam, at the indicated concentrations and durations of incubation. To prepare membranes for Na+‐K+‐ATPase activity assay, RPT cells cultured in 21‐cm2 plastic culture dishes were collected and centrifuged at 3000g for 10 minutes. The cells were then placed on ice and lysed in 2 mL of lysis buffer (1 mmol/L NaHCO3, 2 mmol/L CaCl2, and 5 mmol/L MgCl2). Cellular lysates were centrifuged at 3000g for 2 minutes to remove intact cells, debris, and nuclei. The resulting supernatant was suspended in an equal volume of 1 mol/L sodium iodide, and the mixture was centrifuged at 48 000g for 25 minutes. The pellet (membrane fraction) was washed 2 times and suspended in 10 mmol/L Tris containing 1 mmol/L EDTA (pH 7.4). Protein concentrations were determined by the Bradford assay (Bio‐Rad Laboratories, Hercules, CA) and adjusted to 1 mg/mL. The membranes were stored at −70°C until use.
To measure Na+‐K+‐ATPase activity, 100‐μL aliquots of membrane fraction were added to an 800‐μL reaction mixture (75 mmol/L NaCl, 5 mmol/L KCl, 5 mmol/L MgCl2, 6 mmol/L sodium azide, 1 mmol/L Na4EGTA, 37.5 mmol/L imidazole, 75 mmol/L Tris‐HCl, and 30 mmol/L histidine; pH 7.4), with or without 1 mmol/L ouabain (final volume=1 mL), and preincubated for 5 minutes in a water bath at 37°C. Reactions were initiated by adding Tris‐ATP (4 mmol/L) and terminated after 15 minutes of incubation at 37°C by adding 50 μL of 50% trichloroacetate. For determination of ouabain‐insensitive ATPase activity, NaCl and KCl were omitted from the reaction mixtures containing ouabain. To quantify the amount of phosphate produced, 1 mL of coloring reagent (10% ammonium molybdate in 10N sulfuric acid and 10N ferrous sulfate mix buffer) was added to the reaction mixture. The mixture was then mixed thoroughly and centrifuged at 3000g for 10 minutes. Formation of phosphomolybdate was determined spectrophotometrically at 740 nm, against a standard curve prepared from K2HPO4. Na+‐K+‐ATPase activity was estimated as the difference between total and ouabain‐insensitive ATPase activity and expressed as percentage change of control.
To eliminate the effect of proteases and phosphatases, protease inhibitors (1 mmol/L phenylmethylsulfonyl fluoride and 10 μg/mL each leupeptin and aprotinin) and a phosphatase inhibitor (50 μmol/L sodium orthovanadate) were added in all solutions used after drug/vehicle incubations.
Fenoldopam‐Induced Diuresis and Natriuresis
Twelve rats were selected from each group randomly and used for fenoldopam infusion study. A total of 36 rats from control, PM2.5‐L, and PM2.5‐H groups were anesthetized with pentobarbital (50 mg/kg IP), placed on a heated board to maintain body temperature at 37°C, and tracheotomized (PE‐100). Catheters (PE‐50) were placed into the external jugular and femoral veins and left carotid artery for fluid administration and blood pressure monitoring. Systemic blood pressure was monitored electronically using Cardiomax II (Columbus Instruments, Columbus, OH). Laparotomy was performed to expose the left and right ureters, which were then separately catheterized for urine collection. The right suprarenal artery (which originates from the right renal artery) was located and catheterized (PE‐10; heat stretched to 180 μm). After a 60‐minute stabilization period, a normal saline load equivalent to 5% of body weight was infused intravenously for 30 minutes. After the short‐term saline load, a 60‐minute stabilization period was allowed before starting the urine collection.37
Five consecutive 40‐minute urine samples were collected: Vehicle was given during the first collection period (control). In the next 3 periods, the mice received fenoldopam, a D1‐like receptor agonist, at 3 different dose rates of 0.1, 0.5, and1 μg/kg per minute, each dose that has been shown not to affect blood pressure in previous studies.30, 31 During the last collection period (recovery), only the vehicle, normal saline, was infused. This was considered as the recovery phase of the experiment. The change in all infusates (vehicle and reagents) was commenced 10 minute before each period to account for the dead space in the delivery catheter. All infusions (vehicle and reagents) were given at a rate of 40 μL/h. Fluid losses throughout the experiment were replaced intravenously with 5% albumin at 1% body weight over 30 minutes. The rats were euthanized with an overdose of pentobarbital (100 mg/kg) at the end of the experiment right after the last sample was collected. Urine samples were stored at −80°C until use.
Biochemical Markers of Oxidative Stress
To assess total oxidative stress, plasma samples from rats were used to measure SOD activity using an SOD Assay Kit (Dojindo Laboratories), following the manufacturer's instructions. Malondialdehyde and glutathione peroxidase in the plasma were quantified using a commercially available kit (Nanjing Jianchen Bioengineering Institute, Nanjing, China). For in vitro study, the RPT cell culture medium supernatants were collected to determine SOD, glutathione peroxidase, and malondialdehyde levels.
Statistical Analysis
The data are expressed as mean±SEM. A repeated‐measures ANOVA (dosages of PM2.5×time points) was used to determine the differences of blood pressure, among 3 groups (control, PM2.5‐L, and PM2.5‐H) at 0, 2, 4, 6, and 8 weeks. To analyze the effect of fenoldopam infusion on urine flow and urinary sodium excretion in different dosages of PM2.5‐treated groups (control, PM2.5‐L, and PM2.5‐H), another repeated‐measures (dosages of PM2.5×infusion periods) ANOVA was used. Dosages of PM2.5 were the predictor in these analyses. The Mauchly test of sphericity was performed using Greenhouse‐Geisser adjustment. Moreover, to determine the difference between groups that have been split on 2 dependent variables (whether fenoldopam treatment in cells and the dosage of PM2.5 [0, 10, and 100 μg/mL]) and their interaction, the 2‐way ANOVA was also performed. Besides, other comparisons among groups (or t test when only 2 groups were compared) was made by 1‐way ANOVA with Holm‐Sidak test. Post hoc multiple comparisons were performed by least significant difference method. P<0.05 was considered significant.
Results
Effects of PM2.5 Exposure on Blood Pressure and D1 Receptor–Mediated Sodium Excretion in SD Rats
Long‐term exposure to PM2.5 caused a significant increase in both systolic and diastolic blood pressures, which were measured by the tail‐cuff method, whether by the high dose (PM2.5‐H) or lower dose (PM2.5‐L) of PM2.5 (Figure 1A and 1B). The differences in blood pressure between the control and PM2.5‐treated rats progressively increased with time (Figure 1A and 1B). The high blood pressure might be ascribed to impaired sodium excretion, because the basal levels of sodium excretion were lower in PM2.5‐treated SD rats. Moreover, the higher dose of PM2.5 decreased the sodium excretion to a greater degree compared with the lower dose (Figure 1C and 1D).
Figure 1.
