

Abstract
Background
Although statins reduce cardiovascular events, residual risk remains. Therefore, additional modalities are needed to reduce risk. We evaluated the effect of eicosapentaenoic acid and docosahexaenoic acid in pharmacologic doses added to statin treatment on coronary artery plaque volume.
Methods and Results
A total of 285 subjects with stable coronary artery disease on statins were randomized to omega‐3 ethyl‐ester (1.86 g of eicosapentaenoic acid and 1.5 g of docosahexaenoic acid daily) or no omega‐3 (control) for 30 months. Coronary plaque volume was assessed by coronary computed tomographic angiography. Mean (SD) age was 63.0 (7.7) years; mean low‐density lipoprotein cholesterol ≤80 mg/dL. In the intention‐to‐treat analysis, our primary endpoint, noncalcified plaque volume, was not different between groups (P=0.14) but approached significance in the per protocol analysis (P=0.07). When stratified by age in the intention‐to‐treat analysis, younger omega‐3 subjects had significantly less progression of the primary endpoint, noncalcified plaque (P=0.013), and fibrous, calcified and total plaque. In plaque subtype analysis, controls had significant progression of fibrous plaque compared to no change in the omega‐3 ethyl‐ester group (median % change [interquartile range], 5.0% [−5.7, 20.0] versus −0.1% [−12.3, 14.5], respectively; P=0.018). Among those on low‐intensity statins, omega‐3 ethyl‐ester subjects had attenuation of fibrous plaque progression compared to controls (median % change [interquartile range], 0.3% [−12.8, 9.0] versus 4.8% [−5.1, 19.0], respectively; P=0.032). In contrast, those on high‐intensity statins had no difference in plaque change in either treatment arm.
Conclusions
High‐dose eicosapentaenoic acid and docosahexaenoic acid provided additional benefit to statins in preventing progression of fibrous coronary plaque in subjects adherent to therapy with well‐controlled low‐density lipoprotein cholesterol levels. The benefit on low‐intensity statin, but not high‐intensity statin, suggests that statin intensity affects plaque volume.
Clinical Trial Registration
URL: http://www.ClinicalTrials.gov. Unique identifier: NCT01624727.
Keywords: coronary computed tomography angiography, coronary plaque subtype, eicosapentaenoic acid, omega‐3 fatty acids, plaque progression
Subject Categories: Coronary Artery Disease, Diet and Nutrition, Computerized Tomography (CT), Clinical Studies, Secondary Prevention
Clinical Perspective
What Is New?
High dose eicosapentaenoic acid and docosahexaenoic acid (3.36 g daily) provided additional benefit to statins in preventing progression of fibrous coronary plaque over 30 months measured by coronary computed tomographic angiography in coronary artery disease subjects adherent to therapy with low‐density lipoprotein cholesterol <80 mg/dL.
Subjects aged <64.2 years had significantly less progression of noncalcified plaque (primary endpoint) and fibrous, calcified, and total plaque volume compared with older subjects.
Eicosapentaenoic acid and docosahexaenoic acid prevented plaque progression in those on low‐intensity statin, but not high‐intensity statin.
Musculoskeletal events, infectious diseases, and joint replacement were significantly lower with eicosapentaenoic acid and docosahexaenoic acid.
What Are the Clinical Implications?
The benefit in younger subjects suggests that eicosapentaenoic acid and docosahexaenoic acid earlier in the course of disease may be beneficial.
If high‐intensity statin is not tolerated, adding high‐dose eicosapentaenoic acid and docosahexaenoic acid to low‐intensity statin may still prevent plaque progression.
Past studies suggest that plaque volume predicts cardiovascular events, supporting the potential clinical importance of lack of plaque progression in the eicosapentaenoic acid/docosahexaenoic acid arm.
The dose of 3.36 g is higher than can be achieved by consuming regular foods such as fish and higher than used in past trials with clinical end points.
Eicosapentaenoic acid and docosahexaenoic acid may reduce musculoskeletal events and limit joint replacement.
Despite the significant reduction in cardiovascular events with statin therapy, those on statins still suffer from a high level of residual risk manifested as atherosclerotic plaque progression and cardiovascular events despite statin treatment to achieve target low‐density‐lipoprotein cholesterol (LDL‐C) reduction ≤70 mg/dL.1, 2, 3 Therefore, additional modalities to reduce residual risk are needed. In the JELIS (Japan Eicosapentaenoic Lipid Intervention Study), addition of 1800 mg/day of open‐label eicosapentaenoic acid (EPA) to low‐intensity statin therapy conferred a significant 19% reduction in major coronary events compared with statins alone, a finding suggesting that EPA added to low‐intensity statin therapy can reduce residual risk.4
Omega‐3 fatty acids are approved by the US Food and Drug Administration for lowering elevated levels of triglyceride, but their beneficial effect was independent of triglyceride lowering in the JELIS trial4; therefore, other mechanisms need to be explored. The aim of the current study was to determine whether high‐dose, very‐long‐chain omega‐3 polyunsaturated fatty acids—EPA and docosahexaenoic acid (DHA)—conferred additional benefit to statin treatment in preventing progression of coronary plaque volume compared with statin alone in patients with coronary artery disease (CAD). Coronary plaque volume was measured by coronary computed tomographic angiography (CCTA). We chose CCTA because it is noninvasive, allows for examination of all coronary arteries in contrast to intravascular ultrasound, which examines only a culprit artery, and has been validated to detect progression of coronary plaque volume.5, 6, 7, 8 Moreover, CCTA has shown attenuation of progression of coronary plaque by statin drugs.9, 10, 11, 12, 13
Methods
Imaging methods, analytical methods, and study materials have been made available to other researchers for purposes of reproducing the results or replicating the procedures.14, 15
Study Design
The HEARTS (Slowing HEART Disease With Lifestyle and Omega‐3 Fatty Acids) trial was a randomized, controlled, parallel clinical trial conducted at Beth Israel Deaconess Medical Center (Boston, MA). The protocol was approved by the Beth Israel Deaconess Medical Center Institutional Review Board, and all subjects signed informed consent. The hypothesis was that omega‐3 ethyl‐ester supplementation plus statin is superior to statin alone over 30 months in reducing progression of noncalcified coronary artery plaque measured by CCTA in patients with stable CAD.
Participants
Eligible participants were aged 21 to 80 years and had stable CAD defined as at least 1 of the following: ≥50% stenosis in at least 1 coronary artery at cardiac catheterization; previous myocardial infarction (≥6 months previously) or percutaneous coronary intervention (≥6 months previously); coronary bypass surgery (≥12 months previously); abnormal exercise treadmill test (defined as at least 1 mm of horizontal or downsloping ST depression in at least 2 contiguous ECG leads) or an area of reversible ischemia on nuclear imaging; and pharmacologic stress or stress echocardiography with subsequent revascularization. Inclusion criteria also included a body mass index (BMI; weight in kilograms divided by height in meters squared) ≥27 kg/m2 or a BMI of 25 to 26.9 with either an increased waist circumference or a history of at least 2 components of metabolic syndrome, which included triglyceride ≥150 mg/dL, high‐density lipoprotein cholesterol <40 mg/dL if male or <50 mg/dL if female, glucose ≥100 mg/dL, or treated hypertension or blood pressure ≥130/85 mm Hg.16 Additional inclusion criteria included stable use of 3‐hydroxy‐3‐methyl‐glutaryl‐CoA reductase inhibitor (statin) and estimated creatinine clearance as measured by the Cockcroft‐Gault equation ≥60 mL/min per 1.73 m2. Exclusion criteria for CCTA were BMI >35 kg/m2 (females) or >40 kg/m2 (males), contraindication to iodinated contrast agents, and serum creatinine >1.5 mg/dL.
Randomization and Study Intervention
Randomization was computer allocated in blocks of 4 and stratified by presence or absence of diabetes mellitus. The trial protocol included a screening visit, baseline visit, and follow‐up visits at 6 weeks and 3, 6, 9, 12, 18, 24, and 30 months. A detailed history, physical examination, height, weight, and waist measurement, blood pressure measurement, ECG, and ascertainment of medications were obtained at each visit. Blood samples were obtained after a 12‐hour fast, and plasma and serum were prepared. Glucose, hemoglobin A1c, chemical profile, total white blood cell count, absolute neutrophil, lymphocyte, monocyte, and platelet counts and lipid panel were measured at Quest Diagnostics (Cambridge, MA). High‐sensitivity C‐reactive protein was measured by immunoturbimetric assays using an automated, standardized, high‐throughput method with coefficient of variation below 5% at Boston Heart Diagnostics (a College of American Pathologists‐accredited, Clinical Laboratory Improvement Amendments–certified clinical laboratory; Framingham, MA). CCTA was performed at baseline and repeated at 30 months. Adverse events were assessed by questionnaires at follow‐up visits.
Participants were randomly assigned to receive either open‐label, omega‐3 ethyl esters prepared from fish oil (Lovaza) in the form of 4 capsules daily or no Lovaza (termed control) for 30 months. All subjects were on a stable dose of a statin for at least 3 months. Subjects in the omega‐3 ethyl‐ester group received 3.36 g of Lovaza as 4 soft gels. Each 1000‐mg capsule of Lovaza contains at least 900 mg of the ethyl esters of omega‐3 fatty acids with EPA (465 mg) and DHA (375 mg) for a total daily dose of 1.86 g of EPA and 1.5 g of DHA, a dose higher than the amount of omega‐3 fatty acids that can be derived from dietary intake. The GISSI (Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto Miocardico)‐prevention study17 and the GISSI heart failure trial18 used 1 g of omega‐3 fatty acid daily (≈850 mg of EPA and DHA) and had a reduction in cardiovascular events. Subsequent trials used the same dose, but had negative outcomes possibly attributed to increased use of cardioprotective medications that were not available when GISSI was done.19, 20, 21 Therefore, we used a higher dose of 3.36 g of EPA and DHA daily. Controls were instructed not to take over‐the‐counter omega‐3 fatty acid supplementation. A detailed medication list was obtained during each visit to screen for compliance with omega‐3 ethyl‐ester and the use of over‐the‐counter omega‐3 fatty acids. Lovaza was provided by GlaxoSmithKline (Research Triangle Park, NC).
