

Abstract
Background
Kruppel‐like factor 2 (KLF2) is an important zinc‐finger transcription factor that maintains endothelial homeostasis by its anti‐inflammatory, ‐thrombotic, ‐oxidative, and ‐proliferative effects in endothelial cells. In light of the potent vasoprotective effects of KLF2, modulating KLF2 expression or function could give rise to new therapeutic strategies to treat cardiovascular diseases.
Methods and Results
High‐throughput drug screening based on KLF2 promoter luciferase reporter assay was performed to screen KLF2 activators. Real‐time PCR and western blot were used to detect gene and protein expression. Identified KLF2 activator was orally administered to ApoE−/− mice to evaluate anti‐atherosclerotic efficacy. By screening 2400 compounds in the Spectrum library, we identified suberanilohydroxamic (SAHA) acid, also known as vorinostat as a pharmacological KLF2 activator through myocyte enhancer factor 2. We found that SAHA exhibited anti‐inflammatory effects and attenuated monocyte adhesion to endothelial cells inflamed with tumor necrosis factor alpha. We further showed that the inhibitory effect of SAHA on endothelial inflammation and ensuing monocyte adhesion was KLF2 dependent using KLF2‐deficient mouse lung endothelial cells or KLF2 small interfering RNA– depleted human endothelial cells. Importantly, we observed that oral administration of SAHA reduced diet‐induced atherosclerotic lesion development in ApoE−/− mice without significant effect on serum lipid levels.
Conclusions
These results demonstrate that SAHA has KLF2‐dependent anti‐inflammatory effects in endothelial cells and provide the proof of concept that KLF2 activation could be a promising therapeutic strategy for treating atherosclerosis.
Keywords: atherosclerosis, endothelial cell, inflammation, SAHA
Subject Categories: Inflammation, Atherosclerosis
Clinical Perspective
What Is New?
Histone deacetylase inhibitor suberanilohydroxamic acid activates Kruppel‐like factor 2 by myocyte enhancer factor 2.
Oral administration of suberanilohydroxamic acid represses vascular inflammation and atherosclerosis in ApoE−/− mice.
What Are the Clinical Implications?
Kruppel‐like factor 2 activators have therapeutic potential to treat atherosclerosis.
Suberanilohydroxamic acid could be repurposed as a potential anti‐inflammatory and ‐atherosclerotic drug.
The endothelium plays an important role in maintaining vascular homeostasis and regulating blood vessel tone, inflammation, vessel permeability, thrombosis, and vasculogenesis/angiogenesis processes.1, 2 Endothelial dysfunction, typically characterized by impaired vasoreactivity, enhanced permeability to plasma lipoproteins, and selective adhesiveness for mononuclear leukocytes, is implicated in atherosclerosis, diabetes mellitus, hypertension, and coronary artery disease.2, 3
Kruppel‐like factor 2 (KLF2) is a zinc‐finger transcription factor preferentially expressed in vascular endothelium, lung, and lymphocytes.1, 4, 5 It functions as a master regulator to maintain endothelial homeostasis5 and control multiple genes critical for anti‐inflammatory, ‐thrombotic, ‐oxidative, and ‐proliferative effects in endothelial cells (ECs).1, 4, 5, 6, 7, 8, 9, 10 For example, KLF2 potently induces the expression of vasodilatory and anti‐inflammatory genes such as endothelial nitric oxide synthase, inhibits vasoconstrictive factors such as endothelin‐1 expression, and represses expression of adhesion molecules, such as vascular cell adhesion molecule 1 (VCAM1).1, 8, 10 Experimental studies have demonstrated that shear stress11 along with small molecules such as lipid‐lowering statins10, 12 and resveratrol are capable of exerting vasoprotective effects by promoting KLF2 activation.4, 12, 13 The molecular mechanisms involved in the regulation of endothelial KLF2 expression includes the AMP‐activated protein kinase/dual specificity mitogen‐activated protein kinase kinase 5/extracellular‐signal related kinase 5/MEF2 (myocyte enhancing factor 2) pathway and the histone deacetylase (HDAC) 5 pathway.1, 14, 15 Recent studies have demonstrated that deletion of 1 KLF2 allele in mice increases atherosclerosis,16 and KLF2 controls endothelial and vascular integrity.17 These important findings suggest that KLF2 could serve as a promising therapeutic target for the treatment of atherosclerosis.
In light of the potent vasoprotective effects mediated by KLF2, modulating KLF2 expression or function might be a novel therapeutic strategy for a wide range of human diseases, including cardiovascular diseases.1 Therefore, we aimed to identify novel KLF2 activators using a luciferase assay based high‐throughput drug screening and hoped that these activators would have endothelial protective functions. Interestingly, we have identified a US Food and Drug Administration–approved small molecule, suberoylanilide hydroxamic acid (SAHA, also known as Vorinostat or MK0683), as a novel KLF2 activator, that prevents endothelial inflammation in vitro and atherosclerosis development in vivo. Our study suggests that the anticancer drug SAHA could be repurposed as an effective therapeutic agent for atherosclerosis.
Materials and Methods
Reagents
The library of small‐molecule drugs (NCI Spectrum Compound Library 2400 chemical compounds dissolved at 1 mmol/L in DMSO) was purchased from the University of Rochester Pathway Discovery Resource. SAHA was purchased from L C Laboratories (#V‐8477, purity >99%; Woburn, MA). iQ SYBR Green Supermix (#1708886) was purchased from Bio‐Rad Laboratories (Hercules, CA). A High‐Capacity cDNA Reverse Transcription Kit (#4374966) was obtained from Thermo Fisher Scientific (Waltham, MA). Recombinant human tumor necrosis factor alpha (TNFα) was purchased from R&D Systems (#210‐TA; Minneapolis, MN). Lipofectamine 2000 Transfection Reagent was purchased from Thermo Fisher Scientific. The wild‐type and MEF2 binding site defective mutant KLF2‐luc promoter‐driven luciferase reporter (KLF2‐luc) plasmids were gifted by Prof Mukesh Jain.10 KLF2 adenovirus was from ViGene Biosciences, Inc (#VH849868; Rockville, MD).
