

Abstract
Background
Guanylyl cyclase, a heme‐containing α1β1 heterodimer (GC1), produces cGMP in response to Nitric oxide (NO) stimulation. The NO‐GC1‐cGMP pathway negatively regulates cardiomyocyte contractility and protects against cardiac hypertrophy–related remodeling. We recently reported that the β1 subunit of GC1 is detected at the intercalated disc with connexin 43 (Cx43). Cx43 forms gap junctions (GJs) at the intercalated disc that are responsible for electrical propagation. We sought to determine whether there is a functional association between GC1 and Cx43 and its role in cardiac homeostasis.
Methods and Results
GC1 and Cx43 immunostaining at the intercalated disc and coimmunoprecipitation from membrane fraction indicate that GC1 and Cx43 are associated. Mice lacking the α subunit of GC1 (GCα1 knockout mice) displayed a significant decrease in GJ function (dye‐spread assay) and Cx43 membrane lateralization. In a cardiac‐hypertrophic model, angiotensin II treatment disrupted the GC1‐Cx43 association and induced significant Cx43 membrane lateralization, which was exacerbated in GCα1 knockout mice. Cx43 lateralization correlated with decreased Cx43‐containing GJs at the intercalated disc, predictors of electrical dysfunction. Accordingly, an ECG revealed that angiotensin II–treated GCα1 knockout mice had impaired ventricular electrical propagation. The phosphorylation level of Cx43 at serine 365, a protein‐kinase A upregulated site involved in trafficking/assembly of GJs, was decreased in these models.
Conclusions
GC1 modulates ventricular Cx43 location, hence GJ function, and partially protects from electrical dysfunction in an angiotensin II hypertrophy model. Disruption of the NO‐cGMP pathway is associated with cardiac electrical disturbance and abnormal Cx43 phosphorylation. This previously unknown NO/Cx43 signaling could be a protective mechanism against stress‐induced arrhythmia.
Keywords: arrhythmia, cardiac function, cardiovascular disease, cGMP, connexin 43, guanylyl cyclase, Nitric oxide
Subject Categories: Cell Signalling/Signal Transduction, Myocardial Biology, Contractile function, Basic Science Research, Arrhythmias
Clinical Perspective
What Is New?
The interaction between NO signaling through guanylyl cyclase and connexin 43 modulates cardiac electrical function.
The NO–guanylyl cyclase–cGMP pathway is involved in proper connexin 43 location and gap junction function at the intercalated disc.
Disruption of guanylyl cyclase signaling is associated with connexin 43 lateralization, abnormal phosphorylation, and stress‐induced ventricular electrical defects.
What Are the Clinical Implications?
Interactions between connexin 43 and NO–guanylyl cyclase signaling may be disrupted under pathological conditions, such as renin‐angiotensin system hyperactivation, potentially leading to arrhythmia.
Pharmacological increases of cardiac cGMP at the intercalated disc might be able to correct or prevent cardiac electrical defects.
The NO sensor, guanylyl cyclase (GC1), is a heterodimeric enzyme that produces the second messenger cGMP. Binding of NO to the heme of GC1 increases the catalytic activity >50‐fold. The NO‐GC1‐cGMP pathway regulates numerous cardiovascular functions, including vascular tone and cardiac homeostasis, mostly through activation of protein kinase G. Specifically in the heart, the NO‐cGMP signaling has a well‐established cardioprotective effect against hypertrophy, acute ischemia/reperfusion injury, and heart failure.1 In isolated cardiomyocytes from systemic GCα1 knockout (GCα1‐KO) mice, a model used herein, it was shown that GC1 modulates cardiac contractility by mediating both negative and positive ionotropic effects, under β3 adrenergic stimulation and basal condition, respectively.2 Other specific cardiac roles for the pathway are not as well understood or remain controversial, mostly because disruption or stimulation of the NO‐cGMP pathway affects many cell types in the heart.3
In most cell types, GC1 is found in the cytosol, but in cardiomyocytes, it is also found at the plasma membrane (PM) and is active.4, 5 Moreover, in a model of cardiac hypertrophy induced by pressure overload, the GCβ1 subunit was shown to redistribute from the caveolin 3–enriched microdomain at the PM to the cytosol with a concomitant decrease in NO‐stimulated activity.6 We recently reported the detection of neuronal NO synthase and the GCβ1 subunit at the intercalated disc (ID),7 which could suggest that a functional NO‐cGMP pathway is present at the ID, the site of the mechanical and electrical conduction between cardiomyocytes. Electrical coupling depends on connexin 43 (Cx43)–containing gap junctions (GJs).8 Formation of Cx43‐containing GJs at the ID involves trafficking from the endoplasmic reticulum to the Golgi and the PM, then assembly and stabilization in the ID structure.9 Each step of this cycle is controlled by a complex, not fully understood sequence of kinase activation that leads to Cx43 phosphorylation events and by interaction with several additional binding partners. The cAMP‐activated protein kinase A (PKA) is proposed to be involved in the Cx43‐GJ assembly step.10, 11
In cardiomyocytes, the activity of phosphodiesterases allows the compartmentalization of cyclic nucleotide signaling.12 In specific microdomains, cGMP can modulate cAMP signaling by activation or inhibition of cAMP breakdown by phosphodiesterases 2 and 3, respectively. The organization in microdomains and the cAMP/cGMP interplay are areas under intense investigation because disruption of the cAMP/cGMP spatiotemporal distribution is associated with cardiac dysfunction.13, 14 Our initial observation that GC1 was located at the ID and the novel finding of its proximity to Cx43 prompted us to investigate the potential function of the NO‐cGMP pathway in relation to Cx43 function. In addition to the GCα1‐KO mouse model, we used an angiotensin II (AngII)–induced cardiac hypertrophy mouse model because AngII‐treated animals have decreased NO‐stimulated GC1 activity in the cardiovascular system.15 In our specific model, we observed cardiac hypertrophy, but the cardiac remodeling associated with arrhythmic sudden death and heart failure has not yet taken place, unlike other renin‐angiotensin mouse models.16, 17
We describe herein the exploration of the GC1 cardiac role, its association with Cx43, and its involvement in regulating cardiac rhythm under basal and stress conditions.
Methods
Materials
The following reagents were purchased or otherwise obtained: AngII (Sigma); miniosmotic pump (Alzet, Cupertino, CA; model 2002); Tissue‐Tek O.C.T compound (Sakura; Finetek; Torrance, CA); M.O.M (Mouse on Mouse Detection kit; Vector Laboratory); protease inhibitor cocktail (Sigma); protein A magnetic beads (Dynabeads protein A; Thermo Fisher Scientific); Laemmli Sample Buffer (Bio Rad); glass slides (Superfrost; VWR); Amicon Ultra‐0.5 centrifugation filter devices (Millipore); anti‐GCα1 (rabbit; Sigma); anti‐GCβ1 (rabbit; Cayman); anti‐Cx43 against the C‐terminus of Cx43 (mouse; Sigma) and against the N‐terminus of Cx43 (rabbit; Abcam); phosphospecific antibody against serine 365 of Cx43 (from Dr P. Lampe; Iowa city, IA.); anti–N‐cadherin (rat; DSHB; Iowa city, IA.); anti–β‐actin (mouse; DSHB); rabbit IgG (Alpha Diagnostic Intl Inc; San Antonio, TX.); antibodies AlexaFluor (488/594) goat anti‐mouse, anti‐rabbit, and anti‐rat, wheat germ agglutinin AlexaFluor647, and Prolong antifade reagent with 4′,6‐diamidino‐2‐phenylindole (all from Thermo Fisher Scientific).
Animals
The use of 3.5‐ to 6‐month‐old male mice in the study conformed to standards in the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health, and was approved by the IACUC of Rutgers University. Wild‐type (WT; C57Bl6) mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in our animal facility. GCα1‐KO18 were backcrossed for >15 generations in a C57Bl6 background, bred, and housed in our facility. Animals were kept in a 12‐hour light/dark cycle, and food and water were given ad libitum.
AngII Treatment
Mice were treated with AngII (0.8 mg/kg per day) for 13 days via miniosmotic pump implanted subcutaneously on animals anesthetized with rodent cocktail (90 mg/kg ketamine+10 mg/kg xylazine, IP), as previously described.15 Buprenorphine hydrochloride (0.03 mg/kg, SC) was administered as an analgesic after surgery. Thirteen days after pump implantation, mice were anesthetized and their hearts were removed, washed in PBS, and processed accordingly for each protocol.
