

Abstract
Background
Congestive heart failure (CHF) is a common cardiovascular disease that is often accompanied by ventricular arrhythmias. The decrease of the slow component of the delayed rectifier potassium current (IK s) in CHF leads to action potential (AP) prolongation, and the IK s is an important contributor to the development of ventricular arrhythmias. However, the molecular mechanisms underlying ventricular arrhythmias are still unknown.
Methods and Results
Kcna2 and Kcna2 antisense RNA (Kcna2 AS) transcript expression was measured in rat cardiac tissues using quantitative real‐time reverse transcription–polymerase chain reaction and Western blotting. There was a 43% reduction in Kcna2 mRNA in the left ventricular myocardium of rats with CHF. Kcna2 knockdown in the heart decreased the IKs and prolonged APs in cardiomyocytes, consistent with the changes observed in heart failure. Conversely, Kcna2 overexpression in the heart significantly attenuated the CHF‐induced decreases in the IKs, AP prolongation, and ventricular arrhythmias. Kcna2 AS was upregulated ≈1.7‐fold in rats with CHF and with phenylephrine‐induced cardiomyocyte hypertrophy. Kcna2 AS inhibition increased the CHF‐induced downregulation of Kcna2. Consequently, Kcna2 AS mitigated the decrease in the IKs and the prolongation of APs in vivo and in vitro and reduced ventricular arrhythmias, as detected using electrocardiography.
Conclusions
Ventricular Kcna2 AS expression increases in rats with CHF and contributes to reduced IK s, prolonged APs, and the occurrence of ventricular arrhythmias by silencing Kcna2. Thus, Kcna2 AS may be a new target for the prevention and treatment of ventricular arrhythmias in patients with CHF.
Keywords: IKs, Kcna2 antisense RNA, long noncoding RNA, QTc, ventricular arrhythmias
Subject Categories: Arrhythmias, Heart Failure
Clinical Perspective
What Is New?
Our present study demonstrated that a newly discovered 2.52‐kb native long noncoding RNA, Kcna2 antisense RNA, which is complementary to Kcna2 RNA, is stably expressed in the heart.
Our data showed that ventricular Kcna2 antisense RNA expression was increased in rats with heart failure, and this may contribute to the reduced slow component of the delayed rectifier potassium current, prolonged Aps, and the promotion of ventricular arrhythmias through silencing Kcna2 during heart failure.
What Are the Clinical Implications?
Our results indicate that Kcna2 antisense RNA might be a novel endogenous trigger of ventricular arrhythmias during heart failure and might be a potential target for the treatment of this disorder.
Introduction
With the increasing incidence of heart failure around the world, more and more patients are at risk of sudden cardiac death, and ventricular arrhythmias are a common cause of sudden cardiac death in patients with heart failure.1 Congestive heart failure (CHF) is a common cardiovascular disease associated with lethal ventricular arrhythmias.2 However, the molecular mechanisms underlying arrhythmias are still unknown.3 Delayed rectifier potassium channels are important for controlling the repolarization of several ion species in the heart, and decreases in the delayed rectifier potassium current (IK) in heart failure results in action potential (AP) prolongation, which is an important contributor to the development of ventricular arrhythmias.4, 5, 6 Kcna2, also known as Kv1.2, which is a subunit of the voltage‐gated shaker channel family, is one of the dominant ion channels in cardiac muscle and encodes IK.7, 8 The expression levels of myocardial Kcna2 are decreased in hypertrophic cardiac tissue, senescent hearts, diabetic hearts, and patients with obesity‐induced cardiomyopathy,9, 10, 11, 12 indicating that Kcna2 might be a key regulator of ventricular arrhythmias in CHF.
Recent studies have revealed that long noncoding RNAs (lncRNAs) play roles in gene regulation, and several lncRNAs have been identified in mammals and found to play regulatory roles in gene transcription and translation.13, 14 In particular, a few lncRNAs have been linked to the development of cardiovascular diseases.15, 16 We previously reported a newly discovered 2.52‐kb native lncRNA, Kcna2 antisense RNA (Kcna2 AS), which is complementary to Kcna2 RNA. Kcna2 AS may act as a biologically active regulator and participate in the induction and maintenance of neuropathic pain by specifically silencing Kcna2 expression in the neurons of rat dorsal root ganglia.17
In the present study, we investigated the role of Kcna2 AS in regulating the development of ventricular arrhythmias via alterations in the slow component of IK (IKs) density and APs in rats with CHF. Our data show that ventricular Kcna2 AS expression increased in rats with CHF, and this may contribute to a reduced IKs, prolonged APs, and the promotion of ventricular arrhythmias by silencing Kcna2 during heart failure. Thus, Kcna2 AS may be a new target for the prevention and treatment of ventricular arrhythmias in patients with heart failure.
Methods
The data, analytic methods, and study materials will be made available on request to other researchers for purposes of reproducing the results or replicating the procedure.
Experimental Animals
Male Sprague‐Dawley rats (10–12 weeks old), weighing 200 to 250 g, were obtained from the Animal Core Facility of Nanjing University. Rats were kept in a temperature‐controlled room with a 12:12‐hour light/dark cycle and maintained with access to food and water ad libitum. To minimize intraindividual and interindividual variability in behavioral outcome measures, animals were acclimated for 2 to 3 days before experiments were performed. All procedures were blind to treatment condition. Institutional review board approval was obtained from the ethical committee at the Nanjing Medical University, and this study is consistent with the established guidelines on the use and care of laboratory animals for biomedical research, published by the National Institutes of Health (No. 85‐23, revised 1996).
Human Heart Samples
Samples of human heart failure were collected from patients with end‐stage CHF. Control samples were obtained from the left ventricles of the patients who died without cardiac disease. Written informed consent was obtained from the family of prospective heart donors. The samples were obtained according to the regulations of the hospital ethics committee of the First Affiliated Hospital of Nanjing Medical University (2017‐SRFA‐081).
Recombinant Adeno‐Associated Virus 9 Constructs and Injection
Adeno‐associated virus 9 (AAV9) viral particles carrying 4 cDNAs, including full‐length Kcna2 cDNA, full‐length Kcna2 AS cDNA (AS), a Kcna2‐siRNA cDNA fragment (sense siRNA, SE siRNA, sequence 1, 5′‐GCACATCCACTTCTGCTAGAGACCTGGCT‐3′; sequence 2, 5′‐GGAACATGGAGGCTCGGGTACCCATCTGC‐3′; sequence 3, 5′‐CACCATTATTTCCAGCTCAAGTGAGAAGA‐3′), and a Kcna2 AS‐siRNA cDNA fragment (AS siRNA, sequence 1, 5′‐ATGCAGGCTATTATGCAATATCTGCATTA‐3′; sequence 2, 5′‐TTCATGAAAGCCATGATAAATATTCTGTG‐3′; sequence 3, 5′‐ACATAGTTTGTGTTAGCCAAGGTACAGTT‐3′) were amplified using nested reverse transcription–polymerase chain reaction (RT‐PCR). The restriction enzyme recognition sites were used at the 5′ and 3′ ends of the 3 fragments. The PCR products were cloned using the pGEM‐T Easy cloning kit (Invitrogen, Carlsbad, CA). The positive products were identified through restriction enzyme analysis (BbsI) and clone sequencing. Four fragments were ligated into the BbsI sites of the proviral plasmids (Applied Biological Materials Inc, Richmond, BC, Canada) to replace enhanced green fluorescent protein (EGFP). The 4 resulting vectors expressed or inhibited EGFP, Kcna2 AS, and Kcna2 under the control of the cytomegalovirus promoter.
Before the establishment of the animal models, some rats were injected with recombinant AAV9 at 2×1012 viral genomes per animal via the right internal jugular vein for 1 to 2 weeks.18 Fluorescence and quantitative real‐time RT‐PCR analyses were performed to determine whether the microinjection of AAV9 resulted in increased or decreased myocardial expression levels of Kcna2 and Kcna2 AS in vivo.