Image Acquisition and Reconstruction
Imaging was performed at Beth Israel Deaconess Medical Center using a 320‐row detector scanner (Aquilion ONE; Toshiba Medical Systems, Otawara, Japan) with prospective ECG gating. The protocol for performance of CCTA, plaque identification, and quantification has been previously published14, 15 with image acquisition details and references in Appendix 1 in Supplement 2 of Hauser et al.15
Coronary Plaque Analysis
CCTA images underwent 3‐dimensional reconstruction for coronary segment plaque volume analysis using semiautomated software (SUREPlaque, version 6.3.2; Vital Images, Minnetonka, MN).7, 22, 23, 24 Representative images have been previously published (Supplement 2 in Hauser et al).15 Segments with past revascularization or significant calcification causing calcium‐bloom artifact were excluded. Branches or focal calcification served as fiducial markers to ensure measurement of the same segment at 30‐month follow‐up. Plaque analysis was performed independently by 2 readers blinded to treatment allocation. Noncalcified plaque, the sum of fatty and fibrous plaque, was based on Hounsfield unit densities of fatty (−100 to 49 Hounsfield units) and fibrous plaque (50–150 Hounsfield units). Calcified plaque was ≥150 Hounsfield units. Plaque volumes were indexed to the length of the plaque lesion; indexed plaque volume was defined as plaque volume (mm3) divided by artery segment length (mm).
Statistical Analysis
The primary outcome was percent change in indexed volume of noncalcified coronary plaque, the sum of fatty and fibrous plaque, at 30‐month follow‐up compared with baseline. A prespecified per‐protocol analysis of primary and secondary outcomes was done in those who adhered to treatment. Given that biological mechanisms for formation of fatty and fibrous plaque may differ, prespecified secondary outcomes included assessing changes in fatty and fibrous plaques individually as well as calcified and total plaque volumes. Sample size for the primary outcome was estimated based on data from the single longitudinal CCTA study at the time showing a mean (SD) 24% (13%) reduction in coronary plaque volume with statin treatment in 27 patients.9 Given the lack of data on the effect of omega‐3 fatty acid on coronary plaque, we powered the study to detect a 4% reduction (one sixth that of statins) in plaque volume in the omega‐3 ethyl‐ester group compared with no change in controls. Therefore, a total of 111 subjects completing the trial in each treatment group were required to achieve 80% power with a type I error of 0.05. The analysis was performed following intention‐to‐treat and per‐protocol principles. Subgroup analyses stratified by age, BMI, triglyceride, and LDL‐C ≤ and > than the median was performed. Subgroup analyses were also performed for sex, diabetes mellitus status, and statin intensity. Categorical variables were expressed as counts and percentages. Normality tests were conducted using the Shapiro–Wilk test. Continuous variables were reported as the mean and SD or 95% confidence interval (CI) for normally distributed variables or median and interquartile range [IQR] for non‐normally distributed variables. Plaque volumes and % change were not normally distributed and therefore are reported as median [IQR]. Continuous variables were compared using unpaired (between‐group comparisons) and paired t tests (within‐group comparisons) for normally distributed variables or the Mann–Whitney U test (between‐group comparisons) and Wilcoxon signed‐rank test (within group comparisons) for non‐normally distributed variables. Correlations were determined using Pearson correlation coefficient for normally distributed variables or Spearman's rank correlation coefficient for non‐normally distributed variables. A 2‐sided P≤0.05 was considered statistically significant. Ninety‐five percent CIs around estimated mean differences between treatment groups for change in baseline biochemical and anthropometric data are reported. Comparisons of coronary plaque subtypes were a prespecified secondary analysis; therefore, P values were not adjusted. Data analyses were performed using SPSS software (version 20.0; IBM Corp, Armonk, NY).
Results
Demography and Study Adherence
Of 336 subjects screened, 285 underwent baseline CCTA evaluation and were randomized to omega‐3 ethyl‐ester (n=143) or no omega‐3 ethyl‐ester (n=142; Figure). Fourteen of those receiving omega‐3 ethyl‐ester and 21 of those not receiving omega‐3 ethyl‐ester discontinued the intervention and did not have 30‐month follow‐up. Reasons for discontinuation are shown in Figure. Of those 250 subjects with 30‐month follow‐up, 10 did not have a 30‐month CCTA scan, the majority attributed to developing an allergy to iodinated contrast material over the 30‐month follow‐up. Of the remaining 240 subjects, 126 in the omega‐3 ethyl‐ester group and 114 in control had paired CCTA scans and were included in the intention‐to‐treat analysis for the primary end point. In the per‐protocol analysis, we excluded 17 controls who took ≥900 mg of omega‐3 fatty acids and 4 subjects in the omega‐3 ethyl‐ester group who did not take daily omega‐3 ethyl‐ester.
Figure 1.
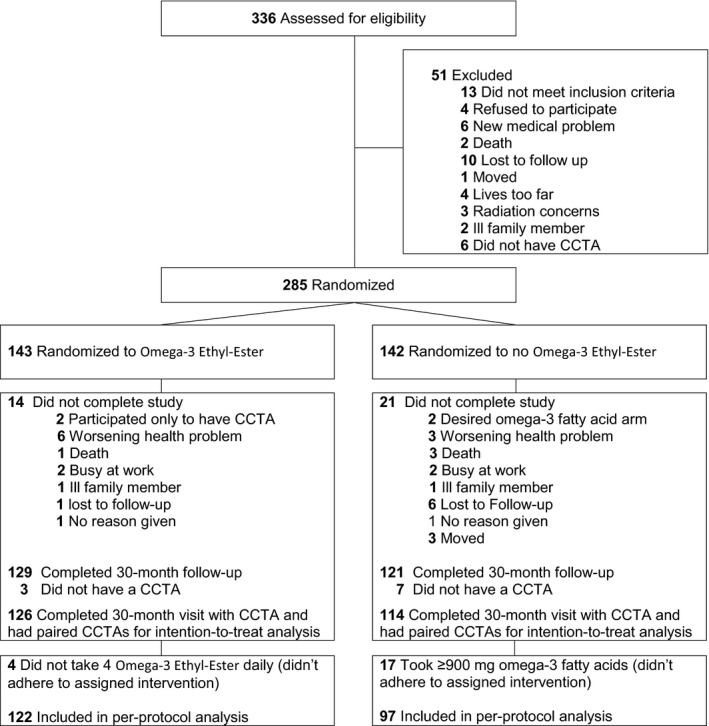
Consolidated standards of reporting trials diagram. CCTA indicates coronary computed tomographic angiography.
Clinical and Laboratory Outcomes
Baseline characteristics are shown in Table 1. Mean (SD) age was 63.0 (7.7) years and 15% were women. Tables 2 and 3 show baseline values of clinical and laboratory variables and % change from baseline at 30‐month follow‐up in the control and omega‐3 ethyl‐ester groups according to the intention‐to‐treat and per‐protocol analyses, respectively. In both analyses at 30‐month follow‐up (Tables 2 and 3), mean LDL‐C was similar between the 2 groups (for intention‐to‐treat: mean [SD], 77.5 mg/dL [28.3] versus 80.5 mg/dL [33.7]; P=0.46). In the intention‐to‐treat analysis (Table 2), those receiving omega‐3 ethyl‐ester had a significant reduction in triglyceride level (median [IQR], 123.0 mg/dL [81.5, 178.5] to 102.0 mg/dL [67.5, 146.8]; paired P<0.001) whereas controls had no significant change (median [IQR], 117.0 mg/dL [83.0, 161.5] to 113.0 mg/dL [78.5, 169.8]; paired P=0.70); the % change between the 2 groups was significant (median % change [IQR], −14.3% [−32.1, 9.5] versus −2.2% [−19.4, 16.1], respectively; P=0.003). No significant difference was observed in the % change of high‐sensitivity C‐reactive protein in the controls compared with the omega‐3 ethyl‐ester group (mean difference, 53.4%; 95% CI, −42.2 to 149.1; P=0.27). There were no other significant between‐group differences.
Table 1.
