

Abstract
Background
The healthy heart has a dynamic capacity to respond and adapt to changes in nutrient availability. Diabetes mellitus disrupts this metabolic flexibility and promotes cardiomyopathy through mechanisms that are not completely understood. Phosphofructokinase 2 (PFK‐2) is a primary regulator of cardiac glycolysis and substrate selection, yet its regulation under normal and pathological conditions is unknown. This study was undertaken to determine how changes in insulin signaling affect PFK‐2 content, activity, and cardiac metabolism.
Methods and Results
Streptozotocin‐induced diabetes mellitus, high‐fat diet feeding, and fasted mice were used to identify how decreased insulin signaling affects PFK‐2 and cardiac metabolism. Primary adult cardiomyocytes were used to define the mechanisms that regulate PFK‐2 degradation. Both type 1 diabetes mellitus and a high‐fat diet induced a significant decrease in cardiac PFK‐2 protein content without affecting its transcript levels. Overnight fasting also induced a decrease in PFK‐2, suggesting it is rapidly degraded in the absence of insulin signaling. An unbiased metabolomic study demonstrated that decreased PFK‐2 in fasted animals is accompanied by an increase in glycolytic intermediates upstream of phosphofructokianse‐1, whereas those downstream are diminished. Mechanistic studies using cardiomyocytes showed that, in the absence of insulin signaling, PFK‐2 is rapidly degraded via both proteasomal‐ and chaperone‐mediated autophagy.
Conclusions
The loss of PFK‐2 content as a result of reduced insulin signaling impairs the capacity to dynamically regulate glycolysis and elevates the levels of early glycolytic intermediates. Although this may be beneficial in the fasted state to conserve systemic glucose, it represents a pathological impairment in diabetes mellitus.
Keywords: autophagy, diabetic cardiomyopathy, glycolysis, insulin action, insulin resistance, metabolism
Subject Categories: Metabolism, Myocardial Biology, Cell Signalling/Signal Transduction, Metabolic Syndrome, Mechanisms
Clinical Perspective
What Is New?
Phosphofructokinase‐2 (PFK‐2), a central regulator of cardiac glycolysis that is normally activated in response to adrenergic stimulation and/or insulin signaling, is decreased in mouse models of reduced insulin signaling, including type 1 diabetes mellitus, high‐fat feeding, and overnight fasting.
Using isolated adult mouse cardiomyocytes, we demonstrate that, in the absence of insulin signaling, PFK‐2 is degraded by mechanisms that include both proteasome‐ and chaperone‐mediated autophagy.
What Are the Clinical Implications?
Cardiac PFK‐2 levels normally fluctuate dynamically between the fed and fasted states, but they are persistently decreased with diabetes mellitus and a high‐fat diet.
A short‐term decrease of PFK‐2 during fasting would contribute to limited glucose use basally and in response to β‐adrenergic signaling.
A long‐term decrease in PFK‐2 could increase cardiac metabolic inflexibility and contribute to diabetic heart disease.
Introduction
Diabetes mellitus has reached epidemic proportions, and cardiovascular complications are the predominant cause of mortality. Coronary artery disease is a major factor, but heart failure in the absence of other contributing factors is also prevalent.1, 2 This condition, termed diabetic cardiomyopathy, is particularly problematic because it can exacerbate comorbidities and increase the occurrence and progression of heart failure.3 It is, thus, critical to identify the mechanisms that promote heart failure in diabetes mellitus.
Although numerous factors are implicated, prominent drivers of diabetic cardiomyopathy are metabolic alterations that affect the import and use of fatty acids and glucose by cardiomyocytes.4 The healthy heart uses fatty acids, glucose, and a mix of other substrates, adjusting substrate preference dynamically with availability and energy demands.5 The fatty acid/glucose balance is disrupted in diabetes mellitus, resulting in metabolic inflexibility and nearly complete reliance on lipids for energy. This results in increased fatty acid use, insubstantial glucose use, myocardial lipid accumulation, lipotoxicity, and mitochondrial dysfunction.6 Furthermore, diabetic substrate use becomes unresponsive to factors, such as insulin and adrenergic signaling, that normally increase glucose use in the healthy myocardium.7, 8
The impairment of glucose use is a critical step in the metabolic phenotype of diabetes mellitus, and multiple contributing factors are involved in this process in the heart. Both membrane glucose transporter type 4 and mitochondrial pyruvate oxidation are reduced, thereby decreasing glucose import and inhibiting glucose oxidation, respectively.9, 10 However, it is less well understood how diabetes mellitus affects glycolytic enzymes. This is important to determine because cardiac glucose uptake is largely maintained in diabetics, especially under conditions of increased workload, even though it is not readily used as an oxidizable energy source.11 Remedying metabolic inflexibility requires glucose uptake, glycolysis, and pyruvate oxidation to be increased in concert.
In the heart, phosphofructokinase 1 (PFK‐1) is the rate‐limiting enzyme of glycolysis.12, 13 The bifunctional enzyme, 6‐phosphofructo‐2‐kinase/fructose 2,6‐bisphosphatase (PFK‐2), either produces or consumes the potent allosteric activator of PFK‐1, fructose 2,6‐bisphosphate. The kinase activity of the enzyme is activated by insulin and adrenergic signaling via phosphorylation, thus allowing the heart to normally alter its substrate selection depending on discrete external cues.14, 15 For example, protein kinase A (PKA)–mediated phosphorylation of PFK‐2 is sufficient to increase glucose use, even in the presence of lipids.16 However, we have found that PFK‐2 is unresponsive to β‐adrenergic pathway agonists in a diabetic model.8 Despite its pivotal role in cardiac metabolism and metabolic flexibility, changes that occur to PFK‐2 abundance, phosphorylation, and activity with diabetes mellitus have not been examined.
In this work, we demonstrate that PFK‐2 content decreases in type 1 diabetic and fasted mice. Using an unbiased metabolomic approach, we show that hearts from fasted animals have an increase in glycolytic intermediates upstream of PFK‐1, implicating the importance of PFK‐2 in this process. Mechanistic studies with primary adult cardiomyocytes establish that insulin signaling regulates PFK‐2 content. Specifically, in the absence of insulin, PFK‐2 is rapidly degraded via both proteasomal and lysosomal mechanisms. This work highlights that PFK‐2 is a central mediator for metabolic changes induced by diabetes mellitus.
Methods
The data that support the findings of this study are available to other researchers on request. All of the materials used in this study are available commercially from the indicated vendors.
Animal Models
Streptozotocin treatments were performed by The Jackson Laboratory (Bar Harbor, ME) on 7‐week‐old C57BL/6J mice using a 50 mg/kg treatment for 5 days. Elevated glucose levels were confirmed to be at least 300 to 400 mg/dL ≈16 days after injection. Age‐matched C57BL/6J mice were used as controls. Mice were fed standard laboratory chow ad libitum. Four months after streptozotocin treatments, mice were euthanized by cervical dislocation for biochemical analysis. For PFK‐2 content studies, 5 control and 5 streptozotocin‐treated diabetic mice were used. For the high‐fat diet (HFD) study: 8‐week‐old C57BL/6J mice were fed either a low‐fat diet (70% carbohydrate, 20% protein, and 10% fat, by kcal) or an HFD (20% carbohydrate, 20% protein, and 60% fat, by kcal) ad libitum for a total of 1 week (n=6 for low‐fat diet, and n=4 for HFD) or 21 weeks (n=4 for both groups; products D12450J and D12492; Research Diets Inc, New Brunswick, NJ). For fasting studies, 3‐month‐old C57BL/6J mice were placed in a fresh cage with access to water but without food for 24 hours (n=4). Age‐matched control C57BL/6J mice were fed standard laboratory chow ad libitum. Treatment groups, as indicated, were given 50 μg/kg isoproterenol hydrochloride (Sigma, St Louis, MO) or 330 mg/kg 8‐bromo‐cAMP (Tocris Bioscience, Bristol, UK) by IP injection. A corresponding volume of saline was used for control treatments. Hearts were excised under 2% isoflurane anesthesia, quickly washed in ice‐cold distilled water, blotted dry, and flash frozen, as recommended for metabolomic analysis.17 All mouse experiments were performed with approval from the Oklahoma Medical Research Foundation (Oklahoma City, OK) Institutional Animal Care and Use Committee.