Immunofluorescence
The apex of hearts was embedded in Tissue‐Tek and progressively frozen to −50°C before storage at −80°C. Transversal cryosections (7–10 µm thick) were mounted onto glass slides and processed, as described.7 Briefly, cryosections were incubated with appropriate primary and fluorescent secondary antibodies (GCα1, 1:1000; GCβ1, 1:200; Cx43, 1:200; N‐cadherin, 1:100; secondary antibodies, 1:200) diluted in M.O.M blocking reagent. Membranes were stained with wheat germ agglutinin, and slides were covered with a glass coverslip with antifade reagent. Fluorescence signals were imaged using a 200 Axiovert fluorescence microscope (Zeiss, Germany) equipped with ×10, ×20, ×40, and an oil ×63 objectives and a confocal‐like system (ApoTome; Zeiss), with sections imaged every 0.5 to 0.7 µm. 3‐Dimensional reconstitution and optical sectioning were done with Zeiss Axovision software. Fluorescence was standardized across conditions. A ×60 oil objective was used to obtain pictures of Figure 1B. For quantification of Cx43 immunofluorescence signal outside of the ID, 3 to 5 images per heart (4–5 animals per group from 4 independent experiments) were used. For each experiment, the same exposure times were used for imaging and, using ImageJ software, the digitized images were corrected with the same threshold for analysis of Cx43 and N‐cadherin signals (pixels). For each frame, the totality of Cx43 signal area was measured, as was N‐cadherin signal area (specific marker of the ID). The total area of Cx43 overlapping with N‐cadherin signal was subtracted to obtain the area of Cx43 outside of the ID (pixels unit). The percentage was determined by the ratio of Cx43 area outside the ID/total Cx43 signal area.
Figure 1.

Guanylyl cyclase (GC) α1 and β1 subunits are localized in proximity to connexin 43 (Cx43) at the intercalated discs. A, Representative image (n=4) of wild‐type (WT) heart cryosections immunostained for GCα1 and GCβ1 (left panels, red), Cx43 (green, middle panels), and merged signal (right panels). Cardiomyocyte membranes are stained with wheat germ agglutinin (WGA; white). Yellow arrowheads indicate intercalated discs (IDs). White bar=20 μm. Negative controls are shown in Figure S1. GC1 represents the heterodimer formed by association of GCα1 and GCβ1 subunits (the other GCα subunit, GCα2, was not included in the present study). B, Representative 3‐dimensional reconstitution of Z‐stack images (0.5–0.7 µm slices) of heart cryosections. GCα1 (top panel) and GCβ1 (bottom panel) are immunostained in red, Cx43 in green, and cardiomyocyte membranes in white with WGA (n=3). Magnification of ID area (boxes, right panels) indicates a close proximity between Cx43 and GCα1 or GCβ1.
Fractionation
Mouse hearts were washed in PBS and ground with an Ultra‐Turrax dispenser (IKA, Wilmington, NC.) in cold lysis buffer without Triton X‐100 (50 mmol/L Tris HCl, pH 7.5, 150 mmol/L NaCl, 1 mmol/L EGTA, 1 mmol/L EDTA, 50 mmol/L NaF, 1 mmol/L Na orthovanadate, and protease inhibitor cocktail). Lysate was centrifuged (4°C, 2 minutes, 500g) to remove debris. Lysate without debris was centrifuged for 10 minutes at 21 000g at 4°C, and the supernatant (cytosolic fraction) was collected. Pellet (total membrane) was washed and resuspended in lysis buffer containing 1% Triton, X‐100 then centrifuged at 9400g for 5 minutes at 4°C. Supernatant (Triton‐soluble membranes, including nonjunctional Cx43) was collected, and the pellet (junctional plaques/ID fraction) was dissolved in lysis buffer containing 1% Triton X‐100 and 4 mol/L urea and then desalted with an Amicon filter (as done by Bruce et al,19 with modifications). The 3 fractions (cytosolic, Triton‐soluble membrane, and junctional plaques) were solubilized in Laemmli buffer, separated by SDS‐PAGE on 7.5% acrylamide gel, and processed for Western blot analysis.
Immunoprecipitation
Harvested hearts were homogenized in 2 to 3 mL of cold lysis buffer with Triton X‐100 (described previously) and centrifuged at 3400g, 4°C, for 2 minutes to remove debris. Protein of the supernatant (1 mg) in 1.5 mL of lysis buffer was precleared with rabbit IgG antibodies bound to protein A. Precleared samples were incubated overnight at 4°C with 3 μg of antibodies anti‐GCβ1 or with 3 μg of rabbit IgG antibodies as a negative control. Samples were rotated for 1 hour at room temperature in the presence of protein A beads. Supernatant was collected (unbound fraction), and immunoprecipitated proteins were eluted at 90°C for 5 minutes in 1× final Laemmli sample buffer with 5% β‐mercaptoethanol and processed for Western blot analysis, as described above.
Dye Spread Assay
A modified protocol from Sovari et al17 was used to evaluate GJ function in WT and GCα1‐KO mice hearts. Hearts were harvested and washed in PBS, as described previously. The apex and top sliced part of ventricles were placed in PBS at 37°C, and a 27‐gauge needle was used to infuse droplets of 0.5% Lucifer Yellow (LY; Thermo Fisher Scientific, Waltham, MA.) and 0.5% Texas Red Dextran (molecular weight, 10 000; Thermo Fisher Scientific) prepared in 150 mmol/L LiCl solution. After 15 minutes of incubation, the samples were fixed in 4% formaldehyde for 30 minutes, washed in PBS, and cryofrozen. Cryosections (14‐µm thick) were mounted onto microscope slides and examined using a 200 Axiovert Zeiss microscope, ×10 objective. The filters were set at Excitation470/Emission509 for LY and Excitation540/Emission580 for Texas Red Dextran. The area of distribution of LY fluorescence and the area of distribution of Texas Red Dextran fluorescence were quantified with Axiovision software. A total of 3 to 7 areas were imaged and analyzed for each heart from 5 WT and 5 GCα1‐KO mice (3 months old). The difference between LY spread area and Texas Red Dextran spread area was calculated to determine GJ permeability. LY acts as a “tracer” for diffusion through GJ, but the molecules of Texas Red Dextran are too large to go through GJ and will stain cardiomyocytes at the site of injection.
Electrocardiography
Mouse ECGs were recorded using needle electrodes in a lead II configuration on mice anesthetized with 2,2,2‐tribromoethanol (Avertin; 290 mg/kg, IP) and kept at a constant 37°C using a heating pad for the duration of the recording (60 minutes). ECG signals were acquired using a DSI Ponemah amplifier and pClamp Axoscope 10 software (Molecular Devices). P‐wave duration, QRS duration, and QT intervals were determined during the first 15 minutes after anesthesia. The QT interval was corrected as a function of the R‐R interval (QT‐corrected, ), as done by Mitchell et al.20 AV blocks (impaired conduction between atria and ventricles) and sinus atrial (SA) node dysfunction (absence of P waves) were assessed during the 60 minutes of recording.
Statistical Analysis
Normality of samples was verified by Shapiro‐Wilk test. Unpaired t tests were used for comparison between 2 groups. Two‐way ANOVA with Tukey post hoc tests were used for comparison between >2 groups. Data are expressed as mean±SEM, and P<0.05 was considered statistically significant.
Results
GC1 Is in Close Proximity to Cx43 at the ID in Mouse Heart
Immunofluorescence imaging of heart ventricle cryosections showed both α1 and β1 subunits of GC1 together with Cx43 at the ID (yellow arrowheads, fiber longitudinal orientation; Figure 1A). The potential association to Cx43 is illustrated by a 3‐dimensional reconstitution of Z‐stack confocal images (optical sectioning; Figure 1B). As negative control, we omitted the primary antibodies (Figure S1A). Weak overlapping signals between Cx43 and GC1 at the ID are also observed in orthogonal sections of the Z‐stack images (Figure S1B).
GCαβ and Cx43 Form a Complex in the Mouse Heart
Ventricle homogenates were prepared by separating cytosol from membrane fraction or by lysis with 1% Triton X‐100 (“total”; Figure 2), as described in Methods. Inputs (starting material, left panel) showed that cytosol contains a high level of GCα and GCβ and no Cx43, as expected. High levels of Cx43 are detected in the membrane fraction. GCα1 and GCβ1 are also detected in the membrane fraction, as previously reported.5 More important, coimmunoprecipitation (right panel) of Cx43 with anti‐GCβ1 antibody yields robust signals in both membrane fraction and total homogenate; the α1 subunit is coimmunoprecipitated with anti‐GCβ1 but to a lesser extent than the β1 subunit. There is no immunoprecipitation of Cx43 or GC1 when nonspecific IgG is used or in the cytosolic fraction. The reverse coimmunoprecipitation using an antibody against Cx43 (directed against the C‐ or N‐terminal peptide of Cx43) did not pull down GC1 in Triton‐soluble homogenates or membranes (not shown). The reason is unknown but could be related to solubilization, masking of the Cx43 recognition site in the GC1‐Cx43 complex, or an intercalated factor between Cx43 and GCαβ1.
Figure 2.