Cell Culture
COS‐7 cells (ATCC, Rockville, MD) were cultured in DMEM (cellgro; Corning Incorporated, Corning, NY) with 10% Fetal Bovine Serum (FBS) (Thermo Fisher Scientific). Human umbilical vein ECs were isolated and cultured as previously described.18 Human coronary artery ECs (HCAECs) were from Cell Applications Inc (#300K‐05A; San Diego, CA) and Lifeline Cell Technologies (#FC‐0032; Fredrick, MD). HCAECs were cultured in MesoEndo Cell Growth Media (Gobico) containing 10% FBS and Growth Supplement (#212K‐500; Cell Applications Inc.).18 Passage 3 to 6 of HCAEC cells were used for experiments. THP‐1 cells (ATCC) were grown in RPMI 1640 containing 10% FBS. All cells were maintained in a 37°C incubator with 5% CO2.
Transfection and Luciferase Assay
COS‐7 cells plated in 100‐mm dishes were transfected with 5.4 μg of KLF2‐Luc using Lipofectamine 2000 in Opti‐MEM (Gibco, Grand Island, NY). After 6 hours of transfection, the transfected cells were then plated in 96‐well plates at a density of 3.5×105 cells/well in normal DMEM containing 10% FBS (100 μL/well). When cells were attached to the bottom of plate, the drugs (1.0 μL/well; final concentration was 5 μmol/L, 200 μL medium/well) were added into the 96‐well plate for 24 hours. Activity of the luciferase reporter gene was assayed using Dual‐Luciferase Reporter Assay System (Promega, Madison, WI) and detected using a microplate spectrophotometer (BMG Labtech, Ortenberg, Germany). The fold of KLF2 activity of the tested compound is calculated as compound firefly luciferase value/DMSO firefly luciferase value.
Real‐Time PCR
Total RNA was extracted from cultured HCAECs and lung ECs using an RNeasy Mini kit (Qiagen, Hilden, Germany).18 In brief, cells were plated in 24‐well plates and treated with SAHA (0.1, 1, and 10 μmol/L) for 24 hours. After treatment, cells were washed with PBS. Total RNA was extracted using a QIAGEN RNeasy Mini kit (Qiagen). Mouse aorta EC RNA was extracted using QiaZol lysis reagent (Qiagen).19 Quantitative real‐time PCR was performed using iQ SYBR Green Supermix (Bio‐Rad) in a Bio‐Rad iQ5 real‐time PCR thermal cycler. Relative mRNA expression of target genes was normalized to the housekeeping gene, glyceraldehyde 3‐phosphate dehydrogenase, and expressed as fold change over control.20, 21 Sequences of the specific mouse primers were designed using IDT PrimerQuest software, and the oligo sequences are listed in Table S1.
Western Blot
Western blot was performed as previously described.18 Blots were incubated overnight at 4°C with appropriate primary antibodies, including VCAM‐1 (#sc‐1504, 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), ICAM‐1 (#sc‐8439, 1:1000; Santa Cruz Biotechnology), and glyceraldehyde 3‐phosphate dehydrogenase (#AB2302, 1:1000; , Burlington, MA). After being washed 3 times with 1× Tris Buffered Saline with 0.1% Tween‐20 (TBST), membranes were incubated with IRDye 680RD Goat anti‐Mouse IgG (H+L), IRDye 800CW Goat anti‐Rabbit IgG (H+L) or IRDye 680RD Goat anti‐Chicken IgG (H+L) (1:10 000 dilution in 1× TBST; LI‐COR) at room temperature for 30 minutes. Images were visualized by using the LI‐COR Odyssey Infrared Imaging System. Densitometric analysis of blots was performed with NIH ImageJ software (NIH, Bethesda, MD).
Assay of Monocyte Adhesion to ECs
Assay of monocyte adhesion to ECs were essentially performed as previously described.22, 23, 24, 25 In brief, confluent HCAECs were plated in 6‐well plates the day before pretreatment with vehicle (0.1% DMSO) or SAHA (10 μmol/L) of indicated concentrations for 12 hours. TNFα (at a final concentration of 10 ng/mL) was added and incubated in a CO2 incubator for 6 hours. Then, 5 to 7×106 of the THP‐1 cells were added to the HCAECs in a final volume of 2.5 mL and incubated in a CO2 incubator for 30 minutes. After treatment, cells were gently washed 3 times with serum‐free medium to remove nonadherent THP‐1 cells from the plate. Phase‐contrast microphotographs of cells were taken using a Canon A640 digital camera with a Zeiss Axiovert 40C microscope (Carl Zeiss, Jena, Germany). Numbers of adherent cells from each image were counted and plotted.
Approval of Animal Studies
All animal procedures conformed to the Guideline for the Care and Use of Laboratory Animals published by the NIH and were approved by the Institutional Animal Care and Use Committee of the University of Rochester Medical Center (Rochester, NY).
Isolation of Mouse Lung ECs
KLF2+/− mice (B6; 129S4‐Klf2 tm1.1Hhn/J, Stock# 026926) were purchased from JAX Laboratory (Bar Harbor, ME). Mouse lung ECs were isolated as described previously.26 Briefly, lungs from 3 mice of each group were minced into pieces and digested with prewarmed type I collagenase (2 mg/mL) at 37°C for 45 minutes. The suspension was filtered through a 70‐μm disposable cell strainer and then centrifuged at 800 g at 4°C for 5 minutes. Precipitates were resuspended in cold PBS+BSA+Penicillin/Streptomycin. The cell suspension was then incubated with CD31 (#553370; BD Pharmingen, San Jose, CA)‐coated Dynabeads (Invitrogen, Carlsbad, CA) on a rotor at room temperature for 15 minutes, and then washed until the suspension became visibly clear. Cells with beads were resuspended in growth medium (DMEM+20%FBS+penicillin/streptomycin+heparin) and plated on 0.1% gelatin‐coated culture dishes. At 5 to 9 days after plating, cells approached confluence. A second sort of cells were performed with CD102‐coated (#553326; BD Pharmingen) Dynabeads sorting. Then, cells were cultured in growth medium and used for indicated experiments immediately.