Transverse Aortic Constriction Model
Rats were placed in a supine position under anesthesia with 5% chloralic hydras on a temperature‐controlled surgical table and provided with room air using a small animal ventilator (SAR‐830/A; CWE, Inc, Ardmore, PA) for the transverse aortic constriction (TAC).19 The chest was opened at the level of the second rib, and the transverse aorta, between the innominate artery and the left common carotid artery, was ligated by tying a 6‐0 silk suture around the vessel and a 17‐gauge needle. The needle was gently removed after ligation. The rats were returned to a preheated chamber after surgery until they recovered from anesthesia. The rats were euthanized after 9 weeks.
Echocardiography
Eight weeks after TAC surgery, echocardiography was performed in rats anesthetized with 1.0% to 1.5% inhaled isoflurane using a Vevo 2100 system (VisualSonics Inc, Toronto, ON, Canada) with a 30‐MHz central frequency scanning head. The following parameters were measured from M‐mode images taken from the parasternal short‐axis view at the level of the papillary muscles: left ventricular ejection fraction and left ventricular fractional shortening. All measurements were taken from >3 beats and then averaged.
Electrocardiogram
Rats were subjected to ECG at 8 weeks after TAC after injection of AAV9. Cardiac arrhythmia was continuously monitored using a standard lead II ECG for 2 hours under anesthesia. The electrocardiographic measurement parameters were evaluated, and the incidence of arrhythmias was recorded. To induce arrhythmias, all groups of rats were injected with 1 mg/kg of norepinephrine.
Construction of Adenovirus Vectors
The full‐length Kcna2 AS cDNA (AS) and Kcna2 AS‐siRNA cDNA fragment (AS siRNA, sequence 1, 5′‐ATGCAGGCTATTATGCAATATCTGCATTA‐3′; sequence 2, 5′‐TTCATGAAAGCCATGATAAATATTCTGTG‐3′; sequence 3, 5′‐ACATAGTTTGTGTTAGCCAAGGTACAGTT‐3′) were inserted into adenovirus vectors (Applied Biological Materials Inc) in a reverse orientation. Adenovirus vectors were packaged in Human Embryonic Kidney 293 cells, and adenovirus vector–GFP (Applied Biological Materials Inc) was used as the control vector.
Primary Cardiomyocyte Culture and Treatment
Primary neonatal rat ventricular cardiomyocytes (NRVMs) were prepared, as described previously.20 Isolated NRVMs were purified via Percoll gradient centrifugation. The cells were maintained at a density of 1×106/mL in high‐glucose DMEM (Gibco, Pasadena, CA) supplemented with 10% fetal bovine serum (Gibco), 5% horse serum (HyClone, Logan, UT), 1% penicillin‐streptomycin, and 0.1 mmol/L 5′‐bromo‐2′‐deoxyuridine (Sigma, St Louis, MO) at 37°C for use in different experiments. After 48 hours, the medium was replaced by serum‐free high‐glucose DMEM to starve the cells. To induce cardiomyocyte hypertrophy that mimicked CHF, cells were treated for 48 hours with 50 μmol/L of phenylephrine (Sigma). In addition, cardiomyocytes were transfected with adenoviral vectors encoding GFP or Kcna2 AS‐siRNA at a multiplicity of infection of 50, according to the manufacturer's instructions. Using this approach, the transfection efficiency was 80% to 90% (Figure S1C).
RNA Extraction, RT‐PCR, and Quantitative Real‐Time RT‐PCR
Total RNA was extracted from cardiomyocytes or myocardial tissue using TRIzol reagent (Invitrogen) and treated with RNase‐free DNase I (1/20 μL; Promega Corp, Madison, WI). RNA was reverse transcribed using the ThermoScript reverse transcriptase with strand‐specific reverse transcription primers targeting the unique sequences for KCNA or Kcna2 AS RNA (Figure S2A). RT and PCR primers listed in Table S1 were determined from the University of California, Santa Cruz, genome database and our previous studies.17, 21 Template (1 μL) was amplified by PCR with TaKaRa Taq DNA polymerase (Clontech Laboratories, Inc) in 20 μL total reaction volume containing 0.5 μmol/L of PCR primer. PCR amplification consisted of 30 seconds at 94°C, 20 seconds at 56°C, and 20 seconds at 72°C for 35 cycles.
For quantitative real‐time RT‐PCR, cDNA was prepared as described previously. Each sample was run in triplicate in a 20 μL reaction containing 250 nmol/L forward and reverse primers, 10 μL of Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), and 20 ng of cDNA. Reactions were performed using the ABI‐7900 Real‐Time PCR Detection System. The primer sequences used in the study are listed in Table S1. The cycle parameters were set as follows: an initial 10‐minute incubation at 95°C, followed by 40 cycles of 95°C for 10 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. All data were normalized to GAPDH. The relative expression level of each gene was calculated using the 2−ΔΔCt method.
Western Blotting
Cardiac tissues were homogenized, and the cultured cells were ultrasonicated in radioimmunoprecipitation assay buffer (P0013C; Beyotime) with 1 mmol/L phenylmethylsulfonyl fluoride (ST505; Beyotime). After protein concentrations were measured, the samples were heated at 99°C for 5 minutes. After electrophoresis on 10% SDS‐PAGE gels, proteins were transferred to polyvinylidene difluoride membranes and blocked with 5% BSA in TBST buffer (10 mmol/L Tris [pH 7.5], 150 mmol/L NaCl, and 0.05% Tween‐20) for 2 hours at room temperature. The following primary antibodies were used: anti‐rabbit Kcna2 (1:200; Alomone Labs), anti‐rabbit kvβ1 (1:1000; Abcam, Cambridge, UK), and anti‐GAPDH (1:1000; Cell Signaling Technology, Boston, MA) at 4°C overnight. Proteins were detected using horseradish peroxidase–conjugated anti‐rabbit (Biosynthesis, Beijing, China) or anti‐mouse (Santa Cruz, Dallas, TX) antibodies and were visualized using the ChemiDoc XRS Imaging System (Bio‐Rad, Hercules, CA). Blot intensity was quantified via densitometry.
Cardiac Myocyte and Fibroblast Isolation From Adult Rat Hearts
Adult rat hearts were removed and mounted onto a modified Langendorff perfusion system for retrograde perfusion through the coronary circulation. The preparation was perfused with Ca2+‐containing Tyrode's solution, made up of the following (in mmol/L): NaCl 126, KCl 5.4, MgCl2 1, CaCl2 1.8, NaH2PO4 0.33, glucose 10, and HEPES 10 (pH 7.4) at 37°C until the effluent was clear of blood. The perfusion solution was then switched to Ca2+‐free Tyrode's solution for 20 minutes at a constant rate of 12 mL/min, followed by perfusion with the same solution with 100 to 150 kU/L collagenase type II (Worthington, Lakewood, NJ) and 1% BSA. The left ventricular epicardial layers were excised from the softened hearts, minced, and then placed in a Krebs‐Henseleit medium for whole‐cell patch‐clamp recording, containing the following (in mmol/L): glutamic acid 70, taurine 15, KCl 30, KH2PO4 10, MgCl2 0.5, EGTA 0.5, glucose 10, and HEPES.
For the isolation of cardiomyocytes and fibroblasts, the perfusion solution was prepared as described previously. After filtering, cell solution was settled to sedimentation for several minutes in a Falcon tube. The cell pellet and supernatant were transferred to individual Falcon tubes for further separation. The cell pellet was resuspended in transfer buffer and settled to precipitation. The initial supernatant was centrifuged first at 50g (3 minutes) and then 300g (5 minutes) before confirmation of noncardiomyocyte identity of pelleted cells by quantitative real‐time PCR for cardiomyocyte and fibroblast markers.