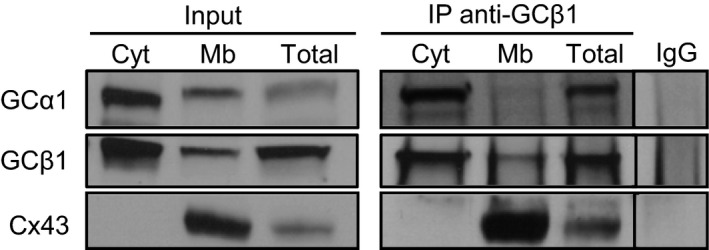
Guanylyl cyclase, a heme‐containing α1β1 heterodimer (GC1), and connexin 43 (Cx43) coimmunoprecipitate. Representative Western blot showing pull down of Cx43 by anti‐GCβ1 (n=3). Cytosolic (Cyt) and membrane (Mb) fractions and whole heart homogenate (total) were immunoprecipitated with anti‐GCβ1 and probed with anti‐GCα1, anti‐GCβ1, or anti‐Cx43. Input indicates the starting material (30 μg lysates). In the negative control (wild‐type membrane fraction pulled down with IgG), no Cx43 could be detected. Unlike the subsequent experiments, homogenates were prepared in the absence of phosphatase inhibitors, which could explain the lack of phosphorylated forms of Cx43.
GJ Permeability Is Impaired in GCα1‐KO Mice
To determine whether there is a physiological link between GC1 and Cx43, we used the systemic GCα1‐KO model21 to assess GJ function. For this, we compared GJ permeability in the heart of WT and GCα1‐KO mice by measuring, in the ventricle, the diffusion of LY. LY goes through GJ, whereas red dextran (which does not go through GJ because of its size) is used to mark the site of injection. Figure 3 showed that the diffusion of LY is significantly impaired in the GCα1‐KO mice, indicating that the permeability of GJ is affected in mice lacking GCα1.
Figure 3.

Gap junction permeability is impaired in guanylyl cyclase α1 knockout (GCα1‐KO) mice heart. A, Ventricle slices from wild‐type (WT) or GCα1‐KO mice hearts were locally injected with Lucifer yellow (LY; gap junction [GJ] permeable) and Red Dextran (RD; GJ not permeable) and incubated for 15 minutes at 37°C. After fixation, fluorescent areas were measured using Axiovision software on 14‐µm cryosections with a 200 Axiovert Zeiss fluorescent microscope (see Methods). Bar=100 μm. B, The difference between areas stained with LY (dye diffusion through GJ) and with RD (site of injection) indicates that diffusion of LY is impaired in GCα1‐KO mice heart compared with WT (GCα1‐KO vs WT, 48 937±8837 vs 91 645±8834 μm2; n=5 for each group). Data are expressed as mean±SEM (B). BF indicates bright field. **P<0.01.
Mice Lacking the GCα1 Subunit Have Mislocalized Cx43, as Do Mice Treated With AngII
Lateralization of Cx43 (eg, accumulation of Cx43 hemichannels at the membranes along the longitudinal axis) is linked to cardiac diseases related to Cx43‐containing GJ dysfunction.19, 22, 23 Thus, to further establish a correlation between GC1‐Cx43 association and GJ function, we assessed the localization of Cx43 in the GCα1‐KO model and under AngII‐induced stress conditions. We used a model of AngII‐induced cardiac hypertrophy because we previously showed that in this model, GC1 is desensitized to NO stimulation (eg, reduced NO‐stimulated activity).15 We assessed localization of Cx43 by measuring the amount of Cx43 outside the ID in 4 groups: WT, WT treated with AngII, GCα1‐KO, and GCα1‐KO treated with AngII, as described in Methods. We observed that the lack of GCα1 induces abnormal localization of Cx43, which accumulates primarily at the lateral membrane of the cardiomyocytes in the ventricle (Figure 4A). Figure 4B showed that this lateralization is statistically significant, as is Cx43 lateralization of WT and GCα1‐KO mice treated with AngII, compared with untreated WT mice. Interestingly, the percentage of Cx43 lateralization is not further increased by AngII treatment in GCα1‐KO mice, which could mean that the same Cx43 location mechanism is disrupted by AngII treatment and deletion of GCα1 or that 30% lateralization represents a plateau. The abnormal localization of Cx43 indicates that one or more of the processes that guide Cx43 trafficking and/or assembly at the ID are impaired by the lack of GCα1 or AngII treatment.
Figure 4.
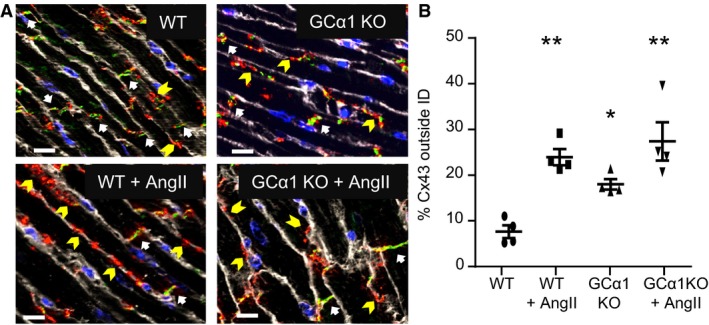
Connexin 43 (Cx43) lateralization is increased by angiotensin II (AngII) treatment and in guanylyl cyclase α1 knockout (GCα1‐KO) mice. A, Representative images of Cx43 localization in heart cryosections from wild‐type (WT) and GCα1‐KO mice untreated or treated with AngII. Cx43 is immunostained in red, and N‐cadherin is immunostained in green. Nuclei (blue) and membranes (white) are stained with 4′,6‐diamidino‐2‐phenylindole and wheat germ agglutinin, respectively. N‐cadherin is specific to intercalated discs (IDs; white arrows). Some of Cx43 labeling outside the ID is indicated by yellow arrowheads. White bar=10 μm. B, Statistical analysis of the percentage of Cx43 signal detected outside the ID (determined by nonoverlapping signals of Cx43 and N‐cadherin), as described in Methods. Compared with WT mice (7.6±1.4% of Cx43 outside the ID), there is a significant increase in lateralization of Cx43 in WT and GCα1‐KO mice treated with AngII (WT+AngII, 24.0±1.8%; and GCα1‐KO+AngII, 30.4±4.4%) and in untreated GCα1‐KO mice (GCα1‐KO, 18.0±1.2%). n=4 for each group. Data are expressed as mean±SEM (B). *P<0.05, **P<0.01.
A Significant Decrease in Cx43‐Containing GJs at the ID Is Observed in WT and GCα1‐KO Mice Treated With AngII
We next wanted to determine whether the lateralization of Cx43 together with the decreased GC permeability were reflected in the decrease of Cx43‐containing GJ specifically in the junctional plaques. We conducted fractionation of the heart to isolate the junctional plaques at the ID from the membrane fraction in the WT and GCα1‐KO mice untreated or treated with AngII. After separation from the cytosolic fraction, the membrane fraction was solubilized with Triton X‐100. The fraction that is not Triton‐soluble corresponds to the ID and contains the junctional plaques that are extracted with 4 mol/L urea (Methods). As expected, the cardiac cytosolic fraction does not have detectable Cx43 (Figure 5A). GCα1‐KO mice, in addition to lacking the α1 subunit, have decreased levels of the GCβ1 subunit. AngII has no effect on the levels of GC (left panel; Figure 5A). The membrane fraction (minus the junctional plaques, middle panel) displays a trend of increased Cx43 in WT mice treated with AngII and in GCα1‐KO mice±AngII compared with WT control that was not significant between the 4 groups (Figure S2A). Interestingly, there is a significant decrease in the amount of Cx43 at the ID (urea‐treated/Triton‐insoluble membrane fraction) in WT and GCα1‐KO mice treated with AngII (Figure 5A, right panel, and Figure 5B). There is also a trend of decreased detection of Cx43 at the ID in GCα1‐KO compared with WT that did not reach significance (Figure 5B; loading control for each fraction was estimated by Ponceau Red staining of the blotted membranes; Figure S2B). This decrease in Cx43‐GJ abundance at the ID in WT and GCα1‐KO mice treated with AngII correlates with the increased Cx43 lateralization at the membrane in the same groups of mice. All together, these results suggest that Cx43‐GJ remodeling takes place in AngII‐treated WT and GCα1‐KO mice.
Figure 5.

Connexin 43 (Cx43) levels are significantly decreased in the intercalated disc (ID) fraction of angiotensin II (AngII)–treated wild‐type (WT) and guanylyl cyclase α1 knockout (GCα1‐KO) mice. A, Representative Western blot of GCα1, GCβ1, and Cx43 from cytosol, Triton‐soluble membrane, and Triton‐insoluble fractions (junctional plaques/IDs) from WT and GCα1‐KO mouse hearts±AngII (10–50 μg of proteins loaded). We did not expect to detect GC1 at the junctional plaques because the treatment with Triton X‐100 and urea probably removes/degrades it. B, Densitometry analysis of Cx43 level in the ID fraction. A decreased Cx43 level at the ID was significant in AngII‐treated WT (WT+AngII) and GCα1‐KO (GCα1‐KO+AngII) mice compared with untreated WT (WT and WT+AngII, n=5; GCα1‐KO and GCα1‐KO+AngII, n=4). Data are expressed as mean±SEM (B). Ctrl indicates control (untreated). *P<0.05, ***P<0.001.