TNFα‐Stimulated Endothelial Inflammation Assay
Isolated lung ECs from 3 mice of each group were treated with SAHA and TNFα the same as in HCAECs. RNA and protein samples were used for western blot and qPCR experiments. In vivo, SAHA (50 mg/kg/day) or vehicle (0.5% carboxymethyl cellulose‐sodium) was orally administered to KLF2+/+ and KLF2+/− mice for 10 days. Then, mice were injected with mTNFα (0.5 μg/mouse) for 6 hours. Mice were euthanatized by CO2 euthanasia. Aortic ECs from 3 mice were pooled by lysed with 1× SDS sample buffer by flushing the vessel lumen. Western blot was performed to detect mouse VCAM1 (#AF643, 1:1000; R&D Systems) and ICAM1 (#AF796, 1:1000; R&D Systems) expression.
Analysis of Atherosclerotic Lesions and Lipid Profile
To evaluate the potential antiatherosclerotic efficacy of SAHA, we administered vehicle (0.5% carboxymethyl cellulose‐sodium; n=9), or SAHA (50 mg/kg body weight/day, by oral gavage; n=10) into 6‐week‐old ApoE−/− mice, which were fed a western‐type diet (#TD88137; Harlan Teklad, North Easton, MA) for an additional 12 weeks to accelerate plaque formation. After treatment, mice were euthanatized, and whole aorta were isolated, fixed, and subject to en face Oil Red O staining, as previously described.18 The ratio of Oil Red O–positive lesion area to total lumen area was calculated for each mouse using NIH ImageJ software and expressed as percentage of total area. Serum from vehicle‐ and SAHA‐treated mice (n=8/each group) was prepared at the end point, and levels of total cholesterol, triglyceride, low‐density‐lipoprotein cholesterol/very‐low‐density‐lipoprotein cholesterol were analyzed with commercial colorimetric kits from BioVision, Inc. (#K622, #K613; Milpitas, CA). After reaction, absorbance at 562 nm was recorded and lipid concentration was calculated using total cholesterol and triglyceride standard curves. One microliter of undiluted serum was used for measuring total cholesterol, 2 μL of undiluted serum for measuring triglyceride, and 2 μL of diluted serum for measuring low‐density‐lipoprotein cholesterol/very‐low‐density‐lipoprotein cholesterol (and multiply dilution factor).
Statistical Analysis
Data obtained from cell‐culture experiments were derived from different batches of HCAECs from different donors. Data analysis was performed using GraphPad Prism 5 software (GraphPad Software Inc, La Jolla, CA). Statistical comparisons and analyses between 2 groups were performed by 2‐tailed, paired Student t test or nonparametric Wilcoxon rank‐sum test, as indicated in figure legends. When comparing more than 2 groups, 1‐way ANOVA with Bonferroni or Kruskal–Wallis post‐hoc test analysis was used. Data are presented as mean±SEM, and P<0.05 was considered statistically significant.
Results
High‐Throughput Compound Screening Identifies SAHA as a Potent Inducer of KLF2
We designed a high‐throughput compound screening method using a human −1.7‐kb KLF2‐luc in COS‐7 cells. To verify our screening system, we first tested the responsiveness of reporter vectors to reported compounds (simvastatin10, 12) that activated KLF2 or KLF2‐upstream activators (dual specificity mitogen‐activated protein kinase kinase 527 and MEF222). As shown in Figure S1, compared with control treatment (DMSO or Ad‐lacZ), simvastatin, adenoviral dual specificity mitogen‐activated protein kinase kinase 5, and MEF2 infection could dramatically increase KLF2 luciferase activity in COS‐7 cells transfected with KLF2‐luc plasmid. These data demonstrated that KLF2‐luc screening model was successfully established and could be used for subsequent screening of potential KLF2 activators.
The Spectrum library is a small collection of 2400 compounds that contains natural products (25%), marketed drugs (in the United States, Europe, and Japan, 60%) and other bioactive compounds (15%). Among the 2400 compounds tested, 25 compounds were discovered to increase KLF2‐luc activity ≥2‐fold (Figure 1A). Among the 25 positive‐hit compounds, known KLF2‐activating compounds, including simvastatin, rosuvastatin, mevastatin, atorvastatin, and resveratrol, were identified and labeled (Figure 1A). These results further validated the successful establishment of our KLF2‐luc screening system.
Figure 1.
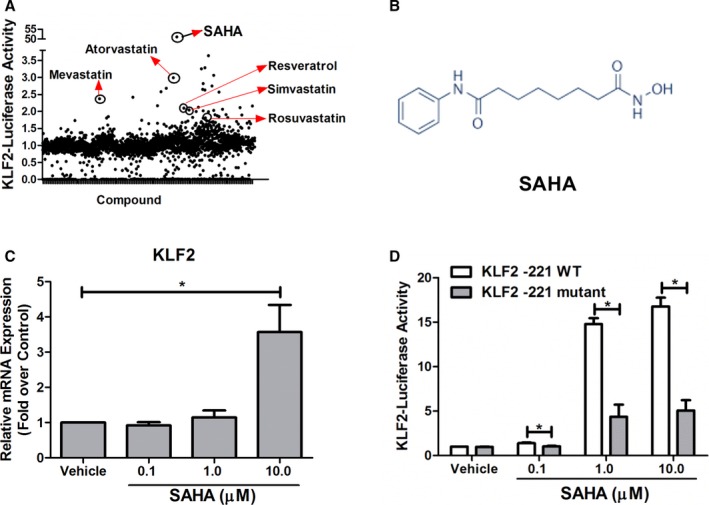
Identification of SAHA as a pharmacological KLF2 activator. A, Results of KLF2‐luc‐based drug screening. B, The chemical structure of SAHA. C, SAHA upregulates KLF2 mRNA expression in HCAECs. HCAECs were treated with vehicle (0.1% DMSO) or indicated concentration of SAHA (0.1, 1, and 10 μmol/L), and then the levels of KLF2 mRNA expression were detected by qPCR using GADPH as the loading control. *P<0.05 (SAHA vs vehicle), nonparametric Wilcoxon rank‐sum test, 2‐tailed, n=4. D, SAHA upregulates KLF2 promoter activity by MEF2. COS7 cells were transfected with KLF2‐wild type (−221 bp, KLF2‐221 WT) or KLF2‐mutant (−221 bp, mutation of MEF2 binding site, KLF2‐221 mutant) before stimulation with indicated concentration of SAHA for 24 hours. Then, luciferase activity was read and expressed as fold over control, nonparametric Wilcoxon rank‐sum test, 2‐tailed, n=4, *P<0.05. KLF2 indicates Kruppel‐like factor 2; SAHA, suberoylanilide hydroxamic acid; WT, wild type.