Measuring Changes in Protein Expression With Targeted Quantitative Mass Spectrometry
These experiments use the tandem mass spectrometry (MS) method of selected reaction monitoring (SRM) to quantify representative peptides, formed by digestion of the protein homogenate with trypsin, from each protein being assayed. For the measurement of PFK‐2 expression, a method was developed using protein expressed in H9c2 cells to select the representative peptides that are detectable by liquid chromatography–tandem MS. Briefly, H9c2 cells were grown to 75% confluence and then treated with an adenoviral vector (200 multiplicity of infection) that expresses the full‐length mouse pfkfb2 gene (Vector Biolabs, Malvern, PA). At 48 hours after transduction, cells were washed with PBS, scraped, and spun for 5 minutes at 500g. The resulting pellet was resuspended in NP40 lysis buffer containing Halt phosphatase and protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA). Samples were then subjected to in‐gel digestion and MS analysis, as described later for heart samples. Results of these experiments revealed that the PFK‐2 peptides YPGGESYQDLVQR, VFNLGVYR, and SYQSYDFFR were the 3 best flyers, and they were used in the subsequent cardiac analysis.18
Heart samples were prepared on the basis of our standard targeted proteomics approach.19, 20, 21, 22 Whole heart homogenates were used for the analysis. Total proteins (50‐µg amounts) were taken for analysis, and 8 pmol BSA was added as an internal standard. The proteins were precipitated with acetone. The dried protein pellet was reconstituted in Laemmli sample buffer at 1 μg/μL, and 20 μL (20 μg total protein) was run into a short (1.5‐cm) SDS‐PAGE gel. The gels were fixed and stained. Each sample was cut from the gel as the entire lane and divided into smaller pieces. The gel pieces were washed to remove the Coomassie blue and then reduced, alkylated, and digested overnight with trypsin. The mixture of peptides was extracted from the gel, evaporated to dryness in a SpeedVac, and reconstituted in 150 μL 1% acetic acid for analysis.
The analyses were performed on a ThermoScientific TSQ Quantiva triple‐quadrupole MS system. The high‐performance liquid chromatograph was an Ultimate 3000 nanoflow system with a 10‐cm×75‐μm internal diameter. A C18 reversed‐phase capillary column was used. Aliquots (5 µL) were injected, and the peptide was eluted with a 60‐minute gradient of acetonitrile in 0.1% formic acid. The data were analyzed with the program skyline to integrate the peak area of the respective peptides. Multiple peptides are measured for each protein. The geomean of the response of those peptides was used for the protein response. The protein responses were normalized to the BSA response to determine the concentration in the homogenate.
Metabolomics
Metabolomics analysis was performed by the West Coast Metabolomics Center at University of California, Davis. In brief, unbiased profiling was performed using automatic linear exchange and cold injection gas chromatography–time‐of‐flight MS. Metabolite values were quantified by peak heights and listed as normalized relative intensities. Additional targeted metabolomics studies were performed by the University of Michigan (Ann Arbor, MI) to measure glycolytic intermediates.
Preparation of Cardiac Homogenates for SDS/PAGE and Western Blot Analysis
After treatments, mice were immediately euthanized by cervical dislocation and hearts were quickly perfused by left ventricle puncture with 3 mL of ice‐cold buffer containing 210 mmol/L mannitol, 70 mmol/L sucrose, 5 mmol/L MOPS, and 1 mmol/L EDTA (perfusion buffer). Hearts were excised and immediately flash frozen. Frozen hearts were homogenized by a Potter‐Elvehjem homogenizer in perfusion buffer with the addition of 20 mmol/L NaF and protease inhibitor cocktail (homogenization buffer). The volume of homogenization buffer was standardized to heart weight. SDS‐PAGE sample buffer (4×) was added to standardized homogenates with 25 mmol/L dithiothreitol. Samples were heated at 95°C for 5 minutes and centrifuged at 16 873g for 5 minutes. After SDS‐PAGE (4%–12% NuPage Bis‐Tris gel; Life Technologies, Carlsbad, CA), gels were transferred to nitrocellulose membranes and blocked for 30 minutes with 2% BSA in PBS with 0.1% Tween‐20. Blots were incubated overnight at 4°C in 2% BSA in PBS with 0.1% Tween‐20 with primary antibodies. All antibodies, including phosphorylated S483 PFK‐2, PFK‐1, PFKFB3, fructose‐2,6‐bisphosphatase (TIGAR), hexokinase II, protein kinase B (Akt), phosphorylated Akt, p70 S6 kinase (S6K), and phosphorylated p70 S6K (Thr389), were from Cell Signaling Technologies (Danvers, MA). The exceptions were antibodies against PFKFB2 (OriGene, Rockville, MD), p62 (Abcam, Cambridge, UK), and horseradish peroxidase–conjugated actin (Santa Cruz Biotechnology, Dallas, TX). Blots were developed with Super‐Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) and imaged with an Alpha Innotech Fluorchem HD2 imaging system. The exposure times were adjusted so that the bands of interest were not saturated. Densitometry analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD).
Quantitative Reverse Transcription–Polymerase Chain Reaction
RNA was isolated using the TriPure Isolation Reagent method (Roche, Basel, Switzerland). cDNA libraries were generated using a QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). Quantitative polymerase chain reactions were performed in duplicate by the CFX96 Real Time System (Bio‐Rad Laboratories Inc, Hercules, CA) in 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds using iQ SYBR Green Supermix (Bio‐Rad Laboratories Inc). Data were analyzed using Biorad CFX manager.
Cell Culture of Adult Mouse Ventricular Cardiomyocytes
Adult primary cardiomyocytes from C57BL/6J mice were isolated and cultured, as previously described.8, 23 In brief, each mouse was treated with 50 IU heparin under isoflurane anesthesia. After 3.0 minutes, the mouse was quickly euthanized by an isoflurane overdose. The heart was immediately excised, the aorta was cannulated within 1 to 2 minutes, and the aorta was perfused with Ca2+‐free buffer (37°C). The perfusion buffer was then switched to a digestion buffer containing type II collagenase, and the heart was digested for 11 minutes or until the heart became soft. Ventricles were removed and teased apart in a calf serum containing buffer into a single‐cell suspension using progressively smaller pipettes. Cells were not used if they consisted of <50% rod‐shaped cells, and they usually consisted of 65% to 75% rods. Calcium was gradually reintroduced before cells were plated on laminin‐coated plates in plating media. After 1 hour, the medium was switched to a serum‐free culture medium (minimal essential medium with Hanks’ balanced salt solution; Thermo Fisher Scientific) supplemented with penicillin‐G, BSA, glutamine, 0.4 mg/mL NaHCO3, and insulin‐transferrin‐selenium (Gibco), as indicated, and butanedione monoxime. Cells were incubated overnight at 37°C and 5% CO2 and used the next day.