Baseline Characteristics in the Control and Omega‐3 Ethyl‐Ester Groups
| Intention‐to‐Treat | Per‐Protocol | |||
|---|---|---|---|---|
| Controls (n=114) | Omega‐3 Ethyl‐Ester (n=126) | Controls (n=97) | Omega‐3 Ethyl‐Ester (n=122) | |
| Demographic characteristics | ||||
| Age, mean±SD, y | 63.5±7.6 | 62.5±7.8 | 62.6±7.5 | 62.4±7.8 |
| Male sex, n (%) | 97 (85.1) | 107 (84.9) | 84 (86.6) | 104 (85.2) |
| Inclusion criteria (may have more than 1), n (%) | ||||
| History of MI | 50 (43.9) | 62 (49.2) | 47 (48.5) | 60 (49.2) |
| History of PCI | 69 (60.5) | 81 (64.3) | 59 (60.8) | 77 (63.1) |
| History of CABG | 35 (30.7) | 24 (19.0) | 26 (26.8) | 23 (18.9) |
| Cardiovascular risk factors, n (%) | ||||
| Hypertension | 101 (88.6) | 99 (78.6) | 87 (89.7) | 96 (78.7) |
| Diabetes mellitus | 34 (29.8) | 34 (27.0) | 27 (27.8) | 33 (27.0) |
| Anthropometric and blood pressure, mean±SD | ||||
| Weight, kg | 90.8±14.0 | 92.2±13.8 | 91.2±13.9 | 92.5±13.7 |
| Body mass index, kg/m2 a | 30.5±3.5 | 30.8±3.7 | 30.5±3.5 | 30.9±3.8 |
| Waist circumference, cm | 106.2±10.2 | 106.9±10.6 | 106.6±10.3 | 106.9±10.7 |
| Systolic BP, mm Hg | 124.5±14.0 | 124.2±14.7 | 124.1±13.9 | 124.3±14.8 |
| Diastolic BP, mm Hg | 73.3±9.6 | 73.3±10.0 | 73.3±9.8 | 73.3±10.1 |
| Biochemical profile, mean±SD | ||||
|
Glucose, mg/dL (mmol/L) |
107.4±35.4 (6.0±2.0) |
104.7±27.2 (5.8±1.5) |
106.2±32.2 (5.9±1.8) |
104.6±27.3 (5.8±1.5) |
|
HbA1c, % (mmol/mol) |
6.2±1.0 (44.0±10.0) |
6.1±0.8 (43.0±9.0) |
6.2±1.0 (44.0±10.0) |
6.1±0.8 (43.0±9.0) |
|
hs‐CRP, mg/L (nmol/L) |
1.7±2.4 (16.2±22.9) |
1.9±2.4 (18.1±22.9) |
1.8±2.5 (17.1±23.8) |
2.0±2.4 (19.1±22.9) |
| Lipids, mean±SD | ||||
|
Total cholesterol, mg/dL (mmol/L) |
151.0±37.5 (3.9±1.0) |
153.2±34.5 (4.0±0.9) |
152.2±38.4 (3.9±1.0) |
153.7±34.6 (4.0±0.9) |
|
Triglyceride, median [IQR], mg/dL (mmol/L) |
117.0 [83.0, 161.5] (1.3 [0.9, 1.8]) |
123.0 [81.5, 178.5] (1.4 [0.9, 2.0]) |
116.0 [84.0, 163.5] (1.3 [1.0, 1.9]) |
122.0 [81.5, 176.5] (1.4 [0.9, 2.0]) |
|
HDL‐C, mg/dL (mmol/L) |
46.9±14.9 (1.2±0.4) |
47.3±14.2 (1.2±0.4) |
47.2±15.5 (1.2±0.4) |
47.6±14.3 (1.2±0.4) |
|
LDL‐C, mg/dL (mmol/L) |
77.5±27.5 (2.0±0.7) |
78.5±27.0 (2.0±0.7) |
78.1±28.4 (2.0±0.7) |
79.1±27.2 (2.1±0.7) |
| Complete blood count, mean±SD | ||||
| WBC, 109 cells/L | 6.5±1.8 | 6.8±2.4 | 6.6±1.8 | 6.8±2.4 |
| Monocytes, cells/μL | 536.7±169.2 | 521.4±165.3 | 538.3±176.7 | 516.0±157.3 |
| Neutrophils, cells/μL | 4125.0±1592.9 | 4179.8±1289.4 | 4153.2±1608.1 | 4155.2±1275.6 |
| Lymphocytes, cells/μL | 1620.7±509.2 | 1880.9±1882.6 | 1649.4±531.1 | 1884.3±1912.2 |
| Platelets, cells/μL | 191.5±45.6 | 191.0±53.9 | 194.7±44.5 | 190.5±54.5 |
| Medication, n (%) | ||||
| Statin | 107 (93.9) | 121 (96.0) | 91 (93.8) | 117 (95.9) |
| Aspirin | 110 (96.5) | 121 (96.0) | 93 (95.9) | 117 (95.9) |
| ACE‐I | 63 (55.3) | 68 (54.0) | 53 (54.6) | 66 (54.1) |
| ARB | 25 (21.9) | 21 (16.7) | 21 (21.6) | 20 (16.4) |
| Hydrochlorothiazide | 23 (20.2) | 22 (17.5) | 21 (21.6) | 21 (17.2) |
| Furosemide | 12 (10.5) | 7 (5.6) | 9 (9.3) | 7 (5.7) |
| Calcium‐channel blocker | 28 (24.6) | 30 (23.8) | 23 (23.7) | 30 (24.6) |
| Beta blockers | 85 (74.6) | 88 (69.8) | 73 (75.3) | 86 (70.5) |
ACE‐I indicates angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; BP, blood pressure; CABG, coronary artery bypass grafting; HbA1c, hemoglobin A1c; HDL‐C, high‐density lipoprotein cholesterol; hs‐CRP, high‐sensitivity C‐reactive protein; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; MI, myocardial infarction; PCI, percutaneous coronary intervention; WBC, white blood cell count.
Calculated as weight in kilograms divided by height in meters squared.
Table 2.
Percent Change From Baseline at 30‐Month Follow‐Up in the Control and Omega‐3 Ethyl‐Ester Groups in the Intention‐to‐Treat Analysis
| Control (n=114) | Omega‐3 Ethyl‐Ester (n=126) | Differences in % Change | ||||
|---|---|---|---|---|---|---|
| Baseline Value Mean±SD | % Change From Baseline Mean (95% CI) | Baseline Value Mean±SD | % Change From Baseline Mean (95% CI) | Mean (95% CI) | P Valuea | |
| Clinical parameters | ||||||
| Systolic BP, mm Hg | 124.5±14.0 | −2.7 (−5.2, −0.1) | 124.2±14.7 | −3.0 (−5.2, −0.9) | 0.4 (−2.9, 3.7) | 0.82 |
| Diastolic BP, mm Hg | 73.3±9.6 | −4.4 (−6.5, −2.2) | 73.3±10.0 | −4.3 (−6.4, −2.2) | −0.1 (−3.1, 2.9) | 0.96 |
| Waist circumference, cm | 106.2±10.2 | 0.3 (−0.6, 1.3) | 106.9±10.6 | 7.6 (−6.6, 21.8) | −7.3 (−22.1, 7.5) | 0.92 |
| Weight, kg | 90.8±14.0 | −1.4 (−2.5, −0.3) | 92.2±13.8 | −0.9 (−2.2, 0.4) | −0.5 (−2.2, 1.2) | 0.58 |
| Body mass index, kg/m2 | 30.5±3.5b | −0.7 (−1.8, 0.4) | 30.8±3.7b | 1.9 (−2.4, 6.2) | −2.6 (−7.2, 2.1) | 0.28 |
| Biochemical profile | ||||||
|
Glucose, mg/dL (mmol/L) |
107.4±35.4 (6.0±2.0) |
7.5 (1.2, 13.8) |
104.7±27.2 (5.8±1.5) |
8.2 (0.0, 16.4) | −0.7 (−11.2, 9.7) | 0.89 |
|
HbA1c, % (mmol/mol) |
6.2±1.0 (44.0±10.0) |
1.7 (−0.1, 3.5) |
6.1±0.8 (43.0±9.0) |
1.4 (−0.2, 3.0) | 0.3 (−2.1, 2.7) | 0.82 |
|
hs‐CRP, mg/L (nmol/L) |
1.7±2.4 (16.2±22.9) |
119.9 (35.8, 204.0) |
1.9±2.4 (18.1±22.9) |
66.4 (13.5, 119.5) | 53.4 (−42.2, 149.1) | 0.27 |
| Lipid profile | ||||||
|
Total cholesterol, mg/dL (mmol/L) |
151.0±37.5 (3.9±1.0) |
0.9 (−2.3, 4.2) |
153.2±34.5 (4.0±0.9) |
0.7 (−3.7, 5.2) | 0.2 (−5.3, 5.6) | 0.26 |
|
Triglyceride, mg/dLc
(mmol/L) |
117.0 [83.0, 161.5] (1.3 [0.9, 1.8]) |
−2.2 [−19.4, 16.1] |
123.0 [81.5, 178.5] (1.4 [0.9, 2.0]) |
−14.3 [−32.1, 9.5] | 17.2 (6.3, 28.0) | 0.003 |
|
HDL‐C, mg/dL (mmol/L) |
46.9±14.9 (1.2±0.4) |
−2.3 (−5.4, 0.9) |
47.3±14.2 (1.2±0.4) |
1.9 (−1.6, 5.4) | −4.2 (8.9, 0.6) | 0.22 |
|
LDL‐C, mg/dL (mmol/L) |
77.5±27.5 (2.0±0.7) |
3.7 (−1.8, 9.1) |
78.5±27.0 (2.0±0.7) |
7.8 (0.1, 15.5) | −4.1 (−13.5, 5.3) | 0.76 |
| Complete blood count | ||||||
| WBC, 109 cells/L | 6.5±1.8 | −2.5 (−6.7, 1.6) | 6.81±2.4 | −4.7 (−8.5, −0.9) | 2.2 (−3.4, 7.8) | 0.64 |
| Monocytes, cells/μL | 536.7±169.2 | −5.3 (−9.9, −0.8) | 521.4±165.3 | −2.1 (−6.9, 2.7) | −3.2 (−9.8, 3.4) | 0.20 |
| Neutrophils, cells/μL | 4125.0±1592.9 | 2.2 (−4.4, 8.8) | 4179.8±1289.4 | −3.7 (−9.2, 1.7) | 6.0 (−2.5, 14.4) | 0.23 |
| Lymphocytes, cells/μL | 1620.7±509.3 | −5.6 (−9.5, −1.8) | 1880.9±1882.6 | −2.9 (−6.7, 0.9) | −2.8 (−8.2, 2.7) | 0.23 |
| Platelets, cells/μL | 191.5±45.6 | −6.1 (−9.2, −3.0) | 191.0±53.9 | 22.7 (−11.9, 57.4) | −28.8 (−63.6, 6.0) | 0.57 |
BP indicates blood pressure; CI, confidence interval; HbA1c, hemoglobin A1c; HDL‐C, high‐density lipoprotein cholesterol; hs‐CRP, high‐sensitivity C‐reactive protein; LDL‐C, low‐density lipoprotein cholesterol; WBC, white blood cell count.
P values calculated using t test (except triglycerides where a Mann–Whitney U test was used).
Calculated as weight in kilograms divided by height in meters squared.
Values represented as median [interquartile range].
Table 3.
