

Abstract
Background
The prevalence of atrial fibrillation (AF) is significantly higher in rheumatoid arthritis (RA) patients, but the underlying mechanisms remain poorly understood. The goal of this study was to assess the effects of RA on AF susceptibility and atrial arrhythmogenic remodeling in a rat model of RA.
Methods and Results
Collagen‐induced arthritis was induced in rats by immunization with type II collagen in Freund's incomplete adjuvant. Among the rats that developed arthritis, AF susceptibility and atrial remodeling were examined 8 weeks after the primary immunization. AF inducibility and duration were substantially increased in collagen‐induced arthritis rats, and AF duration was significantly and positively correlated with the serum IL‐6 and TNF‐α levels. Rats with collagen‐induced arthritis showed prolonged atrial conduction time with no changes in the atrial effective refractory period. Atrial conduction delay was accompanied by significantly increased atrial fibrosis. In addition, atrial structural and autonomic remodeling, including left atrial dilation, apoptosis and autophagy of atrial myocytes, and atrial heterogeneous sympathetic hyperinnervation, was observed. Interestingly, we found that collagen‐induced arthritis had no significant effects on connexins, Nav1.5, and the main ion channels' protein expressions in atria.
Conclusions
We demonstrated that RA increased AF susceptibility by inducing AF‐promoting atrial remodeling. This study may provide insights into mechanisms underlying RA‐induced AF and validate a model that is suitable for further mechanistic and therapeutic exploration.
Keywords: atrial fibrillation, atrial remodeling, autonomic remodeling, fibrosis, rheumatoid arthritis
Subject Categories: Atrial Fibrillation, Electrophysiology
Clinical Perspective
What Is New?
The present study demonstrated that rheumatoid arthritis increased susceptibility to atrial fibrillation by inducing atrial fibrillation–promoting atrial remodeling, including significant atrial fibrosis, apoptosis, autophagy, heterogeneous sympathetic hyperinnervation, and atrial conduction delay in a rat model of collagen‐induced arthritis.
What Are the Clinical Implications?
This study may provide insights into the mechanisms underlying clinical rheumatoid arthritis–induced atrial fibrillation and validate a model that is suitable for further therapeutic exploration.
Introduction
Rheumatoid arthritis (RA) is a chronic immunomediated disease primarily affecting the joints and leading to extra‐articular manifestations, characterized by persistent high‐grade systemic inflammation. RA affects 0.5% to 1% of adults in developed countries and causes significant disability and preterm mortality.1 The morbidity of cardiovascular disease such as ischemic heart disease and congestive heart failure are significantly increased in RA patients.1, 2 Atrial fibrillation (AF) is the most common clinically relevant arrhythmia, with a wide range of potential complications, and contributes significantly to population morbidity and mortality.3 Large population‐based studies indicate that the prevalence of AF is significantly higher in RA patients than in the general population.4, 5, 6 The meta‐analysis of the pooled data from these studies demonstrated ≈30% statistically significant increased risk of AF in RA patients compared with participants without RA.7 However, the effects of RA on AF vulnerability and atrial remodeling have not been studied in experimental models.
Collagen‐induced arthritis (CIA) is a classic animal model of RA and is widely used to study the pathophysiology of human RA.8 To clarify the exact role and the underlying mechanism of RA in AF, we assessed the effects of RA on AF susceptibility and atrial arrhythmogenic remodeling in a CIA rat model.
Materials and Methods
The data, analytic methods, and study materials have been made available to other researchers for purposes of reproducing the results or replicating the procedure. The authors declare that all supporting data are available within the article.
Induction of CIA in Rats
Previous study suggested that female Wistar rats were greatly susceptible to CIA and had low variability in clinical signs.9 Therefore, the CIA model was induced in female Wistar rats in this study.
Female Wistar rats (weighing 180–200 g and aged 8 weeks) from Beijing Vital River Laboratory Animal Technology Co, Ltd (Beijing, China) were used. The present study was performed in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the institutional animal care and use committee at Harbin Medical University. The CIA model was induced as described previously.10 In brief, 30 rats were immunized twice, intradermally at the tail base with 1:1 (vol/vol) 200 μg bovine type II collagen and Freund's incomplete adjuvant at a volume of 200 μL with a 7‐day interval. The control group rats (n=20) were injected with 0.2 mL of sterilized physiological saline only. Arthritic signs in the paws of the rats, characterized by edema and erythema, were inspected daily following the second immunization, and the onset of arthritis was monitored. Among the 25 rats that developed arthritis, 20 were randomly selected for the subsequent experiment (the CIA group). At 8 weeks after the primary bovine type II collagen injection, echocardiography and electrophysiological studies were performed, and atrial tissues were sampled.
Echocardiography
Transthoracic echocardiographic studies were performed with the Philips S12‐4 phased‐sector ultrasound transducer in Philips CX50 ultrasound systems, with rats under sedation with 2% isoflurane. Cross‐sectional echocardiogram was obtained in apical 4‐chamber view to measure left atrial diameter, right atrial (RA) up–down and left–right diameter at end diastole. M‐mode echocardiogram was obtained in parasternal long‐axis views to measure right ventricular diameter at end diastole and in parasternal short‐axis views to measure left ventricular diameter at end diastole and systole. Left ventricular ejection fraction was recorded. Peak velocity of E‐wave in transmitral flow was also measured by pulsed‐wave Doppler. The average of 3 consecutive cardiac cycles was used for each measurement.
Electrophysiological Study
Ten rats in each group were randomly selected to undergo in vivo open‐chest electrophysiological programmed stimulation under 2% isoflurane anesthesia. The 1.9‐F octapolar catheter used for programmed stimulation was placed on the right atrium. To assess AF inducibility, 25‐Hz burst pacing was applied for 30 seconds. AF was defined as >1 second of irregular atrial electrograms (>800 beats/min) with irregular ventricular response. The RA effective refractory period was measured with 8‐beat basic pacing‐stimulus trains (cycle length: 120 milliseconds) followed by a premature extrastimulus with the coupling interval reduced in 2‐millisecond decrements and was defined as the longest coupling interval failing to produce a propagated response. In addition, intra‐atrium conduction time was measured during pacing at a cycle length of 120 milliseconds and defined as the stimulus conduction time between electrodes from the distal dipole (the first dipole) to the third dipole (the distance between the 2 dipoles is 2 mm).