Statistical Analysis
Data were analyzed using Graphpad Prism 7. Results are expressed as mean±SEM. Pairwise comparison between groups was performed using an unpaired 2‐tailed Student t test. Comparison between multiple groups was performed using 1‐way ANOVA with Dunnett or Tukey post hoc analysis, as indicated. P≤0.05 was considered statistically significant.
Results
PFK‐2 Content Is Decreased in the Diabetic Mouse Heart
PFK‐2, the gene product of pfkfb2, regulates flux through glycolysis, and changes in its content or activity may be a driver of diabetic cardiomyopathy. Initial experiments were performed to determine how type 1 diabetes mellitus affects cardiac PFK‐2 content. As shown in Figure 1A, PFK‐2 content is significantly decreased (62.6±7.5%) in hearts of streptozotocin‐induced diabetic mice. Furthermore, short‐term administration of the PKA agonist 8‐bromo‐cAMP revealed that PFK‐2 is robustly phosphorylated in control hearts but not in the hearts of diabetic mice (Figure 1B). Thus, the PFK‐2 that remains is unresponsive to β‐adrenergic signaling, a result consistent with our previous report.8 The decrease in PFK‐2 content is because of changes in protein expression or stability, because transcript levels are not significantly decreased (Figure 1C).
Figure 1.
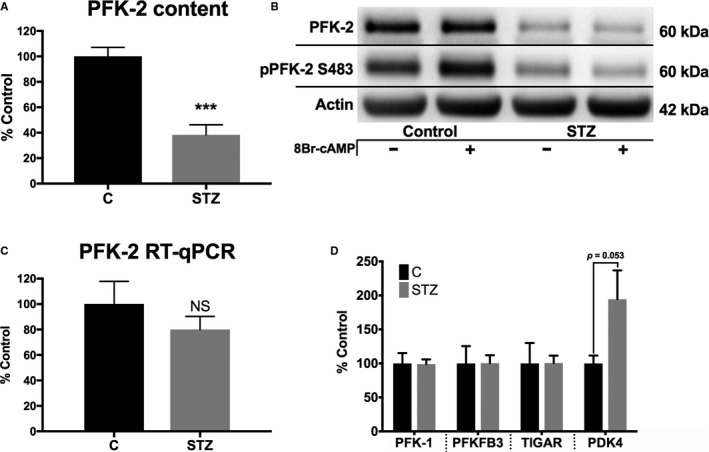
Phosphofructokinase 2 (PFK‐2) content is reduced in diabetic mice without reduction of mRNA. A and B, Cardiac homogenates were analyzed from control and diabetic (4 months after streptozotocin treatment) mice for PFK‐2 and phosphorylated PFK‐2 (pPFK‐2; Ser483). C, PFK‐2 mRNA was measured by quantitative reverse transcription–polymerase chain reaction (RT‐qPCR). D, Metabolic enzymes related to PFK‐2 were analyzed by Western blot analysis. E, PFK‐2 was measured in cardiac homogenates of mice fed either a low‐fat control or a high‐fat diet for 21 weeks. Representative blots on the left and quantified on the right. F, Mice were fed either a low‐fat control or a high‐fat diet for 7 days. Protein homogenates were analyzed by a selected reaction monitoring technique. Data are presented as mean±SEM. NS indicates not significant (P=0.368); PDK4, pyruvate dehydrogenase kinase; PFKFB3, phospho fructokinase 2 isoform 3; and TIGAR, fructose‐2,6‐bisphosphatase. ***P<0.005, unpaired Student t test (n=4–6 for all groups).
Other proteins functionally related to PFK‐2 and its enzymatic product, fructose 2,6‐bisphosphate, were examined in streptozotocin‐treated diabetic mice to determine if changes were global or selective. The content of PFK‐1, the rate‐limiting enzyme of glycolysis that is activated by PFK‐2, was unaltered (Figure 1D). PFKFB3, another less abundantly expressed isozyme of PFK‐2 in the heart, and fructose‐2,6‐bisphosphatase (TIGAR) had similar content in control and diabetic mice (Figure 1D). In contrast, pyruvate dehydrogenase kinase 4, a protein known to be elevated in fasting and diabetic mice,9 was increased. Thus, the decrease that occurs to PFK‐2 is unique compared with the other related glycolytic proteins PFK‐1, PFKFB3, and TIGAR.
We next examined whether a decrease in PFK‐2 is also observed in the hearts of mice fed an HFD, a model that approximates type 2 diabetes mellitus.24, 25 As shown in Figure 2A, PFK‐2 levels are decreased by ≈25% in mice fed an HFD for 21 weeks. Thus, the decrease in cardiac PFK‐2 is common to both insulin deficiency (streptozotocin induced) and insulin resistance (HFD) mouse models. It is possible that the observed decrease in PFK‐2 is a result in limited proteolysis, thereby affecting PFK‐2 detection by antibodies. However, PFK‐2 fragments were not detected by either of the antibodies used in this study (not shown). The antibody used to detect total PFK‐2 was raised against residues 448 to 476 (human pfkfb2 numbering), strongly supporting that the kinase‐activating phosphorylation site at S483 is absent. In addition, we performed an additional experiment to do the following: (1) determine if PFK‐2 is decreased in as short as 1 week in the hearts of mice fed an HFD; and (2) detect the protein by an alternative means that does not depend on antibody recognition. For this, we developed an MS‐based SRM assay to detect PFK‐2 in heart homogenates. The assay was based on the detection of 3 distinct peptides (see Methods). The SRM assay revealed that PFK‐2 is significantly decreased (20% loss) in the hearts of animals fed an HFD for 7 days (Figure 2B). This result supports that PFK‐2 levels are dynamically regulated, even in the short‐term, to dietary conditions that affect insulin signaling.
Figure 2.
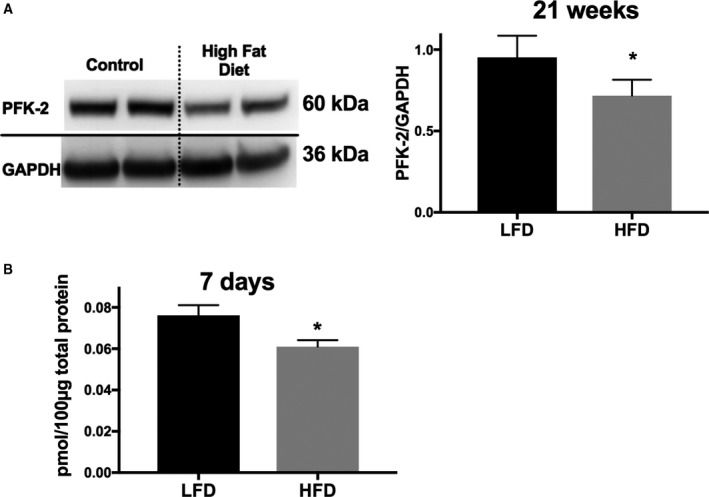
Phosphofructokinase 2 (PFK‐2) content is reduced in mice fed a high‐fat diet (HFD). A, PFK‐2 was measured by Western blot analysis in cardiac homogenates of mice fed either a low‐fat control diet (LFD) or an HFD diet for 21 weeks. Representative blots on the left and quantified on the right. B, Mice were fed either an LFD or an HFD for 7 days, and cardiac homogenates were analyzed by a selected reaction monitoring technique, described in Methods. Data are presented as mean±SEM. *P<0.05, unpaired Student t test (n=4–6 for all groups).