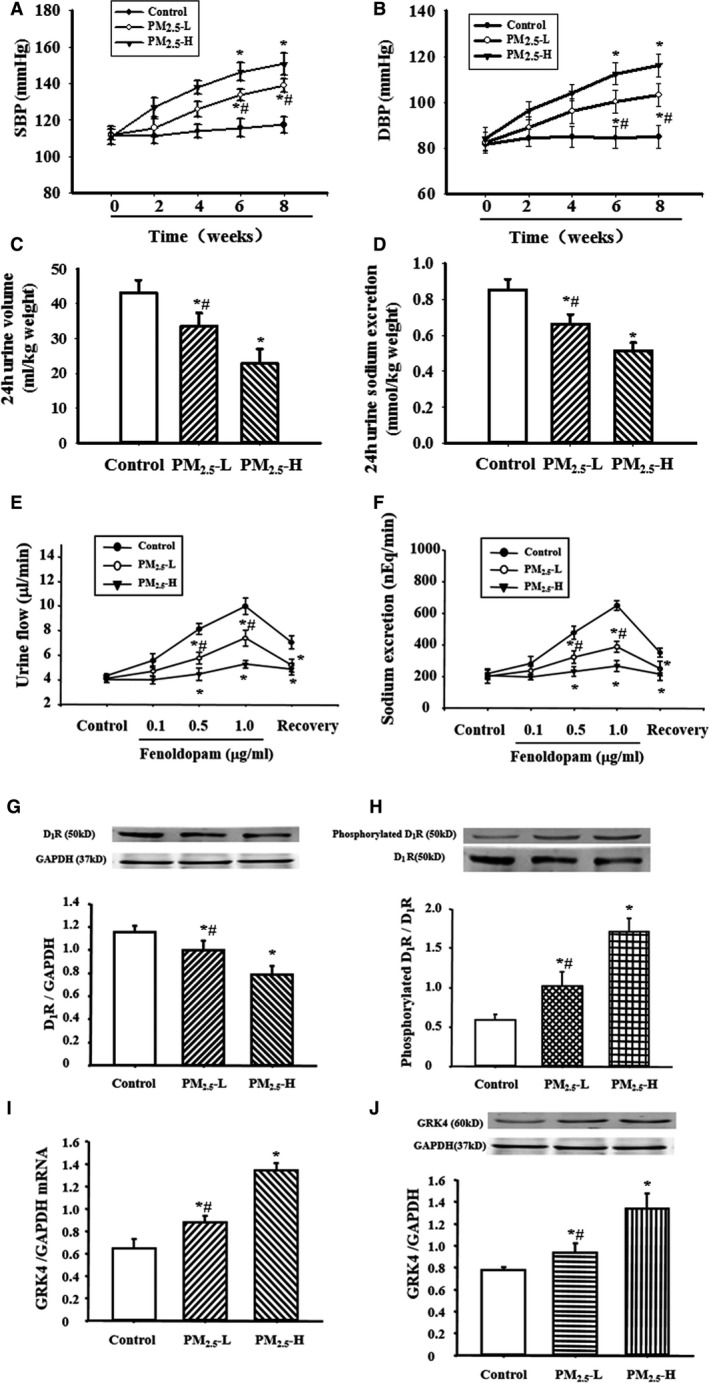
Effect of fine particulate matter (PM 2.5) exposure on blood pressure and sodium excretion in Sprague‐Dawley (SD) rats. A and B, Effect of PM 2.5 exposure on blood pressure. SD rats received lower (3 μg PM 2.5 per time [PM 2.5‐L]) or higher (30 μg PM 2.5 per time [PM 2.5‐H]) dosage of PM 2.5. Systolic blood pressure (SBP) (A) and diastolic blood pressure (DBP) (B) were recorded by the tail‐cuff method (n=12 in each group). C and D, Effect of PM 2.5 exposure on urine volume (C) and urine sodium excretion (D) in SD rats. Urine was collected in SD rats after exposure with PM 2.5 or vehicle for 8 weeks (n=20 in each group). E and F, Effect of PM 2.5 exposure on renal dopamine D1 receptor (D1R) function in SD rats. Varying dosages of the D1‐like receptor agonist, fenoldopam (0.1, 0.5, and 1.0 μg/kg per minute), were infused into the right suprarenal artery in SD rats after 8‐week exposure with PM 2.5 or vehicle. The fenoldopam‐associated urine volume (E) and urine sodium excretion (F) were measured (n=12 in each group). G and H, Effect of PM 2.5 exposure on renal D1R expression (G) and phosphorylation (H) in SD rats. Renal cortical membranes were prepared in SD rats exposed to PM 2.5 or vehicle for 8 weeks. D1R phosphorylation was normalized by total D1R expression (n=10 in each group). I and J, Effect of PM 2.5 exposure on renal G‐protein–coupled receptor kinase 4 (GRK4) mRNA (I) and protein (J) expressions in SD rats (n=10 in each group). *P<0.0001 vs control rats, # P<0.0001 vs PM 2.5‐H group.
Because of the important role of the dopamine receptor on sodium excretion, we also determined the effect of fenoldopam, a D1‐like receptor agonist, on sodium excretion. Consistent with previous reports,41, 42 fenoldopam induced natriuresis (Figure 1E) and diuresis (Figure 1F) in SD rats. However, after the treatment of SD rats by PM2.5 for 8 weeks, the fenoldopam‐induced natriuresis and diuresis were significantly decreased (Figure 1E and 1F). Moreover, the higher dose of PM2.5 worsened the D1 receptor–mediated sodium excretion to a higher degree compared with the lower dose of PM2.5 (Figure 1E and 1F). Our previous studies indicated that hyperphosphorylation of D1 receptor leads to the dysfunction of the renal D1 receptor, which is ascribed to the higher GRK4 expression and activity in spontaneously hypertensive rats or hypertensive patients.17, 18, 19, 20 Our additional study found that D1 receptor expression was lower and GRK4 expression and D1 receptor phosphorylation were higher in PM2.5‐treated SD rats. The degrees of change were greater with the higher dose of PM2.5 than the lower dose (Figure 1G through 1J), indicating GRK4 might be involved in the dysfunction of D1 receptor in PM2.5‐induced hypertension.
Role of GRK4 on PM2.5‐Impaired D1 Receptor Expression and Function in RPT Cells
We used RPT cells to uncover the mechanism of PM2.5‐induced hypertension. We first determined the optimal concentration of PM2.5 required, and it showed that 200 μg/mL PM2.5 decreased RPT cell viability; PM2.5 concentrations from 10 to 100 μg/mL had no effect (Figure 2A). Therefore, we used 10 and 100 μg/mL as lower and higher concentrations, respectively, in the in vitro study.
Figure 2.
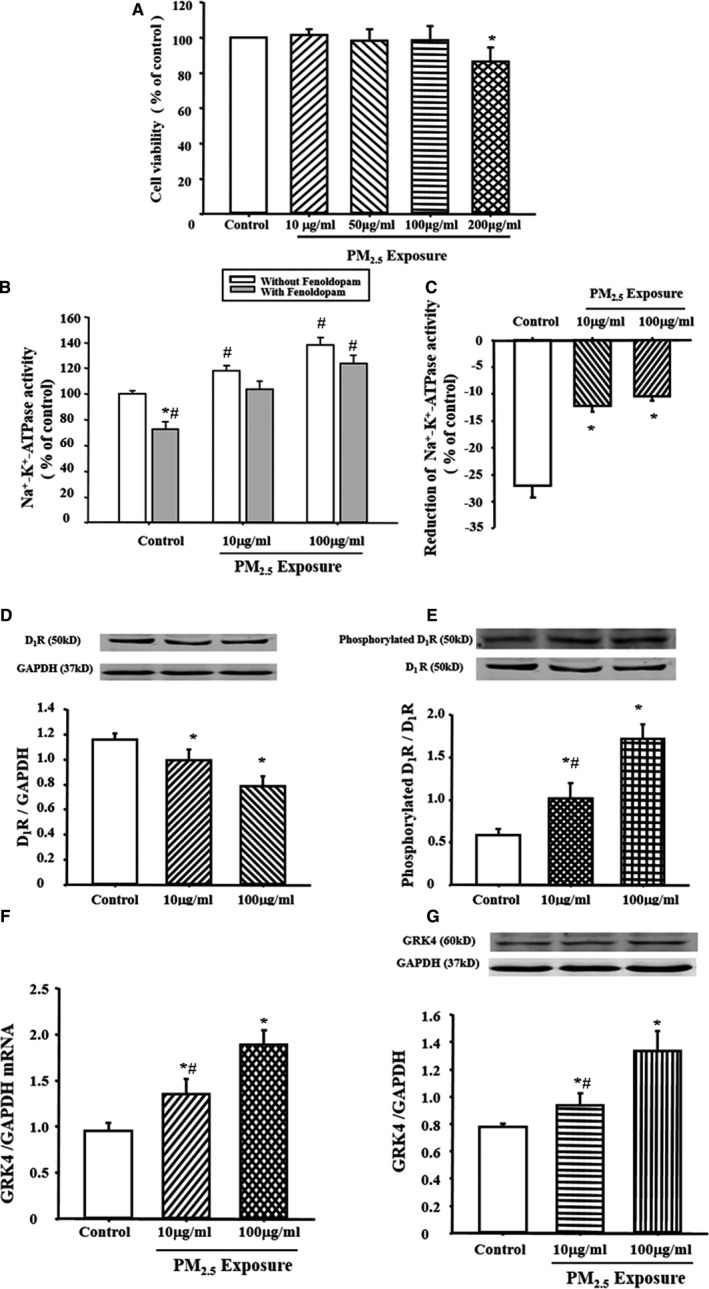
Effect of fine particulate matter (PM 2.5) exposure in in vitro study in renal proximal tubule (RPT) cells. A, Effect of PM 2.5 exposure on cell viability in RPT cells. Cell viability was assayed using Cell Counting Kit‐8 after exposure to vehicle (distilled water [dH2O]) or PM 2.5 for 24 hours. *P<0.0001 vs control (n=9 in each group). B and C, Effect of PM 2.5 exposure on Na+‐K+‐ATPase activity in RPT cells. The cells were treated with PM 2.5 or vehicle (dH 2O) for 24 hours. Results are expressed as percentage change of control (B). *P<0.0001 vs nonfenoldopam treatment in each group, # P<0.0001 vs control without fenoldopam and PM 2.5 treatment (n=10 in each group). C showed the fenoldopam‐induced reduction of Na+‐K+‐ATPase activity in each group. *P<0.0001 vs control in C (n=10 in each group). D and E, Effect of PM 2.5 exposure on dopamine D1 receptor (D1R) protein expression (D) and serine phosphorylation (E) in RPT cells. RPT cells were exposed with 10 or 100 μg/mL concentration of PM 2.5 for 24 hours. Results were expressed as ratio of D1R phosphorylation/total D1R expression. *P<0.05 vs control, # P<0.05 vs PM 2.5‐H group (n=9 in each group). F and G, Effect of PM 2.5 exposure on G‐protein–coupled receptor kinase 4 (GRK4) mRNA (F) and protein (G) expressions in RPT cells. RPT cells were exposed with 10 or 100 μg/mL concentration of PM 2.5 for 24 hours. *P<0.01 vs control, # P<0.05 vs PM 2.5‐H group (n=9 in each group).