Quantitation of Serum Cytokines
After electrophysiological study, the serum was collected, and concentrations of IL‐6 (interleukin 6) and TNF‐α (tumor necrosis factor‐α) were measured using ELISA kits according to the manufacturer's instructions (BlueGene Biotech Co Ltd, Shanghai, China).
Hematoxylin and Eosin and Masson's Trichrome Staining
RA tissues were stained with hematoxylin and eosin or Masson's trichrome. The interstitial fibrotic areas were calculated using Image‐Pro Plus software (version 4.0; Media Cybernetics LP). Collagen volume fraction was calculated as collagen area/total area×100%.
Terminal Deoxynucleotidyl Transferase Mediated dUTP Nick‐End Labeling Staining
TUNEL (Terminal Deoxynucleotidyl Transferase Mediated dUTP Nick‐End Labeling) staining was performed using the ApopTag kit (Roche) according to the manufacturer's instructions. The apoptosis content was calculated as apoptotic cells per field.
Immunohistochemistrical Studies
The nerve sprouting marker GAP43 (growth associated protein 43) and the sympathetic nerve marker TH (tyrosine hydroxylase) were stained on 5‐μm transmural sections of atrial tissues. The sections were incubated overnight at 4°C with primary antibody and with peroxidase‐conjugated second antibody at 37°C for 20 minutes. Then the sections were visualized with a DAB (3,3′‐diaminobenzidine)–based colorimetric method. The density of stained nerves was determined by Image‐Pro Plus software. The nerve density was expressed as the nerve area divided by the total area examined (μm2/mm2).
Transmission Electron Microscopy
Briefly, atrial samples were fixed with 4% glutaraldehyde, followed by 1% osmium tetroxide after fixation. After dehydration in a graded series of ethanol, the samples were embedded in Epon‐Araldite resin. Ultrathin sections with 50‐nm thickness were prepared and then stained with uranyl acetate and lead citrate. A JEM‐1220 transmission electron microscope (JEOL) was used to observe autophagosomes.
Western Blotting
Proteins were separated by electrophoresis on 8% to 12% SDS‐polyacrylamide gels and transferred moist to polyvinylidene difluoride membranes. Membranes were blocked by 5% nonfat dry milk in PBS and incubated overnight at 4°C. Membranes were washed with PBS containing 0.5% Tween 20 and incubated with primary antibodies overnight, then incubated with horseradish peroxidase–conjugated secondary antibody for 1 hour. Antibodies against IL‐6, TNF‐α, collagen I, collagen III, Kir2.1, Kir3.1, Kir3.4, Kv1.5, Nav1.5, Cx40 (connexin 40), and Cx43 (connexin 43) were purchased from Biosynthesis Biotechnology Co, Ltd. Antibodies against α‐SMA (α–smooth muscle actin), Bcl‐2, Bax, CASP‐3 (caspase 3), GAP43, TH, NGF (nerve growth factor), and Cav1.2 were obtained from Abcam. Antibodies against LC3B and ATG5 were purchased from Cell Signaling Technology. Antibody against TGF‐β1 (transforming growth factor‐β1) was provided by Sigma‐Aldrich. The images were captured on the Bio‐Rad system, and GAPDH was used as an internal control.
Statistical Analyses
Data are expressed as mean±SD except for AF duration, which is expressed as median and interquartile range (25–75%). The Fisher exact test was applied to compare AF inducibility. AF duration was compared using the Wilcoxon rank‐sum test. The paired t test was used in the comparison of the serum IL‐6 and TNF‐α levels before and after bovine type II collagen injection within each group. Other data were compared using an unpaired Student t test. Natural logarithmic transformation was performed if the data did not satisfy statistical criteria for normal distribution, and the data were presented on the original scale. Pearson product‐moment correlation analysis was used to determine the relationship between the levels of serum IL‐6 and TNF‐α and AF duration. Analyses were processed with GraphPad Prism version 5.0 (GraphPad Software, Inc). P<0.05 was considered statistically significant.
Results
Systemic and Atrial Inflammation Induced by CIA
At 8 weeks after the primary bovine type II collagen injection, the serum levels of IL‐6 and TNF‐α in the CIA group were significantly higher compared with the control group and baseline (Figure 1A and 1B). Moreover, IL‐6 and TNF‐α protein expressions in atrial tissues of the CIA group were significantly higher than those of control group (Figure 1C and 1D). In addition, hematoxylin and eosin staining showed accumulations of inflammatory cells in the atrial myocardium in the CIA group (Figure 1E and 1F). The results showed that CIA not only induced systemic inflammation but also caused local inflammatory response within the atrial myocardium.
Figure 1.
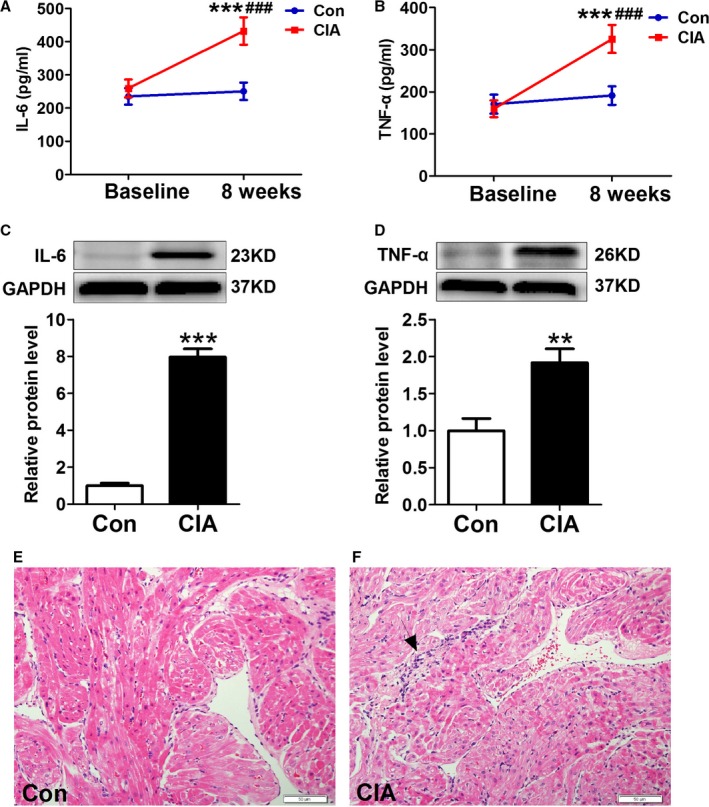
CIA‐induced systemic and atrial inflammation. A and B, Serum levels of IL‐6 (interleukin 6) and TNF‐α (tumor necrosis factor α) 8 weeks after primary collagen II and Freund's incomplete adjuvant injection (n=10 per group). C and D, Representative Western blots and quantification of IL‐6 and TNF‐α protein expression in atrial tissues (n=6 per group). E and F, Examples of hematoxylin and eosin staining (n=6 per group). **P<0.01, ***P<0.001, vs the CON group; ### P<0.001, vs baseline. Scale bar=50 μm. Arrow indicates the accumulation of inflammatory cells. CIA indicates collagen‐induced arthritis; CON, control.