One of the hit drugs we have identified is SAHA (Figure 1B), a US Food and Drug Administration–approved anticancer drug. In our system, SAHA could significantly increase KLF2 promoter luciferase activity (Figure 1A) more than 50‐fold compared with vehicle control. We next evaluated the effect of SAHA on KLF2 expression in HCAECs. Our qPCR assay indicated that SAHA could increase KLF2 mRNA expression at 10 μmol/L (Figure 1C). Meanwhile, SAHA (10 μmol/L) also significantly decreased the mRNA expression of known KLF2‐target gene endothelin‐128, 29 (Figure S2). Taken together, our data suggest that SAHA is a bona‐fide KLF2 activator.
To identify the mechanism for SAHA‐mediated activation of the KLF2 promoter, we performed dual‐luciferase activity assay by transfecting COS7 cells with wild‐type, KLF2‐promoter–driven luciferase reporter plasmid (−221 bp, KLF2‐221‐wt) and mutant plasmids with point mutation in the MEF2 binding site of the KLF2 promoter region (gifted by Prof Mukesh Jain,10 KLF2‐221‐mutant). Transfected cells were incubated with or without SAHA for 24 hours, then luciferase activity was determined. We observed that mutation of the MEF2 binding site significantly reduced the SAHA‐mediated increase of the KLF2 promoter activity (Figure 1D). These data implicate that SAHA induces the KLF2 promoter activity through MEF2 binding.
SAHA Exhibited Anti‐Inflammatory Effects and Attenuated Monocyte Adhesion to ECs
KLF2 is a potent anti‐inflammatory transcription factor that maintains normal endothelial function.5 To determine whether SAHA‐mediated KLF2 upregulation improves EC function, the effect of SAHA on TNFα‐induced monocyte adhesion to HCAECs was evaluated. Our data showed that, at concentrations of 0.1 to 10 μmol/L, SAHA did not significantly affect EC viability (Figure S3). Compared with the vehicle control, TNFα‐induced THP‐1 monocyte adhesion to HCAECs was greatly blocked by SAHA (Figure 2A and 2B).
Figure 2.
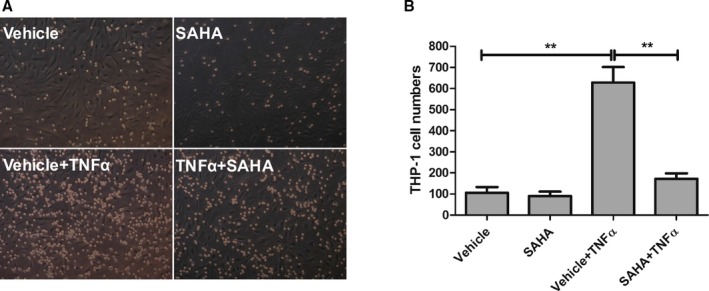
SAHA attenuates THP‐1 monocyte adhesion to HCAECs. HCAECs were plated in 6‐well plates the day before pretreatment with DMSO (vehicle) or SAHA (10 μmol/L) for 12 hours. Then, TNFα (10 ng/mL, final concentration) or vehicle (PBS) was added for an additional 6 hours. Then, THP‐1 monocytes (0.5 mL/well) were added for 30 minutes and then nonadherent monocytes were washed out. A, Representative images of monocyte adhesion assay. Cobblestone‐shaped cells are endothelial cells whereas small and round cells are adherent THP‐1 monocytes. B, Quantification of (A). Adherent THP‐1 cells were counted from 6 independent experiments. One‐way ANOVA with post‐hoc Bonferroni test; **P<0.01. SAHA indicates suberoylanilide hydroxamic acid; TNFα, tumor necrosis factor alpha.
To investigate the mechanism by which SAHA attenuates monocyte adhesion to ECs, expression of KLF2 and adhesion molecules including VCAM1 and ICAM1 were examined. Figure 3A shows that TNFα significantly increased VCAM1 and ICAM1 mRNA expression. Figure 3B shows that TNFα increased VCAM1 and ICAM1 protein expression. However, SAHA treatment significantly decreased TNFα‐induced VCAM1 (but not ICAM1) mRNA. SAHA treatment significantly decreased TNFα‐induced VCAM1 and ICAM1 protein expression (Figure 3A and 3B). Figure 3A shows that TNFα decreased KLF2 mRNA expression whereas SAHA significantly increased KLF2 expression in the presence or absence of TNFα. We also show that adenoviral KLF2 overexpression blocked TNFα‐induced VCAM1, but not ICAM1, expression in ECs (Figure S4). Taken together, these data demonstrated that SAHA had a potent anti‐inflammatory effect in ECs.
Figure 3.
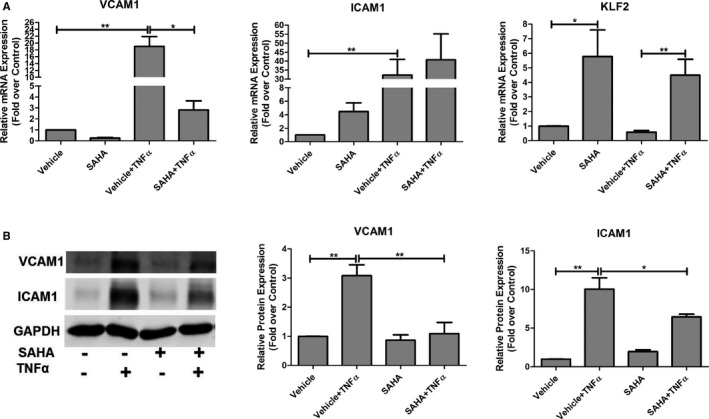
SAHA decreases TNFα‐stimulated vascular inflammation in HCAECs. A, HCAECs were plated in 6‐well plates the day before pretreatment with DMSO (vehicle) or SAHA (10 μmol/L) for 12 hours. Then, TNFα (10 ng/mL, final concentration) or vehicle (PBS) was added for an additional 3 hours, and then qPCR was performed to assess VCAM1 and ICAM1 mRNA expression. Nonparametric Kruskal–Wallis test; *P<0.05; **P<0.01, n=6. B, HCAECs were treated as described above, and then western blot assays were performed to examine VCAM1 and ICAM1 protein expression. Representative images are shown, and the protein levels of VCAM1 and ICAM1 were normalized to GAPDH. One‐way ANOVA with post‐hoc Bonferroni test; *P<0.05; **P<0.01, n=5. GAPDH indicates glyceraldehyde 3‐phosphate dehydrogenase; ICAM1, intercellular adhesion molecule 1; KLF2, Kruppel‐like factor 2; SAHA, suberoylanilide hydroxamic acid; TNFα, tumor necrosis factor alpha; VCAM1, vascular cell adhesion molecule 1.