Percent Change From Baseline at 30‐Month Follow‐Up in the Control and Omega‐3 Ethyl‐Ester Groups in the Per‐Protocol Analysis
| Control (n=97) | Omega‐3 Ethyl‐Ester (n=122) | Differences in % Change | ||||
|---|---|---|---|---|---|---|
| Baseline Value Mean±SD | % Change From Baseline Mean (95% CI) | Baseline Value Mean±SD | % Change From Baseline Mean (95% CI) | Mean (95% CI) | P Valuea | |
| Clinical parameters | ||||||
| Systolic BP, mm Hg | 124.1±13.9 | −2.7 (−5.5, 0.1) | 124.3±14.8 | −3.0 (−5.2, −0.8) | 0.3 (−3.2, 3.8) | 0.86 |
| Diastolic BP, mm Hg | 73.3±9.8 | −4.2 (−6.7, −1.8) | 73.3±10.1 | −4.3 (−6.5, −2.2) | 0.1 (−3.1, 3.3) | 0.95 |
| Waist circumference, cm | 106.6±10.3 | 0.4 (−0.6, 1.5) | 106.9±10.7 | 8.0 (−6.7, 22.6) | −7.5 (−23.7, 8.6) | 0.88 |
| Weight, kg | 91.2±13.9 | −1.4 (−2.7, −0.2) | 92.5±13.7 | −0.9 (−2.2, 0.5) | −0.6 (−2.4, 1.3) | 0.54 |
| BMI, kg/m2 | 30.5±3.5b | −0.7 (−2.0, 0.5) | 30.8±3.7b | 2.0 (−2.4, 6.5) | −2.8 (−7.8, 2.3) | 0.28 |
| Biochemical profile | ||||||
|
Glucose, mg/dL (mmol/L) |
106.2±32.2 (5.9±1.8) |
9.2 (2.0, 16.4) |
104.6±27.3 (5.8±1.5) |
8.3 (−0.1, 16.7) | 0.9 (−10.5, 12.3) | 0.88 |
|
HbA1c, % (mmol/mol) |
6.2±1.0 (44.0±10.0) |
1.7 (−0.4, 3.7) |
6.1±0.8 (43.0±9.0) |
1.6 (−0.1, 3.2) | 0.1 (−2.5, 2.7) | 0.80 |
|
hs‐CRP, mg/L (nmol/L) |
1.8±2.5 (17.1±23.8) |
80.1 (22.0, 138.1) |
2.0±2.4 (19.1±22.9) |
60.8 (7.0, 114.7) | 19.2 (−60.4, 98.9) | 0.63 |
| Lipid profile | ||||||
|
Total cholesterol, mg/dL (mmol/L) |
152.2±38.4 (3.9±1.0) |
0.8 (−2.9, 4.5) |
153.7±34.6 (4.0±0.9) |
0.5 (−4.1, 5.0) | 0.3 (−5.7, 6.3) | 0.29 |
|
Triglyceride, mg/dLc
(mmol/L) |
116.0 [85.0, 163.0] (1.3 [1.0, 1.9]) |
0.9 [−18.3, 17.2] |
122.0 [82.0, 175.0] (1.4 [0.9, 2.0]) |
−13.9 [−31.2, 10.4] | 18.9 (7.2, 30.6) | 0.003 |
|
HDL, mg/dL (mmol/L) |
47.2±15.5 (1.2±0.4) |
−3.5 (−6.9, −0.1) |
47.6±14.3 (1.2±0.4) |
1.6 (−1.9, 5.1) | −5.1 (−10.0, −0.2) | 0.13 |
|
LDL, mg/dL (mmol/L) |
78.1±28.4 (2.0±0.7) |
3.8 (−2.3, 9.9) |
79.1±27.2 (2.1±0.7) |
6.6 (−1.2, 14.5) | −2.9 (−12.7, 7.0) | 0.65 |
| Complete blood count | ||||||
| WBC, 109 cells/L | 6.6±1.8 | −3.7 (−7.5, 0.2) | 6.8±2.4 | −4.8 (−8.7, −0.9) | 1.1 (−4.5, 6.6) | 0.74 |
| Monocytes, cells/μL | 538.3±176.7 | −7.0 (−11.7, −2.2) | 516.0±157.3 | −1.8 (−6.7, 3.1) | −5.1 (−12.1, 1.8) | 0.078 |
| Neutrophils, cells/μL | 4153.2±1608.1 | 1.7 (−5.1, 8.5) | 4155.2±1275.6 | −3.7 (−9.3, 1.9) | 5.3 (−3.3, 14.0) | 0.26 |
| Lymphocytes, cells/μL | 1649.4±531.1 | −6.9 (−10.6, −3.2) | 1884.3±1912.2 | −3.0 (−6.9, 0.9) | −3.9 (−9.3, 1.5) | 0.16 |
| Platelets, cells/μL | 194.7±44.5 | −6.5 (−9.4, −3.5) | 190.5±54.5 | 23.6 (−12.2, 59.4) | −30.1 (−66.0, 5.8) | 0.68 |
BMI indicates body mass index; BP, blood pressure; CI, confidence interval; HbA1c, hemoglobin A1c; HDL‐C, high‐density lipoprotein cholesterol; hs‐CRP, high‐sensitivity C‐reactive protein; LDL‐C, low‐density lipoprotein cholesterol; WBC, white blood cell count.
P values calculated using t test (except triglycerides where a Mann–Whitney U test was used).
Calculated as weight in kilograms divided by height in meters squared.
Values represented as median [interquartile range].
Coronary Plaque Outcomes
The intraobserver and interobserver agreement indexes were 0.99 and 0.98, respectively, showing excellent correlation between readings. Table 4 reports the changes in coronary plaque volume. No significant differences were observed at baseline in the volume of fatty, fibrous, noncalcified, calcified, and total coronary plaque between the 2 treatment groups. For our primary end point of noncalcified plaque, which is the sum of fatty and fibrous plaque, the difference between control and omega‐3 ethyl‐ester groups was not significant at 30‐month follow‐up in the intention‐to‐treat analysis (median % change [IQR], 4.5% [−6.1, 15.8] versus −2.4% [−9.8, 16.7], respectively; between‐group, P=0.14), but approached significance in the per‐protocol analysis (median % change [IQR], 4.4% [−5.9, 16.0] versus −2.7% [−10.3, 16.4], respectively; between‐group, P=0.072). Because the mechanism for development of fatty and fibrous plaque may differ, we also analyzed results for these subtypes. The difference between control and omega‐3 ethyl‐ester groups for fibrous plaque approached significance for the intention‐to‐treat analysis (median % change [IQR], 4.6% [−8.0, 18.5] versus 0.1% [−12.2, 14.9], respectively; between‐group, P=0.063) and was significant for the per‐protocol analysis (median % change [IQR], 5.0% [−5.7, 20.0] versus −0.1% [−12.3, 14.5], respectively; between‐group, P=0.018). There was no difference in % change in total, fatty, or calcified plaque between the omega‐3 ethyl‐ester and control groups.
Table 4.
Percent Change in Plaque Volume at 30‐Month Follow‐Up Compared to Baseline in the Control and Omega‐3 Ethyl‐Ester Groups for the Intention‐to‐Treat and Per‐Protocol Analyses
| Plaque Volumea | Controls | Omega‐3 Ethyl‐Ester | % Change From Baseline | ||||
|---|---|---|---|---|---|---|---|
| Baseline Value Median [IQR] | 30‐Month Value Median [IQR] | Baseline Value Median [IQR] | 30‐Month Value Median [IQR] | Controls Median [IQR] | Omega‐3 Ethyl‐Ester Median [IQR] | P Valueb | |
| Intention‐to‐treat | |||||||
| (n=114) | (n=126) | ||||||
| Fatty | 8.6 [5.1, 14.0] | 8.6 [5.3, 13.7] | 9.4 [4.9, 14.7] | 9.3 [5.5, 14.8] | 2.9 [−9.8, 15.1] | 0.8 [−10.4, 20.1] | 0.94 |
| Fibrous | 15.1 [8.7, 23.0] | 15.9 [9.2, 23.5] | 17.5 [9.5, 25.5] | 16.1 [9.7, 24.3] | 4.6 [−8.0, 18.5] | 0.1 [−12.2, 14.9] | 0.063 |
| Noncalcified | 23.7 [14.3, 36.8] | 24.7 [14.5, 36.6] | 26.4 [14.3, 39.7] | 25.7 [15.0, 39.9] | 4.5 [−6.1, 15.8] | −2.4 [−9.8, 16.7] | 0.14 |
| Calcified | 3.6 [1.3, 7.3] | 6.2 [2.6, 10.3] | 5.0 [2.4, 8.7] | 6.4 [3.3, 10.3] | 57.4 [4.3, 146.6] | 39.1 [−5.2, 118.1] | 0.18 |
| Total | 28.1 [16.6, 44.3] | 33.8 [18.1, 46.5] | 33.2 [17.9, 47.0] | 33.4 [19.1, 50.5] | 10.0 [−3.1, 25.9] | 6.5 [−6.9, 19.2] | 0.11 |
| Per‐protocol | |||||||
| (n=97) | (n=122) | ||||||
| Fatty | 9.6 [5.1, 14.1] | 9.6 [5.3, 14.2] | 9.7 [4.9, 15.0] | 9.3 [5.5, 14.8] | 0.2 [−8.9, 13.0] | 0.4 [−10.6, 20.0] | 0.99 |
| Fibrous | 16.0 [9.0, 23.5] | 17.1 [9.3, 23.9] | 17.7 [9.5, 25.7] | 16.1 [9.7, 24.3] | 5.0 [−5.7, 20.0] | −0.1 [−12.3, 14.5] | 0.018 |
| Noncalcified | 26.9 [14.3, 37.2] | 26.0 [14.9, 36.9] | 27.1 [14.3, 40.0] | 25.7 [15.0, 39.9] | 4.4 [−5.9, 16.0] | −2.7 [−10.3, 16.4] | 0.072 |
| Calcified | 3.7 [1.5, 7.6] | 6.3 [3.0, 10.5] | 5.1 [2.6, 8.7] | 6.4 [3.7, 10.3] | 59.2 [6.2, 154.1] | 39.1 [−3.8, 121.4] | 0.16 |
| Total | 30.9 [16.4, 45.7] | 34.6 [18.6, 47.2] | 33.9 [17.9, 47.4] | 33.4 [19.3, 51.3] | 9.9 [−3.3, 27.1] | 6.3 [−7.4, 18.7] | 0.087 |
Plaque volume expressed as mm3/mm.
P values calculated using a Mann–Whitney U test.
IQR indicates interquartile range.
We next did a post‐hoc subgroup analysis according to statin intensity. High‐intensity statin includes atorvastatin ≥40 mg, rosuvastatin ≥20 mg, and simvastatin 80 mg. Table 5 shows the baseline characteristics in those on low‐intensity statin compared with those on a high‐intensity statin. In Table 6, in the intention‐to‐treat analysis, those on omega‐3 ethyl‐ester on low‐intensity statins had attenuation of fibrous plaque progression compared with controls on low‐intensity statin (median % change [IQR], 0.3% [−12.8, 9.0] versus 4.8% [−5.1, 19.0], respectively; P=0.032). This difference remained significant in the per‐protocol analysis (P=0.023). In contrast, in the high‐intensity statin group, no difference in plaque change was observed in either treatment arm.
Table 5.