Cx43‐GC1 Association in the Heart Is Disrupted by AngII Treatment
We next wanted to assess, by coimmunoprecipitation (as in Figure 2), whether the increased Cx43 mislocalization correlated with a disruption of the Cx43‐GC1 association. The left panel of Figure 6A (input, starting material) showed that there is no decrease of Cx43 or GC1 in the membrane fraction of heart treated with AngII and, as expected, no GCα1 is detected in the input of the GCα1‐KO mice. On the other hand, the middle panel (coimmunoprecipitation) indicated that the amount of Cx43 pulled down by anti‐GCβ1 is significantly decreased in the heart of AngII‐treated mice compared with control (WT untreated), whereas the level of GCβ1 (and GCα1) pulled down by anti‐GCβ1 is not reduced by AngII treatment. The increased amount of unbound Cx43 (eg, not pulled down by anti‐GCβ1) in the AngII‐treated mouse hearts confirms the decreased coimmunoprecipitation in these groups. Likewise, Cx43 is detected in the unbound material of GCα1‐KO and IgG (right panel; Figure 6A). Statistical analysis in Figure 6B indicated that the decreased pull down of Cx43 is significant for WT and GCα1‐KO treated with AngII compared with untreated WT and for GCα1‐KO treated with AngII versus untreated GCα1‐KO (Figure 6B). Interestingly, there is no significant difference in the pull down of Cx43 between WT and GCα1‐KO. This could suggest that the association between GC1 and Cx43 primarily involved the β subunit of GC1. Together, these results indicate that the Cx43‐GC1 association in the heart is disrupted by AngII treatment and is not attributable to a decreased expression of Cx43 (or GCβ1).
Figure 6.

The guanylyl cyclase, a heme‐containing αβ heterodimer (GC1)‐connexin 43 (Cx43) complex is disrupted in the cardiac membrane fraction of wild‐type (WT) and GCα1 knockout (GCα1‐KO) mouse hearts treated with angiotensin II (AngII). A, Western blot of GCα1, GCβ1, and Cx43 showing coimmunoprecipitation of Cx43 by anti‐GCβ1 in the cardiac membrane fraction and decreased pull down of Cx43 by anti‐GCβ1 in AngII‐treated mice. Left panel indicates the starting material (input) for GCα1, GCβ1, Cx43, and β‐actin in heart membranes from WT and GCα1‐KO mice untreated (control [Ctrl]) or treated with AngII. The different bands of Cx43 include unphosphorylated and phosphorylated forms of Cx43.24 β‐Actin is loading control. As expected, no GCα1 is detected in lysates of the GCα1‐KO hearts, whereas Cx43 levels were slightly reduced (input, left panel). Coimmunoprecipitation (middle panel) was done on the membrane fraction with anti‐GCβ1 or rabbit IgG as negative control. AngII treatment decreases pull down of Cx43 in both WT and GCα1‐KO mice. Unbound fraction (right panel) contains the Cx43 not pulled down by anti‐GCβ1 or IgG. B, Densitometry analysis of Cx43 pull down. Results are expressed as the ratio of coimmunoprecipitation of Cx43/input Cx43/immunoprecipitation of GCβ1 (fold WT control). There is a significant decrease of Cx43 coimmunoprecipitation in AngII‐treated vs untreated WT mice, in AngII‐treated vs untreated GCα1‐KO mice, and in AngII‐treated GCα1‐KO mice compared with untreated WT mice. n=7 for each WT and WT+AngII group. n=4 for each GCα1‐KO and GCα1‐KO+AngII group. Two‐way ANOVA. Data are expressed as mean±SEM (B). *P<0.05, **P<0.01.
A Significant Decrease in Serine 365 Phosphorylation in Cx43‐Containing GJs at the ID Is Observed in WT and GCα1‐KO Mice Treated With AngII
Cx43 trafficking, channel assembly, and gating, which are highly dynamic, are modulated by protein kinases: each specific step of the Cx43‐GJ life cycle corresponds to phosphorylation of specific residues by specific kinases, including PKA.11, 25 Although phosphorylation by protein kinase G (activated by cGMP) remains controversial, cGMP is known to modulate cAMP‐hydrolysis by phosphodiesterases in cardiomyocytes. As such, we investigated whether the Cx43 mislocalization seen in our different models was because of alteration of cAMP‐PKA–dependent phosphorylation of Cx43. We focused on phosphorylation at serine 365 of Cx43 because PKA activity enhances Cx43 trafficking to the PM and assembly of Cx43‐containing GJs through increased phosphorylation at this residue.26, 27 Using an antibody specific for phosphoserine 365 of Cx43, we measured the phosphorylation signal in the ID fraction from WT and GCα1‐KO mice untreated or treated with AngII (Figure 7A). As shown in Figure 7B, phosphoserine 365 detection levels are significantly decreased in both WT and GCα1‐KO mice treated with AngII, compared with the WT control group.
Figure 7.

Treatment with angiotensin II (AngII) decreases serine 365 phosphorylation of connexin 43 (Cx43) in junctional plaques of heart from wild‐type (WT) and guanylyl cyclase α1 knockout (GCα1‐KO) mice. A, Representative Western blot of intercalated disc (ID) fraction from WT and GCα1‐KO mice untreated (control [Ctrl]) or treated with AngII and probed for “total” Cx43 (using Cx43 N‐terminal antibody) and Cx43 phosphorylation specific for serine 365 antibodies. B, Densitometry analysis of the ratio of phosphoserine 365–Cx43 signal/total Cx43 and expressed as fold WT control indicates a significant decrease in WT and GCα1‐KO mice treated with AngII compared with WT control. n=3 for each group. Data are expressed as mean±SEM (B). *P<0.05, **P<0.01.
Abnormal Electrical Propagation in GCα1‐KO Mice Treated With AngII
We performed ECGs in WT and GCα1‐KO mice treated or not with AngII to evaluate electrical function. We observed abnormal ventricular electrical propagation in GCα1‐KO mice+AngII, which is significant compared with WT, WT+AngII, or GCα1‐KO mice (Figure 8). In particular, recording traces of WT±AngII and GCα1‐KO±AngII (Figure 8A) showed that GCα1‐KO mice treated with AngII display a significant prolonged QRS duration and QTc interval (Figure 8B‐C), which indicate a slower electrical propagation and delayed repolarization in the ventricles, respectively. The abnormal ventricular electrical propagation is consistent with the GJ decreased abundance and Cx43 lateralization. We also observed supraventricular electrical disturbance (Figure S3), which is further discussed later.
Figure 8.

Abnormal electrical propagation in the ventricle of guanylyl cyclase α1 knockout (GCα1‐KO) mouse. A, Representative ECG traces recorded from wild‐type (WT) mice untreated (control [Ctrl]; n=6) or treated with angiotensin II (WT+AngII; n=7) in left panels and from GCα1‐KO mice untreated (Ctrl; n=6) or treated with AngII (GCα1‐KO+AngII; n=6) in right panels. B, ECG trace showing scales and parameters used in the analysis (P‐wave duration, QRS duration, and QT‐corrected [QTc] interval). QRS duration was defined as in the study by Li et al28 and evaluates progression of depolarization. An isoelectrical level was used to determine the beginning of the T wave. C, Left panel: QRS duration of GCα1‐KO mice treated with AngII (GCα1‐KO+AngII; n=6) is significantly increased, indicating a slower electrical propagation, compared with WT (n=6) or AngII‐treated (n=7) and GCα1‐KO control mice (n=6). Right panel: QTc interval in GCα1‐KO mice treated with AngII (n=7) is also increased compared with WT (n=6) and GCα1‐KO mice control (n=6), but not different from WT treated with AngII (n=6). QTc=QT/√(R−R/100). n=6 to 7 for each group. Data are expressed as mean±SEM (C). *P<0.05, **P<0.01, ***P<0.001.
Discussion
Cx43 has long been recognized as a critical player in cardiac function. Cx43 forms GJs located at the ID that are responsible for electrical propagation in the ventricular myocardium. In mice, cardiac‐specific KO of Cx43 leads to ventricular arrhythmia and sudden cardiac death in the first 2 months.29 On the other hand, NO signaling is also an important modulator of cardiac function. NO stimulates GC1 to produce cGMP, which negatively regulates cardiomyocyte contractility by modulation of calcium homeostasis via protein kinase G–dependent phosphorylation.14, 30
Cx43‐GC1 Association
To our knowledge, a direct link between Cx43 function and NO signaling has not been reported in cardiomyocytes. There is one report in mesangial cells, showing that stimulation of the NO‐cGMP pathway increases Cx43‐containing GJs and enhances GJ function.31 Our findings that Cx43 and the NO receptor (eg, GC1) are associated in the heart is novel and is based on biochemical evidence showing that GC1 and Cx43 coprecipitate in the membrane fraction of the heart and on immunocytochemistry, showing Cx43 and GC1 at the ID. Although not in adult cardiac tissues, a proximity ligation assay in neonatal cardiomyocytes also indicates an interaction between endogenous Cx43 and GC1 (Figure S4). Because the neonatal cardiomyocytes are not adult cardiomyocytes, it is difficult to conclude whether the few complexes detected at the junction between 2 neonatal cardiomyocytes represent GJ.