Anti‐Inflammatory Effects of SAHA Is KLF2 Dependent
To test whether the anti‐inflammatory effects of SAHA is KLF2 dependent, we evaluated vascular inflammation markers in KLF2‐deficient mice treated with SAHA. A previous study has shown that systemic knockout of KLF2 was embryonically lethal16; therefore, in this study, KLF2+/− mice were used to validate KLF2 dependency of SAHA.
Genotypes of KLF2+/+ and KLF2+/− mice were confirmed by PCR assays (Figure 4A). KLF2 deficiency in KLF2+/− mice was confirmed by real‐time PCR.24
Figure 4.
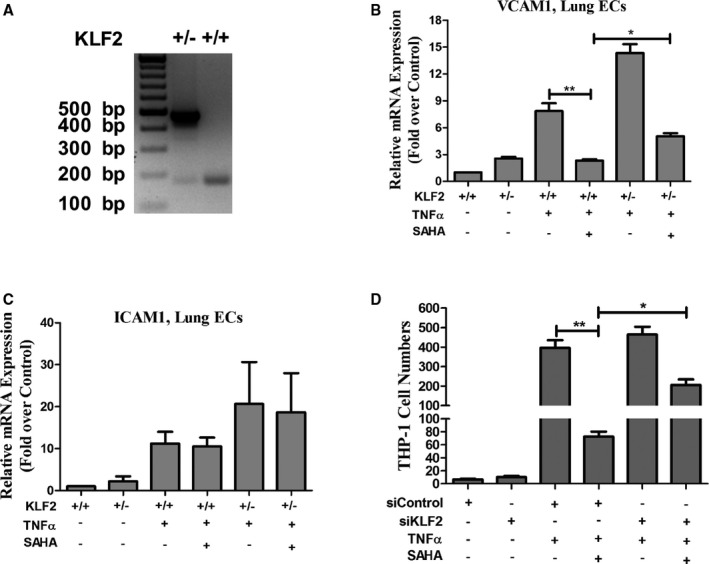
SAHA inhibits vascular inflammation by KLF2. A, Representative genotyping results of KLF2+/+ and KLF2+/− mouse tail DNA. B and C, Lung ECs from KLF2+/+ and KLF2+/− mice were treated with or without SAHA for 12 hours, and then TNFα was added for an additional 3 hours. qPCR was performed to examine VCAM1 and ICAM1 mRNA expression. One‐way ANOVA with post‐hoc Bonferroni test; *P<0.05; **P<0.01, n=4. D, HUVECs were treated with control siRNA or KLF2 siRNA (siKLF2) for 48 hours, then treated with SAHA for 12 hours before stimulation with TNFα for monocyte adhesion assay. One‐way ANOVA with post‐hoc Bonferroni test; *P<0.05; **P<0.01, n=6. ECs indicates endothelial cells; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; ICAM1, intercellular adhesion molecule 1; KLF2, Kruppel‐like factor 2; SAHA, suberoylanilide hydroxamic acid; TNFα, tumor necrosis factor alpha; VCAM1, vascular cell adhesion molecule 1.
To ask whether the effects of SAHA were dependent on KLF2 in cultured cells, lung ECs were isolated from KLF2+/+ and KLF2+/− mice. TNFα significantly increased VCAM1 and ICAM1 mRNA and protein expression in mouse lung ECs. However, in lung ECs from KLF2+/− mice, the preventive effects of SAHA on TNFα‐induced VCAM1 expression was partially rescued (Figure 4B and 4C). We next evaluated the effects of SAHA on expression of KLF2 and the inflammatory markers in mouse aortic endothelium. Aortic intimal (endothelium layer) lysates were collected from KLF2+/+ and KLF2+/− mice treated with or without SAHA. Our western blot analysis showed that TNFα greatly increased VCAM1 and ICAM1 protein expression in both KLF2+/+ and KLF2+/− ECs, but SAHA significantly decreased VCAM1 protein expression in KLF2+/+ aortic ECs. Meanwhile, this inhibitory effect of SAHA was reversed in KLF2+/− lung and aortic ECs (Figure S5).
After observing the anti‐inflammatory effects of SAHA was KLF2 dependent, we asked whether KLF2 depletion by siRNA (Figure S6) could partially reverse SAHA‐mediated inhibitory effects on monocyte adhesion to ECs. Human umbilical vein ECs were treated with control siRNA or KLF2 siRNA for 48 hours before the monocyte adhesion assay was performed. We observed that KLF2 depletion partially reversed the effect of SAHA (Figure 4D). Taken together, these data demonstrate that the anti‐inflammatory effect of SAHA was KLF2 dependent.
Oral Administration of SAHA Reduces Atherosclerosis Development in ApoE−/− Mice
The KLF2‐activating and anti‐inflammatory effects of SAHA prompted us to investigate the potential effects of SAHA on the development of atherosclerosis in an ApoE−/− mouse model. Six‐week‐old male ApoE−/− mice were fed a high‐fat diet, concomitantly treated with carboxymethyl cellulose‐sodium (vehicle) or SAHA (50 mg/kg/day) for 12 weeks by oral gavage. When compared with the vehicle group, SAHA treatment profoundly reduced gross lesion formation in the aortic arch (Figure 5A). The whole en face aortas were stained with Oil Red O, and plaque ratios (plaque area/total area) were calculated for each mouse. As shown in Figure 5B and 5C, the average plaque ratio of vehicle group and SAHA group was 10.0% and 6.7% (P<0.01), respectively. Compared with the vehicle group, SAHA treatment significantly inhibited atherosclerotic lesion formation by 33.2% (Figure 5B and 5C). However, there was no significant change in serum lipid levels between the vehicle‐ and SAHA‐treated groups (Figure 5D). Thus, these results indicate that SAHA treatment attenuates atherosclerosis development in ApoE−/− mice independent of lipid levels.