Whole‐Cell Patch‐Clamp Recording
To record the slowly activated IKs and the calcium channel current, the electrode resistance of the micropipettes was set at ranges from 4 to 6 MΩ. Cells were voltage clamped with an Axopatch‐700B amplifier (Molecular Devices, Sunnyvale, CA). The intracellular pipette solution contained the following (in mmol/L): potassium gluconate 120, KCl 20, MgCl2 2, EGTA 10, HEPES 10, and Mg‐ATP 4 (pH 7.3 with KOH, 310 mOsm). To minimize the Na and Ca component of the voltage‐gated IK, an extracellular solution was used, containing the following (in mmol/L): choline chloride 150, KCl 5, CdCl2 1, CaCl2 2, MgCl2 1, HEPES 10, and glucose 10 (pH 7.4 with Tris base, 320 mOsm). Signals were filtered at 1 kHz and digitized using a DigiData 1440A system with pCLAMP 10.6 software (Molecular Devices). Series resistance was compensated for by 60% to 80%. Cell membrane capacitances were acquired by reading the value for whole‐cell capacitance compensation directly from the amplifier. An online P/4 leak subtraction was performed to eliminate contributions from leak currents. Data were stored via a DigiData 1440A interface and analyzed using the pCLAMP 10.6 software package (Molecular Devices).
We then switched the recording to the current clamp mode to record APs. The internal pipette solution for the AP recordings contained the same components used for the IK recordings, except that the EGTA concentration was 0.05 mmol/L. 4‐Aminopyridine (1 mmol/L) was used to inhibit the transient outward potassium current, and external glyburide (10 mol/L) plus internal Mg‐ATP (5 mmol/L) were used to prevent the ATP‐sensitive IK. The sodium channel current and calcium channel current were inactivated by holding the membrane at −20 mV. All experiments were performed at room temperature.
Statistical Analysis
Kolmogorov‐Smirnov test was used to examine the distributions of the data. When the data fitted normal distribution, the results were given as the mean±SEM and statistical significance was assessed using 1‐way ANOVAs or Student t tests. Differences between groups were analyzed using unpaired 2‐tailed Student t test. When ANOVAs showed significant differences, pairwise comparisons between means were tested using the post hoc Tukey method or the Fisher protected least significant difference post hoc tests (SigmaStat, San Jose, CA). When the data did not have a normal distribution, the results were given as medians (25th–75th percentile) and the between‐group difference was analyzed using Mann‐Whitney U test. P<0.05 was considered statistically significant. The Fisher exact test was used to check for the significance of frequency data.
Results
Identity of Kcna2 AS and Kcna2 mRNA in Rat Hearts
We previously identified the expression of Kcna2 AS in the dorsal root ganglion of several species, including rats, mice, monkeys, and humans.17 Herein, we also detected this transcript in the cardiac myocytes and fibroblasts of rats (Figure S2B). There were no significant differences in Kcna2 AS expression among the left atrium, right atrium, left ventricle, and right ventricle (Figure S2C). Kcna2 mRNA and protein levels were higher in the atrium than in the ventricle (P<0.05), but there was no difference between the left and right ventricles (Figure S2D and S2E).
Changes in Kcna2 AS and Kcna2 Expression After Heart Failure
We then examined whether the expression of cardiac Kcna2 AS is altered in rats with CHF using the TAC model (Figure S3A). Notably, the ventricular Kcna2 AS levels were increased (≈1.7‐fold), whereas the Kcna2 mRNA levels were decreased (41%), in the rats with heart failure compared with the levels in the control group (Figure 1A). Consistent with these results, the Kcna2 protein levels were also reduced in the ventricles of the rats with heart failure compared with the levels in the control group (Figure 1B). Similar results were found in association with phenylephrine‐induced cardiomyocyte hypertrophy and a human heart failure sample (Figure 1C and 1D, Figures S3B and S4).
Figure 1.

Changes in Kcna2 antisense RNA (AS) and Kcna2 expression after congestive heart failure (CHF). A, Kcna2 AS levels were increased, and Kcna2 mRNA levels were reduced, in the hearts of rats with CHF (n=6). Control indicates sham surgery. B, Kcna2 protein levels were decreased in rat hearts with CHF (n=6). C, Quantitative real‐time RT‐PCR results for Kcna2 AS and Kcna2 mRNA expression levels in phenylephrine‐induced cardiomyocyte hypertrophy (n=6). Control indicates untreated group; hypertrophy, treated with 50 μmol/L phenylephrine. D, Western blot showing Kcna2 expression levels in phenylephrine‐induced cardiomyocyte hypertrophy (n=6). *P<0.05 vs control, **P<0.01 vs control, ***P<0.001 vs control.
The Role of Kcna2 in Regulating IKs and APs in Rats With CHF
Previous studies have shown that decreased IKs and prolonged APs contribute to the development of ventricular arrhythmias in association with CHF and that Kcna2 is a key regulator of the IK in the heart.22, 23 To verify the role of Kcna2 in regulating the IKs and APs in the ventricles, rats were injected with AAV9‐Kcna2 and AAV9‐Kcna2‐siRNA to overexpress or knock down myocardial Kcna2, respectively, and were then subjected to the TAC procedure. Whole‐cell current‐clamp recording was performed from 8 to 12 weeks after injection. As expected, the IKs was reduced in rats with CHF compared with the IKs in the controls. Compared with the control group, IKs density was increased and APs were shortened significantly in the AAV9‐Kcna2–treated group. In contrast, Kcna2 knockdown resulted in a reduced IKs density and prolonged APs in rats with CHF. The AAV9‐EGFP injections did not affect the current density and there was no difference between the EGFP group and the heart failure group (Figure 2A through 2C). In addition, AAV9 injections did not alter the calcium channel current density (Figure 2D), indicating that the alterations in the APs were not caused by changes in the calcium current. Meanwhile, we also found that the IKs was significantly decreased and APs were prolonged in adult rat cardiomyocytes after Kcna2 knockdown compared with the values in the control and healthy rats injected with AAV9‐EGFP, which is consistent with the changes observed in association with heart failure (Figure 2E and 2F).
Figure 2.
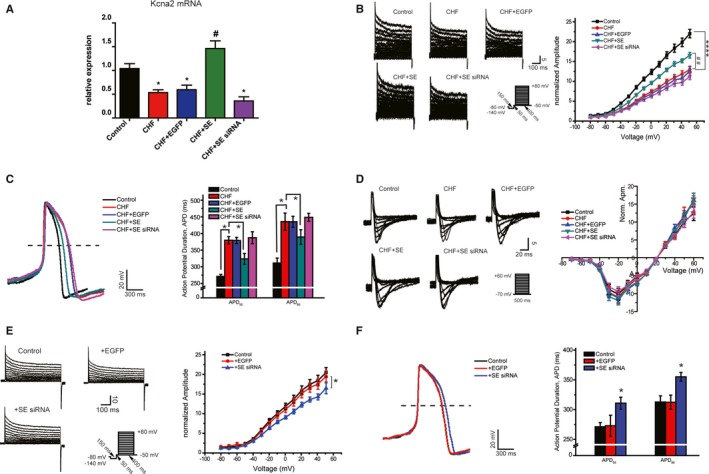
The role of Kcna2 in regulating the decreased slow component of the delayed rectifier potassium current (IKs) and prolonged action potentials (APs) in congestive heart failure (CHF). A, Quantitative real‐time reverse transcription–polymerase chain reaction results for Kcna2 in heart samples from adult rats treated with various constructs (n=6). B and E, IKs in cardiomyocytes isolated from adult rats treated with various constructs (n=6). Control indicates sham surgery; SE siRNA, knockdown of Kcna2 expression in healthy rat hearts; CHF+EGFP, enhanced green fluorescent protein (EGFP) expressing control in rat hearts with CHF; CHF+SE, overexpression of Kcna2 in rat hearts with CHF; CHF+SE siRNA, knockdown of Kcna2 in rat hearts with CHF. C and F, Representative traces of APs in adult rat cardiomyocytes (n=6). D, Representative traces of calcium channel current density (n=6). Data are mean±SEM (B and E). APD indicates AP duration. *P<0.05 vs control, ****P<0.0001 vs control, # P<0.05 vs CHF, ## P<0.01 vs CHF.