Impaired GJ Function in Mice With an Altered NO‐GC‐cGMP Pathway
After these initial observations, our first aim was to investigate the function of this association and the impact of a decreased functional NO‐cGMP pathway on Cx43 location and function. To impair the NO‐cGMP pathway, we first used a systemic knockout of the GCα1 subunit.18 Functionally, we observed that GJ permeability assayed by dye spread in ventricle slices is significantly reduced in the heart of mice lacking the GCα1 subunit, supporting a link between GC1 and Cx43 (Figure 3). This supports the novel idea that a decrease in the NO‐cGMP pathway affects GJ function in the heart and specifically in the ventricle.
Altered NO‐GC‐cGMP Pathway Correlates With Mislocalization of Cx43
We next asked whether this decreased function was attributable to a decrease in Cx43‐containing GJ at the ID. In addition to the GCα1‐KO mice model, we used an AngII‐induced hypertrophy mouse model for 2 reasons. First, we previously showed that AngII treatment desensitizes GC1 activity to NO stimulation in the vasculature15 and in the heart (Figure S5), giving us another model of the impaired NO‐GC‐cGMP pathway. Second, we wanted to assess whether stress‐induced disruption of the GC1‐Cx43 association could have a pathological relevance. The AngII treatment and the lack of GCα in GCα1‐KO mice induce a significant increase of Cx43 outside the ID and apparent Cx43 accumulation in the lateral membranes along the longitudinal axis of the ventricular cardiomyocytes. This is an important observation because it is proposed that activity of these unapposed hemichannels at the lateral membrane results in a “leaky” current that could lead to abnormal electrical propagation.32, 33 The increased lateralization of Cx43 in AngII‐treated GCα1‐KO is similar in untreated GCα1‐KO and in AngII‐treated WT (no additive effect), which could suggest that the GCα1‐KO and AngII treatment disrupts the same pathway involved in Cx43 localization or that the 30% lateralization corresponds to a plateau of Cx43 mislocalization under our conditions (13‐day AngII treatment or partial KO of GC activity). We favor the latter explanation because it is consistent with the results of our fractionation experiment, as follows. The abnormal localization of Cx43 is also detectable through a reduction of Cx43 within GJs at the ID in the ventricles of AngII‐treated WT mice, and this depletion is significantly exacerbated in GCα1‐KO mice when treated with AngII. This would suggest that the lack of the GCα1 subunit and the treatment with AngII have an additive effect on Cx43 mislocalization. We could not detect GC1 in the ID fraction, but this is not surprising because the isolation of junctional plaques from the Triton‐insoluble fraction with 4 mol/L urea would be expected to “damage” GC1.
The significant increase in Cx43 lateralization in GCα1‐KO compared with WT mice is consistent with the decreased, albeit nonsignificant, level of Cx43 present at the ID. The significant versus nonsignificant discrepancy could be because of differences in sensitivity of the 2 methods (immunofluorescent pixel analysis via ImageJ versus densitometry analysis of anti‐Cx43 blots after fractionation) or because the immunofluorescence signal for lateralized Cx43 is the sum of the Cx43 attributable to the disassembly of GJ plus Cx43 that is trafficked from the Golgi to the PM before the assembly of any GJ.9, 34 Independently of the potential mechanism, the correlation between decreased GJ permeability and increased percentage of Cx43 outside the ID in GCα1‐KO mice strongly supports a link between GJ function and the NO‐cGMP pathway.
Potential Molecular Mechanism
Cyclic nucleotide signaling is highly compartmentalized, mostly through the activity of phosphodiesterases,12 and proper cardiac physiological function relies on the organization, in microdomains, of these intracellular signaling pathways.13, 14 Disruption of the cAMP‐related signaling could be induced, for example, by dysregulation of the cGMP‐inhibited phosphodiesterase 3, which preferentially hydrolyzes cAMP.13, 35 As such, disruption of the NO‐cGMP pathway would impair PKA activation through increased cAMP hydrolysis by phosphodiesterase 3. PKA activity enhances Cx43 trafficking to the PM and maintenance of Cx43‐containing GJs, through increased phosphorylation at serine 364/365.26, 27 This led to our mechanistic hypothesis that Cx43 mislocalization could be because of alteration of PKA‐dependent upregulation of serine 364/365 phosphorylation of Cx43 in the GC1 activity‐impaired mouse models. We observed a decrease in Cx43 phosphorylation at serine 365 in the junctional plaques (ID, Triton‐insoluble fraction) of WT treated with AngII and GCα1‐KO mice±AngII (Figure 7A). This decrease was significant in WT and GCα1‐KO mice treated with AngII (not in GCα1), compared with untreated WT (Figure 7B). To our knowledge, this is the first evidence that AngII treatment of WT and GCα1‐KO mice induces a change in phosphoserine 365 of Cx43. The nonsignificant decrease in phosphoserine 365 in GCα1‐KO suggests that phosphorylation of another residue(s) involved in Cx43 trafficking could be under the control of the NO‐GC1 pathway. Overall, our biochemical data indicate that there is a strong correlation between the decreased serine 365 phosphorylation, the decreased GJ abundance, and increased Cx43 lateralization on treatment with AngII of WT and that Cx43 mislocalization is exacerbated if the AngII treatment is applied to mice lacking the GCα1 subunit. This suggests that the AngII treatment and the lack of GCα1 could affect Cx43 trafficking and assembly at 2 different steps (such as differential Cx43 phosphorylation). Further study will be conducted to investigate the involvement of phosphodiesterase 3, PKA, and an unbalanced cAMP/cGMP pool at the ID in NO‐cGMP–dependent Cx43 trafficking. We cannot rule out that other pathways of Cx43 trafficking and GJ life cycle, besides phosphorylation, are affected, including microtubule‐dependent forward trafficking to the junctional plaques.36
Abnormal Electrical Propagation in AngII‐Treated GCα1‐KO Mice
We investigated further the functional implications of decreased NO‐cGMP signaling by recording an ECG in WT and GCα1‐KO mice with and without AngII treatment, because Cx43 lateralization and GJ abundance reduction should translate into cardiac electrical dysfunction37 in the AngII‐treated GCα1‐KO mouse model. Our biochemical data, obtained from the ventricle of GCα1‐KO mice treated with AngII, showing decreased GJ abundance and Cx43 lateralization, may explain the abnormal ECG in these mouse models. In particular, the prolonged QRS duration and longer QTc interval are indicative of a slower ventricular electrical propagation and repolarization, respectively. The longest QRS duration and largest QTc interval were observed in the AngII‐treated GCα1‐KO model, which was the model in which the most significant decrease of Cx43 at the ID and the largest Cx43 lateralization were observed, suggesting a strong correlation between the electrical dysfunction and the Cx43 mislocalization. However, the disruption of the Cx43 and GC1 association at the ID could indirectly affect other key players of electrical conduction, notably sodium channels whose dysfunction also accounts for ventricular arrhythmias.38 Moreover, sodium channels are associated with the ID,39 and Cx43 KO is associated with decreased Nav1.5.40 Thus, we analyzed the potential effect of sodium current in ventricular cardiomyocytes isolated from WT and AngII‐treated GCα1‐KO mice by patch‐clamp recording of their action potentials (Figure S6). We observed that action potential amplitude remained the same in AngII‐treated GCα1‐KO myocytes compared with WT myocytes. Furthermore, no difference was observed in the maximal change rate in membrane potential phase 0, which is the steepest portion of the action potential upstroke representing the maximal sodium ion flow (ie, sodium channel conductance).41, 42 These results suggest that the conduction disturbance in GCα1‐KO mice treated with AngII is most likely not mediated by the sodium current. Action potential duration (Figure S6) was prolonged in AngII‐treated GCα1‐KO mice compared with WT, which is consistent with the prolonged QTc interval found in the ECG recording of Figure 8. We also observed the occurrence of early afterdepolarizations in some cardiac cells isolated from the AngII‐treated GCα1‐KO mice (top right panel of Figure S6). These results suggest that AngII‐treated GCα1‐KO mouse hearts exhibit both substrate and trigger that could favor cardiac arrhythmogenesis.
Interestingly, we also observed abnormal supraventricular (SA node and atrium) electrical propagation (Figure S3). Although the mechanisms are still poorly understood, several lines of evidence suggest that disruption of the NO‐cGMP pathway affects supraventricular function. For example, it is proposed that the NO‐cGMP pathway modulates the L‐type calcium current in atrial myocytes43, 44 and the hyperpolarization‐activated current in the SA node.45, 46, 47 Thus, the observed absence of P waves and AV block could be explained by SA dysfunction in the GCα1‐KO, independently of its association with Cx43. The significant prolonged duration of P waves in GCα1‐KO mice treated with AngII is consistent with a slower electrical conduction in the atrium. Although Cx43 expression in the SA node is limited, suggesting that the Cx43‐GC1 association might not be at work in the observed SA node dysfunction, atrial GJs are formed of both Cx40 and Cx43,48 and we detected GC1 in the atriums (Figure S7, which, to our knowledge, has not been previously reported). As such, we cannot exclude that the supraventricular dysfunction could be linked partially to a disrupted Cx43‐GC1 association in the atrium. Whether this is one of the underlying mechanisms of abnormal supraventricular propagation will require further investigation.