Baseline Characteristics for the Total Group Stratified by Statin Intensity
| Low‐Intensity Statin (n=114) | High‐Intensity Statin (n=126) | P Value | |
|---|---|---|---|
| Controls | 53 (46.5%) | 61 (48.4%) | 0.766 |
| Omega‐3 ethyl‐ester | 61 (53.5%) | 65 (51.6%) | |
| Demographic characteristics | |||
| Age, mean±SD, y | 62.8±7.9 | 63.1±7.5 | 0.819 |
| Male sex, n (%) | 97 (85.1%) | 107 (84.9%) | 0.971 |
| Inclusion criteria (may have more than 1), n (%) | |||
| History of MI | 45 (39.5%) | 67 (53.2%) | 0.034 |
| History of PCI | 62 (54.4%) | 88 (69.8%) | 0.014 |
| History of CABG | 32 (28.1%) | 27 (21.4%) | 0.233 |
| Cardiovascular risk factors, n (%) | |||
| Hypertension | 91 (79.8%) | 109 (86.5%) | 0.165 |
| Diabetes mellitus | 33 (28.9%) | 35 (27.8%) | 0.841 |
| Anthropometric and blood pressure, mean±SD | |||
| Weight, kg | 91.4±13.2 | 91.6±14.5 | 0.898 |
| Body mass index, kg/m2 a | 30.6±3.3 | 30.7±3.9 | 0.951 |
| Waist circumference, cm | 106.0±9.9 | 107.1±10.7 | 0.436 |
| Systolic BP, mm Hg | 124.1±14.2 | 124.5±14.5 | 0.826 |
| Diastolic BP, mm Hg | 73.7±9.8 | 72.9±9.8 | 0.519 |
| Biochemical profile, mean±SD | |||
|
Glucose, mg/dL (mmol/L) |
104.4±31.2 (5.8±1.7) |
107.5±31.5 (6.0±1.8) |
0.440 |
|
HbA1c, % (mmol/mol) |
6.1±0.8 (43.2±6.4) |
6.2±1.0 (44.3±8.5) |
0.345 |
|
hs‐CRP, mg/L (nmol/L) |
1.8±2.5 (17.1±23.8) |
1.8±2.3 (1.8±17.1) |
0.983 |
| Lipids, mean±SD | |||
|
Total cholesterol, mg/dL (mmol/L) |
154.3±36.1 (4.0±0.9) |
150.3±35.7 (3.9±0.9) |
0.390 |
|
Triglyceride, median [IQR], mg/dL (mmol/L) |
120.0 [83.0, 171.0] (1.4 [0.9, 1.9]) |
120.0 [79.0, 167.0] (1.4 [0.9, 1.9]) |
0.941 |
|
HDL‐C, mg/dL (mmol/L) |
46.9±14.8 (1.2±0.4) |
47.2±14.3 (1.2±0.4) |
0.890 |
|
LDL‐C, mg/dL (mmol/L) |
80.1±27.3 (2.1±0.7) |
76.1±27.0 (2.0±0.7) |
0.259 |
| Complete blood count, mean±SD | |||
| WBC, 109 cells/L | 6.4±1.4 | 6.9±2.6 | 0.088 |
| Monocytes, cells/μL | 521.7±160.6 | 535.0±173.0 | 0.538 |
| Neutrophils, cells/μL | 4031.9±1202.5 | 4264.0±1620.1 | 0.213 |
| Lymphocytes, cells/μL | 1644.5±526.4 | 1859.4±1881.1 | 0.240 |
| Platelets, cells/μL | 192.7±44.1 | 189.9±55.0 | 0.658 |
| Medication, n (%) | |||
| Statin | 103 (90.4%) | 126 (100%) | 0.002 |
| Aspirin | 111 (97.4%) | 120 (95.2%) | 0.386 |
| ACE‐I | 59 (51.8%) | 72 (57.1%) | 0.402 |
| ARB | 16 (14.0%) | 30 (23.8%) | 0.055 |
| Hydrochlorothiazide | 21 (18.4%) | 24 (19.0%) | 0.901 |
| Furosemide | 9 (7.9%) | 10 (7.9%) | 0.990 |
| Calcium‐channel blocker | 25 (21.9%) | 33 (26.2%) | 0.441 |
| Beta blockers | 82 (71.9%) | 91 (72.2%) | 0.960 |
ACE‐I indicates angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; BP, blood pressure; CABG, coronary artery bypass grafting; HbA1c, hemoglobin A1c; HDL‐C, high‐density lipoprotein cholesterol; hs‐CRP, high‐sensitivity C‐reactive protein; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; MI, myocardial infarction; PCI, percutaneous coronary intervention; WBC, white blood cell count.
Calculated as weight in kilograms divided by height in meters squared.
Table 6.
Percent Change in Plaque Volume at 30‐Month Follow‐Up in Control and Omega‐3 Ethyl‐Ester Groups According to Intensity of Statin
| On Low‐Intensity Statin | On High‐Intensity Statin | |||||
|---|---|---|---|---|---|---|
| Control | Omega‐3 Ethyl‐Ester | P Valuea | Control | Omega‐3 Ethyl‐Ester | P Valuea | |
| % Change Median [IQR] | % Change Median [IQR] | % Change Median [IQR] | % Change Median [IQR] | |||
| Intention‐to‐treat | ||||||
| (n=53) | (n=61) | (n=61) | (n=65) | |||
| Fatty | 4.6 [−7.6, 15.3] | −2.3 [−10.4, 13.2] | 0.289 | 0.2 [−10.0, 15.1] | 5.0 [−9.8, 24.8] | 0.286 |
| Fibrous | 4.8 [−5.1, 19.0] | 0.3 [−12.8, 9.0] | 0.032 | 3.9 [−9.8, 18.5] | 0.0 [−11.8, 20.1] | 0.596 |
| Noncalcified | 5.0 [−4.9, 15.3] | −2.9 [−11.1, 10.7] | 0.062 | 3.1 [−7.3, 19.7] | 0.6 [−9.3, 21.6] | 0.820 |
| Calcified | 65.1 [3.4, 154.7] | 37.5 [−6.9, 118.1] | 0.261 | 54.7 [4.6, 130.7] | 40.9 [−2.7, 122.2] | 0.456 |
| Total | 10.1 [−3.1, 24.3] | 5.0 [−11.1, 16.4] | 0.066 | 8.9 [−1.9, 28.6] | 8.4 [−3.9, 22.7] | 0.556 |
| Per‐protocol | ||||||
| Control (n=44) | Omega‐3 Ethyl‐Ester (n=60) | Control (n=53) | Omega‐3 Ethyl‐Ester (n=62) | |||
| Fatty | 2.9 [−7.6, 11.3] | −2.9 [−10.5, 12.9] | 0.453 | 0.0 [−9.8, 16.0] | 3.6 [−11.1, 24.3] | 0.497 |
| Fibrous | 4.6 [−2.8, 20.5] | 0.0 [−12.9, 8.6] | 0.023 | 7.3 [−8.9, 20.0] | −0.1 [−12.2, 18.7] | 0.297 |
| Noncalcified | 3.9 [−5.3, 14.3] | −3.5 [−11.5, 8.8] | 0.078 | 4.4 [−6.2, 24.0] | −2.4 [−9.7, 20.6] | 0.459 |
| Calcified | 67.5 [9.0, 161.5] | 34.6 [−7.3, 118.3] | 0.156 | 54.7 [4.6, 139.3] | 41.8 [0.2, 128.4] | 0.591 |
| Total | 10.0 [−3.4, 24.3] | 4.7 [−12.0, 16.3] | 0.068 | 8.9 [−2.9, 30.8] | 8.7 [−5.6, 22.6] | 0.473 |
P value compares control with omega‐3 ethyl‐ester group.
IQR indicates interquartile range.
In subgroup analyses, there was no significant difference stratified by BMI, triglyceride, and LDL‐C ≤ and > than the median and for analyses stratified by sex and diabetes mellitus status. Table 7 shows that omega‐3 subjects with age ≤ the median had significantly lower plaque volume, including the primary end point, noncalcified plaque.
Table 7.
Percent Change in Plaque Volume at 30‐Month Follow‐Up Compared With Baseline in the Control and Omega‐3 Ethyl‐Ester Groups for the Intention‐to‐Treat and Per‐Protocol Analyses Stratified by Age (≤ or > Than the Median)
| Plaque Volumea | Age ≤ Median | Age > Median | ||||
|---|---|---|---|---|---|---|
| Control | Omega‐3 Ethyl‐Ester | P Valueb | Control | Omega‐3 Ethyl‐Ester | P Valueb | |
| % Change Median [IQR] | % Change Median [IQR] | % Change Median [IQR] | % Change Median [IQR] | |||
| Intention‐to‐treatc | ||||||
| (n=55) | (n=65) | (n=59) | (n=61) | |||
| Fatty | 2.6 [−7.7, 23.2] | −0.2 [−12.2, 10.8] | 0.339 | 4.9 [−9.9, 13.3] | 4.5 [−10.2, 28.3] | 0.190 |
| Fibrous | 7.3 [−5.8, 20.3] | −1.8 [−16.1, 8.4] | 0.010 | 3.9 [−10.7, 17.7] | 2.9 [−9.8, 23.2] | 0.948 |
| Noncalcified | 7.6 [−6.5, 18.2] | −4.9 [−11.7, 8.7] | 0.013 | 2 [−5.8, 13.9] | 3.9 [−8.1, 25.6] | 0.591 |
| Calcified | 63.6 [0.8, 156.5] | 18.3 [−20.7, 111] | 0.044 | 52.2 [8.1, 133.4] | 56 [12.8, 135.2] | 0.698 |
| Total | 10.1 [−0.5, 29.5] | 1.4 [−12.3, 14.9] | 0.003 | 9.9 [−3.5, 23] | 10.8 [0.1, 26.4] | 0.341 |
| Per‐protocold | ||||||
| (n=49) | (n=61) | (n=48) | (n=61) | |||
| Fatty | 3 [−7.1, 23.4] | −2 [−12.2, 9.9] | 0.135 | −0.7 [−10.1, 8.4] | 3.1 [−10.7, 28.3] | 0.117 |
| Fibrous | 7.3 [−5.5, 22.3] | −3.2 [−16.5, 8.4] | 0.003 | 4.2 [−10.3, 18.4] | 1.2 [−9.8, 22.2] | 0.723 |
| Noncalcified | 8.5 [−6.1, 19.7] | −5.1 [−12.5, 8.7] | 0.004 | 1.9 [−5.9, 13.3] | −0.1 [−8.8, 25.6] | 0.665 |
| Calcified | 45 [−7, 142.7] | 18.3 [−19.4, 111] | 0.120 | 64.8 [18, 161.5] | 59.6 [12.8, 138.9] | 0.750 |
| Total | 10.1 [0, 42.8] | 1.4 [−13.3, 14.9] | 0.004 | 8.9 [−3.9, 25.1] | 10 [−0.9, 25.2] | 0.534 |
Plaque volume expressed as mm3/mm.