The Decrease in PFK‐2 Is Accompanied by an Increase in Early Glycolytic Intermediates
Diabetes mellitus has a multitude of effects on whole body metabolism and can impair normal cardiac physiological characteristics. We, therefore, examined whether fasting, which also decreases insulin signaling, cardiac glucose use, and increased reliance on lipids are sufficient to decrease PFK‐2 content independently of other diabetic complications and pathological features.26 As shown in Figure 3A, control mice fasted for 24 hours had a significant decrease in cardiac PFK‐2 content (37.0±8.1% decrease). This, thus, identifies PFK‐2 as a labile protein that responds to nutrient stresses. We next determined how a decrease in PFK‐2 content in fasted animals affects cardiac metabolism. An unbiased metabolomics approach was used to examine the differences between fed and fasted animals of glycolytic intermediates and related molecules, citric acid cycle intermediates, fatty acids, and amino acids.
Figure 3.
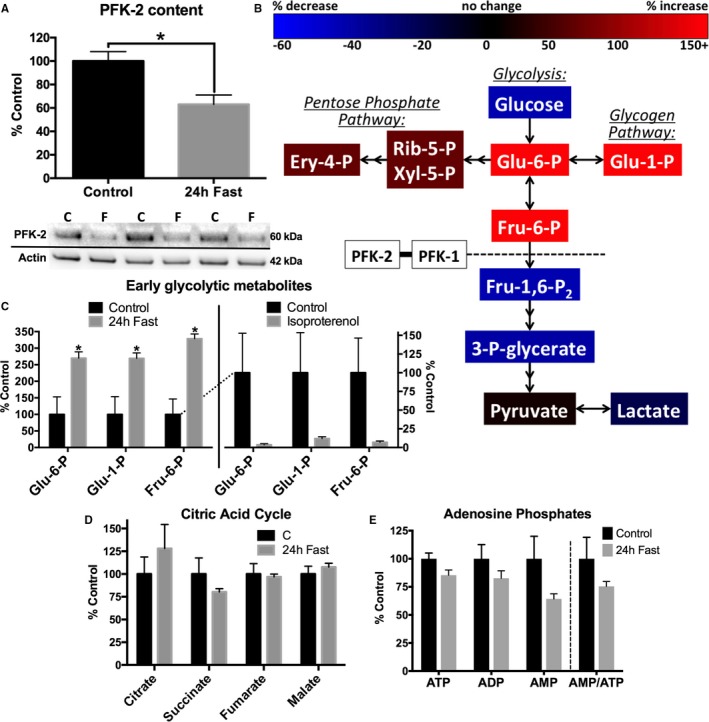
Fasting decreases cardiac phosphofructokinase 2 (PFK‐2) content and causes an accumulation of early glycolytic intermediates. A, Cardiac homogenates from control and 24‐hour fasted mice were analyzed by Western blotting. Densitometric values were standardized to actin. Representative blots shown below (n=5 for both groups). B, Untargeted metabolomics from flash‐frozen heart tissue of control and 24‐hour fasted mice. C, Five minutes before cardiac excision, saline or 50 μg/kg isoproterenol IP injections were administered. Changes in early glycolytic intermediates are shown. D and E, Citric acid intermediates and adenosine phosphates were not significantly affected by fasting (n=3 for all groups). Data are presented as mean±SEM. Ery‐4‐P indicates erythrose 4‐phosphate; Fru‐1,6‐P2, fructose 1,6‐bisphosphate; Fru‐6‐P, fructose 6‐phosphate; Glu‐1‐P, glucose 1‐phosphate; Glu‐6‐P, glucose 6‐phosphate; 3‐P‐glycerate, glycerate 3‐phosphate; Rib‐5‐P, ribulose 5‐phosphate; and Xyl‐5‐P, xylulose 5‐phosphate. *P<0.05, unpaired Student t test.
The changes in metabolites in fasted animals are shown schematically in Figure 3B, and the changes in early glycolytic intermediates (EGIs) are quantified in Figure 3C. More important, the decrease in PFK‐2 content in fasted animals was accompanied by an increase in glycolytic intermediates upstream of PFK‐1 and included an increase in metabolites within the pentose phosphate pathway. However, metabolites downstream of PFK‐1 were significantly decreased in fasted animals. Collectively, these changes in metabolites are consistent with a decrease in PFK‐1 activity. In contrast to glycolytic intermediates, citric acid cycle intermediates (Figure 3D) and adenosine phosphates (Figure 3E) were relatively unaffected by fasting. The lack of change in citrate and adenosine phosphates, regulators of PFK‐1 activity, support that loss of PFK‐2 content may be a primary promoter of EGIs.
To further demonstrate how activation of PFK‐2 affects glycolytic intermediates, control animals were administered the β‐agonist, isoproterenol, to stimulate PKA activity. As shown in Figure 3C, this treatment dramatically decreased glycolytic intermediates upstream of PFK‐1. This result is consistent with β‐adrenergic stimulation activating PFK‐2, increasing glycolysis, and decreasing EGIs.
Insulin Signaling Regulates PFK‐2 Content
Having established the labile nature of cardiac PFK‐2 and its potential functional significance, we next sought to determine whether insulin signaling, which is decreased in type 1 diabetes mellitus and fasting, is coupled to PFK‐2 content. Cardiomyocytes were isolated from control adult mice, and the culture conditions were varied to influence insulin signaling. As shown in Figure 4A, cells cultured overnight in the absence of insulin exhibit a 40% decrease in PFK‐2 content. A similar decrease was seen in cells cultured in the presence of lipids to mimic high fat. In contrast, high glucose had no effect on PFK‐2 content. The decrease in PFK‐2 closely follows alterations in insulin signaling. For example, the conditions that decreased PFK‐2 content also decreased AKT phosphorylation (Figure 4B). Supplementation with insulin‐like growth factor 1 was also able to maintain high levels of PFK‐2 (Figure 4C). However, cosupplementation with both insulin and insulin‐like growth factor 1 showed no further benefit, suggesting these pathways maintain PFK‐2 content by a similar mechanism.
Figure 4.

Phosphofructokinase 2 (PFK‐2) is rapidly degraded in the absence of insulin signaling. A, Primary adult mouse cardiomyocytes from control C57B6/J mice were cultured overnight with standard conditions (C), lacking insulin, high glucose (HG; 450 mg/dL), or a high‐fat (HF) diet (100 μmol/L oleate/100 μmol/L palmitate conjugated to 0.02% BSA, HG). Cells were then analyzed by Western blot analysis for PFK‐2 (A) or phosphorylated protein kinase B (pAKT)/AKT (B). C, Primary adult mouse cardiomyocytes from control C57B6/J mice were cultured overnight with 10 mg/L insulin or 200 μg/L insulin‐like growth factor 1 (IGF‐1), as indicated. D, Primary adult mouse cardiomyocytes from control C57B6/J mice were cultured with insulin and treated with either 50 μg/mL cycloheximide (black) or cycloheximide with 500 nmol/L wortmannin (red). The dotted line represents a theoretical degradation curve for the listed half‐life. Densitometry from Western blots of cardiac homogenates was standardized to actin (A and C), Akt (B), or cardiac PFK‐2 (D). Data are presented as mean±SEM. *P<0.05, **P<0.01, and **** P≤0.0001 by ANOVA with the Dunnett post hoc test (A [n=5], B [n=5], and C [n=3]) or an unpaired Student t test (D [n=4]).