Potential Function of the Cx43‐GC1 Association
A link between AngII‐induced hypertrophy/remodeling, Cx43 dysfunction, and arrhythmogenicity has been proposed, but the molecular mechanism remains largely unknown.16, 49 In humans and animals subjected to a prolonged state of cardiac hypertrophy, the initial compensatory mechanisms and their failure lead to maladaptive structural remodeling associated with decreased Cx43 assembly in GJs at the ID and increased Cx43 membrane lateralization.19, 22, 23, 50 Mouse models of pressure overload–induced hypertrophy/remodeling showed that protein kinase G and GC1 activity is downregulated and that the localization of GCβ1 is directly disrupted.6, 51 We observed significant hypertrophy compared with WT in all studied groups (Figure S8), but no other obvious structural damage was seen (not shown). Conversely, cGMP is a negative regulator of the cardiac hypertrophy–related remodeling process,52 and it was recently shown that stimulation of GC1 activity protects from pathological remodeling and heart failure after myocardial infarction.53 With our observation that Cx43 mislocalization correlates with decreased biochemical detection of the Cx43‐GC1 association in WT and GCα1‐KO treated with AngII, this leads us to propose that the NO‐cGMP pathway protects against hypertrophy‐induced remodeling and Cx43‐GJ dysfunction via a functional Cx43‐GC1 association, potentially at the ID. However, because the GCα1‐KO is systemic (as is AngII treatment), we cannot rule out that some effects are attributable to GC1 activity impairment in other tissues, particularly the vascular system. Future work using an inducible cardiomyocyte‐specific GC1 KO model will provide more specific information. Yet, this study clearly establishes that an intact NO‐GC‐cGMP pathway is critical, directly or indirectly, for Cx43 proper location and function under oxidative stress.
Sources of Funding
This work was supported by the National Institute of General Medical Sciences (GM112415 and GM067640 to Beuve and GM055632 to Lampe), the National Heart, Lung, and Blood Institute (HL97979 and HL133294 to Xie), and the American Heart Association (Grant‐in‐Aid to Xie).
Disclosures
None.
Supporting information
Figure S1. A, Negative control for immunofluorescence. Representative image of immunofluorescence of heart cryosection from WT mice performed with secondary AlexaFluor488 goat anti‐mouse and AlexaFluor594 goat anti‐rabbit (1:200) and without primary antibodies with same exposure time as in Figure 1. No red or green signal is detected. Nuclei and membranes are stained in blue with DAPI and in white with Wheat Germ Agglutinin (WGA), respectively. (n=4). B, Overlapping of GC and Cx43 signals. The yellow signals (black arrows) detected in ID of the right orthogonal section (blue box) and the top orthogonal section (red box) of the optical sectioning indicate an overlap of immunolabeled GCβ1 (red) and Cx43 (green). This picture is representative of n=3. There is a low signal overlap between the Cx43 and GC under these conditions, reminiscent of the moderate colocalization observed between ZO‐1 and Cx43 at the ID (Barker RJ et al, Circulation Research 2002; 90:317–334).
Figure S2. Probing of Cx43 and total protein in cytosolic, membranes (triton soluble) and junctional plaques (triton insoluble) fractions of WT and GCα1 KO mice±AngII. A, Densitometry analysis of Cx43 level in the membrane fraction (triton soluble). There was no significant change compare to WT control (P>0.05, n=4). B, Representative image of nitrocellulose membranes probed with anti‐Connexin 43 or stained with Ponceau red after transfer of 10 to 50 μg of proteins from WT Ctrl, WT+AngII, GCα1 KO Ctrl, and GCα1 KO+AngII. (n=3).
Figure S3. Supraventricular abnormality in electrical propagation in GCα1KO mice heart. A, Representative traces recorded from WT (n=6) and GCα1 KO mice (n=6) show that GCα1 KO mice have (a) irregular RR interval (dotted double arrows) and atrioventricular (AV) block (3rd degree, *uncoupled P‐waves); (b) absence of P‐waves, which are indicative of dysfunction in sinoatrial (SAN) conduction. B, The incidence of AV block and SA node impairment was significantly higher in GCα1 KO mice compared to WT (6 GCα1‐KO mice out of 10 and 0 WT mice out of 6; *P<0.05, Fisher exact test). The duration of AV blocks or SAN dysfunction was analyzed in a 10‐minute recording period and the supraventricular abnormalities were observed for 4.4±0.9 minutes. C, Significant prolonged duration of P‐wave in GCα1 KO treated with AngII (n=6), compared to WT (n=6), WT+AngII (n=6) and untreated GCα1 KO (n=7), which reflects a slower electrical conduction in the atrium. **P<0.01 and ***P<0.001. Data are expressed as mean±SEM.
Figure S4. A GC1–Cx43 complex is detected in rat neonatal cardiomyocytes (NCM) by proximity ligation assay. Representative fluorescent image (n=3, independent cultures) of GCα1‐Cx43 (top left) and GCβ1‐Cx43 association (top right) detected by proximity ligation assay (PLA). Red dots indicate endogenous complex. Black arrows indicate GC‐Cx43 complex potentially located at the junction between two NCM. As a negative control, cells were incubated with anti‐Cx43 only (no anti‐GC antibodies, bottom left). White bar scale: 20 μm. Images were taken with 200 Axiovert Zeiss fluorescent microscope equipped with ApoTome (confocal‐like system) at ×63 magnification and with DIC.
Figure S5. NO‐stimulated GC activity is significantly decreased in cardiac membrane fraction by AngII treatment. cGMP production after stimulation with 100 μmol/L S‐Nitroso‐N‐acetyl‐D‐penicillamine (SNAP, Nitric Oxide donor) of 10 to 50 μg protein for 10 minutes at 37°C was determined by Enzyme Immuno Assay in cytosolic and membrane fractions of hearts from control (Ctrl, n=3) or AngII‐treated (n=3) mice. GC activity is significantly decreased in heart membrane fraction of mice treated with AngII (*P<0.05). The decreased trend in cytosol was strong but not significant.
Figure S6. Action potentials (AP) recorded from ventricular myocytes isolated from WT and AngII‐treated GCα1KO mice. A, Representative traces and dV/dt values recorded from a ventricular myocyte in each group as indicated. Note the occurrence of early afterdepolarizations (EADs) in three APs shown in different colors (upper right panel). B, Summarized data for maximal dV/dt of AP upstroke. C, Summarized data for action potential amplitude (APA). D, Summarized data for AP duration at 50% repolarization (APD50). The number of cells is indicated above each bar.
Figure S7. GC1 and Cx43 are expressed in mice atrium. Representative images of immunostaining of GC, N‐Cadherin, Cx43 and Cx40 in atrium cryosection from WT mice (n=3). GCα1 (red, top panel) and GCβ1 (red, Middle panel) are localized at the ID (white arrow) identified with anti N‐Cadherin (green). GC is also found around the nucleus and is highly expressed in vascular smooth muscle cells of vessels (white arrowhead, middle panel). Cx43 (red) and Cx40 (green) are both expressed in mice atrium and show similar location at the membrane (bottom panel). Negative control (bottom right image) was processed with secondary antibodies (IIary Ab, AlexaFluor488 goat anti‐mouse and AlexaFluor594 goat antirabbit at 1:200) and no primary antibody. Nuclei and membranes are stained in blue with DAPI and in white with WGA, respectively. White bar scales: 20 μm to ×20 objective. Immunofluorescence signals are merged on Differential Interface Contrast (DIC) images of the atrium.
Figure S8. GCα1 KO mice develop cardiac hypertrophy that is exacerbated after AngII treatment. A, Ratio of heart weight (HW, mg) over tibia length (TL, cm) in AngII‐treated WT mice, and GCα1 KO mice±AngII indicates significant cardiac hypertrophy compared to WT control (n=5–10). B, The width of >8 cm (longitudinal view) per field of heart cryosections was measured and averaged showing significant enlargement in WT treated with AngII, and in GCα1 KO mice±AngII compared to control WT (n=5–7). *P<0.05; **P<0.01; ***P<0.001. Ratio and width values are indicated as mean±SEM in the tables below.