P values calculated using a Mann–Whitney U test comparing % change in the 2 treatment groups.
Median [IQR] age is 64.2 [58.6, 68.3] years for intention‐to‐treat.
Median [IQR] age is 63.7 [58.5, 68.0] years for per‐protocol.
IQR indicates interquartile range.
In the omega‐3 ethyl‐ester group, there was no correlation between triglyceride reduction and % change in fibrous plaque in either the intention‐to‐treat (r=0.094; P=0.29) or per‐protocol analyses (r=0.108; P=0.24).
Adverse Events
No significant differences were observed in incidence of adverse events between the 2 treatment groups, with the exception of a significantly lower incidence of serious musculoskeletal events in the omega‐3 ethyl‐ester group compared with control (5 versus 14 events, respectively; P=0.034; Table 8). Of these, 11 control subjects had total knee or hip replacements compared with only 1 in the omega‐3 ethyl‐ester group. Furthermore, the incidence of nonserious musculoskeletal adverse events was also significantly lower in the omega‐3 ethyl‐ester group compared with control (43 versus 64 events; P=0.008). No significant differences in serious and nonserious musculoskeletal adverse events were observed in those on low‐ versus high‐intensity statins. Finally, a significantly lower incidence of nonserious infectious disease events was observed in those taking omega‐3 ethyl‐ester.
Table 8.
Adverse Events
| Omega‐3 Ethyl‐Ester (n) | Control (n) | P Valuea | |
|---|---|---|---|
| Serious adverse events | |||
| Cardiac | 41 | 35 | 0.424 |
| Atypical chest pain | 5 | 6 | 0.768 |
| Vascular | 4 | 4 | 1.000 |
| Neurological | 5 | 8 | 0.409 |
| Pulmonary | 0 | 3 | 0.100 |
| Gastrointestinal | 17 | 17 | 1.000 |
| Endocrine | 2 | 0 | 0.498 |
| Musculoskeletal | 5 | 14 | 0.034 |
| Infectious disease | 6 | 8 | 0.595 |
| Genitourinary | 5 | 1 | 0.214 |
| Psychiatric | 2 | 1 | 1.000 |
| HEENT | 1 | 0 | 1.000 |
| Nonserious adverse events | |||
| Cardiac | 27 | 19 | 0.262 |
| Atypical chest pain | 9 | 8 | 1.000 |
| Dental | 10 | 9 | 1.000 |
| Vascular | 3 | 1 | 0.623 |
| Neurological | 8 | 9 | 0.807 |
| Rheumatological | 4 | 5 | 0.748 |
| Pulmonary | 15 | 16 | 0.851 |
| Gastrointestinal | 32 | 25 | 0.377 |
| Musculoskeletal | 43 | 64 | 0.008 |
| Infectious disease | 47 | 63 | 0.049 |
| Genitourinary | 16 | 6 | 0.063 |
| Hematological | 2 | 2 | 1.000 |
| Skin | 25 | 14 | 0.117 |
| HEENT | 23 | 29 | 0.360 |
| Endocrine | 8 | 5 | 0.572 |
| Psychiatric | 1 | 2 | 0.620 |
HEENT indicates head, eye, ear, nose, and throat.
P values calculated using chi‐square or Fisher's exact, where appropriate.
Discussion
In the current randomized controlled trial, we examined whether omega‐3 ethyl‐ester added to statin therapy in patients with CAD reduced progression of coronary plaque volume compared with statin alone. In the intention‐to‐treat analysis, no difference was observed in our primary endpoint, noncalcified plaque volume, between the 2 treatment groups. However, when stratified by age, younger subjects had significant benefit, a finding suggesting that initiation of treatment earlier in the course of disease may be beneficial. Because this is a post‐hoc analysis, it is hypothesis generating and should be examined further in future studies.
Because we were interested in the biological effect of omega‐3 ethyl‐ester, we also performed a per‐protocol analysis in which our primary end point approached significance (P=0.072), a finding suggesting that adherence to treatment assignment is important. Only 4 subjects randomized to omega‐3 ethyl‐ester did not tolerate and stopped; however, 17 controls took ≥900 mg of over‐the‐counter omega‐3 fatty acid daily. Thus, a limitation of our trial is the open‐label nature, which gave subjects knowledge of their assignment and allowed controls to “drop‐in” and perhaps affected the outcome.
Noncalcified plaque is the sum of fatty and fibrous plaque. Because the mechanisms for development of these 2 plaque subtypes may differ, we also analyzed results for subtypes in a prespecified subtype analysis. In the intention‐to‐treat analysis, the difference in fibrous plaque approached significance (P=0.063). Interestingly, in the per‐protocol analysis, control subjects had significant progression of fibrous plaque compared with no change in those taking omega‐3 ethyl‐ester (P=0.018), a finding supporting a potential biological effect of omega‐3 ethyl‐ester with adherence to treatment assignment. This difference was observed in the setting of statin therapy, with a mean LDL‐C of 77.5 mg/dL in the control group and 80.5 mg/dL in the omega‐3 ethyl‐ester group.
In a post hoc subgroup analysis of statin intensity, among those on low‐intensity statin, controls had significant progression of fibrous plaque volume whereas those receiving high‐dose omega‐3 ethyl‐esters had no change (median % change [IQR], 4.8% [−5.1, 19.0] versus 0.3% [−12.8, 9.0], respectively; P=0.032). Similar changes were noted in noncalcified plaque, with a trend toward significance (P=0.062). In contrast, in the high‐intensity statin group, no difference in plaque change was observed in either treatment arm. Therefore, omega‐3 ethyl‐ester supplementation prevented progression of fibrous plaque in those on low‐intensity statin therapy, but not in those on high‐intensity statin. Clinical trials of the effect of omega‐3 fatty acids on cardiovascular events have shown conflicting results with those preceding the statin era showing benefit,17, 18 whereas those conducted during the statin era, with the exception of JELIS, have not (reviewed in de Lorgeril et al).19 The Omega and Alpha Omega trials were conducted in post–myocardial infarction patients, but showed no benefit.20, 21 A total of 81.5% of patients were receiving statins in the Omega Trial20 and 85% of patients were receiving statins in the Alpha Omega Trial.21 In an analysis of statin users and nonusers in the Alpha Omega Trial, no benefit of omega‐3 fatty acids was observed on reduction of cardiovascular events among statin users (13% versus 15%, respectively). In contrast, among statin nonusers, 9% of those receiving omega‐3 fatty acids had cardiovascular events compared with 18% of those not receiving omega‐3 fatty acids (hazard ratio, 0.46: 95% CI, 0.21–1.01; P=0.051).25 As noted earlier, the JELIS trial reported that addition of 1800 mg/day of open‐label EPA to low‐intensity statin therapy conferred a significant 19% reduction in major coronary events compared with low‐intensity statin alone, a finding suggesting that high‐dose EPA added to low‐intensity statin therapy can reduce residual risk.4 These findings, coupled with our results, suggest that statin use and dose may be important factors affecting clinical outcomes and plaque volume. Further studies should investigate the mechanism of how statins interact with omega‐3 fatty acids. This may be an important pharmacological issue because higher intensity statin therapy is more likely to have adverse side effects, particularly on muscle and joint symptoms, whereas omega‐3 fatty acids actually reduce such symptoms as shown by the significantly lower incidence of both serious and nonserious musculoskeletal adverse events in the present study. Remarkably, 11 of the 14 serious musculoskeletal events in the control group were total knee or hip replacements attributed to progressive pain and/or arthritis whereas there was only 1 in the omega‐3 ethyl‐ester group. This was an unexpected finding, which may be a consequence of more‐limited symptoms and/or decreased progression of arthritis. Joint replacement is an expensive procedure; therefore, further studies should examine the effect of high‐dose omega‐3 ethyl‐ester on joint symptoms given that prevention of joint replacement could be a huge cost‐savings to the healthcare system.
Elevated levels of triglyceride are associated with increasing risk of coronary heart disease.26 As expected, triglyceride levels were significantly reduced in the omega‐3 ethyl‐ester group compared with the control group. This reduction was not correlated with change in volume of fibrous plaque, and our subgroup analysis showed no difference in outcome when stratified by triglyceride levels above and below the median, findings suggesting that other mechanisms account for the benefit. Atherosclerosis is a disease of chronic inflammation in the arterial wall characterized by upregulation of vascular cell adhesion molecules, which promote monocyte infiltration into the subendothelial space where differentiation to macrophages occurs.27 These macrophages take up oxidized apolipoprotein B–containing LDL particles, a process leading to foam cell and fatty streak formation, the precursor of advanced atherosclerotic lesions. Potential beneficial effects of EPA on atherosclerotic plaque include anti‐inflammatory and antioxidant effects, decreased adhesion of monocytes to the endothelium, decreased macrophage and foam cell accumulation in fatty streaks, and increased thickness of the fibrous cap overlying lipid‐rich plaque28, 29, 30 (reviewed in Nelson et al).31 EPA and DHA are also converted to specialized proresolving lipid mediators, with EPA and DHA being the respective precursors of the E‐series and D‐series resolvins.32 The resolvins have been shown to stimulate the resolution of acute inflammation by stopping further neutrophil recruitment to inflamed tissues and stimulating nonphlogistic infiltration of monocytes, which differentiate into resolution macrophages.33 These resolution macrophages then phagocytize and clear apoptotic neutrophils and debris, a process termed efferocytosis, which is a key step in resolution and prevention of chronic inflammation.33, 34 Recent research has shown that omega‐3 fatty acids may hasten the resolution of inflammation when converted to resolvins.32, 33 Formation of fibrous tissue is an end stage of the chronic inflammatory process with the recruitment of collagen to form a scar. The formation of resolvins from EPA and DHA leads to resolution of inflammation and, by hastening resolution, may cause less fibrosis. Therefore, the conversion of EPA and DHA to their downstream products may account for a beneficial effect on inflammation, leading to a decrease in fibrotic plaque. In a recent study, EPA and DHA have been shown to reduce myocardial fibrosis. EPA and DHA supplementation for 6 months in post–myocardial infarction patients was associated with a favorable effect on left ventricular remodeling and a significant reduction in noninfarct myocardial fibrosis measured by cardiac magnetic resonance imaging (mean reduction, −5.6%; 95% CI, −10.4% to −0.9%; P=0.022) compared with placebo.35 In a subset of 6 patients from the current study, several resolvins, which have been shown to be present in healthy subjects,36, 37, 38 were absent or present at low levels in CAD patients not on omega‐3 ethyl‐ester; the resolvins were restored or their level increased in those receiving omega‐3 ethyl‐ester.39 The stimulation of resolution of infection and inflammation by resolvins (the downstream products of EPA and DHA) may be responsible for the significantly lower rate of nonserious infectious disease events in the omega‐3 group.40, 41 Future studies should examine whether conversion of EPA and DHA to resolvins is responsible for these beneficial effects.