Changes in AF Inducibility and Electrophysiological Parameters
Figure 2 showed changes in AF inducibility and main electrophysiological parameters. Burst pacing rarely induced AF in control rats (Figure 2A), whereas it commonly induced AF in CIA rats (Figure 2B). AF inducibility was substantially increased in CIA rats, with 9 of 10 CIA rats (90%) becoming inducible versus 2 of 10 control rats (20%; Figure 2C). AF duration was also significantly increased in CIA rats (Figure 2D). There were no significant differences in RA effective refractory period between the 2 groups (Figure 2E); however, atrial conduction time was significantly longer in CIA group (Figure 2F). Moreover, AF duration was significantly and positively correlated with the serum IL‐6 (r=0.82, P<0.01; Figure 2G) and TNF‐α levels (r=0.84, P<0.01; Figure 2H).
Figure 2.
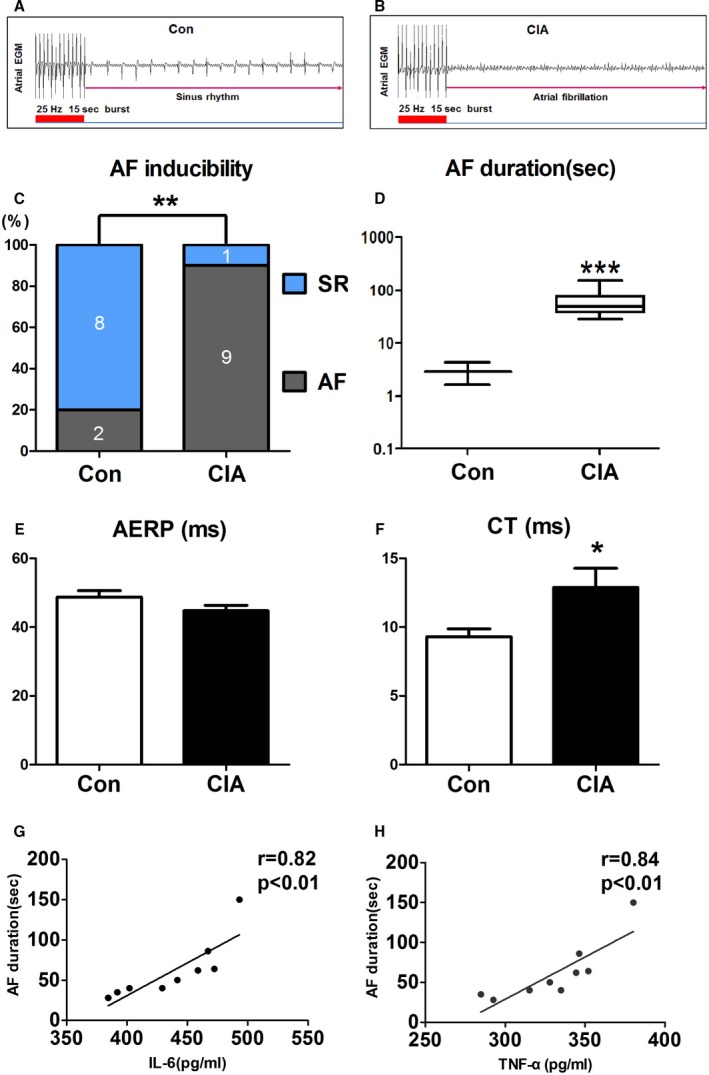
AF susceptibility and electrophysiological parameters changes. A and B, Examples of AF induction attempts in (A) a control rat and (B) a CIA rat. C, AF inducibility. D, AF duration. E, AERP. F, Intra‐atrium CT. G and H, AF duration was significantly and positively correlated with the serum levels of IL‐6 (interleukin 6; r=0.82, P<0.01) and TNF‐α (tumor necrosis factor α; r=0.84, P<0.01). *P<0.05, **P<0.01, ***P<0.001, vs the CON group, n=10 per group. AERP indicates atrial effective refractory period; AF, atrial fibrillation; CIA, collagen‐induced arthritis; CON, control; CT, conduction time; EGM, electrogram; SR, sinus rhythm.
Atrial Structural Remodeling Produced by CIA
To understand the mechanisms underlying CIA‐induced AF promotion, we analyzed cardiac structural changes. As shown in Figure 3, echocardiography revealed mild but statistically significant increases in left atrial diameter but no differences in RA diameter. Left ventricular diameter, right ventricular diameter, and left ventricular systolic function (as indicated by left ventricular ejection fraction) did not change; however, the peak velocity of E‐wave decreased, indicating left ventricular diastolic dysfunction. Moreover, atrial fibrosis, apoptosis, and autophagy were examined. Masson's trichrome staining showed greater atrial collagen deposition (Figure 4A) and significantly increased collagen volume fraction (Figure 4B) in CIA rats compared with control rats. The protein expression of collagen I, collagen III, and fibrosis‐related molecules TGF‐β1 and α‐SMA were also increased significantly in atria from CIA rats (Figure 4C through 4F). The results demonstrated CIA could induce atrial interstitial fibrosis. Furthermore, CIA increased the apoptosis of atrial cells (Figure 5A). The apoptotic cells sharply increased in the atria of the CIA group compared with the control group (Figure 5B). The antiapoptotic Bcl‐2 protein level was decreased, whereas proapoptotic Bax and CASP‐3 protein levels were increased in atrial samples from CIA rats compared with control rats (Figure 5C through 5E). In this study, we found that CIA also induced autophagy in atrial myocytes. As shown in Figure 6A and 6B, the electron microphotographs of atria samples from control rats showed few autophagic vacuoles, whereas CIA rats exhibited several autophagic vacuoles, and most of them were late autophagic vacuoles. Western blot showed that the ratio of LC3II/I (an established marker of autophagy) and ATG5 (a key autophagy‐regulatory gene) expression were increased (Figure 6C and 6D). These findings suggested that autophagy was activated in the CIA group.