The Downregulation of Kcna2 May Lead to Ventricular Arrhythmias in CHF
The electrocardiographic measurement parameters for all rat groups are presented in the Table. The heart rates were higher and the QT intervals were longer in the CHF group than in the control group and in the CHF+SE group, whereas no significant differences were observed among the groups in the P duration, PR interval, or QRS duration. The norepinephrine‐induced incidences of premature ventricular beats, ventricular tachycardia, and ventricular fibrillation in arrhythmias are shown in Figure 3A. We found that AAV9‐Kcna2 transfection in vivo into the hearts of rats with CHF significantly suppressed the incidence of norepinephrine‐induced arrhythmias. In contrast, AAV9‐Kcna2‐siRNA injections in the CHF group promoted norepinephrine‐induced arrhythmias (Figure 3B).
Table 1.
Electrocardiographic Parameters Derived From ECG Traces of Rat Groups
| Group | Heart Rate (bpm) | P Duration (ms) | PR Interval (ms) | QRS Duration (ms) | QT Interval (ms) | QTc Interval (ms) |
|---|---|---|---|---|---|---|
| Control | 290 (262–300) | 20.59 (19.60–22.08) | 55.05 (52.75–56.34) | 21.26 (20.62–22.47) | 68.77 (60.19–73.16) | 140.60 (132.10–158.40) |
| CHF+SE siRNA | 346 (321–368) | 24.91 (10.16–42.12) | 53.11 (47.24–58.75) | 24.53 (17.00–28.50) | 98.46 (92.35–109.60) | 232.62 (216.20–250.50) |
| CHF+SE | 328 (320–336)a | 18.17 (16.25–20.98) | 52.64 (51.56–54.14) | 21.57 (19.65–22.50) | 79.33 (73.07–88.46)a | 184.40 (177.50–188.30)a |
| CHF | 352 (315–383)b | 22.94 (12.16–35.38) | 55.85 (43.25–64.30) | 26.92 (19.18–30.92) | 89.01 (82.55–92.10)b | 205.10 (197.40–222.50)b |
| CHF+EGFP | 343 (340–376) | 23.98 (14.21–36.88) | 56.75 (53.62–60.75) | 23.50 (18.50–28.51) | 87.92 (80.28–98.14) | 203.74 (191.70–233.10) |
| CHF+AS | 372 (335–398)a | 31.11 (18.15–43.44) | 56.48 (48.62–61.80) | 24.50 (16.30–32.40) | 95.67 (89.51–106.80)a | 230.70 (218.20–237.00)a |
| CHF+AS siRNA | 331 (319–339)a | 26.53 (11.07–37.23) | 62.85 (53.82–62.60) | 19.50 (16.97–26.50) | 75.13 (69.68–82.13)a | 178.10 (165.40–187.80)a |
Data are given as median (25th–75th percentile). AS indicates antisense RNA; bpm, beats/min; CHF, congestive heart failure; EGFP, enhanced green fluorescent protein; QTc, corrected QT; and SE, sense, Kcna2.
Significant difference at P<0.05 when compared with the CHF group (n=10).
Significant difference at P<0.05 when compared with the control group (n=10).
Figure 3.
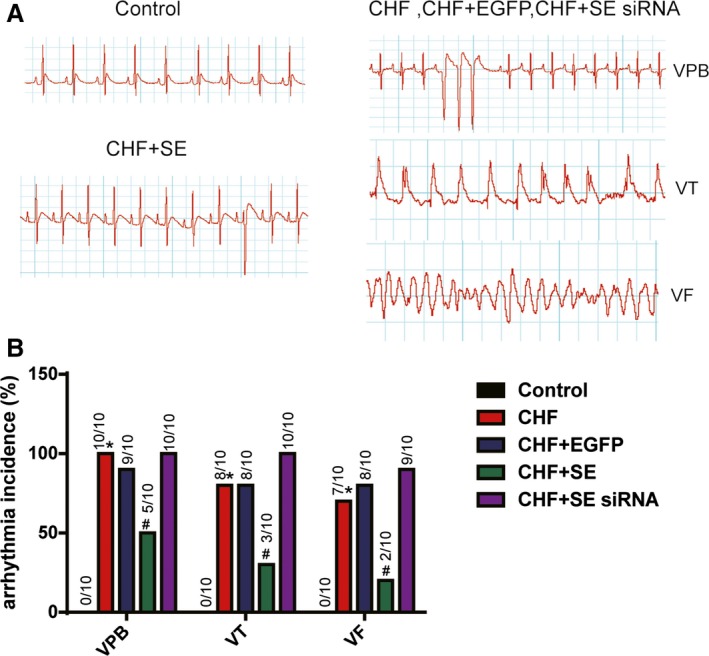
The role of Kcna2 in ventricular arrhythmias in congestive heart failure (CHF). A, Examples of norepinephrine‐induced arrhythmias in hearts from rats with CHF, and normal ECGs from control hearts (n=10). Arrhythmias included ventricular premature beat (VPB), ventricular tachycardia (VT), and ventricular fibrillation (VF). B, Data are expressed as the percentage incidence, and the actual incidence is indicated by the numbers above each bar. EGFP indicates enhanced green fluorescent protein; and SE, sense, Kcna2. *P<0.05 vs control, # P<0.05 vs CHF.
Kcna2 AS Contributes to Ventricular Arrhythmias in CHF
Next, we investigated the effects of Kcna2 AS on the IK and APs in rats with CHF. We injected AAV9‐Kcna2 AS‐siRNA and found that blocking Kcna2 AS markedly attenuated the CHF‐induced downregulation of IKs and prolongation of APs (P<0.05). Conversely, the overexpression of this transcript, resulting from the injection of AAV9‐Kcna2 AS, promoted the decrease of the IKs and prolonged repolarization (Figure 4A through 4C). Similarly, a decreased IKs and prolonged APs were also observed in association with phenylephrine‐induced cardiomyocyte hypertrophy, which were mitigated via the knockdown of Kcna2 AS (Figure 4D through 4F).
Figure 4.

Kcna2 antisense RNA (AS) controls slow component of the delayed rectifier potassium current (IKs) and action potentials (APs) in congestive heart failure (CHF). A, Quantitative real‐time reverse transcription–polymerase chain reaction (qRT‐PCR) results for Kcna2 AS in heart samples from adult rats treated with various constructs (n=6). IKs (B) and AP (C) records from cardiomyocytes isolated from adult rats treated with various constructs (n=6). Control indicates sham surgery; CHF, rats with CHF; CHF+EGFP, enhanced green fluorescent protein (EGFP)–expressing control in rat hearts with CHF. D, qRT‐PCR results for Kcna2 AS in cultured cardiomyocytes treated with various constructs (n=6).IKs (E) and AP (F) recordings from freshly isolated neonatal rat ventricular myocytes (n=6). Control indicates untreated group; phenylephrine, treated with 50 μmol/L phenylephrine; phenylephrine+AS siRNA, transfection of adenovirus vector (ADV)–Kcna2 AS‐siRNA into phenylephrine‐treated cells; phenylephrine+NC, transfection of ADV‐GFP into phenylephrine‐treated cells. Data are mean±SEM (B and C). APD indicates AP duration; and Iend indicates the last 10ms of tail current. *P<0.05 vs control, **P<0.01 vs CHF, ***P<0.01 vs phenylephrine, ****P<0.0001 vs control, # P<0.05 vs CHF.
To further investigate whether Kcna2 AS is an arrhythmogenic factor, standard lead II ECG tests were performed in this group (Table). Kcna2 AS knockdown markedly suppressed the norepinephrine‐induced arrhythmias, whereas Kcna2 AS overexpression promoted norepinephrine‐induced arrhythmogenesis, in rats with CHF (Figure 5A and 5B).