The phosphatidylinositol 3‐kinase (PI3K) signaling pathway is activated by both insulin and insulin‐like growth factor 1, suggesting its activity is necessary in maintaining PFK‐2 content. We, therefore, examined the importance of PI3K activity on the half‐life of PFK‐2. Cells were cultured with insulin and cycloheximide, a protein translation inhibitor, and in the presence or absence of the PI3K inhibitor, wortmannin. Treatment with cycloheximide alone had a minimal effect on PFK‐2 content (Figure 4D), suggesting it is a relatively long‐lived protein. However, wortmannin induced a significant and rapid decrease in PFK‐2 content, with a calculated half‐life of 2.2 hours. Taken together, PI3K activity, which is decreased by both fasting and diabetes mellitus, dynamically controls PFK‐2 content.
PFK‐2 Stability Is Affected by Its Phosphorylation State, and Its Degradation Is Mediated by Both Proteasome‐ and Chaperone‐Mediated Autophagy
We next sought the mechanisms that lead to the rapid degradation of PFK‐2 in the absence of insulin signaling. We first examined whether PFK‐2 is degraded in a proteasome‐dependent manner. As shown in Figure 5A, adult cardiomyocytes cultured in the absence of insulin for 4 hours had a significant decrease in PFK‐2 content, and this was largely prevented by the proteasome inhibitor, MG132. Furthermore, treatment of cells with the PKA agonist, 8‐bromo‐cAMP, also sustained PFK‐2 levels in the absence of insulin. These results support that PFK‐2 is degraded via the proteasome. However, degradation is prevented when the protein is phosphorylated and activated by PKA.
Figure 5.
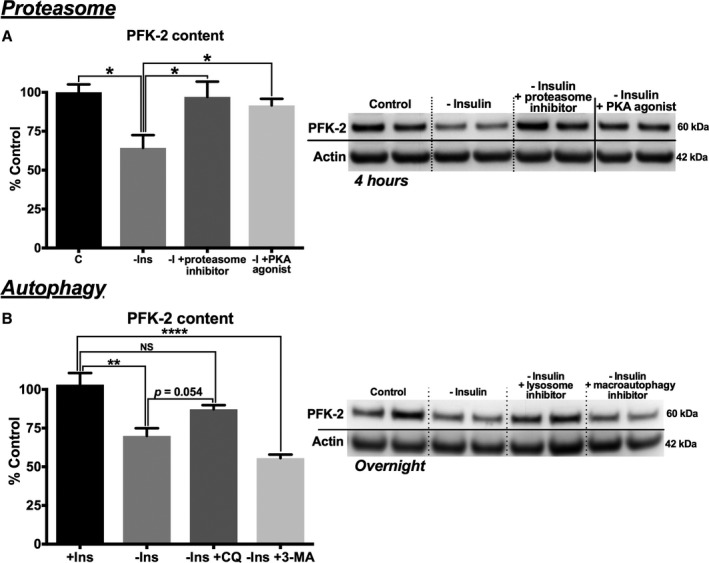
Phosphofructokinase 2 (PFK‐2) degradation is mediated by both proteasomal and lysosomal mechanisms. A, Control adult cardiomyocytes were cultured under control conditions with insulin (C) or in the absence of insulin and the proteasome inhibitor (MG132) or a protein kinase A (PKA) agonist (8‐bromo‐cAMP), as indicated for 4 hours (left, quantification; and right, representative blots). B, Control adult cardiomyocytes were cultured under C or in the absence of insulin and the lysosomal inhibitor (chloroquine) or macroautophagy inhibitor (3‐methyladenine), as indicated overnight (left, quantification; and right, representative blots). Data are presented as mean±SEM (P=0.202). NS indicates not significant. *P<0.05, number of stars specifies degree of significance P≤0.0001 by ANOVA with the Tukey post hoc test (A [n=4] and B [n=5]).
We next tested whether other proteostasis mechanisms also regulate PFK‐2 levels. As shown in Figure 5B, the lysosomal inhibitor, chloroquine, largely prevented the loss of PFK‐2 in cardiomyocytes cultured in the absence of insulin for 24 hours. However, chloroquine had no effect on PFK‐2 levels in cells cultured under similar conditions for 4 hours (data not shown). This suggests proteasomal degradation may mediate PFK‐2 degradation in the short‐term, whereas a slower lysosomal‐mediated process may contribute to repressed levels in the long‐term. Macroautophagy is one mechanism whereby proteins are targeted to lysosomes for degradation. However, the nonspecific macroautophagy inhibitor, 3‐methyladenine, was unable to prevent PFK‐2 degradation (Figure 5B). This suggests that lysosomal‐mediated degradation of PFK‐2 occurs but is independent of macroautophagy.
An alternative mechanism of lysosomal degradation is mediated by chaperone‐mediated autophagy (CMA), whereby targeted proteins bind hsc70, a member of the heat shock protein 70 family, and are shuttled to the lysosomes. PFK‐2 has a canonical CMA motif (Dr Ann Maria Cuervo, written communication, May 2016; Figure 6A) that can potentially bind hsc70. It has been recently shown that CMA is regulated by lysosomal‐targeted Akt1 activity in a mammalian target of rapamycin complex (mTORC) 2– and PH domain leucine‐rich repeat protein phosphatase (PHLPP1)–dependent manner (Figure 6B).27, 28 Specifically, mTORC2 phosphorylates and activates Akt1, thereby decreasing CMA. In contrast, PHLPP1 is recruited to lysosomes to dephosphorylate Akt1 and promote CMA. To determine if CMA contributes to PFK‐2 degradation, control adult cardiomyocytes were cultured in the presence of insulin and the mTORC1/2 inhibitor, AZD8055. As shown in Figure 6C, AZD8055 decreased PFK‐2 content by 65%. Likewise, cells cultured with insulin and an Akt inhibitor also had a significant loss of PFK‐2 (40% decrease; Figure 6D). Reciprocally, addition of a selective inhibitor against the catalytic site of PHLPP1 (National Service Center identification 117079)29 prevented the decrease in PFK‐2 in cells cultured in the absence of insulin (Figure 6E). Finally, the effects of the PHLPP1 inhibitor and chloroquine were not additive (Figure 6E), suggesting that PHLPP1 inhibition itself was preventing PFK‐2 degradation in a lysosomal‐dependent manner. In conclusion, these results support that PFK‐2 is degraded in an Akt1‐dependent manner, which is consistent with a CMA‐mediated mechanism.
Figure 6.
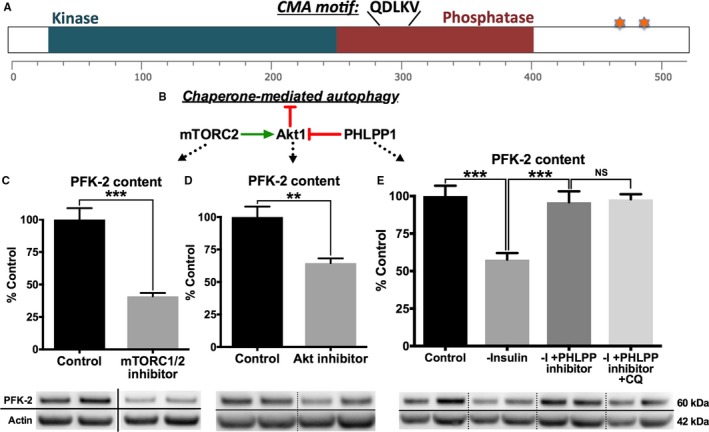
Phosphofructokinase 2 (PFK‐2) is degraded by chaperone‐mediated autophagy (CMA). A, A schematic of PFK‐2 indicating a putative CMA consensus sequence, catalytic cores, and known phosphorylation sites (starred). B, Lysosomal localized protein kinase B (Akt) 1 is phosphorylated and dephosphorylated by mammalian target of rapamycin complex (mTORC) 2 and PH domain leucine‐rich repeat protein phosphatase (PHLPP) 1, respectively.28 Phosphorylated Akt1 prevents CMA. C, Control adult cardiomyocytes were cultured overnight with insulin and with or without 100 nmol/L AZD8055 (an mTORC1/2 inhibitor). D, Control adult cardiomyocytes were cultured overnight with insulin and with or without an Akt inhibitor. E, Control adult cardiomyocytes were cultured overnight in the absence of insulin (control) and with or without a PHLPP1 inhibitor and chloroquine. C through E, Quantification of Western blots (top) and representative blots (bottom). Data are presented as mean±SEM. **P<0.005, ***P<0.0005 by unpaired Student t test (C and D [n=4]) or by ANOVA with the Tukey post hoc test (n=5) in E.