Figure 5.
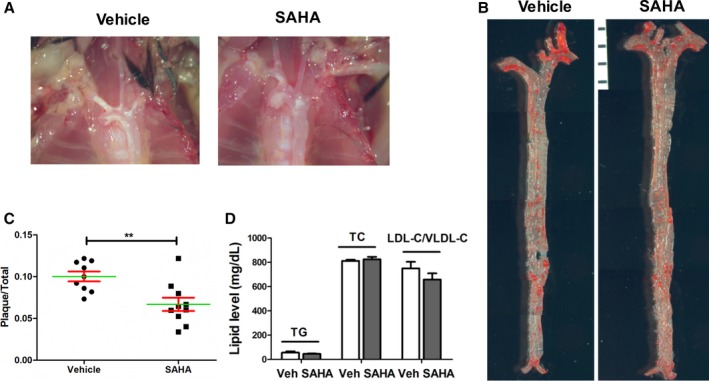
SAHA attenuates atherosclerosis development in ApoE−/− mice. A, 6‐week‐old male ApoE−/− mice were fed a high‐fat diet, concurrent with the treatment of vehicle (CMC‐Na) or SAHA (50 mg/kg/day, oral gavage) for 12 weeks. Representative gross images of aortic arch of each group of mice were shown. B, Representative images (composite of 5‐6 images of the same aorta) of en face Oil Red O (ORO) staining for the lesions of each group mice are shown. C, Quantitative analysis of en face ORO staining lesions. Student t test, n=9 to 10. **P<0.01. D, Effect of SAHA on serum lipid profile. P>0.05 for all comparisons, Student t test, n=8. LDL‐C indicates low‐density lipoprotein cholesterol; SAHA, suberoylanilide hydroxamic acid; TC, total cholesterol; TG, triglyceride; Veh, vehicle; VLDL‐C, very‐low‐density lipoprotein cholesterol.
Discussion
Atherosclerosis, as a chronic inflammatory disease,30, 31, 32, 33 plays a major role in the pathogenesis of cardiovascular diseases.34 Therapeutic targeting of different inflammatory pathways might be effective in preventing and treating cardiovascular diseases.35, 36, 37 The endothelium, as the key regulator of vascular homeostasis, has vasoprotective properties, including anti‐inflammatory response and vasodilation.38 Endothelial dysfunction is regarded as the primary driver of atherosclerotic disease.39 Previous studies have shown that KLF2 is protective molecule in vascular diseases.10 Therefore, designing drugs that specifically modulate endothelial KLF2 expression could result in novel therapies to promote vascular health and thus prevent endothelial dysfunction–associated diseases.12 The aim of this study was designed to identify potential KLF2 activators that have therapeutic potential to treat atherosclerosis. In this regard, our high‐throughput drug screening identified dozens of compounds that can increase KLF2 expression/activity. Among these compounds, SAHA, a well‐known US Food and Drug Administration–approved drug that is used to treat various cancers, was identified as a KLF2 activator that could limit atherosclerosis by inhibiting vascular inflammation.
Our study shows that SAHA is a KLF2 activator that blocks monocyte adhesion to ECs and inhibits the development of atherosclerosis. Based on our in vitro and in vivo data, SAHA reduces monocyte adhesion mainly by inhibiting TNFα‐stimulated upregulation of VCAM1. Because VCAM1 is a well‐known target of KLF2,40 we investigated whether the downregulation of VCAM1 by SAHA is KLF2 dependent. Given that KLF2 overexpression inhibits TNFα‐mediated induction of the proinflammatory molecule, VCAM1,41 in ECs, it is conceivable that the mechanism for the antiatherosclerotic effect of SAHA is partially exerted through KLF2‐dependent anti‐inflammatory action. The consequent antiatherosclerotic effects accompanying KLF2 upregulation by SAHA may include promoting a set of genes with demonstrated anti‐thrombotic, ‐inflammatory, and ‐proliferative functions.10, 35, 42 SAHA, as a pan‐HDAC inhibitor, was approved by the US Food and Drug Administration as an effective anticancer agent.43 Our recent study has shown that HDAC5 associates with KLF2 and represses KLF2 transcriptional activation.44 In light of the fact that SAHA is a pan‐inhibitor for multiple HDACs, therefore it could conceptually increase expression of many genes by increasing transcriptional accessibility of epigenetic marks to gene promoters. However, the contributory role of SAHA on HDAC isoforms to KLF2 upregulation need to be further elucidated. Although we have shown that SAHA reduces monocyte adhesion under static conditions, it would be important to validate the antiadhesive effects of SAHA in a more physiologically relevant model by applying shear flow and TNFα.45, 46 We also recognize that atherosclerosis is a complex, chronic inflammatory disease with the functional interplay among different cell types.47 Given that KLF2 is expressed in multiple cell types involved in atherosclerosis,16, 48, 49 including monocyte/macrophage, ECs, smooth muscle cells, and T cells, it is possible that the atheroprotective effects of SAHA observed in ApoE−/− mouse models could result from the combined effects of SAHA on multiple cellular events involved in atherosclerotic lesion formation. Therefore, the specific contribution of different cell types in SAHA‐mediated atheroprotection warrants further studies.
Consistent with our results, previous studies have shown that Scriptaid or trichostatin A as pan‐HDAC inhibitors could successfully prevent neointima formation from balloon injury,50 and trichostatin A also can attenuate TNFα‐induced monocyte adhesion through suppression of VCAM1.51 Preclinical atherosclerosis models have suggested that drugs modulating histone acetylation may be utilized to treat cardiovascular disease in the future.52 It is noteworthy that, to date, there are some debates on the effects of HDAC inhibitors in treating experimental atherosclerosis.50 Moreover, the side effects of HDAC inhibitor on the vascular system need to be systematically and closely monitored.50 Therefore, HDAC inhibitors with a high efficacy and safety profile might be exploited as promising therapeutic agents of atherosclerotic vascular diseases.
In summary, the present study demonstrates that SAHA is a novel KLF2 activator and suggests that the anticancer drug, SAHA, could be repurposed as an effective KLF2‐dependent anti‐inflammatory and ‐atherosclerotic drug, which merits further clinical evaluation.
Sources of Funding
This project is partially supported by National Institutes of Health RO1 grants (HL09502, HL114570, HL116386, HL128363, HL130167 to Z.G. Jin) and China Scholarship Council (to Y. Xu).