Several randomized controlled trials have reported a beneficial relationship between EPA and DHA levels and atherosclerosis (reviewed in Nelson et al).31 In a study of 81 subjects with type 2 diabetes mellitus, those randomized to open‐label, purified EPA (1800 mg/day) had a significant annual decrease in mean and maximum carotid intima‐media thickness compared with those on no EPA.42 In the Estrogen Replacement and Atherosclerosis Trial, 228 women were randomized to either 0.625 mg of conjugated equine estrogen, 0.625 mg of conjugated equine estrogen plus 2.5 mg of medroxyprogesterone acetate, or placebo.43 After 3.2‐year follow‐up, those with phospholipid DHA levels above the median had less atherosclerosis progression as measured with coronary angiography. Two studies in Japanese patients have shown a significant additional benefit of EPA (1800 mg/day) added to statin therapy compared with statin alone on coronary plaque volume, as measured with integrated backscatter intravascular ultrasound.44, 45 In a post‐hoc analysis of 50 diabetics who were randomized to pravastatin or pitavastatin after percutaneous intervention and followed for 8 months, only low DHA levels remained significantly associated with atheroma progression measured by intravascular ultrasound after multivariate regression analysis.46 Moreover, subjects with plaque regression had regression of only the fibrous component. None of the other plaque components changed significantly, findings similar to the current study, which detected a significant difference in fibrous plaque. This study did not randomize subjects to EPA and/or DHA. To our knowledge, the current study is the first randomized trial showing a benefit of high‐dose EPA and DHA added to statin therapy on the progression of coronary plaque volumes measured by CCTA. Taken together, the results of these past studies, along with our results, suggest that EPA and DHA affect plaque composition and volume. Although our trial was not designed to assess cardiovascular events, it is worth noting that several studies assessing coronary plaque subtypes with CCTA have shown that higher volume of total noncalcified plaque and total plaque volume are associated with higher rates of cardiac death, myocardial infarction, and coronary revascularization47 and higher rates of acute coronary syndrome.48, 49 Furthermore, evidence from intravascular ultrasound studies shows that progression of plaque atheroma volume is independently associated with higher rates of a composite of cardiac death, myocardial infarction, and coronary revascularization (P<0.002) and regression is associated with fewer events.3 Taken together, these findings suggest that plaque composition and volume predict cardiovascular events and support the potential clinical importance of lack of progression of plaque volume observed in the omega‐3 ethyl‐ester arm in the current trial.
No significant differences in adverse events were observed in those receiving omega‐3 ethyl‐ester compared with control, with the exception of a significantly lower rate of both serious and nonserious musculoskeletal adverse events and nonserious infectious diseases in those receiving omega‐3 ethyl‐ester. These findings support the safety of the intervention. In past studies, DHA raised LDL‐C levels whereas EPA did not.50, 51 In the current study of EPA plus DHA, there was no difference in LDL‐C levels in the omega‐3 versus no omega‐3 groups; therefore, high‐dose EPA plus DHA had no effect on LDL‐C.
Major strengths of the current study are the randomized controlled design and high‐dose of EPA and DHA (3.36 g). A limitation is that the 30‐month duration may not be long enough to observe the full effect of EPA and DHA supplementation given that atherosclerosis is a long‐term process. We hypothesize that longer duration of follow‐up would result in a larger difference between the 2 treatment groups. Limitations of the study also include the open‐label study design as noted earlier; however, our primary outcome, plaque ascertainment, was blinded and therefore should not be affected by knowledge of treatment assignment. Although we consider it unlikely, we cannot rule out subtle effects on the randomized treatments attributed to the knowledge of treatment assignment by the principal investigator and participants.
In conclusion, high‐dose omega‐3 ethyl‐ester provided additional benefit to statin treatment in preventing progression of fibrous coronary plaque volume over 30 months compared with a statin alone in subjects who adhered to treatment assignment and in the setting of well‐controlled LDL‐C levels. Moreover, the benefit was not correlated with triglyceride reduction. The benefit on low‐intensity statin, but not high‐intensity statin, suggests that statin intensity affects plaque volume. These findings support future trials examining mechanisms for beneficial effects of high‐dose omega‐3 fatty acids on plaque progression beyond triglyceride lowering.
Sources of Funding
This work was supported by a National Heart, Lung, and Blood Institute (NHLBI) Specialized Centers of Clinically Oriented Research (SCCOR) program grant to Dr Welty: P50 HL083813 and supported by the Harvard Clinical and Translational Science Center Award, NIH UL1 TR001102. GlaxoSmithKline donated Lovaza.
Disclosures
None.
Acknowledgments
We thank the study participants for contributing their time and participating in the trial and the external data safety monitoring board members for their assistance in the successful completion of this trial. We thank Boston Heart Diagnostics for the CRP measurement.
(J Am Heart Assoc. 2017;6:e006981 DOI: 10.1161/JAHA.117.006981.)29246960
The data have been presented at the Robert Levy Young Investigator Award Competition at the American Heart Association Scientific Sessions, November 13, 2016, in New Orleans, LA.
References
- 1. Sampson UK, Fazio S, Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep. 2012;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bayturan O, Kapadia S, Nicholls SJ, Tuzcu EM, Shao M, Uno K, Shreevatsa A, Lavoie AJ, Wolski K, Schoenhagen P, Nissen SE. Clinical predictors of plaque progression despite very low levels of low‐density lipoprotein cholesterol. J Am Coll Cardiol. 2010;55:2736–2742. [DOI] [PubMed] [Google Scholar]
- 3. Nicholls SJ, Hsu A, Wolski K, Hu B, Bayturan O, Lavoie A, Uno K, Tuzcu EM, Nissen SE. Intravascular ultrasound‐derived measures of coronary atherosclerotic plaque burden and clinical outcome. J Am Coll Cardiol. 2010;55:2399–2407. [DOI] [PubMed] [Google Scholar]
- 4. Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, Oikawa S, Sasaki J, Hishida H, Itakura H, Kita T, Kitabatake A, Nakaya N, Sakata T, Shimada K, Shirato K; Japan EPA lipid intervention study (JELIS) Investigators . Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open‐label, blinded endpoint analysis. Lancet. 2007;369:1090–1098. [DOI] [PubMed] [Google Scholar]
- 5. Obaid DR, Calvert PA, Gopalan D, Parker RA, West NE, Goddard M, Rudd JH, Bennett MR. Dual‐energy computed tomography imaging to determine atherosclerotic plaque composition: a prospective study with tissue validation. J Cardiovasc Comput Tomogr. 2014;8:230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Obaid DR, Calvert PA, Gopalan D, Parker RA, Hoole SP, West NE, Goddard M, Rudd JH, Bennett MR. Atherosclerotic plaque composition and classification identified by coronary computed tomography: assessment of computed tomography‐generated plaque maps compared with virtual histology intravascular ultrasound and histology. Circ Cardiovasc Imaging. 2013;6:655–664. [DOI] [PubMed] [Google Scholar]
- 7. Rinehart S, Vazquez G, Qian Z, Murrieta L, Christian K, Voros S. Quantitative measurements of coronary arterial stenosis, plaque geometry, and composition are highly reproducible with a standardized coronary arterial computed tomographic approach in high‐quality CT datasets. J Cardiovasc Comput Tomogr. 2011;5:35–43. [DOI] [PubMed] [Google Scholar]
- 8. Sandfort V, Lima JA, Bluemke DA. Noninvasive imaging of atherosclerotic plaque progression: status of coronary computed tomography angiography. Circ Cardiovasc Imaging. 2015;8:e003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burgstahler C, Reimann A, Beck T, Kuettner A, Baumann D, Heuschmid M, Brodoefel H, Claussen CD, Kopp AF, Schroeder S. Influence of a lipid‐lowering therapy on calcified and noncalcified coronary plaques monitored by multislice detector computed tomography: results of the New Age II Pilot Study. Invest Radiol. 2007;42:189–195. [DOI] [PubMed] [Google Scholar]
- 10. Hoffmann H, Frieler K, Schlattmann P, Hamm B, Dewey M. Influence of statin treatment on coronary atherosclerosis visualised using multidetector computed tomography. Eur Radiol. 2010;20:2824–2833. [DOI] [PubMed] [Google Scholar]
- 11. Inoue K, Motoyama S, Sarai M, Sato T, Harigaya H, Hara T, Sanda Y, Anno H, Kondo T, Wong ND, Narula J, Ozaki Y. Serial coronary CT angiography‐verified changes in plaque characteristics as an end point: evaluation of effect of statin intervention. JACC Cardiovasc Imaging. 2010;3:691–698. [DOI] [PubMed] [Google Scholar]
- 12. Shimojima M, Kawashiri MA, Nitta Y, Yoshida T, Katsuda S, Kaku B, Taguchi T, Hasegawa A, Konno T, Hayashi K, Yamagishi M. Rapid changes in plaque composition and morphology after intensive lipid lowering therapy: study with serial coronary CT angiography. Am J Cardiovasc Dis. 2012;2:84–88. [PMC free article] [PubMed] [Google Scholar]
- 13. Zeb I, Li D, Nasir K, Malpeso J, Batool A, Flores F, Dailing C, Karlsberg RP, Budoff M. Effect of statin treatment on coronary plaque progression—a serial coronary CT angiography study. Atherosclerosis. 2013;231:198–204. [DOI] [PubMed] [Google Scholar]