Diabetes Mellitus Is Accompanied by Deficient Macroautophagy With Enhanced CMA
In the heart, both macroautophagy and CMA are elevated by fasting.30 In contrast, macroautophagy is inhibited with diabetes mellitus,30, 31 whereas changes in CMA activity have not been well explored. We, therefore, examined both general autophagy and CMA markers to determine the relative activity of each pathway with diabetes mellitus.
S6K is a downstream substrate of mTORC1.32 Thus, an increase in S6K phosphorylation is indicative of increased mTORC1 activity and decreased autophagy. As shown in Figure 7A, phosphorylation of S6K at the mTORC1 site, Thr389, is significantly elevated in diabetic mice. In contrast, the downstream substrate of mTORC2, Ser473 of Akt, is similarly phosphorylated in control and diabetic mice. This suggests mTORC1 and mTORC2 activities are differentially affected by diabetes mellitus.
Figure 7.
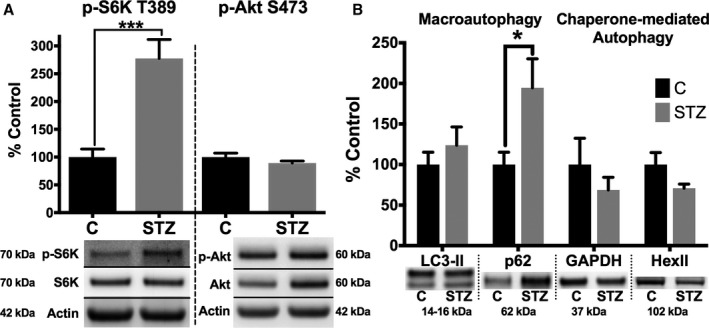
Macroautophagy is impaired, and selective autophagy is overactive, in the hearts of diabetic mice. A, Cardiac homogenates from control and diabetic (4 months after streptozotocin treatment) mice were analyzed by Western blot analysis for phosphorylated S6 kinase (p‐S6K) Thr389 (a substrate of mammalian target of rapamycin complex [mTORC] 1) and phosphorylated protein kinase B (p‐Akt) Ser473 (a substrate of mTORC2). B, LC3‐II and p62, markers of macroautophagy (left), and GAPDH and hexokinase II (HexII), known targets of chaperone‐mediated autophagy) (right), were analyzed by Western blot analysis. Densitometry from Western blots of cardiac homogenates was standardized to actin. Representative blots are shown below quantification. Data are presented as mean±SEM. *P<0.05, ***P<0.001 by unpaired Student t test (n=5 for all groups).
Further differences between macroautophagy and CMA were next explored. LC3‐II, the ethanolamine conjugated form of LC3‐I that is recruited to autophagosomes, showed no significant change in diabetic mice and is, thus, inconclusive. However, p62 (sequestosome‐1), which accumulates when autophagy is inhibited, was significantly elevated in diabetic compared with control mice (Figure 7B). This is consistent with previous reports that autophagy is decreased in type 1 diabetic models.30, 31 In contrast, 2 established CMA targets, GAPDH and hexokinase II,33, 34 trended towards a decrease in diabetic mice. This suggests that CMA may be overactive in diabetes mellitus. The significant decrease in PFK‐2, compared with established CMA targets, may be distinctive because of its shorter half‐life and alternative protein degradation via the ubiquitin proteasome system. In summary, our results support that CMA, unlike macroautophagy, is sustained in the hearts of diabetic mice.
Discussion
The heart is adapted to deriving energy from available nutrients, and metabolic flexibility is essential for proper cardiac function. This ensures that regardless of the body's nutrient status, ATP levels are strictly sustained at steady‐state levels.35, 36 Under the fed state, insulin signaling promotes glucose uptake and glycolysis is increased by phosphorylation of PFK‐2 through Akt and/or PKA signaling (Figure 8). In concert, pyruvate dehydrogenase activity is increased to promote glucose oxidation and prevent lactate production. However, under the fasted state, the increased reliance on fatty acid oxidation and lack of insulin signaling cause a decrease in glucose uptake, PFK‐2 content, and PFK‐1 activity. Decreased glycolysis, coupled with inhibition of pyruvate dehydrogenase, ensures that any lactate produced is returned to the circulation for hepatic glucose production; collectively, these processes contribute to a glucose‐sparing effect. However, despite being sensitive to insulin‐mediated glucose uptake, our metabolomics study revealed that glycolytic intermediates upstream of PFK‐1 are surprisingly elevated in the fasted state. This suggests that insulin‐independent glucose uptake can persist and that the PFK‐1/PFK‐2 nexus is not just a redundant means of decreasing cardiac glucose use, but is a primary means of rerouting glycolytic intermediates to alternative pathways under the fasted state. Furthermore, under diabetic conditions, when PFK‐2 content is decreased in the long‐term, hyperglycemia may overwhelm this endogenous rerouting of intermediates, causing pathological elevation of alternative glucose metabolic pathways.
Figure 8.
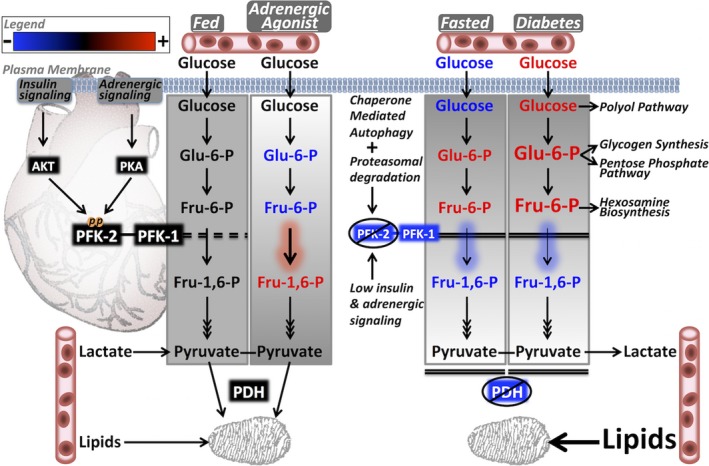
Model showing how phosphofructokinase 2 (PFK‐2) regulation dynamically affects cardiac metabolism in the fed, adrenergically stimulated, fasted, and diabetic states. The fed state can regulate glycolytic flux dynamically on the basis of substrate availability. The normal mild elevation in early glycolytic intermediates (EGIs) during the fed state is abolished by β‐adrenergic activation of PFK‐2, allowing use of the intermediates for energy utilization. In fasting, despite low blood glucose, there is a paradoxical increase in EGIs because of the strong inhibition of PFK‐2 and phosphofructokinase 1 (PFK‐1). In diabetes mellitus, the combination of high circulating glucose levels and diminished PFK‐2/PFK‐1 activities in the long‐term may lead to a highly pathological accumulation of EGIs that promotes overactivation of branching metabolic pathways. AKT indicates protein kinase B; Fru‐1,6‐P, fructose 1,6‐bisphosphate; Fru‐6‐P, fructose 6‐phosphate; Glu‐6‐P, glucose 6‐phosphate; PDH, pyruvate dehydrogenase; and PKA, protein kinase A.