(J Am Heart Assoc. 2017;6:e006397 DOI: 10.1161/JAHA.117.006397.)29269353
References
- 1. Lukowski R, Rybalkin SD, Loga F, Leiss V, Beavo JA, Hofmann F. Cardiac hypertrophy is not amplified by deletion of cGMP‐dependent protein kinase I in cardiomyocytes. Proc Natl Acad Sci U S A. 2010;107:5646–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cawley SM, Kolodziej S, Ichinose F, Brouckaert P, Buys ES, Bloch KD. sGC{alpha}1 mediates the negative inotropic effects of NO in cardiac myocytes independent of changes in calcium handling. Am J Physiol Heart Circ Physiol. 2011;301:H157–H163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Patrucco E, Domes K, Sbroggio M, Blaich A, Schlossmann J, Desch M, Rybalkin SD, Beavo JA, Lukowski R, Hofmann F. Roles of cGMP‐dependent protein kinase I (cGKI) and PDE5 in the regulation of ang II‐induced cardiac hypertrophy and fibrosis. Proc Natl Acad Sci U S A. 2014;111:12925–12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agullo L, Garcia‐Dorado D, Escalona N, Ruiz‐Meana M, Mirabet M, Inserte J, Soler‐Soler J. Membrane association of nitric oxide‐sensitive guanylyl cyclase in cardiomyocytes. Cardiovasc Res. 2005;68:65–74. [DOI] [PubMed] [Google Scholar]
- 5. Zabel U, Kleinschnitz C, Oh P, Nedvetsky P, Smolenski A, Muller H, Kronich P, Kugler P, Walter U, Schnitzer JE, Schmidt HH. Calcium‐dependent membrane association sensitizes soluble guanylyl cyclase to nitric oxide. Nat Cell Biol. 2002;4:307–311. [DOI] [PubMed] [Google Scholar]
- 6. Tsai EJ, Liu Y, Koitabashi N, Bedja D, Danner T, Jasmin JF, Lisanti MP, Friebe A, Takimoto E, Kass DA. Pressure‐overload‐induced subcellular relocalization/oxidation of soluble guanylyl cyclase in the heart modulates enzyme stimulation. Circ Res. 2012;110:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gonzalez JP, Crassous PA, Schneider JS, Beuve A, Fraidenraich D. Neuronal nitric oxide synthase localizes to utrophin expressing intercalated discs and stabilizes their structural integrity. Neuromuscul Disord. 2015;25:964–976. [DOI] [PubMed] [Google Scholar]
- 8. Meens MJ, Pfenniger A, Kwak BR, Delmar M. Regulation of cardiovascular connexins by mechanical forces and junctions. Cardiovasc Res. 2013;99:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thevenin AF, Kowal TJ, Fong JT, Kells RM, Fisher CG, Falk MM. Proteins and mechanisms regulating gap‐junction assembly, internalization, and degradation. Physiology (Bethesda). 2013;28:93–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Solan JL, Lampe PD. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol. 2007;217:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Solan JL, Lampe PD. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014;588:1423–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fischmeister R, Castro LR, Abi‐Gerges A, Rochais F, Jurevicius J, Leroy J, Vandecasteele G. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006;99:816–828. [DOI] [PubMed] [Google Scholar]
- 13. Kokkonen K, Kass DA. Nanodomain regulation of cardiac cyclic nucleotide signaling by phosphodiesterases. Annu Rev Pharmacol Toxicol. 2017;57:455–479. [DOI] [PubMed] [Google Scholar]
- 14. Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2009;122:216–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crassous PA, Couloubaly S, Huang C, Zhou Z, Baskaran P, Kim DD, Papapetropoulos A, Fioramonti X, Duran WN, Beuve A. Soluble guanylyl cyclase is a target of angiotensin II‐induced nitrosative stress in a hypertensive rat model. Am J Physiol Heart Circ Physiol. 2012;303:H597–H604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fischer R, Dechend R, Gapelyuk A, Shagdarsuren E, Gruner K, Gruner A, Gratze P, Qadri F, Wellner M, Fiebeler A, Dietz R, Luft FC, Muller DN, Schirdewan A. Angiotensin II‐induced sudden arrhythmic death and electrical remodeling. Am J Physiol Heart Circ Physiol. 2007;293:H1242–H1253. [DOI] [PubMed] [Google Scholar]
- 17. Sovari AA, Iravanian S, Dolmatova E, Jiao Z, Liu H, Zandieh S, Kumar V, Wang K, Bernstein KE, Bonini MG, Duffy HS, Dudley SC. Inhibition of c‐Src tyrosine kinase prevents angiotensin II‐mediated connexin‐43 remodeling and sudden cardiac death. J Am Coll Cardiol. 2011;58:2332–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buys ES, Sips P, Vermeersch P, Raher MJ, Rogge E, Ichinose F, Dewerchin M, Bloch KD, Janssens S, Brouckaert P. Gender‐specific hypertension and responsiveness to nitric oxide in sGCalpha1 knockout mice. Cardiovasc Res. 2008;79:179–186. [DOI] [PubMed] [Google Scholar]
- 19. Bruce AF, Rothery S, Dupont E, Severs NJ. Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO‐1. Cardiovasc Res. 2008;77:757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q‐T interval in the conscious mouse. Am J Physiol. 1998;274:H747–H751. [DOI] [PubMed] [Google Scholar]
- 21. Nimmegeers S, Sips P, Buys E, Brouckaert P, Van de Voorde J. Functional role of the soluble guanylyl cyclase alpha(1) subunit in vascular smooth muscle relaxation. Cardiovasc Res. 2007;76:149–159. [DOI] [PubMed] [Google Scholar]
- 22. Mayama T, Matsumura K, Lin H, Ogawa K, Imanaga I. Remodelling of cardiac gap junction connexin 43 and arrhythmogenesis. Exp Clin Cardiol. 2007;12:67–76. [PMC free article] [PubMed] [Google Scholar]
- 23. Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DY, Chi NC, Shaw RM. Limited forward trafficking of connexin 43 reduces cell‐cell coupling in stressed human and mouse myocardium. J Clin Invest. 2010;120:266–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giepmans BN. Gap junctions and connexin‐interacting proteins. Cardiovasc Res. 2004;62:233–245. [DOI] [PubMed] [Google Scholar]
- 26. Solan JL, Marquez‐Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down‐regulation by PKC. J Cell Biol. 2007;179:1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. TenBroek EM, Lampe PD, Solan JL, Reynhout JK, Johnson RG. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J Cell Biol. 2001;155:1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J, McLerie M, Lopatin AN. Transgenic upregulation of IK1 in the mouse heart leads to multiple abnormalities of cardiac excitability. Am J Physiol Heart Circ Physiol. 2004;287:H2790–H2802. [DOI] [PubMed] [Google Scholar]
- 29. Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac‐restricted inactivation of connexin43. Circ Res. 2001;88:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hammond J, Balligand JL. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: from contractility to remodeling. J Mol Cell Cardiol. 2012;52:330–340. [DOI] [PubMed] [Google Scholar]
- 31. Yao J, Hiramatsu N, Zhu Y, Morioka T, Takeda M, Oite T, Kitamura M. Nitric oxide‐mediated regulation of connexin43 expression and gap junctional intercellular communication in mesangial cells. J Am Soc Nephrol. 2005;16:58–67. [DOI] [PubMed] [Google Scholar]
- 32. Gonzalez JP, Ramachandran J, Xie LH, Contreras JE, Fraidenraich D. Selective connexin43 inhibition prevents isoproterenol‐induced arrhythmias and lethality in muscular dystrophy mice. Sci Rep. 2015;5:13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang N, De Vuyst E, Ponsaerts R, Boengler K, Palacios‐Prado N, Wauman J, Lai CP, De Bock M, Decrock E, Bol M, Vinken M, Rogiers V, Tavernier J, Evans WH, Naus CC, Bukauskas FF, Sipido KR, Heusch G, Schulz R, Bultynck G, Leybaert L. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2013;108:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chkourko HS, Guerrero‐Serna G, Lin X, Darwish N, Pohlmann JR, Cook KE, Martens JR, Rothenberg E, Musa H, Delmar M. Remodeling of mechanical junctions and of microtubule‐associated proteins accompany cardiac connexin43 lateralization. Heart Rhythm. 2012;9:1133–1140.e1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hambleton R, Krall J, Tikishvili E, Honeggar M, Ahmad F, Manganiello VC, Movsesian MA. Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J Biol Chem. 2005;280:39168–39174. [DOI] [PubMed] [Google Scholar]
- 36. Zhang SS, Shaw RM. Trafficking highways to the intercalated disc: new insights unlocking the specificity of connexin 43 localization. Cell Commun Adhes. 2014;21:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duffy HS. The molecular mechanisms of gap junction remodeling. Heart Rhythm. 2012;9:1331–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tse G, Yeo JM. Conduction abnormalities and ventricular arrhythmogenesis: the roles of sodium channels and gap junctions. Int J Cardiol Heart Vasc. 2015;9:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kucera JP, Rohr S, Rudy Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res. 2002;91:1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M, van Rijen HV. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 2012;9:600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kleber AG. The shape of the electrical action‐potential upstroke: a new aspect from optical measurements on the surface of the heart. Circ Res. 2005;97:204–206. [DOI] [PubMed] [Google Scholar]