Figure 3.
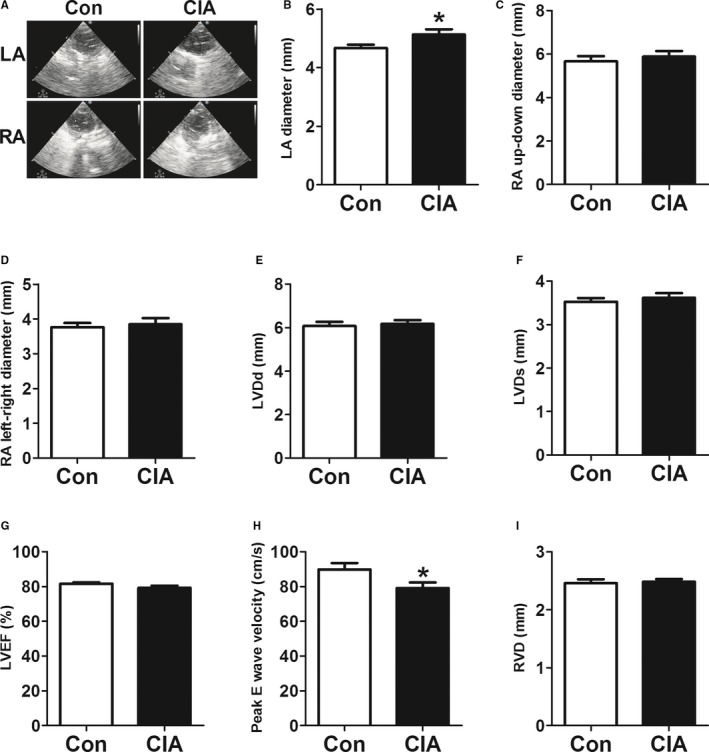
Echocardiographic variables changes. A, Echocardiography images of left atrial (LA) and right atrial (RA) dimensions in the control (CON) and collagen‐induced arthritis (CIA) groups. B through D, LA diameter, RA up–down diameter and RA left–right diameter in CON and CIA rats. E through I, Left ventricular diameter at end diastole (LVDd) and systole (LVDs), left ventricular ejection fraction (LVEF), peak E‐wave velocity, and right ventricular diameter (RVD) in CON and CIA rats. *P<0.05, vs the CON group, n=10 per group.
Figure 4.
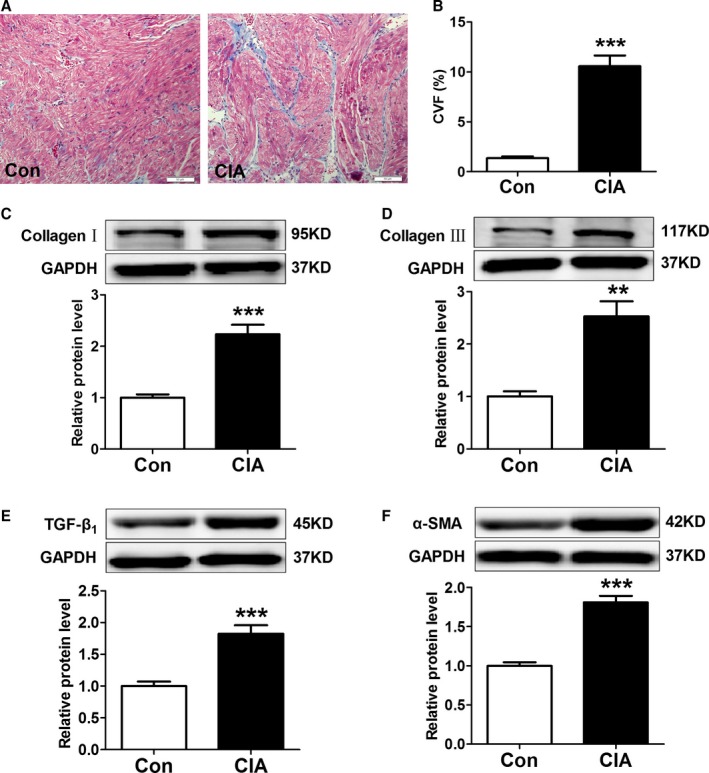
Effect of collagen‐induced arthritis (CIA) on atrial fibrosis. A, Examples of Masson's trichrome staining. B, Collagen volume fraction (CVF) of each group, n=8 per group. C through F, Representative Western blots and quantification of protein expressions of collagen I, collagen III, TGF‐β1 (transforming growth factor‐β1), and α‐SMA (α–smooth muscle actin) in atrial tissues; n=6 per group. **P<0.01, ***P<0.001, vs the control (CON) group. Scale bar=50 μm.
Figure 5.
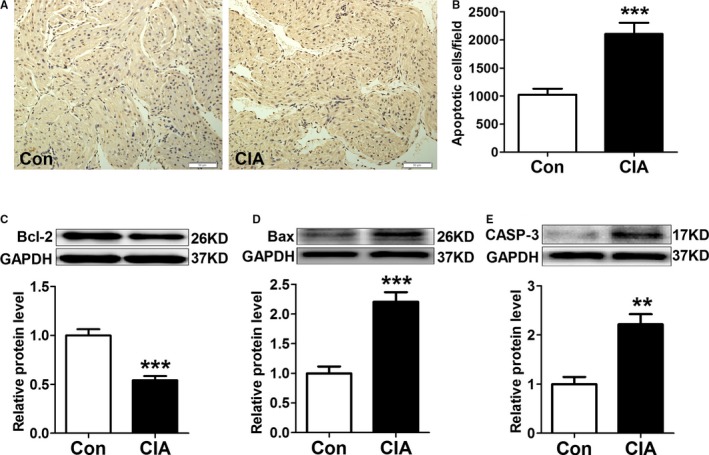
CIA increased atrial myocytes apoptosis. A, Examples of TUNEL (terminal deoxynucleotidyl transferase mediated dUTP nick‐end labeling) staining. B, The number of apoptotic cells per field, n=8 per group. C through E, Representative western blots and quantification of protein expressions of Bcl‐2, Bax, and CASP‐3 (caspase 3) in atrial tissues; n=6 per group. **P<0.01, ***P<0.001, vs the CON group. Scale bar=50 μm. CIA indicates collagen‐induced arthritis; CON, control.