Disclosures
None.
Supporting information
Table S1. Real‐Time PCR Primer Sequence
Figure S1. Confirmation of the screening model.
Figure S2. SAHA decreased KLF2 target gene ET‐1 expression in HCAECs.
Figure S3. SAHA does not significantly affect endothelial cell viability.
Figure S4. KLF2 decreases VCAM1, but not ICAM1 expression, in HUVECs.
Figure S5. Effect of SAHA on VCAM1 and ICAM1 protein expression in endothelial cells from KLF2+/+ and KLF2+/− mice.
Figure S6. Silencing efficiency of KLF2 siRNA.
(J Am Heart Assoc. 2017;6:e007134 DOI: 10.1161/JAHA.117.007134.)29191808
References
- 1. Novodvorsky P, Chico TJ. The role of the transcription factor KLF2 in vascular development and disease. Prog Mol Biol Transl Sci. 2014;124:155–188. [DOI] [PubMed] [Google Scholar]
- 2. Gimbrone MA Jr. Endothelial dysfunction, hemodynamic forces, and atherosclerosis. Thromb Haemost. 1999;82:722–726. [PubMed] [Google Scholar]
- 3. Liao JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. 2013;123:540–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marrone G, Russo L, Rosado E, Hide D, Garcia‐Cardena G, Garcia‐Pagan JC, Bosch J, Gracia‐Sancho J. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial‐stellate cell deactivation induced by statins. J Hepatol. 2013;58:98–103. [DOI] [PubMed] [Google Scholar]
- 5. Atkins GB, Jain MK. Role of Kruppel‐like transcription factors in endothelial biology. Circ Res. 2007;100:1686–1695. [DOI] [PubMed] [Google Scholar]
- 6. Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA Jr, Balasubramanian V, Garcia‐Cardena G, Jain MK. Kruppel‐like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:e48–e57. [DOI] [PubMed] [Google Scholar]
- 7. Lin Z, Natesan V, Shi H, Dong F, Kawanami D, Mahabeleshwar GH, Atkins GB, Nayak L, Cui Y, Finigan JH, Jain MK. Kruppel‐like factor 2 regulates endothelial barrier function. Arterioscler Thromb Vasc Biol. 2010;30:1952–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA Jr, Garcia‐Cardena G. Integration of flow‐dependent endothelial phenotypes by Kruppel‐like factor 2. J Clin Invest. 2006;116:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin Z, Hamik A, Jain R, Kumar A, Jain MK. Kruppel‐like factor 2 inhibits protease activated receptor‐1 expression and thrombin‐mediated endothelial activation. Arterioscler Thromb Vasc Biol. 2006;26:1185–1189. [DOI] [PubMed] [Google Scholar]
- 10. Sen‐Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, Banerjee P, Kumar A, Jain MK. Kruppel‐like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720–726. [DOI] [PubMed] [Google Scholar]
- 11. Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H, Horrevoets AJ. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel‐like factor (KLF2). Blood. 2002;100:1689–1698. [DOI] [PubMed] [Google Scholar]
- 12. Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA Jr, Garcia‐Cardena G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280:26714–26719. [DOI] [PubMed] [Google Scholar]
- 13. Gracia‐Sancho J, Villarreal G Jr, Zhang Y, Garcia‐Cardena G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc Res. 2010;85:514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou Z, Rawnsley DR, Goddard LM, Pan W, Cao XJ, Jakus Z, Zheng H, Yang J, Arthur JS, Whitehead KJ, Li D, Zhou B, Garcia BA, Zheng X, Kahn ML. The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev Cell. 2015;32:168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Young A, Wu W, Sun W, Benjamin Larman H, Wang N, Li YS, Shyy JY, Chien S, Garcia‐Cardena G. Flow activation of AMP‐activated protein kinase in vascular endothelium leads to Kruppel‐like factor 2 expression. Arterioscler Thromb Vasc Biol. 2009;29:1902–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Atkins GB, Wang YM, Mahabeleshwar GH, Shi H, Gao HY, Kawanami DJ, Natesan V, Lin ZY, Simon DI, Jain MK. Hemizygous deficiency of Kruppel‐like factor 2 augments experimental atherosclerosis. Circ Res. 2008;103:690–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sangwung P, Zhou G, Nayak L, Chan ER, Kumar S, Kang DW, Zhang R, Liao X, Lu Y, Sugi K, Fujioka H, Shi H, Lapping SD, Ghosh CC, Higgins SJ, Parikh SM, Jo H, Jain MK. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight. 2017;2:e91700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu S, Liu B, Yin M, Koroleva M, Mastrangelo M, Ture S, Morrell CN, Zhang DX, Fisher EA, Jin ZG. A novel TRPV4‐specific agonist inhibits monocyte adhesion and atherosclerosis. Oncotarget. 2016;7:37622–37635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu S, Koroleva M, Yin M, Jin ZG. Atheroprotective laminar flow inhibits Hippo pathway effector YAP in endothelial cells. Transl Res. 2016;176:18–28.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Le K, Li R, Xu S, Wu X, Huang H, Bao Y, Cai Y, Lan T, Moss J, Li C, Zou J, Shen X, Liu P. PPARalpha activation inhibits endothelin‐1‐induced cardiomyocyte hypertrophy by prevention of NFATc4 binding to GATA‐4. Arch Biochem Biophys. 2012;518:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lan T, Liu W, Xie X, Xu S, Huang K, Peng J, Shen X, Liu P, Wang L, Xia P, Huang H. Sphingosine kinase‐1 pathway mediates high glucose‐induced fibronectin expression in glomerular mesangial cells. Mol Endocrinol. 2011;25:2094–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin ZG. Fluid shear stress stimulates phosphorylation‐dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood. 2010;115:2971–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu S, Yin M, Koroleva M, Mastrangelo MA, Zhang W, Bai P, Little PJ, Jin ZG. SIRT6 protects against endothelial dysfunction and atherosclerosis in mice. Aging (Albany NY). 2016;8:1064–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu Y, Liu P, Xu S, Koroleva M, Zhang S, Si S, Jin ZG. Tannic acid as a plant‐derived polyphenol exerts vasoprotection via enhancing KLF2 expression in endothelial cells. Sci Rep. 2017;7:6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Z, Xu S, Huang X, Wang J, Gao S, Li H, Zhou C, Ye J, Chen S, Jin ZG, Liu P. Cryptotanshinone, an orally bioactive herbal compound from Danshen, attenuates atherosclerosis in apolipoprotein E‐deficient mice: role of lectin‐like oxidized LDL receptor‐1 (LOX‐1). Br J Pharmacol. 2015;172:5661–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao J, Wang W, Ha CH, Kim JY, Wong C, Redmond EM, Hamik A, Jain MK, Feng GS, Jin ZG. Endothelial Grb2‐associated binder 1 is crucial for postnatal angiogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Komaravolu RK, Adam C, Moonen JR, Harmsen MC, Goebeler M, Schmidt M. ERK5 inhibits endothelial migration via KLF2‐dependent down‐regulation of PAK1. Cardiovasc Res. 2015;105:86–95. [DOI] [PubMed] [Google Scholar]