- 42. Berecki G, Wilders R, de Jonge B, van Ginneken AC, Verkerk AO. Re‐evaluation of the action potential upstroke velocity as a measure of the Na+ current in cardiac myocytes at physiological conditions. PLoS One. 2010;5:e15772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rozmaritsa N, Christ T, Van Wagoner DR, Haase H, Stasch JP, Matschke K, Ravens U. Attenuated response of L‐type calcium current to nitric oxide in atrial fibrillation. Cardiovasc Res. 2014;101:533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vandecasteele G, Verde I, Rucker‐Martin C, Donzeau‐Gouge P, Fischmeister R. Cyclic GMP regulation of the L‐type Ca(2+) channel current in human atrial myocytes. J Physiol. 2001;533:329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herring N, Rigg L, Terrar DA, Paterson DJ. NO‐cGMP pathway increases the hyperpolarisation‐activated current, I(F), and heart rate during adrenergic stimulation. Cardiovasc Res. 2001;52:446–453. [DOI] [PubMed] [Google Scholar]
- 46. Musialek P, Lei M, Brown HF, Paterson DJ, Casadei B. Nitric oxide can increase heart rate by stimulating the hyperpolarization‐activated inward current, I(F). Circ Res. 1997;81:60–68. [DOI] [PubMed] [Google Scholar]
- 47. Yoo S, Lee SH, Choi BH, Yeom JB, Ho WK, Earm YE. Dual effect of nitric oxide on the hyperpolarization‐activated inward current (I(F)) in sino‐atrial node cells of the rabbit. J Mol Cell Cardiol. 1998;30:2729–2738. [DOI] [PubMed] [Google Scholar]
- 48. Lin X, Gemel J, Glass A, Zemlin CW, Beyer EC, Veenstra RD. Connexin40 and connexin43 determine gating properties of atrial gap junction channels. J Mol Cell Cardiol. 2010;48:238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kasi VS, Xiao HD, Shang LL, Iravanian S, Langberg J, Witham EA, Jiao Z, Gallego CJ, Bernstein KE, Dudley SC Jr. Cardiac‐restricted angiotensin‐converting enzyme overexpression causes conduction defects and connexin dysregulation. Am J Physiol Heart Circ Physiol. 2007;293:H182–H192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kostin S, Dammer S, Hein S, Klovekorn WP, Bauer EP, Schaper J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res. 2004;62:426–436. [DOI] [PubMed] [Google Scholar]
- 51. Nakamura T, Ranek MJ, Lee DI, Shalkey Hahn V, Kim C, Eaton P, Kass DA. Prevention of PKG1alpha oxidation augments cardioprotection in the stressed heart. J Clin Invest. 2015;125:2468–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Takimoto E. Cyclic GMP‐dependent signaling in cardiac myocytes. Circ J. 2012;76:1819–1825. [DOI] [PubMed] [Google Scholar]
- 53. Fraccarollo D, Galuppo P, Motschenbacher S, Ruetten H, Schafer A, Bauersachs J. Soluble guanylyl cyclase activation improves progressive cardiac remodeling and failure after myocardial infarction: cardioprotection over ACE inhibition. Basic Res Cardiol. 2014;109:421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A, Negative control for immunofluorescence. Representative image of immunofluorescence of heart cryosection from WT mice performed with secondary AlexaFluor488 goat anti‐mouse and AlexaFluor594 goat anti‐rabbit (1:200) and without primary antibodies with same exposure time as in Figure 1. No red or green signal is detected. Nuclei and membranes are stained in blue with DAPI and in white with Wheat Germ Agglutinin (WGA), respectively. (n=4). B, Overlapping of GC and Cx43 signals. The yellow signals (black arrows) detected in ID of the right orthogonal section (blue box) and the top orthogonal section (red box) of the optical sectioning indicate an overlap of immunolabeled GCβ1 (red) and Cx43 (green). This picture is representative of n=3. There is a low signal overlap between the Cx43 and GC under these conditions, reminiscent of the moderate colocalization observed between ZO‐1 and Cx43 at the ID (Barker RJ et al, Circulation Research 2002; 90:317–334).
Figure S2. Probing of Cx43 and total protein in cytosolic, membranes (triton soluble) and junctional plaques (triton insoluble) fractions of WT and GCα1 KO mice±AngII. A, Densitometry analysis of Cx43 level in the membrane fraction (triton soluble). There was no significant change compare to WT control (P>0.05, n=4). B, Representative image of nitrocellulose membranes probed with anti‐Connexin 43 or stained with Ponceau red after transfer of 10 to 50 μg of proteins from WT Ctrl, WT+AngII, GCα1 KO Ctrl, and GCα1 KO+AngII. (n=3).
Figure S3. Supraventricular abnormality in electrical propagation in GCα1KO mice heart. A, Representative traces recorded from WT (n=6) and GCα1 KO mice (n=6) show that GCα1 KO mice have (a) irregular RR interval (dotted double arrows) and atrioventricular (AV) block (3rd degree, *uncoupled P‐waves); (b) absence of P‐waves, which are indicative of dysfunction in sinoatrial (SAN) conduction. B, The incidence of AV block and SA node impairment was significantly higher in GCα1 KO mice compared to WT (6 GCα1‐KO mice out of 10 and 0 WT mice out of 6; *P<0.05, Fisher exact test). The duration of AV blocks or SAN dysfunction was analyzed in a 10‐minute recording period and the supraventricular abnormalities were observed for 4.4±0.9 minutes. C, Significant prolonged duration of P‐wave in GCα1 KO treated with AngII (n=6), compared to WT (n=6), WT+AngII (n=6) and untreated GCα1 KO (n=7), which reflects a slower electrical conduction in the atrium. **P<0.01 and ***P<0.001. Data are expressed as mean±SEM.
Figure S4. A GC1–Cx43 complex is detected in rat neonatal cardiomyocytes (NCM) by proximity ligation assay. Representative fluorescent image (n=3, independent cultures) of GCα1‐Cx43 (top left) and GCβ1‐Cx43 association (top right) detected by proximity ligation assay (PLA). Red dots indicate endogenous complex. Black arrows indicate GC‐Cx43 complex potentially located at the junction between two NCM. As a negative control, cells were incubated with anti‐Cx43 only (no anti‐GC antibodies, bottom left). White bar scale: 20 μm. Images were taken with 200 Axiovert Zeiss fluorescent microscope equipped with ApoTome (confocal‐like system) at ×63 magnification and with DIC.
Figure S5. NO‐stimulated GC activity is significantly decreased in cardiac membrane fraction by AngII treatment. cGMP production after stimulation with 100 μmol/L S‐Nitroso‐N‐acetyl‐D‐penicillamine (SNAP, Nitric Oxide donor) of 10 to 50 μg protein for 10 minutes at 37°C was determined by Enzyme Immuno Assay in cytosolic and membrane fractions of hearts from control (Ctrl, n=3) or AngII‐treated (n=3) mice. GC activity is significantly decreased in heart membrane fraction of mice treated with AngII (*P<0.05). The decreased trend in cytosol was strong but not significant.
Figure S6. Action potentials (AP) recorded from ventricular myocytes isolated from WT and AngII‐treated GCα1KO mice. A, Representative traces and dV/dt values recorded from a ventricular myocyte in each group as indicated. Note the occurrence of early afterdepolarizations (EADs) in three APs shown in different colors (upper right panel). B, Summarized data for maximal dV/dt of AP upstroke. C, Summarized data for action potential amplitude (APA). D, Summarized data for AP duration at 50% repolarization (APD50). The number of cells is indicated above each bar.
Figure S7. GC1 and Cx43 are expressed in mice atrium. Representative images of immunostaining of GC, N‐Cadherin, Cx43 and Cx40 in atrium cryosection from WT mice (n=3). GCα1 (red, top panel) and GCβ1 (red, Middle panel) are localized at the ID (white arrow) identified with anti N‐Cadherin (green). GC is also found around the nucleus and is highly expressed in vascular smooth muscle cells of vessels (white arrowhead, middle panel). Cx43 (red) and Cx40 (green) are both expressed in mice atrium and show similar location at the membrane (bottom panel). Negative control (bottom right image) was processed with secondary antibodies (IIary Ab, AlexaFluor488 goat anti‐mouse and AlexaFluor594 goat antirabbit at 1:200) and no primary antibody. Nuclei and membranes are stained in blue with DAPI and in white with WGA, respectively. White bar scales: 20 μm to ×20 objective. Immunofluorescence signals are merged on Differential Interface Contrast (DIC) images of the atrium.
Figure S8. GCα1 KO mice develop cardiac hypertrophy that is exacerbated after AngII treatment. A, Ratio of heart weight (HW, mg) over tibia length (TL, cm) in AngII‐treated WT mice, and GCα1 KO mice±AngII indicates significant cardiac hypertrophy compared to WT control (n=5–10). B, The width of >8 cm (longitudinal view) per field of heart cryosections was measured and averaged showing significant enlargement in WT treated with AngII, and in GCα1 KO mice±AngII compared to control WT (n=5–7). *P<0.05; **P<0.01; ***P<0.001. Ratio and width values are indicated as mean±SEM in the tables below.