Figure 6.
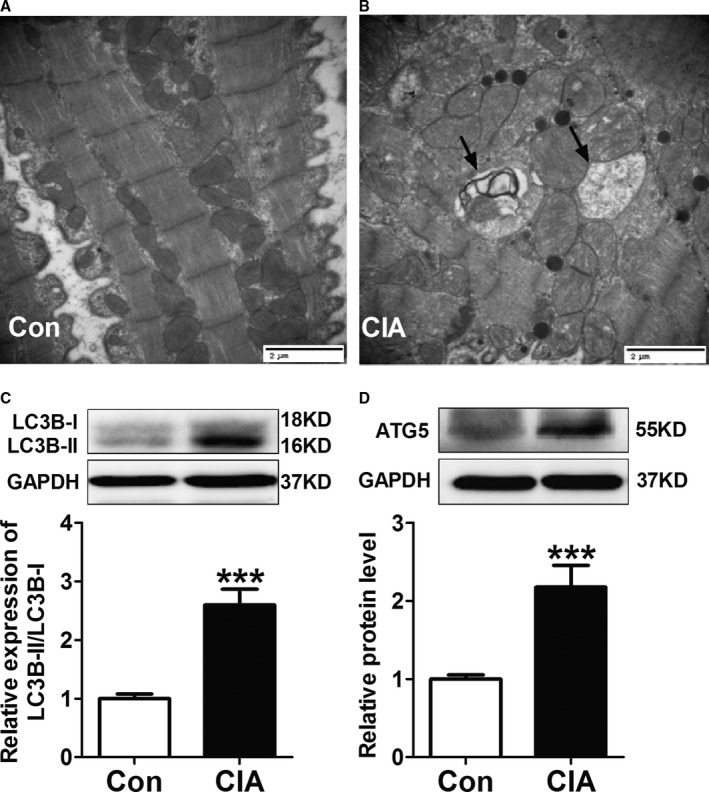
CIA‐induced autophagy in the atria. A and B, Representative electron microphotographs showing autophagic vacuoles in atrial myocytes. C and D, Representative Western blots and quantification of protein expressions of LC3B and ATG5 in atrial tissues. ***P<0.001, vs the CON group, n=6 per group. Scale bar=2 μm. Arrows indicate the autophagic vacuoles. CIA indicates collagen‐induced arthritis; CON, control.
Protein Expression of Connexins, Nav1.5, and the Main Ion Channels
In addition to atrial fibrosis, connexins and Na+‐channel α subunit (Nav1.5) expression alteration also can contribute to atrial conduction abnormalities. Therefore, we studied the changes of connexin and Nav1.5 protein expressions in atria. As shown in Figure 7A and 7B, both Cx40 and Cx43 protein levels showed decreases in CIA rats compared with control rats; however, the observed changes were not significant. In addition, Nav1.5 protein expression showed no significant changes (Figure 7C).
Figure 7.
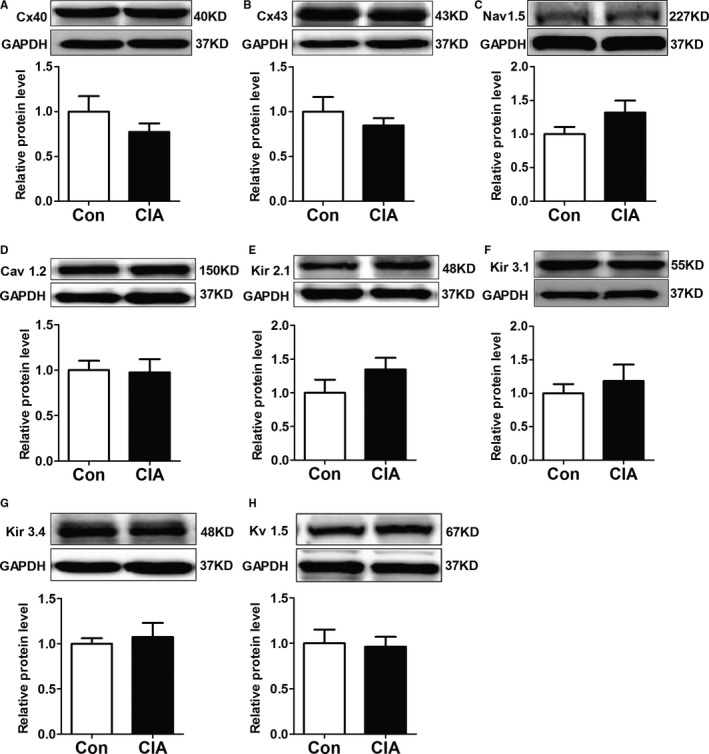
Protein expressions of connexins, Nav1.5, and the main ion channels in the atria. A and B, Representative Western blots and quantification of protein expressions of total Cx40 (connexin 40) (A) and total Cx43 (connexin 43) (B) in control (CON) and collagen‐induced arthritis (CIA) rats. C, Representative Western blots and quantification of protein expressions of Nav1.5 in CON and CIA rats. D through H, Representative Western blots and quantification of protein expressions of Cav 1.2, Kir 2.1, Kir 3.1, Kir 3.4, and Kv 1.5 in atrial tissues of CON and CIA rats. n=6 per group.
We also detected protein expression of the main ion channels in atria. We found that protein levels of Cav 1.2, Kir 2.1, Kir 3.1, Kir 3.4, and Kv 1.5 were not significantly different between the 2 groups (Figure 7D through 7H).
Atrial Sympathetic Remodeling
We further examined atrial sympathetic innervation of CIA rats. Immunohistochemistrical staining showed that nerves that were immunopositive to GAP43 (a marker for nerve sprouting) and TH (the sympathetic nerve marker) were more abundant in rats with CIA than in controls, and the distribution of nerves within the same microscopic field showed significant inhomogeneity (Figure 8A through 8D). The atrial protein expression of GAP43, TH, and NGF was further determined by Western blot analysis, and the results showed that all were dramatically increased in CIA rats compared with control rats (Figure 8E through 8G). These results suggested that CIA induced atrial nerve sprouting and heterogeneous sympathetic hyperinnervation.
Figure 8.