- 28. Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J, de Vries CJ, Biessen EA, van Berkel TJ, Pannekoek H, Horrevoets AJ. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone‐regulating genes. Am J Pathol. 2005;167:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Groenendijk BC, Van der Heiden K, Hierck BP, Poelmann RE. The role of shear stress on ET‐1, KLF2, and NOS‐3 expression in the developing cardiovascular system of chicken embryos in a venous ligation model. Physiology (Bethesda). 2007;22:380–389. [DOI] [PubMed] [Google Scholar]
- 30. Ladeiras‐Lopes R, Agewall S, Tawakol A, Staels B, Stein E, Mentz RJ, Leite‐Moreira A, Zannad F, Koenig W. Atherosclerosis: recent trials, new targets and future directions. Int J Cardiol. 2015;192:72–81. [DOI] [PubMed] [Google Scholar]
- 31. Libby P, Hansson GK. Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res. 2015;116:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu S, Bai P, Little PJ, Liu P. Poly(ADP‐ribose) polymerase 1 (PARP1) in atherosclerosis: from molecular mechanisms to therapeutic implications. Med Res Rev. 2014;34:644–675. [DOI] [PubMed] [Google Scholar]
- 33. Fang J, Little PJ, Xu S. Atheroprotective effects and molecular targets of tanshinones derived from herbal medicine Danshen. Med Res Rev. 2017;133:e38. doi: 10.1002/med.21438. [DOI] [PubMed] [Google Scholar]
- 34. Zhou B, Margariti A, Zeng L, Xu Q. Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc Res. 2011;90:413–420. [DOI] [PubMed] [Google Scholar]
- 35. Ridker PM, Luscher TF. Anti‐inflammatory therapies for cardiovascular disease. Eur Heart J. 2014;35:1782–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ridker PM. Targeting inflammatory pathways for the treatment of cardiovascular disease. Eur Heart J. 2014;35:540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu X, Huang H, Tang F, Le K, Xu S, Liu P. Regulated expression of endothelial lipase in atherosclerosis. Mol Cell Endocrinol. 2010;315:233–238. [DOI] [PubMed] [Google Scholar]
- 38. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–III32. [DOI] [PubMed] [Google Scholar]
- 39. Mudau M, Genis A, Lochner A, Strijdom H. Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr. 2012;23:222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA Jr, Garcia‐Cardena G, Jain MK. KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu M, Kluger MS, D'Alessio A, Garcia‐Cardena G, Pober JS. Regulation of arterial‐venous differences in tumor necrosis factor responsiveness of endothelial cells by anatomic context. Am J Pathol. 2008;172:1088–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bu DX, Griffin G, Lichtman AH. Mechanisms for the anti‐inflammatory effects of statins. Curr Opin Lipidol. 2011;22:165–170. [DOI] [PubMed] [Google Scholar]
- 43. Xu SS, Alam S, Margariti A. Epigenetics in vascular disease—therapeutic potential of new agents. Curr Vasc Pharmacol. 2014;12:77–86. [DOI] [PubMed] [Google Scholar]
- 44. Kwon IS, Wang W, Xu S, Jin ZG. Histone deacetylase 5 interacts with Kruppel‐like factor 2 and inhibits its transcriptional activity in endothelium. Cardiovasc Res. 2014;104:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jones DA, Smith CW, McIntire LV. Leucocyte adhesion under flow conditions: principles important in tissue engineering. Biomaterials. 1996;17:337–347. [DOI] [PubMed] [Google Scholar]
- 46. Nallasamy P, Si H, Babu PV, Pan D, Fu Y, Brooke EA, Shah H, Zhen W, Zhu H, Liu D, Li Y, Jia Z. Sulforaphane reduces vascular inflammation in mice and prevents TNF‐alpha‐induced monocyte adhesion to primary endothelial cells through interfering with the NF‐kappaB pathway. J Nutr Biochem. 2014;25:824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tabas I, Garcia‐Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weinreich MA, Takada K, Skon C, Reiner SL, Jameson SC, Hogquist KA. KLF2 transcription‐factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity. 2009;31:122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das H, Kumar A, Lin Z, Patino WD, Hwang PM, Feinberg MW, Majumder PK, Jain MK. Kruppel‐like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci USA. 2006;103:6653–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yoon S, Eom GH. HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J. 2016;52:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Inoue K, Kobayashi M, Yano K, Miura M, Izumi A, Mataki C, Doi T, Hamakubo T, Reid PC, Hume DA, Yoshida M, Aird WC, Kodama T, Minami T. Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule‐1 expression. Arterioscler Thromb Vasc Biol. 2006;26:2652–2659. [DOI] [PubMed] [Google Scholar]
- 52. Khyzha N, Alizada A, Wilson MD, Fish JE. Epigenetics of atherosclerosis: emerging mechanisms and methods. Trends Mol Med. 2017;23:332–347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Real‐Time PCR Primer Sequence
Figure S1. Confirmation of the screening model.
Figure S2. SAHA decreased KLF2 target gene ET‐1 expression in HCAECs.
Figure S3. SAHA does not significantly affect endothelial cell viability.
Figure S4. KLF2 decreases VCAM1, but not ICAM1 expression, in HUVECs.
Figure S5. Effect of SAHA on VCAM1 and ICAM1 protein expression in endothelial cells from KLF2+/+ and KLF2+/− mice.
Figure S6. Silencing efficiency of KLF2 siRNA.