Figure 5.

Kcna2 antisense RNA (AS) is arrhythmogenic in congestive heart failure (CHF). A, Examples of norepinephrine‐induced arrhythmias in hearts from rats with CHF and normal ECGs in control hearts (n=10). Arrhythmias included ventricular premature beats (VPBs), ventricular tachycardia (VT), and ventricular fibrillation (VF). B, Data are expressed as the percentage incidence, and the actual incidence is indicated by the numbers above each bar. EGFP indicates enhanced green fluorescent protein. *P<0.05 vs control, # P<0.05 vs CHF.
Kcna2 AS Regulates Arrhythmias by Silencing Kcna2
To further investigate the potential molecular mechanism underlying the role of Kcna2 AS in heart failure, we examined cardiac Kcna2 AS‐induced alterations in Kcna2 expression. As shown in Figure 6A and 6C, Kcna2 AS overexpression induced significant decreases in Kcna2 mRNA and protein expression in cultured cardiomyocytes (P<0.05); however, no change was observed in kvβ1 expression. In contrast, Kcna2 AS inhibition increased basal Kcna2 mRNA and protein expression levels in cultured cardiomyocytes (Figure 6B and 6D). More strikingly, silencing Kcna2 AS in the hearts of rats with CHF resulted in a notable upregulation of Kcna2 protein levels compared with the untreated heart failure group. The Kcna2 mRNA and protein levels were significantly downregulated by overexpressing Kcna2 AS with AAV9 in healthy rats compared with controls, which indicated an inverse correlation between the levels of lncRNA Kcna2 AS and Kcna2 expression (Figure 6E and 6F).
Figure 6.
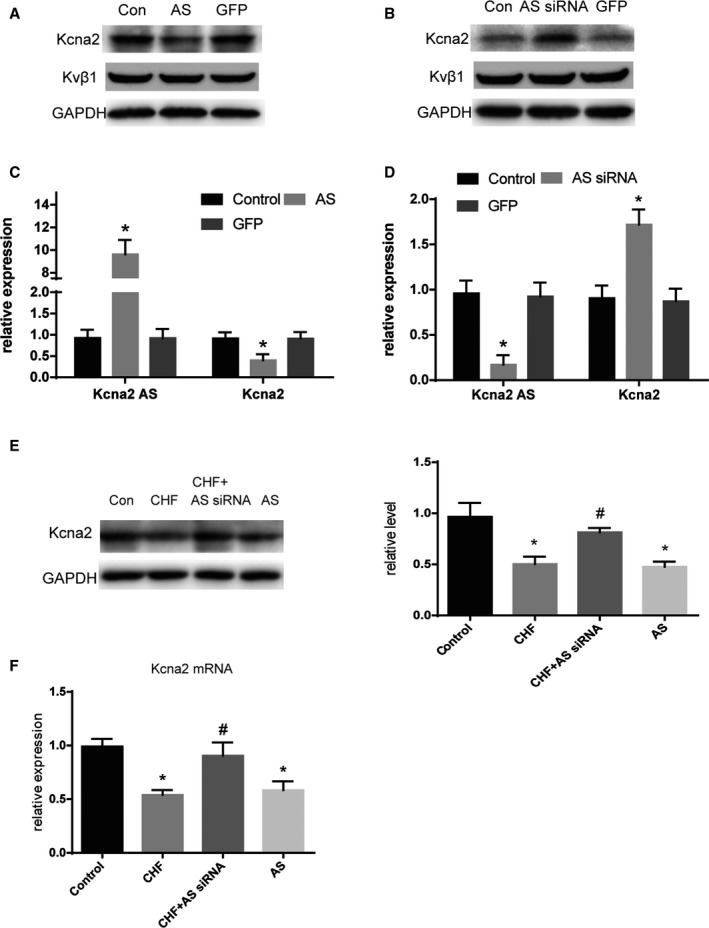
Kcna2 is a target gene of Kcna2 antisense RNA (AS). Western blot analysis for Kcna2 and Kvβ1 (A) and quantitative real‐time reverse transcription–polymerase chain reaction (qRT‐PCR) results for Kcna2 AS and the Kcna2 gene (C) in cultured cardiomyocytes treated with adenovirus vector (ADV)–Kcna2 AS (n=6). Western blot analysis for Kcna2 and Kvβ1 (B) and qRT‐PCR results for Kcna2 AS and the Kcna2 gene (D) in cultured cardiomyocytes treated with ADV‐Kcna2 AS‐siRNA (n=6). E and F, Western blot and qRT‐PCR analysis for Kcna2 in heart samples from normal rats, rats with congestive heart failure (CHF), rats with CHF treated with adeno‐associated virus 9 (AAV9)–Kcna2 AS‐siRNA, and normal rats treated with AAV9‐Kcna2 AS (n=6). GFP indicates green fluorescent protein. *P<0.05 vs control, # P<0.05 vs CHF.
Discussion
Potassium channels play a crucial role in abnormal cardiac electrical function and sudden cardiac death.24 The modulation of potassium channel expression is also a critical factor in controlling cardiac currents and APs during physiological and pathological states. Herein, we demonstrated that ventricular Kcna2 AS levels were increased during heart failure and that this may contribute to the reduction of the IKs and AP prolongation by silencing Kcna2 during heart failure resulting from ventricular arrhythmias. Kcna2 AS may be a new target for the prevention and treatment of ventricular arrhythmias in patients with heart failure.
Potassium channels, a major component responsible for the membrane current, contribute to the electrical activity of the heart.25, 26 There are >15 different potassium channel genes that have been quantitatively demonstrated to exist in rat atrial and ventricular muscles, with Kcna2, kv1.4, kv1.5, kv2.1, and kv4.2 being expressed at significant levels.7, 27 Western blot analyses of atrial and ventricular membrane proteins confirmed the presence of Kcna2 at 75 kDa in adult rat heart membranes.8 Furthermore, Kcna2 has also been observed in human ventricular tissue.5 Herein, Kcna2 RNA and protein expression was detected (Figure S2). Moreover, Kcna2 channels have been shown to play a pivotal role in maintaining the resting membrane potential and, consequently, regulating cellular excitability in neurons.28 There is direct evidence showing that the IKs is generated by Kcna2 in Xenopus oocytes, rabbit vascular myocytes, pulmonary arterial smooth muscle cells, rat mesenteric artery smooth muscle cells, and canine colonic circular smooth muscles.29, 30, 31, 32, 33 Specifically, Kcna2 was demonstrated to contribute to the IKs, rather than the transient outward potassium current, in adult rat myocytes.8 We verified that Kcna2 channels encode IKs, as shown in Figure S5. Furthermore, the molecular basis for changes in K+ current function associated with heart failure has been examined during the past 8 years.34, 35, 36, 37 The expression of Kcna2 channels was significantly decreased in rat hearts with ventricular hypertrophy.11 We found that ventricular Kcna2 levels decreased in rats with heart failure and in association with phenylephrine‐induced cardiomyocyte hypertrophy. These findings support that Kcna2 is the one that causes reduced IKs and prolonged APs. We also observed that the Kcna2 knockdown that mimics the heart failure–induced Kcna2 downregulation decreased the IKs and prolonged APs in normal rats. However, Kcna2 overexpression significantly attenuated heart failure–induced abnormal IK in rats, suggesting that upregulation of Kcna2 might be a potential therapeutic strategy for the prevention of ventricular arrhythmias in patients with heart failure. In fact, Kcna2 channels have been identified as a target for antiarrhythmic and bradycardic drugs (class I, III, and IV) in clinical settings.38, 39
lncRNAs are a class of functional RNAs that do not encode a protein but play a significant role in the occurrence and development of diseases via the control of imprinting, chromatin modification, transcription, splicing, translation, and other processes.40, 41, 42 Emerging evidence has shown that lncRNAs are involved in the regulation of several genes related to the development of several diseases. We previously identified a 2.52‐kb lncRNA, Kcna2 AS, that for most of its sequence, including the 3′ untranslated region and part of the 5′ untranslated region, complemented the Kcna2 gene. Kcna2 AS overexpression reduces Kcna2 mRNA and protein expression. Herein, we first showed that Kcna2 AS was significantly upregulated in rats with heart failure. Interestingly, Kcna2 AS knockdown also ameliorated heart failure–induced decreases in Kcna2 mRNA and protein expression. Increases in endogenous Kcna2 AS expression have been shown to be induced by the overexpression of the myeloid zinc finger protein 1 (MZF1), a transcriptional activator, via specific and selective binding to the Kcna2 AS promoter.17, 21 In addition, MZF1 can inhibit cardiac cell differentiation through the activation of the Nkx2.5 cardiac enhancer during embryonic heart development. MZF1 gene expression is upregulated in cardiac progenitor cells but not in adult cardiomyocytes, indicating that the occurrence of MZF1 in adult cardiomyocytes might be associated with cardiac dysfunction.43 CHF‐induced increases of endogenous Kcna2 AS RNA levels probably were triggered as a result of MZF1 activation in adult rat hearts. It seems that MZF1 may be the regulator of the regulatory network composed. These possibilities have not been investigated and will be explored in future studies.