Consistent with previous research findings,43, 44 the selective D1 receptor agonist fenoldopam caused an inhibition of the Na+‐K+‐ATPase activity, achieving a maximum of ≈30% in the control rats. With PM2.5 exposure, the basal Na+‐K+‐ATPase activity was increased and the fenoldopam‐mediated inhibitory effects on Na+‐K+‐ATPase activity were reduced (Figure 2B and 2C). Moreover, we also found that, compared with control, PM2.5‐treated cells had lower D1 receptor expression and higher D1 receptor phosphorylation (Figure 2D and 2E) and GRK4 expression (Figure 2F and 2G).
To further clarify the role of GRK4 on the Na+‐K+‐ATPase activity after PM2.5 exposure, we used a specific GRK4 siRNA to decrease the expression of GRK4 (Figure 3A and 3B). Downregulation of GRK4 by the siRNA reduced the increase in the basal Na+‐K+‐ATPase activity and reversed the inhibitory effect of fenoldopam on Na+‐K+‐ATPase activity in PM2.5‐treated cells (Figure 3C and 3D). Moreover, the decreased D1 receptor expression (Figure 3E) and increased D1 receptor phosphorylation (Figure 3F) by PM2.5 were also reversed after GRK4 siRNA treatment in RPT cells.
Figure 3.
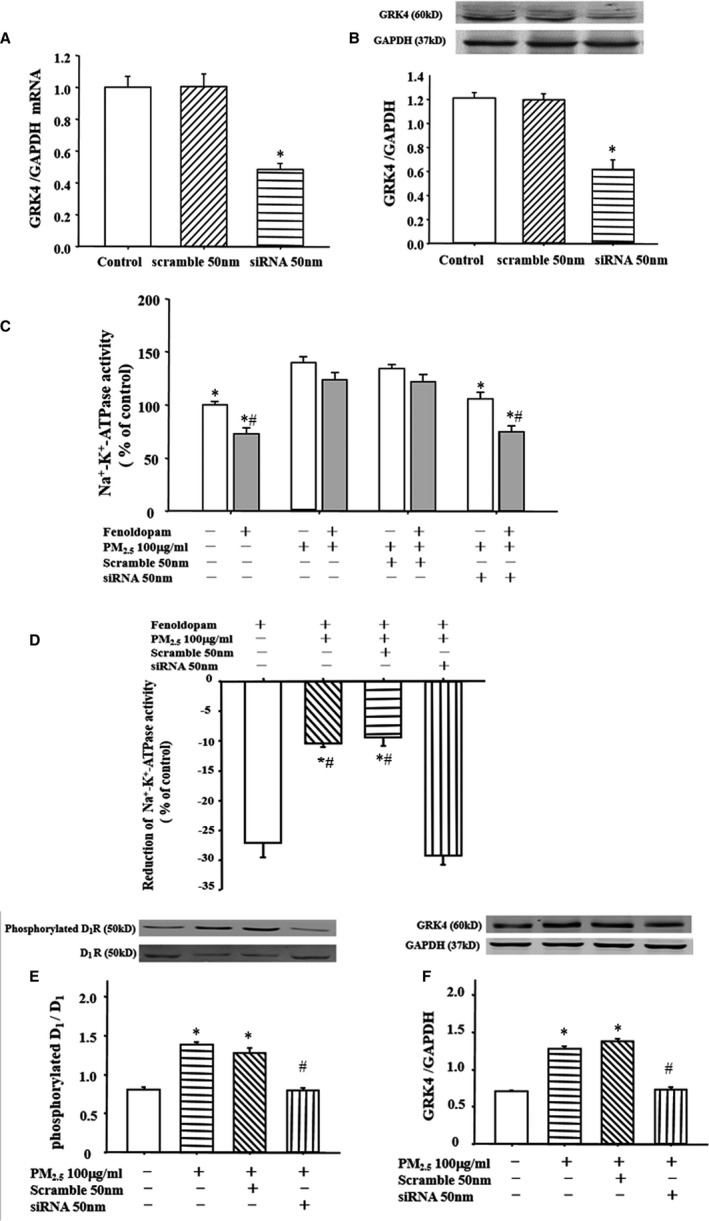
Role of G‐protein–coupled receptor kinase 4 (GRK4) on fine particulate matter (PM 2.5)–mediated impairment in renal proximal tubule (RPT) cells. A and B, Effect of GRK4 small‐interfering RNA (siRNA) on GRK4 expression. RPT cells were transfected with siRNA for 24 hours. GRK4 mRNA expression (A) and protein expression (B) were determined by quantitative polymerase chain reaction or immunoblotting. *P<0.0001 vs control (n=10 in each group). C and D, Effect of GRK4 siRNA on Na+‐K+‐ATPase activity in RPT cells. After transfection with GRK4 siRNA for 24 hours, RPT cells were treated with PM 2.5 (100 μg/mL) or vehicle (distilled water) for another 24 hours. Na+‐K+‐ATPase activity was checked with or without the presence of fenoldopam. C, Results are expressed as percentage change of control. *P<0.0001 vs PM 2.5 only (100 μg/mL), # P<0.0001 vs nonfenoldopam treatment in each group (n=10 in each group). D, Fenoldopam‐induced reduction of Na+‐K+‐ATPase activity in each group. *P<0001 vs fenoldopam only, # P<0001 vs siRNA treatment (n=10 in each group). E and F, Effect of GRK4 siRNA on dopamine D1 receptor (D1R) phosphorylation and GRK4 protein expressions in RPT cells exposed to PM 2.5 (100 μg/mL). After transfection with GRK4 siRNA for 24 hours, D1R phosphorylation (E) and GRK4 expression (F) were checked in PM 2.5‐exposed RPT cells. *P<0001 vs control, # P<0001 vs PM 2.5 only (100 μg/mL) (n=10 in each group).