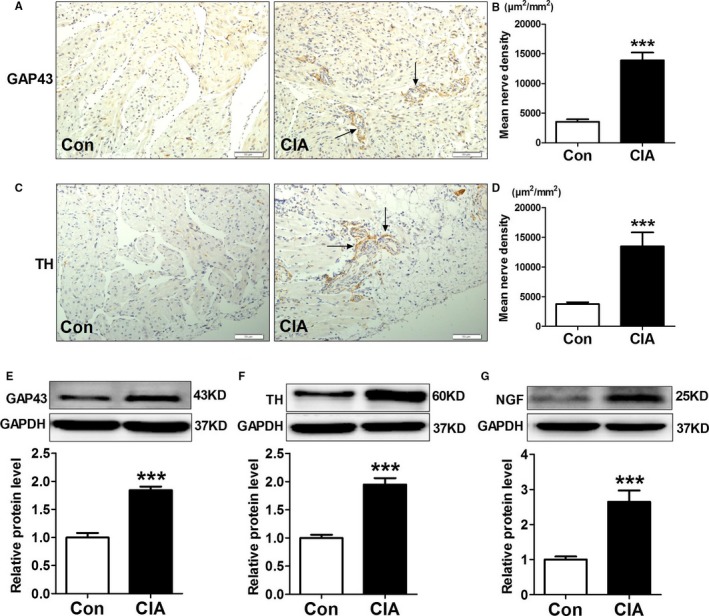
Atrial sympathetic remodeling induced by collagen‐induced arthritis (CIA). A through D, Atrial immunostaining and mean density of GAP43 (growth associated protein 43)–positive nerves (A and B) and TH (tyrosine hydroxylase)–positive nerves (C and D), n=8 per group. E through G, Representative Western blots and quantification of protein expressions of GAP43, TH, and NGF (nerve growth factor) in atrial tissues; n=6 per group. ***P<0.001, vs the control (CON) group. Scale bar=50 μm. Arrows indicate the GAP43‐ and TH‐positive nerves.
Discussion
Major Findings
To our knowledge, the present study was the first experimental analysis of the effects of RA on AF susceptibility and atrial arrhythmogenic remodeling. We found that AF inducibility and duration were substantially increased in CIA rats 8 weeks after primary immunization, and AF duration was significantly and positively correlated with serum IL‐6 and TNF‐α levels. The prolonged atrial conduction time was observed in CIA rats, with no changes in the atrial effective refractory period. Atrial conduction delay was accompanied by significantly increased atrial fibrosis. In addition, atrial structural and autonomic remodeling was observed. Interestingly, we found that CIA had no significant effects on protein expression of connexins and the main ion channels in atria.
Relationship to Clinical Findings
The potential association between RA and AF in humans has been evaluated in 3 large retrospective cohort studies involving ≈40 000 RA patients and >4 million non‐RA controls.4, 5, 6 The meta‐analysis of these studies demonstrated ≈30% higher risk of AF in RA patients compared with non‐RA participants.7 Moreover, recent studies have demonstrated significant atrial remodeling in patients with RA. Prolonged electromechanical delays and impaired left atrial mechanical functions were observed in RA patients and correlated with CRP level.11 P‐wave dispersion was found to be increased in RA and correlated with CRP and disease duration.12 P‐wave dispersion is considered as an electrocardiographic marker of inhomogeneous and discontinuous propagation of sinus impulses in the atrial myocardium, representing a sensitive and specific clinical predictor of AF. The left atrial volume index increased at a higher rate in the RA cohort than in the non‐RA cohort.13 In clinical studies, however, it is difficult to determine whether RA directly causes atrial remodeling or whether the observed associations are due to potential confounders. Consequently, an experimental study is needed to understand the association between RA and AF. Our findings are consistent with the cited clinical studies. We found that CIA is sufficient to induce significant atrial conduction slowing and then increase AF susceptibility. Furthermore, our present study may provide insights into the mechanisms underlying AF promotion by RA, including significant atrial fibrosis, increased atrial cardiomyocyte apoptosis and autophagy, and atrial heterogeneous sympathetic hyperinnervation.
It is also important to consider the differences between experimentally induced arthritis and human RA when interpreting findings from rat arthritis models; therefore, our findings in CIA rats may not be applied directly to human RA.
Atrial Arrhythmogenic Remodeling by CIA and the Possible Underlying Mechanism
Atrial fibrosis is a common feature of clinical AF and a hallmark of arrhythmogenic structural remodeling, which provides a basis for unidirectional conduction block and macro reentry, resulting in an AF substrate.14 Increased apoptosis has been detected in atrial tissues of patients with AF and has been proven to contribute to AF‐promoting atrial contractile abnormalities and dilation associated with heart failure.15, 16 Moreover, apoptosis is thought to cause atrial fibrosis by inducing a reparative fibrosis process to replace the degenerating myocardial cells.17 However, TUNEL is only a qualitative procedure that gives no information about the cell type undergoing apoptosis. Further studies are needed to clarify what kind of cells undergo apoptosis. Atrial autonomic remodeling has been observed in chronic AF patients and in a canine model of rapid atrial pacing, and chronic left ventricular myocardial infarction and rapid ventricular pacing induced heart failure.18, 19, 20 Previous studies demonstrated that atrial autonomic remodeling could promote heterogeneity of atrial refractoriness and provide an important substrate for AF.20 Consequently, RA‐induced atrial fibrosis, apoptosis, and autonomic remodeling could contribute to AF vulnerability.
The precise effect of autophagy on AF in our study remains poorly understood. Our previous study proved that autophagy occurred in atrial myocytes of rapid atrial pacing‐induced experimental AF canines and chronic AF patients.21 Considering the finding that autophagy activation was observed in most cases of atrial cardiomyocytes with severe mitral and tricuspid regurgitation and was closely associated with the degree of myolysis,22 it is reasonable to conclude that autophagy may be a novel mechanistic contributor to AF.
In our present study, in addition to systemic inflammation, local inflammation in atrial myocardium was also observed, as shown by the accumulations of inflammatory cells and increased TNF‐α and IL‐6 protein expression. Previous studies suggested that these inflammatory cytokines play a key stimulatory role in atrial remodeling through several mechanisms. TNF‐α has been demonstrated to induce atrial fibrosis by increasing atrial myofibroblast proliferation and metalloproteinase secretion.23, 24 Saba et al25 found that cardiac‐specific overexpression of TNF‐α could cause atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model. Moreover, a recent study has revealed that enhanced expression of TNF‐α in the murine atrial cardiomyocyte HL‐1 cell line promoted cardiomyocyte apoptosis by activating CASP‐3.26 IL‐6 has also been shown to be an important regulator of cardiac interstitial fibrosis. IL‐6 can induce STAT3 (signal transducer and activator of transcription 3) phosphorylation, increase collagen production in cardiac fibroblasts, and thus promote cardiac fibrosis.27, 28 Deletion of IL‐6 was consistently shown to be beneficial against fibrotic heart disease.29 However, IL‐6 seems to exert pleiotropic effects and control the balance between anti‐ and proapoptotic pathways.30, 31 The exact role of IL‐6 signaling in atrial cardiomyocyte apoptosis induced by RA needs to be further investigated in future studies.