Our results support that the increase in EGIs in the fasted heart is facilitated by the decrease in PFK‐2 protein content. For example, PFK‐1 protein levels and metabolic feedback inhibitors of PFK‐1, such as citrate and adenosine phosphates, are unchanged with fasting. Thus, the decrease in PFK‐1 activity is likely mediated by decreased content of fructose‐2,6‐bisphosphate. Unfortunately, fructose‐2,6‐bisphosphate is not detected by metabolomic analysis because of its low concentration, instability, and the unavailability of metabolic standards that would allow refinement of current analytical approaches. Nevertheless, the critical role of PFK‐2 in modulating EGIs is reciprocally supported by the metabolic analysis of hearts after isoproterenol stimulation. Under these conditions, activation of PFK‐2 results in a 90% decrease in EGIs (Figure 3C). In addition, Gibb et al have shown, through metabolic tracer studies in cardiomyocytes, that a decrease in PFK‐1 activity increases upstream intermediates and flux through pentose phosphate pathway and hexosamine biosynthetic pathway.13 Our results are also consistent with work by Donthi et al, who demonstrated that the cardiac expression of a kinase‐deficient PFK‐2 promotes an increase in glycolytic intermediates upstream of PFK‐1.37 Over time, this mouse model also develops fibrosis and reduced cardiomyocyte function. Thus, the long‐term decrease in PFK‐2 we observe in diabetic mice may likewise be a driver of diabetic cardiomyopathy.
The increase in EGIs with fasting may be part of a metabolic response program that reroutes glucose from glycolysis to other intracellular metabolic pathways. This includes an increase in flux through the pentose phosphate pathway and the production of glycogen. Indeed, nicotinamide‐adenine dinucleotide phosphate, reduced form, produced by the pentose phosphate pathway, can increase antioxidant capacity, and this may be a necessary adaptation that counters the increase in reactive oxygen species production that occurs with fatty acid oxidation.38, 39 However, prolonged rerouting of glucose to alternative pathways, especially when circulating glucose levels are elevated, may be detrimental for cardiac health. For example, glycogen content is counterintuitively elevated during fasting40; during the long‐term, hyperglycemia leads to overt glycogen accumulation, a process associated with cardiomyocyte stress and cardiac pathological features.41, 42 Future experiments will test the hypothesis that sustaining PFK‐2 content and activity may be a means of mitigating long‐term rerouting of EGIs to alternative pathways. Experimentally, and potentially clinically, this may be mediated by administration of metformin, which diminishes diabetic cardiomyopathy in a chronic type 1 diabetic model via activation of AMP kinase.43 A potential, and unexamined, benefit of sustained activation of AMP kinase may be by the phosphorylation and stabilization of PFK‐2.44 Alternatively, targeting the concerted actions of PFK‐2 and PFK‐1 more specifically may be a way to potently reverse diabetic metabolic changes.
This work is the first to demonstrate PFK‐2 content is rapidly degraded in response to decreased insulin signaling. This is true with type 1 diabetes mellitus, high‐fat feeding, and fasting. The development of the PFK‐2 SRM assay occurred late in the study and was used to provide additional confirmation of decreased protein independently of antibody‐based assays. Because 2 different methods were used to measure PFK‐2 content in 1‐ and 20‐week HFD fed animals, we cannot definitively conclude that the loss of PFK‐2 does not progress longitudinally. Future studies using the SRM assay, which is much more quantitative, will address this question. The 2.2‐hour half‐life of PFK‐2, induced by a PI3K inhibitor in the presence of cycloheximide, is unusual for an autophagic mechanism. Proteins turned over by CMA generally have a half‐life >6 hours.45 The potency by which PFK‐2 is preferentially degraded is further illustrated by comparing its content with that of other known CMA targets, GAPDH and hexokinase II (Figure 7B). This accelerated rate of degradation of PFK‐2, relative to these other targets, may be because of its susceptibility to both proteasomal‐ and lysosomal‐mediated degradation (Figures 5, 6 through 7).
The regulation of PFK‐2 likely evolved to support fasting metabolism. In this context, inactivation of PFK‐2 by lipids and low insulin signaling contributes to reduced glucose use by the heart, thus sparing it for the brain.46, 47 The primary means of regulating PFK‐2 activity, which occurs with moment‐to‐moment changes in energy demands, is via phosphorylation. However, we demonstrate herein that a decrease in PFK‐2 content offers another layer of metabolic regulation. By reducing the content of this protein, alternative pathways that activate PFK‐2 via phosphorylation would have less of an effect on glycolytic flux under fasting conditions. For example, β‐adrenergic activation preferentially increases glucose metabolism48 via PKA‐mediated phosphorylation of PFK‐2 and a concurrent increase in the calcium‐mediated activation of pyruvate dehydrogenase in the mitochondria.16, 49 Thus, the decrease in PFK‐2 content would ensure that periods of high sympathetic activity and fasting do not cause dangerously low blood glucose levels. However, the long‐term decrease in PFK‐2 observed herein with diabetes mellitus could be a contributor to the metabolic inflexibility that contributes to diabetic cardiomyopathy.
Sources of Funding
This work was supported by National Institutes of Health (NIH) grant R01HL125625 (Humphries), from the National Heart, Lung, and Blood Institute; by Institutional Development Award P20GM104934 (Humphries), from the National Institute of General Medical Sciences; and by NIH P30AG050911 (Kinter). The TSQ Quantiva was purchased with funding from NIH P20GM103447.
Disclosures
None.
Acknowledgments
We thank Ann Louise Olson (University of Oklahoma Health Sciences Center) for a critical review of the manuscript and Timothy Griffin (Oklahoma Medical Research Foundation [OMRF]) and Albert Batushansky (OMRF) for assistance with statistical analysis.