Role of ROS in PM2.5‐Induced Hypertension and Impaired Renal Function
ROS has been reported to play a role in the dysfunction of D1 receptor in obese Zucker rats,23, 26, 27 and there is evidence showing that PM2.5 increases inflammation and ROS production.28, 31 Therefore, we wondered whether ROS is involved in the pathogenesis of PM2.5‐induced hypertension. We first measured ROS production in the PM2.5‐ and vehicle‐treated (control) rats. We found that plasma malondialdehyde levels were higher, whereas SOD and glutathione peroxidase levels were lower, in PM2.5‐treated rats versus the vehicle‐treated rats (Figure 4A through 4C). The dichloro‐dihydro‐fluorescein diacetate staining showed ROS level was significantly increased after PM2.5 treatment in both RPT alone and RPT/THP‐1 cocultured cells, whereas the extent of ROS elevation was much higher in RPT/THP‐1 cocultured cells than those in RPT cells alone with PM2.5 treatment (Figure S4).
Figure 4.
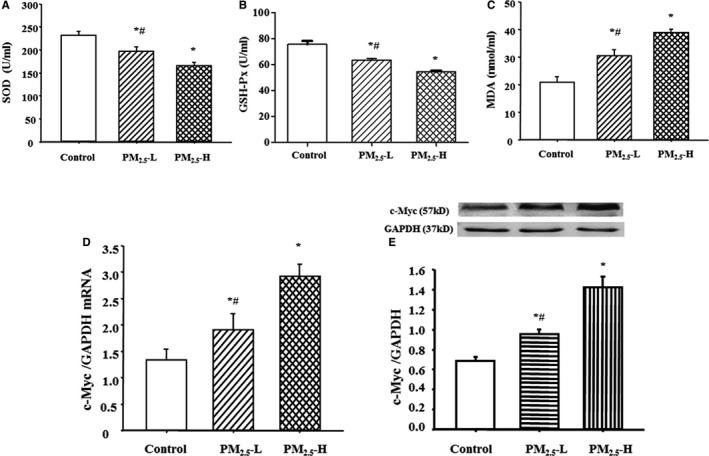
Effect of fine particulate matter (PM2.5) exposure on oxidative stress and c‐Myc expression in Sprague‐Dawley (SD) rats. SD rats were exposed with lower (3 μg PM 2.5 per time [PM 2.5‐L]) or higher (30 μg PM 2.5 per time [PM 2.5‐H]) dosage of PM 2.5 for 8 weeks. Plasma superoxide dismutase (SOD) (A), glutathione peroxidase (GSH‐Px) (B), and malondialdehyde (MDA) (C) levels, and c‐Myc mRNA (D) and protein (E) expressions were determined in those rats. *P<0.005 vs control rats, # P<0.001 vs PM 2.5‐H group (n=12 in each group).
It is known that c‐Myc is a transcription factor for GRK4, and there is evidence that ROS increases c‐Myc expression.45, 46, 47 We, therefore, tried to determine whether PM2.5 increased GRK4 via the c‐Myc pathway. Results showed that c‐Myc expression was higher in PM2.5‐treated than control cells (Figure 4D and 4E), indicating that PM2.5 might increase GRK4 expression via an increased c‐Myc expression.
To determine directly the role of ROS in PM2.5‐induced hypertension, tempol, an SOD mimetic, was used to treat PM2.5 rats, which were exposed to PM2.5 for 8 weeks. After tempol (1.0 mmol/L in drinking water24, 25) treatment for 3 weeks, the increased malondialdehyde and the decreased SOD/ glutathione peroxidase levels were recovered in PM2.5 rats (Figure 5A through 5C). Furthermore, the sodium excretion was increased, and blood pressure was lowered, in PM2.5 rats (Figure 5D through 5G), and the fenoldopam‐induced natriuresis and diuresis were also recovered in PM2.5‐exposed rats (Figure 5H and 5I), accompanied with a recovered renal D1 receptor expression and phosphorylation and GRK4 expression (Figure 5J and 5K). It is interesting to find that the renal c‐Myc expression is also reduced after treatment with tempol in PM2.5‐exposed rats (Figure 5L and 5M), which further indicated the role of c‐Myc in the PM2.5‐induced hypertension.
Figure 5.
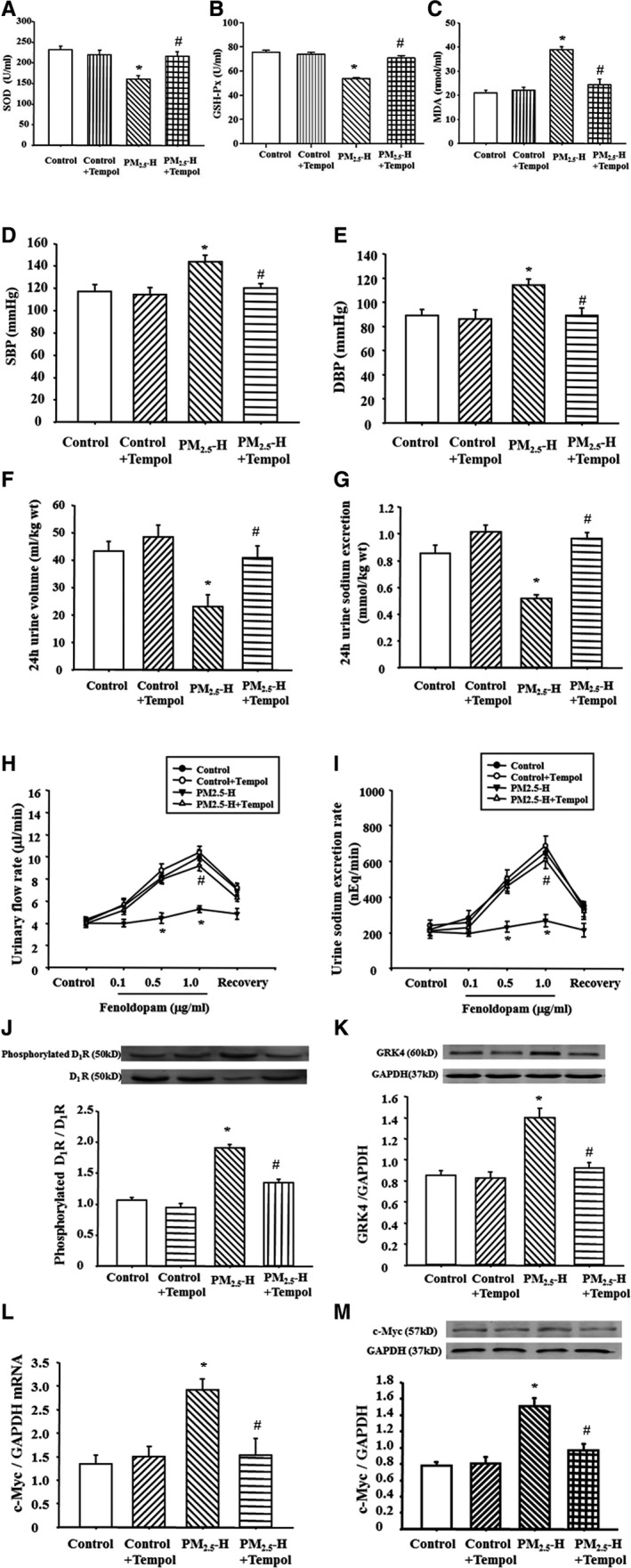
Effect of tempol on blood pressure and sodium excretion in Sprague‐Dawley (SD) rats exposed to fine particulate matter (PM 2.5). SD rats exposed to PM 2.5 were treated with tempol for 3 weeks. Plasma superoxide dismutase (SOD) (A), glutathione peroxidase (GSH‐Px) (B), and malondialdehyde (MDA) (C) levels were checked in those rats (n=12 in each group). The blood pressure (D and E) and sodium excretion (F and G) were recorded (n=12 in each group). Moreover, the fenoldopam‐mediated urinary flow rate (H) and urine sodium excretion rate (I), G‐protein–coupled receptor kinase 4 (GRK4) expression (J), and dopamine D1 receptor (D1R) phosphorylations (K), as well as c‐Myc mRNA (L) and protein expressions (M) were checked (n=10 in each group). DBP indicates diastolic blood pressure; PM2.5‐H, 30 μg PM 2.5 per time; and SBP, systolic blood pressure. *P<0.0001 vs control rats, # P<0.0001 vs PM 2.5‐H rats.