Zhou et al32 showed that NGF, which is critical for sympathetic nerve sprouting, is upregulated after myocardial infarction in animal models, resulting in the regeneration of cardiac sympathetic nerves and heterogeneous innervation. Therefore, CIA‐induced atrial autonomic remodeling in our current study may be mainly attributed to the upregulation of NGF. Previous studies have shown that NGF could be synthesized by multiple cell types within myocardium including macrophages, myofibroblasts, and atrial myocytes.33, 34, 35 Consequently, increased synthesis by the accumulated inflammatory cells and increased myofibroblasts (as shown by upregulation of α‐SMA protein, a marker of transformation from fibroblasts to myofibroblasts) in the atrial myocardium is thought to contribute to the upregulation of NGF in this study.
Limitations
Some limitations should be acknowledged when considering the results of the present study. First, we detected only protein expression of the subunits of potassium and calcium channels. Additional work is needed to examine related currents of these channels. Second, we examined only total Cx40 and Cx43 expression. The changes in connexin phosphorylation and distribution in present study are still unclear and need further investigation. Third, the precise molecular basis of proarrhythmic atrial remodeling in this CIA model remains unknown. Further work will be necessary to establish the precise molecular mechanisms by which RA induces atrial remodeling. Last, only female Wistar rats were used to induce the CIA model in this study; therefore, our findings may not be applicable to males.
Conclusions
In summary, our work provided convincing experimental evidence, for the first time, that RA increased AF susceptibility by inducing atrial remodeling. The AF‐promoting substrate includes atrial conduction abnormalities related to atrial fibrosis, atrial cardiomyocyte apoptosis and autophagy, and atrial autonomic remodeling. This study provides insights into mechanisms underlying RA‐induced AF and validates a model that is suitable for further mechanistic and therapeutic exploration.
Sources of Funding
This work was supported by grants from National Natural Science Foundation of China (81100121 to Gong, 81670297 and 81470462 to Li, 81300133 to Sheng), the Youth Foundation of Heilongjiang Province (QC2010004 to Gong), and the Heilongjiang Province Outstanding Youth Foundation (JC201208 to Li).
Disclosures
None.
(J Am Heart Assoc. 2017;6:e007320 DOI: 10.1161/JAHA.117.007320.)29269354
Contributor Information
Yongtai Gong, Email: gongth@126.com.
Yue Li, Email: ly99ly@vip.163.com.
References
- 1. Klingenberg R, Lüscher TF. Rheumatoid arthritis and coronary atherosclerosis: two cousins engaging in a dangerous liaison. Eur Heart J. 2015;36:3423–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gabriel SE. Cardiovascular morbidity and mortality in rheumatoid arthritis. Am J Med. 2008;121:S9–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iwasaki YK, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology: implications for management. Circulation. 2011;124:2264–2274. [DOI] [PubMed] [Google Scholar]
- 4. Lindhardsen J, Ahlehoff O, Gislason GH, Madsen OR, Olesen JB, Svendsen JH, Torp‐Pedersen C, Hansen PR. Risk of atrial fibrillation and stroke in rheumatoid arthritis: Danish nationwide cohort study. BMJ. 2012;344:e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim SC, Liu J, Solomon DH. The risk of atrial fibrillation in patients with rheumatoid arthritis. Ann Rheum Dis. 2014;73:1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bacani AK, Crowson CS, Roger VL, Gabriel SE, Matteson EL. Increased incidence of atrial fibrillation in patients with rheumatoid arthritis. Biomed Res Int. 2015;2015:809514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ungprasert P, Srivali N, Kittanamongkolchai W. Risk of incident atrial fibrillation in patients with rheumatoid arthritis: a systematic review and meta‐analysis. Int J Rheum Dis. 2017;20:434–441. [DOI] [PubMed] [Google Scholar]
- 8. Williams RO. Collagen‐induced arthritis as a model for rheumatoid arthritis. Methods Mol Med. 2004;98:207–216. [DOI] [PubMed] [Google Scholar]
- 9. Song HP, Li X, Yu R, Zeng G, Yuan ZY, Wang W, Huang HY, Cai X. Phenotypic characterization of type II collagen‐induced arthritis in Wistar rats. Exp Ther Med. 2015;10:1483–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Felcenloben I, Piasecki T, Miller J, Rossowska J, Bańcyr E, Atamaniuk W, Nowak M, Świerkot J, Ratajczak K, Chełmońska‐Soyta A. Adoptively transferred Tregs accumulate in a site‐specific manner and ameliorate signs of less advanced collagen‐induced arthritis progress in rats. Immunotherapy. 2015;7:215–228. [DOI] [PubMed] [Google Scholar]
- 11. Acar G, Sayarlioğlu M, Akçay A, Sökmen A, Sökmen G, Yalçintaş S, Nacar AB, Gündüz M, Tuncer C. Evaluation of atrial electromechanical delay and left atrial mechanical functions in patients with rheumatoid arthritis. Turk Kardiyol Dern Ars. 2009;37:447–453. [PubMed] [Google Scholar]
- 12. Guler H, Seyfeli E, Sahin G, Duru M, Akgul F, Saglam H, Yalcin F. P wave dispersion in patients with rheumatoid arthritis: its relation with clinical and echocardiographic parameters. Rheumatol Int. 2007;27:813–818. [DOI] [PubMed] [Google Scholar]