Furthermore, IKs upregulation and AP shortening were caused by microinjections of Kcna2 AS–siRNA into the hearts of rats with CHF, and enhanced Kcna2 AS expression in rats with heart failure promoted heart failure–induced reductions in the IKs and delayed APs. These results may be related to the functional role of Kcna2 levels in rat cardiac myocytes, which may contribute to ventricular arrhythmias during heart failure. Recently, an increasing number of factors affecting IKs have been reported. A study showed that blockade of the IKs contributes to drug‐induced long‐QT syndrome, particularly when repolarization reversal is inhibited.44 The inhibition of KCNQ1 and KCNE1 (encoding IKs) expression on plasma membranes has been shown to cause QT prolongation.45 In our study, Kcna2 (encoding IKs) knockdown in rat hearts also prolonged QT intervals. We found that QT intervals were significantly increased in rats with heart failure and that Kcna2 overexpression inhibited QT prolongation. The Bazett QT formula was mainly used for corrections. Long‐QT syndrome has a high probability of resulting in life‐threatening cardiac events, particularly increases in the risk of sudden cardiac death,46, 47 suggesting that Kcna2 AS acts as an arrhythmogenic factor promoting QT prolongation, which plays a newly discovered role in ventricular arrhythmias by regulating the IKs and APs.
To investigate the role of Kcna2 AS and Kcna2 in relation to clinical situations, we measured the incidence of norepinephrine‐induced arrhythmias, including premature ventricular beats, ventricular tachycardia, and ventricular fibrillation, using a standard lead II ECG (Figures 3 and 5). We found that the group receiving CHF therapy (injections of AAV9‐Kcna2) showed obviously attenuated risks of arrhythmias. Conversely, inhibited Kcna2 expression in rats with CHF increased the risk of arrhythmias. In patients with CHF, QT interval prolongation is not corrected, leading to torsade de pointes and ventricular fibrillation, which are life threatening.48 Potassium flux has long been recognized as a critical effector of myocardial repolarization via rare mutations that underlie long‐QT syndrome.49 Therefore, the QT interval prolongation and dispersion associated with serious arrhythmias and infusions of potassium can correct QT abnormalities in patients with CHF.50 Studies show that because of island‐shaped M‐cell distribution and low IKs density, increased transmural dispersion of APs and repolarization occur in acquired long‐QT syndrome. These prolonged APs are helpful if early afterdepolarizations occur, which can trigger torsade de pointes.51, 52 This indicates that the lack of Kcna2 may cause malignant ventricular arrhythmias that are regulated by Kcna2 AS. In addition, we also observed an increased expression of Kcna2 AS in human heart failure tissue. This indicates that the Kcna2 AS may have similar function between the rat and the human.
Several limitations should be highlighted for the present study. First, the previously reported target gene of Kcna2 AS, MZF1, should be identified to further clarify the molecular mechanisms by which the increasing ventricular Kcna2 AS levels contribute to ventricular arrhythmias. Second, using AAV in the rat might create a mosaic distribution of Kcna2 and Kcna2 AS expression throughout the heart. We will verify the role of Kcna2 and Kcna2 AS underlying ventricular arrhythmias with transgenic or knockout mice in future studies. Third, patients with myocardial infarction often have endocardial/epicardial scars that are an important anatomical basis for arrhythmia. Although our results indicated that Kcna2 AS might be a novel trigger of ventricular arrhythmias in CHF, the exact role of Kcna2 AS in the ischemic cardiac model, especially left descending artery ligation‐induced myocardial infarction, needs to be further identified.
Conclusion
Our data demonstrate that increased Kcna2 AS levels may lead to the dysregulation of Kcna2 channel expression and APs and an increased incidence of ventricular arrhythmias in association with heart failure. Kcna2 AS might be a novel endogenous trigger of ventricular arrhythmias during heart failure and might be a potential target for the treatment of this disorder.
Sources of Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81270255, 81570363, 81471611, and 81671610), a grant from the Natural Science Foundation of Jiangsu Province (No. BK20171051), the “333 Project” of Jiangsu Province (No. BRA2015326), a grant from the Natural Science Foundation of Nanjing Medical University (No. 2016NJMU060), the Jiangsu Province Health Development Project With Science and Education (No. QNRC2016857), the Postgraduate Research and Innovation Project in Jiangsu Province (grant JX22013360), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Disclosures
None.
Supporting information
Table S1. Sequences of Primers
Figure S1. Transfection efficiency using fluorescence in vivo and in vitro.
Figure S2. Identification and expression of Kcna2 AS and Kcna2 mRNA in rat hearts.
Figure S3. The evaluation of cardiac dysfunction in the respective in vivo and in vitro models.
Figure S4. Expression of Kcna2 AS and Kcna2 in human normal and heart failure tissue.
Figure S5. Kv1.2 channel encoding the Iks current.
Acknowledgments
We thank Prof Yuan‐Xiang Tao (Department of Anesthesiology, New Jersey Medical School, Rutgers, the State University of New Jersey, Newark, NJ) for his kind help. We also thank Prof Dao‐Wu Wang (cardiovascular laboratory, First Affiliated Hospital of Nanjing Medical University).