Consistent with the in vivo study, PM2.5 increased malondialdehyde and ROS concentrations in the supernatants from RPT cells, which were normalized by tempol (200 μmol/L) treatment (Figure 6A and 6B), whereas the high expression of c‐Myc was decreased (Figure 6C and 6D). Moreover, tempol reversed the fenoldopam‐mediated inhibitory effects on Na+‐K+‐ATPase activity in PM2.5‐treated RPT cells (Figure 6E and 6F). Meanwhile, the lowered renal D1 receptor expression and the upregulated renal D1 receptor phosphorylation and GRK4 expression were normalized by tempol in PM2.5‐treated RPT cells (Figure 6G and 6H).
Figure 6.
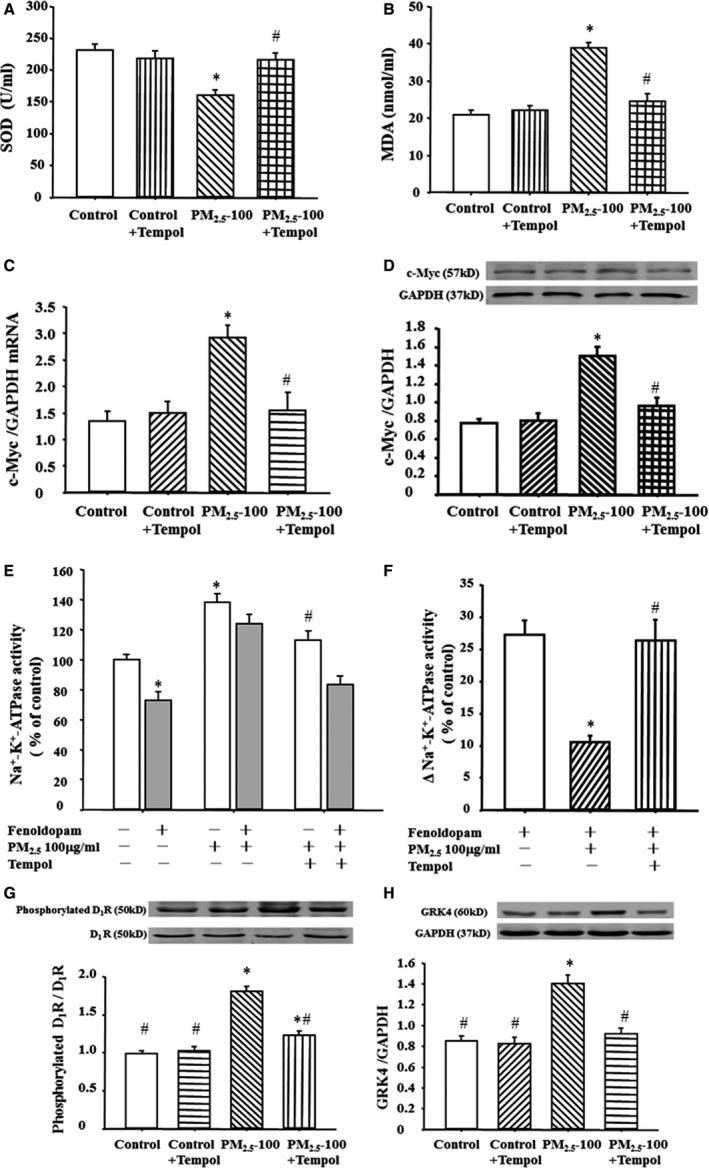
Effect of tempol on Na+‐K+‐ATPase activity and G‐protein–coupled receptor kinase 4 (GRK4) expression in renal proximal tubule (RPT) cells exposed to fine particulate matter (PM 2.5). RPT cells exposed to vehicle (control) or PM 2.5 were treated with tempol (200 μmol/L) for 24 hours. Superoxide dismutase (SOD) (A) and malondialdehyde (MDA) levels (B) were quantified in the cell culture medium supernatant. *P<0.0001 vs control, # P<0.0001 vs PM 2.5 only (100 μg/mL) (n=12 in each group). c‐Myc mRNA (C) and protein (D) expressions were checked in those cells. *P<0.0001 vs control, # P<0.0001 vs PM 2.5 only (100 μg/mL) (n=10 in each group). Moreover, the Na+‐K+‐ATPase activity (E and F) and GRK4 expression (G) and dopamine D1 receptor (D1R) phosphorylations (H) were tested in those cells. *P<0.05 vs control, # P<0.001 vs PM 2.5 treated (100 μg/mL) (n=10 in each group).
Discussion
Exposure to ambient particulate matter has been associated with increased hospital admission and mortality for cardiovascular diseases.4, 6, 7, 29 However, the mechanisms are not completely understood. Hypertension is an established risk factor for cardiovascular disease and may be implicated in the association between ambient particulate matter and cardiovascular morbidity and mortality.11 Epidemiological studies have shown that traffic4, 48 or household49, 50, 51 PM2.5 exposure can increase blood pressure in both the short‐ and long‐term. Many animal experimental studies have validated the view of the above‐mentioned evidence‐based‐studies.8, 31, 52, 53, 54 In this study, PM2.5 was confirmed to be associated with increased blood pressure, and the blood pressure was positively correlated with the exposure dose within a certain range. Even a lower concentration of PM2.5 exposure can cause an increased blood pressure.
Although the mechanisms of PM2.5‐induced hypertension are not clear, some mechanisms, including oxidative stress, inflammatory reaction, changes in sympathetic nerve activity, DNA damage, and apparent genetic changes, have been reported to be involved in the pathogenesis of hypertension.6, 8, 10, 11, 12 Our present study and others show the role of impaired sodium excretion in PM2.5‐induced hypertension. It is known that sodium excretion is regulated by numerous hormones and humoral factors, including dopamine. Renal dopamine, independent of renal nerves, plays an important role in maintaining sodium homeostasis and blood pressure regulation, especially during moderately increased sodium intake. Under conditions of moderate sodium excess, the renal dopaminergic system, mainly the D1R, is responsible for >50% of renal sodium excretion.13 Abnormalities in renal dopamine and D1R response to an increased sodium load have been implicated in the diminished natriuretic response and increase in blood pressure in hypertensive patients and rodents.13, 14, 15, 16, 23 The impaired D1R function is ascribed to the hyperphosphorylation, which leads to the uncoupling of D1R with G protein.13, 14, 15, 16 Our previous study found that the hyperphosphorylated D1R is caused by a constitutively increased GRK4 activity and expression in hypertension.13, 18, 19, 20, 21 Consistent with hypertension, our present study found that D1R‐mediated diuresis and natriuresis are impaired in PM2.5‐exposed rats, accompanied with D1R hyperphosphorylation. The increased phosphorylation of D1R is ascribed to GRK4, because in both in vivo and in vitro studies, GRK4 expression is increased because of PM2.5 exposure; after downregulation of GRK4 with siRNA, the impaired inhibitory effect of D1R on Na+‐K+‐ATPase activity was reversed in RPT cells exposed to PM2.5. Thus, the current studies support the notion that GRK4 is involved in the impaired function of renal D1R in PM2.5‐exposed hypertension.