- 14. Khosa F, Khan AN, Nasir K, Bedayat A, Malik Z, Jon AF, Cheema AR, Clouse ME, Welty FK. Comparison of coronary plaque subtypes in male and female patients using 320‐row MDCTA. Atherosclerosis. 2013;226:428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hauser TH, Salastekar N, Schaefer EJ, Desai T, Goldfine HL, Fowler KM, Weber GM, Welty F, Clouse M, Shoelson SE, Goldfine AB; Targeting Inflammation Using Salsalate in Cardiovascular Disease (TINSAL‐CVD) Study Team . Effect of targeting inflammation with salsalate: the TINSAL‐CVD randomized clinical trial on progression of coronary plaque in overweight and obese patients using statins. JAMA Cardiol. 2016;1:413–423. [DOI] [PubMed] [Google Scholar]
- 16. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC Jr; International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; International Association for the Study of Obesity . Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. [DOI] [PubMed] [Google Scholar]
- 17. Dietary supplementation with n‐3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI‐Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico. Lancet. 1999;354:447–455. [PubMed] [Google Scholar]
- 18. GISSI‐HF Investigators . Effect of n‐3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI‐HF trial): a randomised, double‐blind, placebo controlled trial. Lancet. 2008;372:1223–1230. [DOI] [PubMed] [Google Scholar]
- 19. de Lorgeril M, Salen P, Defaye P, Rabaeus M. Recent findings on the health effects of omega‐3 fatty acids and statins, and their interactions: do statins inhibit omega‐3? BMC Med. 2013;11:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rauch B, Schiele R, Schneider S, Diller F, Victor N, Gohlke H, Gottwik M, Steinbeck G, Del Castillo U, Sack R, Worth H, Katus H, Spitzer W, Sabin G, Senges J; OMEGA Study Group . OMEGA, a randomized, placebo‐controlled trial to test the effect of highly purified omega‐3 fatty acids on top of modern guideline‐adjusted therapy after myocardial infarction. Circulation. 2010;22:2152–2159. [DOI] [PubMed] [Google Scholar]
- 21. Kromhout D, Giltay EJ, Gelejinse JM; Alpha Omega Trial Group . n‐3 Fatty acids and cardiovascular events after myocardial infarction. N Engl J Med. 2010;363:2015–2026. [DOI] [PubMed] [Google Scholar]
- 22. Brodoefel H, Burgstahler C, Sabir A, Yam CS, Khosa F, Claussen CD, Clouse ME. Coronary plaque quantification by voxel analysis: dual‐source MDCT angiography versus intravascular sonography. AJR Am J Roentgenol. 2009;192:W84–W89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Voros S, Rinehart S, Qian Z, Joshi P, Vazquez G, Fischer C, Belur P, Hulten E, Villines TC. Coronary atherosclerosis imaging by coronary CT angiography: current status, correlation with intravascular interrogation and meta‐analysis. JACC Cardiovasc Imaging. 2011;4:537–548. [DOI] [PubMed] [Google Scholar]
- 24. Brodoefel H, Burgstahler C, Heuschmid M, Reimann A, Khosa F, Kopp A, Schroeder S, Claussen CD, Clouse ME. Accuracy of dual‐source CT in the characterisation of non‐calcified plaque: use of a colour‐coded analysis compared with virtual histology intravascular ultrasound. Br J Radiol. 2009;82:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eussen SR, Geleijnse JM, Giltay EJ, Rompelberg CJ, Klungel OH, Kromhout D. Effects of n‐3 fatty acids on major cardiovascular events in statin users and non‐users with a history of myocardial infarction. Eur Heart J. 2012;33:1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626–635. [DOI] [PubMed] [Google Scholar]
- 27. Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res. 2014;114:1867–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis. 2015;242:357–366. [DOI] [PubMed] [Google Scholar]
- 29. Budoff M. Triglycerides and triglyceride‐rich lipoproteins in the causal pathway of cardiovascular disease. Am J Cardiol. 2016;118:138–145. [DOI] [PubMed] [Google Scholar]
- 30. Gdula‐Argasińska J, Czepiel J, Woźniakiewicz A, Wojtoń K, Grzywacz A, Woźniakiewicz M, Jurczyszyn A, Perucki W, Librowski T. n‐3 Fatty acids as resolvents of inflammation in the A549 cells. Pharmacol Rep. 2015;67:610–615. [DOI] [PubMed] [Google Scholar]
- 31. Nelson JR, Wani O, May HT, Budoff M. Potential benefits of eicosapentaenoic acid on atherosclerotic plaques. Vascul Pharmacol. 2017;91:1–9. [DOI] [PubMed] [Google Scholar]
- 32. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti‐inflammatory and pro‐resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Serhan CN. Pro‐resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tabas I, Glass CK. Anti‐inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heydari B, Abdullah S, Pottala JV, Shah R, Abbasi S, Mandry D, Francis SA, Lumish H, Ghoshhajra BB, Hoffman U, Appelbaum E, Feng JH, Blankstein R, Steigner M, McConnell JP, Harris W, Antman EM, Jerosch‐Herold M, Kwong RY. Effect of omega‐3 acid ethyl esters on left ventricular remodeling after acute myocardial infarction: the OMEGA‐REMODEL Randomized Clinical Trial. Circulation. 2016;134:378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barden A, Mas E, Croft KD, Phillips M, Mori TA. Short‐term n‐3 fatty acid supplementation but not aspirin increases plasma proresolving mediators of inflammation. J Lipid Res. 2014;55:2401–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and signature profiles for pro‐resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol. 2014;307:C39–C54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mas E, Croft KD, Zahra P, Barden A, Mori TA. Resolvins D1, D2, and other mediators of self‐limited resolution of inflammation in human blood following n‐3 fatty acid supplementation. Clin Chem. 2012;58:1476–1484. [DOI] [PubMed] [Google Scholar]
- 39. Elajami TK, Colas RA, Dalli J, Chiang N, Serhan CN, Welty FK. Specialized proresolving lipid mediators in patients with coronary artery disease and their potential for clot remodeling. FASEB J. 2016;30:2792–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee CR, Zeldin DC. Resolvin infectious inflammation by targeting the host response. N Engl J Med. 2015;373:2183–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dalli J, Chiang N, Serhan CN. Elucidation of novel 13‐series resolvins that increase with atorvastatin and clear infections. Nat Med. 2015;21:1071–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mita T, Watada H, Ogihara T, Nomiyama T, Ogawa O, Kinoshita J, Shimizu T, Hirose T, Tanaka Y, Kawamori R. Eicosapentaenoic acid reduces the progression of carotid intima‐media thickness in patients with type 2 diabetes. Atherosclerosis. 2007;191:162–167. [DOI] [PubMed] [Google Scholar]
- 43. Erkkilä AT, Matthan NR, Herrington DM, Lichtenstein AH. Higher plasma docosahexaenoic acid is associated with reduced progression of coronary atherosclerosis in women with CAD. J Lipid Res. 2006;47:2814–2819. [DOI] [PubMed] [Google Scholar]
- 44. Watanabe T, Ando K, Daidoji H, Otaki Y, Sugawara S, Matsui M, Ikeno E, Hirono O, Miyawaki H, Yashiro Y, Nishiyama S, Arimoto T, Takahashi H, Shishido T, Miyashita T, Miyamoto T, Kubota I; CHERRY study investigators . A randomized controlled trial of eicosapentaenoic acid in patients with coronary heart disease on statins. J Cardiol. 2017;70:537–544. [DOI] [PubMed] [Google Scholar]
- 45. Niki T, Wakatsuki T, Yamaguchi K, Taketani Y, Oeduka H, Kusunose K, Ise T, Iwase T, Yamada H, Soeki T, Sata M. Effects of the addition of eicosapentaenoic acid to strong statin therapy on inflammatory cytokines and coronary plaque components assessed by integrated backscatter intravascular ultrasound. Circ J. 2016;80:450–460. [DOI] [PubMed] [Google Scholar]
- 46. Nozue T, Yamamoto S, Tohyama S, Fukui K, Umezawa S, Onishi Y, Kunishima T, Sato A, Nozato T, Miyake S, Takeyama Y, Morino Y, Yamauchi T, Muramatsu T, Hibi K, Terashima M, Michishita I. Low serum docosahexaenoic acid is associated with progression of coronary atherosclerosis in statin‐treated patients with diabetes mellitus: results of the treatment with statin on atheroma regression evaluated by intravascular ultrasound with virtual histology (TRUTH) study. Cardiovasc Diabetol. 2014;13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nadjiri J, Hausleiter J, Jähnichen C, Will A, Hendrich E, Martinoff S, Hadamitzky M. Incremental prognostic value of quantitative plaque assessment in coronary CT angiography during 5 years of follow up. J Cardiovasc Comput Tomogr. 2016;10:97–104. [DOI] [PubMed] [Google Scholar]
- 48. Versteylen MO, Kietselaer BL, Dagnelie PC, Joosen IA, Dedic A, Raaijmakers RH, Wildberger JE, Nieman K, Crijns HJ, Niessen WJ, Daemen MJ, Hofstra L. Additive value of semiautomated quantification of coronary artery disease using cardiac computed tomographic angiography to predict future acute coronary syndrome. J Am Coll Cardiol. 2013;61:2296–2305. [DOI] [PubMed] [Google Scholar]
- 49. Motoyama S, Sarai M, Harigaya H, Anno H, Inoue K, Hara T, Naruse H, Ishii J, Hishida H, Wong ND, Virmani R, Kondo T, Ozaki Y, Narula J. Computed tomographic angiography characteristics of atherosclerotic plaques subsequently resulting in acute coronary syndrome. J Am Coll Cardiol. 2009;54:49–57. [DOI] [PubMed] [Google Scholar]
- 50. Jacobson TA, Glickstein SB, Rowe JD, Soni PN. Effects of eicosapentaenoic acid and docosahexaenoic acid on low‐density lipoprotein cholesterol and other lipids: a review. J Clin Lipidol. 2012;6:5–18. [DOI] [PubMed] [Google Scholar]
- 51. Wei MW, Jacobson TA. Effects of eicosapentaenoic acid versus docosahexaenoic acid on serum lipids: a systematic review and meta‐analysis. Curr Atheroscler Rep. 2011;13:474–483. [DOI] [PubMed] [Google Scholar]