(J Am Heart Assoc. 2017;6:e007159 DOI: 10.1161/JAHA.117.007159.)29203581
References
- 1. Nagoshi T, Yoshimura M, Rosano GM, Lopaschuk GD, Mochizuki S. Optimization of cardiac metabolism in heart failure. Curr Pharm Des. 2011;17:3846–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bauters C, Lamblin N, Mc Fadden EP, Van Belle E, Millaire A, de Groote P. Influence of diabetes mellitus on heart failure risk and outcome. Cardiovasc Diabetol. 2003;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aneja A, Tang WH, Bansilal S, Garcia MJ, Farkouh ME. Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am J Med. 2008;121:748–757. [DOI] [PubMed] [Google Scholar]
- 4. Abel ED. Glucose transport in the heart. Front Biosci. 2004;9:201–215. [DOI] [PubMed] [Google Scholar]
- 5. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fukushima A, Lopaschuk GD. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim Biophys Acta. 2016;1861:1525–1534. [DOI] [PubMed] [Google Scholar]
- 7. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57:660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bockus LB, Humphries KM. cAMP‐dependent protein kinase (PKA) signaling is impaired in the diabetic heart. J Biol Chem. 2015;290:29250–29258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu P, Sato J, Zhao Y, Jaskiewicz J, Popov KM, Harris RA. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem J. 1998;329(pt 1):197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hong EG, Jung DY, Ko HJ, Zhang Z, Ma Z, Jun JY, Kim JH, Sumner AD, Vary TC, Gardner TW, Bronson SK, Kim JK. Nonobese, insulin‐deficient Ins2Akita mice develop type 2 diabetes phenotypes including insulin resistance and cardiac remodeling. Am J Physiol Endocrinol Metab. 2007;293:E1687–E1696. [DOI] [PubMed] [Google Scholar]
- 11. Hall JL, Stanley WC, Lopaschuk GD, Wisneski JA, Pizzurro RD, Hamilton CD, McCormack JG. Impaired pyruvate oxidation but normal glucose uptake in diabetic pig heart during dobutamine‐induced work. Am J Physiol. 1996;271:H2320–H2329. [DOI] [PubMed] [Google Scholar]
- 12. Depre C, Vanoverschelde JL, Taegtmeyer H. Glucose for the heart. Circulation. 1999;99:578–588. [DOI] [PubMed] [Google Scholar]
- 13. Gibb AA, Lorkiewicz PK, Zheng YT, Zhang X, Bhatnagar A, Jones SP, Hill BG. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem J. 2017;474:2785–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6‐Phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase: head‐to‐head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004;381:561–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ros S, Schulze A. Balancing glycolytic flux: the role of 6‐phosphofructo‐2‐kinase/fructose 2,6‐bisphosphatases in cancer metabolism. Cancer Metab. 2013;1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Depre C, Ponchaut S, Deprez J, Maisin L, Hue L. Cyclic AMP suppresses the inhibition of glycolysis by alternative oxidizable substrates in the heart. J Clin Invest. 1998;101:390–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Overmyer KA, Thonusin C, Qi NR, Burant CF, Evans CR. Impact of anesthesia and euthanasia on metabolomics of mammalian tissues: studies in a C57BL/6J mouse model. PLoS One. 2015;10:e0117232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ludwig C, Claassen M, Schmidt A, Aebersold R. Estimation of absolute protein quantities of unlabeled samples by selected reaction monitoring mass spectrometry. Mol Cell Proteomics. 2012;11:M111.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kinter CS, Lundie JM, Patel H, Rindler PM, Szweda LI, Kinter M. A quantitative proteomic profile of the Nrf2‐mediated antioxidant response of macrophages to oxidized LDL determined by multiplexed selected reaction monitoring. PLoS One. 2012;7:e50016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kinter CS, Kinter M. Application of Selected Reaction Monitoring to Highly Multiplexed Targeted Quantitative Proteomics: A Replacement for Western Blot Analysis. New York, NY: Springer‐Verlag; 2013. [Google Scholar]
- 21. Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Hu Z, Deberardinis RJ, Schiattarella G, Hill JA, Oz O, Lu Z, Zhang CC, Kimura W, Sadek HA. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222–227. [DOI] [PubMed] [Google Scholar]
- 22. Vadvalkar SS, Matsuzaki S, Eyster CA, Giorgione JR, Bockus LB, Kinter CS, Kinter M, Humphries KM. Decreased mitochondrial pyruvate transport activity in the diabetic heart: role of mitochondrial pyruvate carrier 2 (MPC2) acetylation. J Biol Chem. 2017;292:4423–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–296. [DOI] [PubMed] [Google Scholar]
- 24. Fernandes J, Weddle A, Kinter CS, Humphries KM, Mather T, Szweda LI, Kinter M. Lysine acetylation activates mitochondrial aconitase in the heart. Biochemistry. 2015;54:4008–4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rindler PM, Plafker SM, Szweda LI, Kinter M. High dietary fat selectively increases catalase expression within cardiac mitochondria. J Biol Chem. 2013;288:1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Kahn CR, Abel ED. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002;109:629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arias E. Lysosomal mTORC2/PHLPP1/Akt axis: a new point of control of chaperone‐mediated autophagy. Oncotarget. 2015;6:35147–35148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone‐mediated autophagy. Mol Cell. 2015;59:270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sierecki E, Sinko W, McCammon JA, Newton AC. Discovery of small molecule inhibitors of the PH domain leucine‐rich repeat protein phosphatase (PHLPP) by chemical and virtual screening. J Med Chem. 2010;53:6899–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riehle C, Abel ED. Insulin regulation of myocardial autophagy. Circ J. 2014;78:2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Delbridge LM, Mellor KM, Taylor DJ, Gottlieb RA. Myocardial autophagic energy stress responses: macroautophagy, mitophagy, and glycophagy. Am J Physiol Heart Circ Physiol. 2015;308:H1194–H1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saitoh M, Pullen N, Brennan P, Cantrell D, Dennis PB, Thomas G. Regulation of an activated S6 kinase 1 variant reveals a novel mammalian target of rapamycin phosphorylation site. J Biol Chem. 2002;277:20104–20112. [DOI] [PubMed] [Google Scholar]
- 33. Xia HG, Najafov A, Geng J, Galan‐Acosta L, Han X, Guo Y, Shan B, Zhang Y, Norberg E, Zhang T, Pan L, Liu J, Coloff JL, Ofengeim D, Zhu H, Wu K, Cai Y, Yates JR, Zhu Z, Yuan J, Vakifahmetoglu‐Norberg H. Degradation of HK2 by chaperone‐mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol. 2015;210:705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aniento F, Roche E, Cuervo AM, Knecht E. Uptake and degradation of glyceraldehyde‐3‐phosphate dehydrogenase by rat liver lysosomes. J Biol Chem. 1993;268:10463–10470. [PubMed] [Google Scholar]
- 35. Katz LA, Swain JA, Portman MA, Balaban RS. Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am J Physiol. 1989;256:H265–H274. [DOI] [PubMed] [Google Scholar]
- 36. Yaniv Y, Juhaszova M, Nuss HB, Wang S, Zorov DB, Lakatta EG, Sollott SJ. Matching ATP supply and demand in mammalian heart: in vivo, in vitro, and in silico perspectives. Ann N Y Acad Sci. 2010;1188:133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Donthi RV, Ye G, Wu C, McClain DA, Lange AJ, Epstein PN. Cardiac expression of kinase‐deficient 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J Biol Chem. 2004;279:48085–48090. [DOI] [PubMed] [Google Scholar]
- 38. Rindler PM, Crewe CL, Fernandes J, Kinter M, Szweda LI. Redox regulation of insulin sensitivity due to enhanced fatty acid utilization in the mitochondria. Am J Physiol Heart Circ Physiol. 2013;305:H634–H643. [DOI] [PubMed] [Google Scholar]
- 39. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. [DOI] [PubMed] [Google Scholar]
- 40. Evans G. The glycogen content of the rat heart. J Physiol. 1934;82:468–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mellor KM, Varma U, Stapleton DI, Delbridge LM. Cardiomyocyte glycophagy is regulated by insulin and exposure to high extracellular glucose. Am J Physiol Heart Circ Physiol. 2014;306:H1240–H1245. [DOI] [PubMed] [Google Scholar]
- 42. Shearer J, Ross KD, Hughey CC, Johnsen VL, Hittel DS, Severson DL. Exercise training does not correct abnormal cardiac glycogen accumulation in the db/db mouse model of type 2 diabetes. Am J Physiol Endocrinol Metab. 2011;301:E31–E39. [DOI] [PubMed] [Google Scholar]
- 43. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK‐2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. [DOI] [PubMed] [Google Scholar]
- 45. Patel B, Cuervo AM. Methods to study chaperone‐mediated autophagy. Methods. 2015;75:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty‐acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. [DOI] [PubMed] [Google Scholar]
- 47. Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. 2009;297:E578–E591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Collins‐Nakai RL, Noseworthy D, Lopaschuk GD. Epinephrine increases ATP production in hearts by preferentially increasing glucose metabolism. Am J Physiol. 1994;267:H1862–H1871. [DOI] [PubMed] [Google Scholar]
- 49. McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. [DOI] [PubMed] [Google Scholar]