Many studies have indicated that both hypertensive patients and animals have decreased antioxidant capacity and produce excessive amounts of ROS.27, 55, 56, 57 Antioxidant treatment could mitigate the production of ROS and further increase antioxidant capacity, such as SOD and glutathione in plasma and tissue, consequently decreasing the blood pressure in hypertensive animal models. PM2.5 is known to increase serum tumor necrosis factor‐α level and stimulate macrophages to generate ROS and enhance oxidative stress.28, 29, 30, 31, 33, 58, 59, 60 Multiple studies have shown that an impaired intrarenal D1R signaling in hypertension can cause or be caused by oxidative stress.24, 25, 26, 27, 61 Our present study found that tempol treatment would reduce ROS, lower blood pressure, and reverse the impaired D1R‐mediated natriuresis and diuresis in PM2.5‐exposed rats.
As stated previously and in other reports, ROS is related to the regulation of GRK4 on D1R expression and function and tempol has been shown to reduce GRK4 level, restore D1R expression and function, and normalize blood pressure by decreasing oxidative stress.22, 24, 25 However, the molecular mechanisms involved are not well understood. c‐Myc, a human homolog of the avian myelocytomatosis viral oncogene v‐Myc, is involved in cancer progression, which has been implicated in hypertrophy and fibrosis of the heart, atherosclerosis, and hypertension.45, 46, 47 c‐Myc was shown to bind to the promoter region of GRK4 in Burkitt lymphoma cells. Gildea et al found c‐Myc may be involved in the increase in blood pressure in hypertension that is mediated by increased activity of the renin‐angiotensin system and decreased activity of the renal dopaminergic system.45 Many other studies also report that ROS increased c‐Myc expression.46, 47 Our present study found that ROS associated with increased c‐Myc levels and increased GRK4 expression in PM2.5‐exposed rats and cells.
In the present study, the particulate sampling method is universally accepted,32, 33, 34 and we performed a scanning electron microscope analysis of the samples to determine the reliability of the samples. To ensure the success of the exposure model, we also performed a hematoxylin‐eosin stain of the lung after the rats were euthanized, and a light microscope detection confirmed the presence of PM2.5 in the lung tissue. According to the World Health Organization's Air Quality Guideline, an annual mean PM2.5 concentration of 35 μg/m3 was selected as the level 1 interim target, which corresponds to the highest mean concentrations reported in studies of long‐term health effects. This level has been shown to be associated with significant mortality in the developed world.62 For this reason, in this study, the rats received 2 dosages of PM2.5. A lower dosage represented 35 μg/m3 per 24 hours PM2.5, whereas the higher dosage represented 350 μg/m3 per 24 hours PM2.5 in the air.
In summary, our study shows direct evidence supporting the hypothesis that PM2.5 causes hypertension and, for first time, links the dysfunction of the renal D1R to the hypertension attributable to PM2.5 exposure. Increased oxidative stress, associated with increased GRK4 levels, impairs renal D1R function and leads to hypertension. Restoration of D1R function by antioxidants may be a therapeutic target for PM2.5‐caused hypertension.
Sources of Funding
These studies were supported in part by grants from the National Natural Science Foundation of China (31730043 and 31430043), a grant from the National Basic Research Program of China (2013CB531104), and grants from the National Institutes of Health (7R37HL023081‐37 and 5P01HL074940‐11).
Disclosures
None.
Supporting information
Figure S1. Scanning electron microscopy analysis of PM2.5 samples.
Figure S2. The flowchart of animal studies.
Figure S3. Lung histology from control and exposed rats.
Figure S4. The ROS generation in RPT cells after PM2.5 exposure.
(J Am Heart Assoc. 2018;7:e007185 DOI: 10.1161/JAHA.117.007185.)29307864
Contributor Information
Ken Chen, Email: chenken0205@yahoo.com.
Chunyu Zeng, Email: chunyuzeng01@163.com.
References
- 1. Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair‐Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker‐Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan‐Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng AT, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FG, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD III, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, Lalloo R, Lan Q, Lathlean T, Leasher JL, Leigh J, Li Y, Lin JK, Lipshultz SE, London S, Lozano R, Lu Y, Mak J, Malekzadeh R, Mallinger L, Marcenes W, March L, Marks R, Martin R, McGale P, McGrath J, Mehta S, Mensah GA, Merriman TR, Micha R, Michaud C, Mishra V, Mohd Hanafiah K, Mokdad AA, Morawska L, Mozaffarian D, Murphy T, Naghavi M, Neal B, Nelson PK, Nolla JM, Norman R, Olives C, Omer SB, Orchard J, Osborne R, Ostro B, Page A, Pandey KD, Parry CD, Passmore E, Patra J, Pearce N, Pelizzari PM, Petzold M, Phillips MR, Pope D, Pope CA III, Powles J, Rao M, Razavi H, Rehfuess EA, Rehm JT, Ritz B, Rivara FP, Roberts T, Robinson C, Rodriguez‐Portales JA, Romieu I, Room R, Rosenfeld LC, Roy A, Rushton L, Salomon JA, Sampson U, Sanchez‐Riera L, Sanman E, Sapkota A, Seedat S, Shi P, Shield K, Shivakoti R, Singh GM, Sleet DA, Smith E, Smith KR, Stapelberg NJ, Steenland K, Stockl H, Stovner LJ, Straif K, Straney L, Thurston GD, Tran JH, Van Dingenen R, van Donkelaar A, Veerman JL, Vijayakumar L, Weintraub R, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams W, Wilson N, Woolf AD, Yip P, Zielinski JM, Lopez AD, Murray CJ, Ezzati M, AlMazroa MA, Memish ZA. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380:2224–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brundtland GH. From the World Health Organization: reducing risks to health, promoting healthy life. JAMA. 2002;288:1974. [DOI] [PubMed] [Google Scholar]
- 3. Cesaroni G, Forastiere F, Stafoggia M, Andersen ZJ, Badaloni C, Beelen R, Caracciolo B, de Faire U, Erbel R, Eriksen KT, Fratiglioni L, Galassi C, Hampel R, Heier M, Hennig F, Hilding A, Hoffmann B, Houthuijs D, Jockel KH, Korek M, Lanki T, Leander K, Magnusson PK, Migliore E, Ostenson CG, Overvad K, Pedersen NL, J JP, Penell J, Pershagen G, Pyko A, Raaschou‐Nielsen O, Ranzi A, Ricceri F, Sacerdote C, Salomaa V, Swart W, Turunen AW, Vineis P, Weinmayr G, Wolf K, de Hoogh K, Hoek G, Brunekreef B, Peters A. Long term exposure to ambient air pollution and incidence of acute coronary events: prospective cohort study and meta‐analysis in 11 European cohorts from the ESCAPW Project. BMJ. 2014;348:f7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen H, Burnett RT, Kwong JC, Villeneuve PJ, Goldberg MS, Brook RD, van Donkelaar A, Jerrett M, Martin RV, Kopp A, Brook JR, Copes R. Spatial association between ambient fine particulate matter and incident hypertension. Circulation. 2014;129:562–569. [DOI] [PubMed] [Google Scholar]