(J Am Heart Assoc. 2017;6:e005965 DOI: 10.1161/JAHA.117.005965.)29263036
References
- 1. Thompson BS. Sudden cardiac death and heart failure. AACN Adv Crit Care. 2009;20:356–365. [DOI] [PubMed] [Google Scholar]
- 2. Ajayi OE, Abiodun OO, Akintomide AO, Adebayo RA, Ogunyemi SA, Balogun MO , Bamikole OJ, Ajibare AO, Ajayi AA. Pattern of arrhythmias among Nigerians with congestive heart failure. Int J Gen Med. 2015;8:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Packer M. Lack of relation between ventricular arrhythmias and sudden death in patients with chronic heart failure. Circulation. 1992;85:I50–I56. [PubMed] [Google Scholar]
- 4. Charpentier F, Merot J, Loussouarn G, Baro I. Delayed rectifier K(+) currents and cardiac repolarization. J Mol Cell Cardiol. 2010;48:37–44. [DOI] [PubMed] [Google Scholar]
- 5. Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–1393. [DOI] [PubMed] [Google Scholar]
- 6. Michael G, Xiao L, Qi XY, Dobrev D, Nattel S. Remodelling of cardiac repolarization: how homeostatic responses can lead to arrhythmogenesis. Cardiovasc Res. 2009;81:491–499. [DOI] [PubMed] [Google Scholar]
- 7. Dixon JE, McKinnon D. Quantitative analysis of potassium channel mRNA expression in atrial and ventricular muscle of rats. Circ Res. 1994;75:252–260. [DOI] [PubMed] [Google Scholar]
- 8. Barry DM, Trimmer JS, Merlie JP, Nerbonne JM. Differential expression of voltage‐gated K+ channel subunits in adult rat heart: relation to functional K+ channels? Circ Res. 1995;77:361–369. [DOI] [PubMed] [Google Scholar]
- 9. Ren J, Duan J, Thomas DP, Yang X, Sreejayan N, Sowers JR, Leri A, Kajstura J, Gao F, Anversa P. IGF‐I alleviates diabetes‐induced RhoA activation, eNOS uncoupling, and myocardial dysfunction. Am J Physiol Regul Integr Comp Physiol. 2008;294:R793–R802. [DOI] [PubMed] [Google Scholar]
- 10. Ren J, Li Q, Wu S, Li SY, Babcock SA. Cardiac overexpression of antioxidant catalase attenuates aging‐induced cardiomyocyte relaxation dysfunction. Mech Ageing Dev. 2007;128:276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee JK, Nishiyama A, Kambe F, Seo H, Takeuchi S, Kamiya K, Kodama I, Toyama J. Downregulation of voltage‐gated K(+) channels in rat heart with right ventricular hypertrophy. Am J Physiol. 1999;277:H1725–H1731. [DOI] [PubMed] [Google Scholar]
- 12. Relling DP, Esberg LB, Fang CX, Johnson WT, Murphy EJ, Carlson EC, Saari JT, Ren J. High‐fat diet‐induced juvenile obesity leads to cardiomyocyte dysfunction and upregulation of Foxo3a transcription factor independent of lipotoxicity and apoptosis. J Hypertens. 2006;24:549–561. [DOI] [PubMed] [Google Scholar]
- 13. Gibb EA, Brown CJ, Lam WL. The functional role of long non‐coding RNA in human carcinomas. Mol Cancer. 2001;10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2001;21:354–361. [DOI] [PubMed] [Google Scholar]
- 15. Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, Tao ZF, Song YC, Chen Q, Jiang Q. lncRNA‐MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116:1143–1156. [DOI] [PubMed] [Google Scholar]
- 16. Vausort M, Wagner DR, Devaux Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ Res. 2014;115:668–677. [DOI] [PubMed] [Google Scholar]
- 17. Zhao X, Tang Z, Zhang H, Atianjoh FE, Zhao JY, Liang L, Wang W, Guan X, Kao SC, Tiwari V, Gao YJ, Hoffman PN, Cui H, Li M, Dong X, Tao YX. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat Neurosci. 2013;16:1024–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muller OJ, Schinkel S, Kleinschmidt JA, Katus HA, Bekeredjian R. Augmentation of AAV‐mediated cardiac gene transfer after systemic administration in adult rats. Gene Ther. 2008;15:1558–1565. [DOI] [PubMed] [Google Scholar]
- 19. Yoshioka K, Otani H, Shimazu T, Fujita M, Iwasaka T, Shiojima I. Sepiapterin prevents left ventricular hypertrophy and dilatory remodeling induced by pressure overload in rats. Am J Physiol Heart Circ Physiol. 2015;309:H1782–H1791. [DOI] [PubMed] [Google Scholar]
- 20. Liu X, Xiao J, Zhu H, Wei X, Platt C, Damilano F, Xiao C, Bezzerides V, Bostrom P, Che L, Zhang C, Spiegelman BM, Rosenzweig A. miR‐222 is necessary for exercise‐induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015;21:584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Z, Gu X, Sun L, Wu S, Liang L, Cao J, Lutz BM, Bekker A, Zhang W, Tao YX. Dorsal root ganglion myeloid zinc finger protein 1 contributes to neuropathic pain after peripheral nerve trauma. Pain. 2015;156:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee HC, Rudy Y, Po‐Yuan P, Sheu SH, Chang JG, Cui J. Modulation of KCNQ1 alternative splicing regulates cardiac IKs and action potential repolarization. Heart Rhythm. 2013;10:1220–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishii K, Norota I, Obara Y. Endocytic regulation of voltage‐dependent potassium channels in the heart. J Pharmacol Sci. 2012;120:264–269. [DOI] [PubMed] [Google Scholar]
- 24. Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379–385. [DOI] [PubMed] [Google Scholar]
- 25. Carmeliet E, Biermans G, Callewaert G, Vereecke J. Potassium currents in cardiac cells. Experientia. 1987;43:1175–1184. [DOI] [PubMed] [Google Scholar]
- 26. Anumonwo JMB, Freeman LC, Kwok WM, Kass RS. Potassium channels in the heart: electrophysiology and pharmacological regulation. Cardiovasc Drug Rev. 1991;9:299–316. [Google Scholar]
- 27. Ma ML, Watanabe K, Watanabe H, Hosaka Y, Komura S, Aizawa Y, Yamamoto T. Different gene expression of potassium channels by thyroid hormone and an antithyroid drug between the atrium and ventricle of rats. Jpn Heart J. 2003;44:101–110. [DOI] [PubMed] [Google Scholar]
- 28. Zhang G, Edmundson M, Telezhkin V, Gu Y, Wei X, Kemp PJ, Song B. The role of Kv1.2 channel in electrotaxis cell migration. J Cell Physiol. 2016;231:1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Russell SN, Publicover NG, Hart PJ, Carl A, Hume JR, Sanders KM, Horowitz B. Block by 4‐aminopyridine of a Kv1.2 delayed rectifier K+ current expressed in Xenopus oocytes. J Physiol. 1994;481(pt 3):571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kerr PM, Clement‐Chomienne O, Thorneloe KS, Chen TT, Ishii K, Sontag DP, Walsh MP, Cole WC. Heteromultimeric Kv1.2‐Kv1.5 channels underlie 4‐aminopyridine‐sensitive delayed rectifier K(+) current of rabbit vascular myocytes. Circ Res. 2001;89:1038–1044. [DOI] [PubMed] [Google Scholar]
- 31. Lu Y, Hanna ST, Tang G, Wang R. Contributions of Kv1.2, Kv1.5 and Kv2.1 subunits to the native delayed rectifier K(+) current in rat mesenteric artery smooth muscle cells. Life Sci. 2002;71:1465–1473. [DOI] [PubMed] [Google Scholar]
- 32. Yuan XJ, Wang J, Juhaszova M, Golovina VA, Rubin LJ. Molecular basis and function of voltage‐gated K+ channels in pulmonary arterial smooth muscle cells. Am J Physiol. 1998;274:L621–L635. [DOI] [PubMed] [Google Scholar]
- 33. Hart PJ, Overturf KE, Russell SN, Carl A, Hume JR, Sanders KM, Horowitz B. Cloning and expression of a Kv1.2 class delayed rectifier K+ channel from canine colonic smooth muscle. Proc Natl Acad Sci U S A. 1993;90:9659–9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. [DOI] [PubMed] [Google Scholar]