- 13. Davis JM III, Lin G, Oh JK, Crowson CS, Achenbach SJ, Therneau TM, Matteson EL, Rodeheffer RJ, Gabriel SE. Five‐year changes in cardiac structure and function in patients with rheumatoid arthritis compared with the general population. Int J Cardiol. 2017;240:379–385. pii: S0167‐5273(16)34189‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Everett TH IV, Olgin JE. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007;4:S24–S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aimé‐Sempé C, Folliguet T, Rücker‐Martin C, Krajewska M, Krajewska S, Heimburger M, Aubier M, Mercadier JJ, Reed JC, Hatem SN. Myocardial cell death in fibrillating and dilated human right atria. J Am Coll Cardiol. 1999;34:1577–1586. [DOI] [PubMed] [Google Scholar]
- 16. Cardin S, Li D, Thorin‐Trescases N, Leung TK, Thorin E, Nattel S. Evolution of the atrial fibrillation substrate in experimental congestive heart failure: angiotensin‐dependent and ‐independent pathways. Cardiovasc Res. 2003;60:315–325. [DOI] [PubMed] [Google Scholar]
- 17. Bing OH, Ngo HQ, Humphries DE, Robinson KG, Lucey EC, Carver W, Brooks WW, Conrad CH, Hayes JA, Goldstein RH. Localization of alpha1(I) collagen mRNA in myocardium from the spontaneously hypertensive rat during the transition from compensated hypertrophy to failure. J Mol Cell Cardiol. 1997;29:2335–2344. [DOI] [PubMed] [Google Scholar]
- 18. Nguyen BL, Fishbein MC, Chen LS, Chen PS, Masroor S. Histopathological substrate for chronic atrial fibrillation in humans. Heart Rhythm. 2009;6:454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang CM, Wu TJ, Zhou S, Doshi RN, Lee MH, Ohara T, Fishbein MC, Karagueuzian HS, Chen PS, Chen LS. Nerve sprouting and sympathetic hyperinnervation in a canine model of atrial fibrillation produced by prolonged right atrial pacing. Circulation. 2001;103:22–25. [DOI] [PubMed] [Google Scholar]
- 20. Miyauchi Y, Zhou S, Okuyama Y, Miyauchi M, Hayashi H, Hamabe A, Fishbein MC, Mandel WJ, Chen LS, Chen PS, Karagueuzian HS. Altered atrial electrical restitution and heterogeneous sympathetic hyperinnervation in hearts with chronic left ventricular myocardial infarction: implications for atrial fibrillation. Circulation. 2003;108:360–366. [DOI] [PubMed] [Google Scholar]
- 21. Yuan Y, Zhao J, Yan S, Wang D, Zhang S, Yun F, Zhao H, Sun L, Liu G, Ding X, Liu L, Li Y. Autophagy: a potential novel mechanistic contributor to atrial fibrillation. Int J Cardiol. 2014;172:492–494. [DOI] [PubMed] [Google Scholar]
- 22. Chen MC, Chang JP, Wang YH, Liu WH, Ho WC, Chang HW. Autophagy as a mechanism for myolysis of cardiomyocytes in mitral regurgitation. Eur J Clin Invest. 2011;41:299–307. [DOI] [PubMed] [Google Scholar]
- 23. Liew R, Khairunnisa K, Gu Y, Tee N, Yin NO, Naylynn TM, Moe KT. Role of tumor necrosis factor‐α in the pathogenesis of atrial fibrosis and development of an arrhythmogenic substrate. Circ J. 2013;77:1171–1179. [DOI] [PubMed] [Google Scholar]
- 24. Porter KE, Turner NA, O'Regan DJ, Ball SG. Tumor necrosis factor alpha induces human atrial myofibroblast proliferation, invasion and MMP‐9 secretion: inhibition by simvastatin. Cardiovasc Res. 2004;64:507–515. [DOI] [PubMed] [Google Scholar]
- 25. Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, Feldman AM, Salama G, McTiernan CF, London B. Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac‐specific overexpression of tumor necrosis factor‐{alpha}. Am J Physiol Heart Circ Physiol. 2005;289:H1456–H1467. [DOI] [PubMed] [Google Scholar]
- 26. Koyani CN, Windischhofer W, Rossmann C, Jin G, Kickmaier S, Heinzel FR, Groschner K, Alavian‐Ghavanini A, Sattler W, Malle E. 15‐deoxy‐Δ¹²,¹⁴‐PGJ₂ promotes inflammation and apoptosis in cardiomyocytes via the DP2/MAPK/TNFα axis. Int J Cardiol. 2014;173:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang Z, Chen XJ, Qian C, Dong Q, Ding D, Wu QF, Li J, Wang HF, Li WH, Xie Q, Cheng X, Zhao N, Du YM, Liao YH. Signal transducer and activator of transcription 3/microRNA‐21 feedback loop contributes to atrial fibrillation by promoting atrial fibrosis in a rat sterile pericarditis model. Circ Arrhythm Electrophysiol. 2016;9:e003396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan YT, Girgis M, Elias HK, Zhu Y, Davani A, Yang Y, Chen X, Ye S, Wang OL, Chen L, Hauptman J, Vincent RJ, Dawn B. Deletion of interleukin‐6 attenuates pressure overload‐induced left ventricular hypertrophy and dysfunction. Circ Res. 2016;118:1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Banerjee I, Fuseler JW, Intwala AR, Baudino TA. IL‐6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am J Physiol Heart Circ Physiol. 2009;296:H1694–H1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ponce NE, Cano RC, Carrera‐Silva EA, Lima AP, Gea S, Aoki MP. Toll‐like receptor‐2 and interleukin‐6 mediate cardiomyocyte protection from apoptosis during Trypanosoma cruzi murine infection. Med Microbiol Immunol. 2012;201:145–155. [DOI] [PubMed] [Google Scholar]
- 32. Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, Fishbein MC, Sharifi B, Chen PS. Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res. 2004;95:76–83. [DOI] [PubMed] [Google Scholar]
- 33. Wernli G, Hasan W, Bhattacherjee A, van Rooijen N, Smith PG. Macrophage depletion suppresses sympathetic hyperinnervation following myocardial infarction. Basic Res Cardiol. 2009;104:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mias C, Coatrieux C, Denis C, Genet G, Seguelas MH, Laplace N, Rouzaud‐Laborde C, Calise D, Parini A, Cussac D, Pathak A, Sénard JM, Galés C. Cardiac fibroblasts regulate sympathetic nerve sprouting and neurocardiac synapse stability. PLoS One. 2013;8:e79068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saygili E, Schauerte P, Küppers F, Heck L, Weis J, Weber C, Schwinger RH, Hoffmann R, Schröder JW, Marx N, Rana OR. Electrical stimulation of sympathetic neurons induces autocrine/paracrine effects of NGF mediated by TrkA. J Mol Cell Cardiol. 2010;49:79–87. [DOI] [PubMed] [Google Scholar]