- 5. Dvonch JT, Kannan S, Schulz AJ, Keeler GJ, Mentz G, House J, Benjamin A, Max P, Bard RL, Brook RD. Acute effects of ambient particulate matter on blood pressure: differential effects across urban communities. Hypertension. 2009;53:853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dubowsky SD, Suh H, Schwartz J, Coull BA, Gold DR. Diabetes, obesity, and hypertension may enhance associations between air pollution and markers of systemic inflammation. Environ Health Perspect. 2006;114:992–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coogan PF, White LF, Jerrett M, Brook RD, Su JG, Seto E, Burnett R, Palmer JR, Rosenberg L. Air pollution and incidence of hypertension and diabetes mellitus in black women living in Los Angeles. Circulation. 2012;125:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ying Z, Xu X, Bai Y, Zhong J, Chen M, Liang Y, Zhao J, Liu D, Morishita M, Sun Q, Spino C, Brook RD, Harkema JR, Rajagopalan S. Long‐term exposure to concentrated ambient PM2.5 increases mouse blood pressure through abnormal activation of the sympathetic nervous system: a role for hypothalamic inflammation. Environ Health Perspect. 2014;122:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baddour LM, Epstein AE, Erickson CC, Knight BP, Levison ME, Lockhart PB, Masoudi FA, Okum EJ, Wilson WR, Beerman LB, Bolger AF, Estes NA III, Gewitz M, Newburger JW, Schron EB, Taubert KA; American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee; Council on Cardiovascular Disease in Young; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Nursing; Council on Clinical Cardiology; Interdisciplinary Council on Quality of Care; American Heart Association . Update on cardiovascular implantable electronic device infections and their management: a scientific statement from the American Heart Association. Circulation. 2010;121:458–477. [DOI] [PubMed] [Google Scholar]
- 10. Kim JY, Prouty LA, Fang SC, Rodrigues EG, Magari SR, Modest GA, Christiani DC. Association between fine particulate matter and oxidative DNA damage may be modified in individuals with hypertension. J Occup Environ Med. 2009;51:1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bellavia A, Urch B, Speck M, Brook RD, Scott JA, Albetti B, Behbod B, North M, Valeri L, Bertazzi PA, Silverman F, Gold D, Baccarelli AA. DNA hypomethylation, ambient particulate matter, and increased blood pressure: findings from controlled human exposure experiments. J Am Heart Assoc. 2013;2:e000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brook RD, Rajagopalan S, Pope CA III, Brook JR, Bhatnagar A, Diez‐Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC Jr, Whitsel L, Kaufman JD; American Heart Association Council on Epidemiology and Prevention, Council on the Kidney in Cardiovascular Disease, and Council on Nutrition, Physical Activity and Metabolism . Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–2378. [DOI] [PubMed] [Google Scholar]
- 13. Zeng C, Jose PA. Dopamine receptors: important antihypertensive counterbalance against hypertensive factors. Hypertension. 2011;57:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harris RC, Zhang MZ. Dopamine, the kidney, and hypertension. Curr Hypertens Rep. 2012;14:138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pinto V, Pinho MJ, Soares‐da‐Silva P. Renal amino acid transport systems and essential hypertension. FASEB J. 2013;27:2927–2938. [DOI] [PubMed] [Google Scholar]
- 16. Carey RM. Theodore Cooper lecture: renal dopamine system: paracrine regulator of sodium homeostasis and blood pressure. Hypertension. 2001;38:297–302. [DOI] [PubMed] [Google Scholar]
- 17. Felder RA, Seikaly MG, Cody P, Eisner GM, Jose PA. Attenuated renal response to dopaminergic drugs in spontaneously hypertensive rats. Hypertension. 1990;15:560–569. [DOI] [PubMed] [Google Scholar]
- 18. Watanabe H, Xu J, Bengra C, Jose PA, Felder RA. Desensitization of human renal D1 dopamine receptors by G protein‐coupled receptor kinase 4. Kidney Int. 2002;62:790–798. [DOI] [PubMed] [Google Scholar]
- 19. Harris RC. Abnormalities in renal dopamine signaling and hypertension: the role of GRK4. Curr Opin Nephrol Hypertens. 2012;21:61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeng C, Liu Y, Wang Z, He D, Huang L, Yu P, Zheng S, Jones JE, Asico LD, Hopfer U, Eisner GM, Felder RA, Jose PA. Activation of D3 dopamine receptor decreases angiotensin II type 1 receptor expression in rat renal proximal tubule cells. Circ Res. 2006;99:494–500. [DOI] [PubMed] [Google Scholar]
- 21. Yang J, Villar VA, Jones JE, Jose PA, Zeng C. G protein‐coupled receptor kinase 4: role in hypertension. Hypertension. 2015;65:1148–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chugh G, Lokhandwala MF, Asghar M. Altered functioning of both renal dopamine D1 and angiotensin II type 1 receptors causes hypertension in old rats. Hypertension. 2012;59:1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fardoun RZ, Asghar M, Lokhandwala M. Role of oxidative stress in defective renal dopamine D1 receptor‐G protein coupling and function in old Fischer 344 rats. Am J Physiol Renal Physiol. 2006;291:F945–F951. [DOI] [PubMed] [Google Scholar]
- 24. Banday AA, Marwaha A, Tallam LS, Lokhandwala MF. Tempol reduces oxidative stress, improves insulin sensitivity, decreases renal dopamine D1 receptor hyperphosphorylation, and restores D1 receptor‐G‐protein coupling and function in obese Zucker rats. Diabetes. 2005;54:2219–2226. [DOI] [PubMed] [Google Scholar]
- 25. Marwaha A, Lokhandwala MF. Tempol reduces oxidative stress and restores renal dopamine D1‐like receptor‐G protein coupling and function in hyperglycemic rats. Am J Physiol Renal Physiol. 2006;291:F58–F66. [DOI] [PubMed] [Google Scholar]
- 26. Yu P, Han W, Villar VA, Yang Y, Lu Q, Lee H, Li F, Quinn MT, Gildea JJ, Felder RA, Jose PA. Unique role of NADPH oxidase 5 in oxidative stress in human renal proximal tubule cells. Redox Biol. 2014;2:570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cuevas S, Villar VA, Jose PA, Armando I. Renal dopamine receptors, oxidative stress, and hypertension. Int J Mol Sci. 2013;14:17553–17572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gurgueira SA, Lawrence J, Coull B, Murthy GG, Gonzalez‐Flecha B. Rapid increases in the steady‐state concentration of reactive oxygen species in the lungs and heart after particulate air pollution inhalation. Environ Health Perspect. 2002;110:749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pope CA III, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K, Thurston GD. Lung cancer, cardiopulmonary mortality, and long‐term exposure to fine particulate air pollution. JAMA. 2002;287:1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aakko‐Saksa PT, Rantanen‐Kolehmainen L, Skytta E. Ethanol, isobutanol, and biohydrocarbons as gasoline components in relation to gaseous emissions and particulate matter. Environ Sci Technol. 2014;48:10489–10496. [DOI] [PubMed] [Google Scholar]
- 31. Wang G, Jiang R, Zhao Z, Song W. Effects of ozone and fine particulate matter (PM(2.5)) on rat system inflammation and cardiac function. Toxicol Lett. 2013;217:23–33. [DOI] [PubMed] [Google Scholar]
- 32. Mirowsky JE, Jin L, Thurston G, Lighthall D, Tyner T, Horton L, Galdanes K, Chillrud S, Ross J, Pinkerton KE, Chen LC, Lippmann M, Gordon T. In vitro and in vivo toxicity of urban and rural particulate matter from California. Atmos Environ. 2015;103:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cuevas AK, Niu J, Zhong M, Liberda EN, Ghio A, Qu Q, Chen LC. Metal rich particulate matter impairs acetylcholine‐mediated vasorelaxation of microvessels in mice. Part Fibre Toxicol. 2015;12:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cao XN, Yan C, Liu DY, Peng JP, Chen JJ, Zhou Y, Long CL, He DW, Lin T, Shen LJ, Wei GH. Fine particulate matter leads to reproductive impairment in male rats by overexpressing phosphatidylinositol 3‐kinase (PI3K)/protein kinase b (AKT) signaling pathway. Toxicol Lett. 2015;237:181–190. [DOI] [PubMed] [Google Scholar]
- 35. Rao GV, Tinkle S, Weissman DN, Antonini JM, Kashon ML, Salmen R, Battelli LA, Willard PA, Hoover MD, Hubbs AF. Efficacy of a technique for exposing the mouse lung to particles aspirated from the pharynx. J Toxicol Environ Health A. 2003;66:1441–1452. [DOI] [PubMed] [Google Scholar]