- 35. Akar FG, Wu RC, Juang GJ, Tian Y, Burysek M, Disilvestre D, Xiong W, Armoundas AA, Tomaselli GF. Molecular mechanisms underlying K+ current downregulation in canine tachycardia‐induced heart failure. Am J Physiol Heart Circ Physiol. 2005;288:H2887–H2896. [DOI] [PubMed] [Google Scholar]
- 36. Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, Nattel S. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004;561:735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1‐KCNE1 potassium channel. Science. 2002;295:496–499. [DOI] [PubMed] [Google Scholar]
- 38. Yamagishi T, Ishii K, Taira N. Antiarrhythmic and bradycardic drugs inhibit currents of cloned K+ channels, KV1.2 and KV1.4. Eur J Pharmacol. 1995;281:151–159. [DOI] [PubMed] [Google Scholar]
- 39. Rolf S, Haverkamp W, Borggrefe M, Musshoff U, Eckardt L, Mergenthaler J, Snyders DJ, Pongs O, Speckmann EJ, Breithardt G, Madeja M. Effects of antiarrhythmic drugs on cloned cardiac voltage‐gated potassium channels expressed in Xenopus oocytes. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:22–31. [DOI] [PubMed] [Google Scholar]
- 40. Clark MB, Mattick JS. Long noncoding RNAs in cell biology. Semin Cell Dev Biol. 2011;22:366–376. [DOI] [PubMed] [Google Scholar]
- 41. Mohammad F, Pandey GK, Mondal T, Enroth S, Redrup L, Gyllensten U, Kanduri C. Long noncoding RNA‐mediated maintenance of DNA methylation and transcriptional gene silencing. Development. 2012;139:2792–2803. [DOI] [PubMed] [Google Scholar]
- 42. Marchese FP, Huarte M. Long non‐coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics. 2014;9:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Doppler SA, Werner A, Barz M, Lahm H, Deutsch MA, Dressen M, Schiemann M, Voss B, Gregoire S, Kuppusamy R, Wu SM, Lange R, Krane M. Myeloid zinc finger 1 (Mzf1) differentially modulates murine cardiogenesis by interacting with an Nkx2.5 cardiac enhancer. PLoS One. 2014;9:e113775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, Wilders R, Tan HL. Slow delayed rectifier potassium current blockade contributes importantly to drug‐induced long QT syndrome. Circ Arrhythm Electrophysiol. 2013;6:1002–1009. [DOI] [PubMed] [Google Scholar]
- 45. Taniguchi T, Uesugi M, Arai T, Yoshinaga T, Miyamoto N, Sawada K. Chronic probucol treatment decreases the slow component of the delayed‐rectifier potassium current in CHO cells transfected with KCNQ1 and KCNE1: a novel mechanism of QT prolongation. J Cardiovasc Pharmacol. 2012;59:377–386. [DOI] [PubMed] [Google Scholar]
- 46. Zhang Y, Post WS, Blasco‐Colmenares E, Dalal D, Tomaselli GF, Guallar E. Electrocardiographic QT interval and mortality: a meta‐analysis. Epidemiology. 2011;22:660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, McNitt S, Robinson JL, Benhorin J, Kaufman ES, Towbin JA, Barsheshet A. Risk of life‐threatening cardiac events among patients with long QT syndrome and multiple mutations. Heart Rhythm. 2013;10:378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rautaharju PM, Zhang ZM, Haisty WK Jr, Gregg RE, Warren J, Horacek MB, Kucharska‐Newton AM, Rosamond W, Soliman EZ. Race‐ and sex‐associated differences in rate‐adjusted QT, QTpeak, ST elevation and other regional measures of repolarization: the Atherosclerosis Risk in Communities (ARIC) study. J Electrocardiol. 2014;47:342–350. [DOI] [PubMed] [Google Scholar]
- 49. Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, Koopmann TT, Sotoodehnia N, Rossin EJ, Morley M, Wang X, Johnson AD, Lundby A, Gudbjartsson DF, Noseworthy PA, Eijgelsheim M, Bradford Y, Tarasov KV, Dorr M, Muller‐Nurasyid M, Lahtinen AM, Nolte IM, Smith AV, Bis JC, Isaacs A, Newhouse SJ, Evans DS, Post WS, Waggott D, Lyytikainen LP, Hicks AA, Eisele L, Ellinghaus D, Hayward C, Navarro P, Ulivi S, Tanaka T, Tester DJ, Chatel S, Gustafsson S, Kumari M, Morris RW, Naluai AT, Padmanabhan S, Kluttig A, Strohmer B, Panayiotou AG, Torres M, Knoflach M, Hubacek JA, Slowikowski K, Raychaudhuri S, Kumar RD, Harris TB, Launer LJ, Shuldiner AR, Alonso A, Bader JS, Ehret G, Huang H, Kao WH, Strait JB, Macfarlane PW, Brown M, Caulfield MJ, Samani NJ, Kronenberg F, Willeit J, Smith JG, Greiser KH, Meyer Zu Schwabedissen H, Werdan K, Carella M, Zelante L, Heckbert SR, Psaty BM, Rotter JI, Kolcic I, Polasek O, Wright AF, Griffin M, Daly MJ, Arnar DO, Holm H, Thorsteinsdottir U, Denny JC, Roden DM, Zuvich RL, Emilsson V, Plump AS, Larson MG, O'Donnell CJ, Yin X, Bobbo M, D'Adamo AP, Iorio A, Sinagra G, Carracedo A, Cummings SR, Nalls MA, Jula A, Kontula KK, Marjamaa A, Oikarinen L, Perola M, Porthan K, Erbel R, Hoffmann P, Jockel KH, Kalsch H, Nothen MM, den Hoed M, Loos RJ, Thelle DS, Gieger C, Meitinger T, Perz S, Peters A, Prucha H, Sinner MF, Waldenberger M, de Boer RA, Franke L, van der Vleuten PA, Beckmann BM, Martens E, Bardai A, Hofman N, Wilde AA, Behr ER, Dalageorgou C, Giudicessi JR, Medeiros‐Domingo A, Barc J, Kyndt F, Probst V, Ghidoni A, Insolia R, Hamilton RM, Scherer SW, Brandimarto J, Margulies K, Moravec CE, del Greco MF, Fuchsberger C, O'Connell JR, Lee WK, Watt GC, Campbell H, Wild SH, El Mokhtari NE, Frey N, Asselbergs FW, Mateo Leach I, Navis G, van den Berg MP, van Veldhuisen DJ, Kellis M, Krijthe BP, Franco OH, Hofman A, Kors JA, Uitterlinden AG, Witteman JC, Kedenko L, Lamina C, Oostra BA, Abecasis GR, Lakatta EG, Mulas A, Orru M, Schlessinger D, Uda M, Markus MR, Volker U, Snieder H, Spector TD, Arnlov J, Lind L, Sundstrom J, Syvanen AC, Kivimaki M, Kahonen M, Mononen N, Raitakari OT, Viikari JS, Adamkova V, Kiechl S, Brion M, Nicolaides AN, Paulweber B, Haerting J, Dominiczak AF, Nyberg F, Whincup PH, Hingorani AD, Schott JJ, Bezzina CR, Ingelsson E, Ferrucci L, Gasparini P, Wilson JF, Rudan I, Franke A, Muhleisen TW, Pramstaller PP, Lehtimaki TJ, Paterson AD, Parsa A, Liu Y, van Duijn CM, Siscovick DS, Gudnason V, Jamshidi Y, Salomaa V, Felix SB, Sanna S, Ritchie MD, Stricker BH, Stefansson K, Boyer LA, Cappola TP, Olsen JV, Lage K, Schwartz PJ, Kaab S, Chakravarti A, Ackerman MJ, Pfeufer A, de Bakker PI, Newton‐Cheh C. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet. 2014;46:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Choy AM, Lang CC, Chomsky DM, Rayos GH, Wilson JR, Roden DM. Normalization of acquired QT prolongation in humans by intravenous potassium. Circulation. 1997;96:2149–2154. [DOI] [PubMed] [Google Scholar]
- 51. Viskin S. Long QT syndromes and torsade de pointes. Lancet. 1999;354:1625–1633. [DOI] [PubMed] [Google Scholar]
- 52. Antzelevitch C, Sicouri S. Clinical relevance of cardiac arrhythmias generated by afterdepolarizations: role of M cells in the generation of U waves, triggered activity and torsade de pointes. J Am Coll Cardiol. 1994;23:259–277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences of Primers
Figure S1. Transfection efficiency using fluorescence in vivo and in vitro.
Figure S2. Identification and expression of Kcna2 AS and Kcna2 mRNA in rat hearts.
Figure S3. The evaluation of cardiac dysfunction in the respective in vivo and in vitro models.
Figure S4. Expression of Kcna2 AS and Kcna2 in human normal and heart failure tissue.
Figure S5. Kv1.2 channel encoding the Iks current.