- 36. Miller MR, McLean SG, Duffin R, Lawal AO, Araujo JA, Shaw CA, Mills NL, Donaldson K, Newby DE, Hadoke PW. Diesel exhaust particulate increases the size and complexity of lesions in atherosclerotic mice. Part Fibre Toxicol. 2013;10:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Luo H, Chen C, Chen K, Wang J, Cai Y, Zheng S, Yang X, Zhou L, Jose PA, Zeng C. Prenatal lipopolysaccharide exposure results in dysfunction of the renal dopamine D1 receptor in offspring. Free Radic Biol Med. 2014;76:242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zeng C, Luo Y, Asico LD, Hopfer U, Eisner GM, Felder RA, Jose PA. Perturbation of D1 dopamine and AT1 receptor interaction in spontaneously hypertensive rats. Hypertension. 2003;42:787–792. [DOI] [PubMed] [Google Scholar]
- 39. Ding J, Huang S, Wu S, Zhao Y, Liang L, Yan M, Ge C, Yao J, Chen T, Wan D, Wang H, Gu J, Yao M, Li J, Tu H, He X. Gain of miR‐151 on chromosome 8q24.3 facilitates tumour cell migration and spreading through downregulating RhoGDIA. Nat Cell Biol. 2010;12:390–399. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y, Fu C, Asico LD, Villar VA, Ren H, He D, Wang Z, Yang J, Jose PA, Zeng C. Role of galpha(12)‐ and galpha(13)‐protein subunit linkage of D(3) dopamine receptors in the natriuretic effect of D(3) dopamine receptor in kidney. Hypertens Res. 2011;34:1011–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Y, Asico LD, Zheng S, Villar VA, He D, Zhou L, Zeng C, Jose PA. Gastrin and D1 dopamine receptor interact to induce natriuresis and diuresis. Hypertension. 2013;62:927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen K, Deng K, Wang X, Wang Z, Zheng S, Ren H, He D, Han Y, Asico LD, Jose PA, Zeng C. Activation of D4 dopamine receptor decreases angiotensin II type 1 receptor expression in rat renal proximal tubule cells. Hypertension. 2015;65:153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Horiuchi A, Takeyasu K, Mouradian MM, Jose PA, Felder RA. D1a dopamine receptor stimulation inhibits Na+/K(+)‐ATPase activity through protein kinase a. Mol Pharmacol. 1993;43:281–285. [PubMed] [Google Scholar]
- 44. Gildea JJ, Wang X, Shah N, Tran H, Spinosa M, Van Sciver R, Sasaki M, Yatabe J, Carey RM, Jose PA, Felder RA. Dopamine and angiotensin type 2 receptors cooperatively inhibit sodium transport in human renal proximal tubule cells. Hypertension. 2012;60:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gildea JJ, Tran HT, Van Sciver RE, Bigler Wang D, Carlson JM, Felder RA. A novel role for c‐Myc in g protein‐coupled receptor kinase 4 (GRK4) transcriptional regulation in human kidney proximal tubule cells. Hypertension. 2013;61:1021–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang CY, Fujimura M, Noshita N, Chang YY, Chan PH. SOD1 down‐regulates NF‐kappab and c‐Myc expression in mice after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:163–173. [DOI] [PubMed] [Google Scholar]
- 47. Yu Y, Niapour M, Zhang Y, Berger SA. Mitochondrial regulation by c‐Myc and hypoxia‐inducible factor‐1 alpha controls sensitivity to econazole. Mol Cancer Ther. 2008;7:483–491. [DOI] [PubMed] [Google Scholar]
- 48. Urch B, Silverman F, Corey P, Brook JR, Lukic KZ, Rajagopalan S, Brook RD. Acute blood pressure responses in healthy adults during controlled air pollution exposures. Environ Health Perspect. 2005;113:1052–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shan M, Yang X, Ezzati M, Chaturvedi N, Coady E, Hughes A, Shi Y, Yang M, Zhang Y, Baumgartner J. A feasibility study of the association of exposure to biomass smoke with vascular function, inflammation, and cellular aging. Environ Res. 2014;135:165–172. [DOI] [PubMed] [Google Scholar]
- 50. Painschab MS, Davila‐Roman VG, Gilman RH, Vasquez‐Villar AD, Pollard SL, Wise RA, Miranda JJ, Checkley W; CRONICAS Cohort Study Group . Chronic exposure to biomass fuel is associated with increased carotid artery intima‐media thickness and a higher prevalence of atherosclerotic plaque. Heart. 2013;99:984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chan SH, Van Hee VC, Bergen S, Szpiro AA, DeRoo LA, London SJ, Marshall JD, Kaufman JD, Sandler DP. Long‐term air pollution exposure and blood pressure in the sister study. Environ Health Perspect. 2015;123:951–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nemmar A, Subramaniyan D, Zia S, Yasin J, Ali BH. Airway resistance, inflammation and oxidative stress following exposure to diesel exhaust particle in angiotensin II‐induced hypertension in mice. Toxicology. 2012;292:162–168. [DOI] [PubMed] [Google Scholar]
- 53. Wagner JG, Kamal AS, Morishita M, Dvonch JT, Harkema JR, Rohr AC. PM2.5‐induced cardiovascular dysregulation in rats is associated with elemental carbon and temperature‐resolved carbon subfractions. Part Fibre Toxicol. 2014;11:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wagner JG, Allen K, Yang HY, Nan B, Morishita M, Mukherjee B, Dvonch JT, Spino C, Fink GD, Rajagopalan S, Sun Q, Brook RD, Harkema JR. Cardiovascular depression in rats exposed to inhaled particulate matter and ozone: effects of diet‐induced metabolic syndrome. Environ Health Perspect. 2014;122:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Usui T, Okada M, Hara Y, Yamawaki H. Death‐associated protein kinase 3 mediates vascular inflammation and development of hypertension in spontaneously hypertensive rats. Hypertension. 2012;60:1031–1039. [DOI] [PubMed] [Google Scholar]
- 56. Cuevas S, Zhang Y, Yang Y, Escano C, Asico L, Jones JE, Armando I, Jose PA. Role of renal DJ‐1 in the pathogenesis of hypertension associated with increased reactive oxygen species production. Hypertension. 2012;59:446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wakui H, Dejima T, Tamura K, Uneda K, Azuma K, Maeda A, Ohsawa M, Kanaoka T, Azushima K, Kobayashi R, Matsuda M, Yamashita A, Umemura S. Activation of angiotensin II type 1 receptor‐associated protein exerts an inhibitory effect on vascular hypertrophy and oxidative stress in angiotensin II‐mediated hypertension. Cardiovasc Res. 2013;100:511–519. [DOI] [PubMed] [Google Scholar]
- 58. Sun Q, Yue P, Ying Z, Cardounel AJ, Brook RD, Devlin R, Hwang JS, Zweier JL, Chen LC, Rajagopalan S. Air pollution exposure potentiates hypertension through reactive oxygen species‐mediated activation of Rho/ROCK. Arterioscler Thromb Vasc Biol. 2008;28:1760–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. MacIntyre EA, Brauer M, Melen E, Bauer CP, Bauer M, Berdel D, Bergstrom A, Brunekreef B, Chan‐Yeung M, Klumper C, Fuertes E, Gehring U, Gref A, Heinrich J, Herbarth O, Kerkhof M, Koppelman GH, Kozyrskyj AL, Pershagen G, Postma DS, Thiering E, Tiesler CM, Carlsten C; TAG Study Group . GSTP1 and TNF gene variants and associations between air pollution and incident childhood asthma: the traffic, asthma and genetics (TAG) study. Environ Health Perspect. 2014;122:418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Osornio‐Vargas AR, Bonner JC, Alfaro‐Moreno E, Martinez L, Garcia‐Cuellar C, Ponce‐de‐Leon Rosales S, Miranda J, Rosas I. Proinflammatory and cytotoxic effects of Mexico City air pollution particulate matter in vitro are dependent on particle size and composition. Environ Health Perspect. 2003;111:1289–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Banday AA, Fazili FR, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and hypertension via mechanisms that involve nuclear factor‐kappaB and protein kinase c. J Am Soc Nephrol. 2007;18:1446–1457. [DOI] [PubMed] [Google Scholar]
- 62. World Health Organization . WHO's global air‐quality guidelines. Lancet. 2006;368:1302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Scanning electron microscopy analysis of PM2.5 samples.
Figure S2. The flowchart of animal studies.
Figure S3. Lung histology from control and exposed rats.
Figure S4. The ROS generation in RPT cells after PM2.5 exposure.