

Abstract
Background
It is uncertain whether pharmacological reductions in very‐low‐density lipoproteins (VLDLs), and their component triglyceride and cholesterol could reduce residual risk of atherosclerotic cardiovascular disease (ASCVD) events among individuals in whom low‐density lipoprotein cholesterol (LDL‐C) has been adequately lowered. We examined whether individuals with greater on‐statin reductions in VLDL‐related measures—beyond reductions in LDL‐C—were at further reduced risk of ASCVD.
Methods and Results
In 9423 participants in the JUPITER (Justification for the Use of Statins in Prevention) trial (NCT00239681), at baseline and on statin we measured standard lipids, 400‐MHz proton nuclear magnetic resonance spectroscopy‐measured VLDL particle subclasses (small, medium, and large VLDL lipoprotein particle concentration), and total VLDL cholesterol mass. Compared with individuals allocated to placebo, we examined risk of incident ASCVD (N=211) among statin‐allocated participants who achieved minimal (<median) or greater (≥median) marker reductions using adjusted Cox models. On‐statin changes in VLDL‐related markers were only modestly correlated (Spearman r≤0.29) with change in LDL‐C. On‐statin median LDL‐C was 54 mg/dL and triglyceride was 101 mg/dL. Dose‐response reductions in ASCVD risk were observed for greater reductions in LDL‐C, VLDL cholesterol mass, and small VLDL lipoprotein particle concentration; the latter 2 remained significant after incremental adjustment for change in LDL‐C (P≤0.006). Conversely, there was no further risk reduction with greater reductions in triglycerides or large/medium VLDL lipoprotein particle concentration.
Conclusions
Pharmacological reduction in small, cholesterol‐enriched, triglyceride‐depleted VLDL was associated with reduction in ASCVD risk. Chemically measured triglycerides may not sufficiently capture risk related to VLDL pathways. These findings also support broader profiling of lipid and lipoprotein changes in response to statins as prognostic markers of individual benefit, supporting more precision‐medicine, individualized approaches to cardiovascular risk reduction.
Clinical Trial Registration
URL: https://www.clinicaltrials.gov. Unique identifier: NCT00239681.
Keywords: arteriosclerosis, lipids, lipoproteins, metabolomics, personalized medicine, primary prevention, statin
Clinical Perspective
What Is New?
Among a population intended to represent the growing number of individuals with low low‐density lipoprotein cholesterol, pharmacological reductions in small very‐low‐density lipoproteins (small very‐low‐density lipoprotein lipoprotein particle concentration) and remnant cholesterol (very‐low‐density lipoprotein cholesterol mass) were associated with reductions in atherosclerotic cardiovascular disease risk.
This observed risk reduction occurred independent of changes in low‐density lipoprotein cholesterol.
What Are the Clinical Implications?
Pharmacological reductions in small, cholesterol‐enriched, triglyceride‐depleted very‐low‐density lipoprotein could result in reductions in residual atherosclerotic cardiovascular disease risk, independent of changes in low‐density lipoprotein cholesterol.
Chemically measured triglycerides may not sufficiently capture the risk related to the spectrum of very‐low‐density lipoprotein subtypes.
Broader profiling of changes in lipids and lipoproteins using metabolomics platforms in response to statin therapy could identify prognostic markers of individual benefit, supporting more precision‐medicine, individualized approaches to cardiovascular risk reduction.
Levels of low‐density lipoprotein cholesterol (LDL‐C) in the population are declining in response to lifestyle interventions and pharmacological strategies that target LDL‐C reduction, such as statins.1, 2, 3 However, among individuals with achieved lower levels of LDL‐C, atherosclerotic cardiovascular disease (ASCVD) events continue to occur with unacceptable frequency.4 Such residual risk may reflect aspects of atherogenesis not captured by LDL‐C, including effects of very‐low‐density lipoproteins (VLDLs) and their contents (cholesterol and triglycerides).5, 6, 7, 8, 9 Enzymatic hydrolysis of VLDL results in the formation of smaller, cholesterol‐enriched lipoprotein particles,5, 10, 11 which experimental studies suggest could contribute to the development of ASCVD.5, 7, 10, 12, 13, 14, 15, 16 Clinically, however, there has been uncertainty as to whether ASCVD events could be further reduced by reducing these triglyceride‐rich lipoproteins, with several neutral trials examining this strategy to date.10, 17
To study whether pharmacological reductions in concentrations of VLDL lipoprotein particle (VLDL‐p) subclasses, and their component cholesterol (VLDL‐C) and triglyceride, may be associated with lower ASCVD risk among individuals with low LDL‐C on‐statin, we built on the observation that, in some individuals, statins reduce triglycerides and VLDL (in part, through increased clearance and effects on lipoprotein lipase),18, 19 and that, for uncertain reasons, these effects are variable: Some individuals experience reductions whereas others experience no reduction in VLDL lipoproteins on statin. We thus examined whether individuals achieving larger reductions in VLDL‐p, VLDL‐C, and triglycerides were at incrementally lower risk of ASCVD events than those with smaller or no reductions. Importantly, we examined whether these potential risk reductions conferred benefit beyond statin‐related changes in LDL‐C. Overall, we hypothesized that incrementally greater reductions in small (relatively cholesterol‐enriched) VLDL lipoproteins and VLDL‐C would be associated with incrementally greater reductions in ASCVD events, beyond reductions in LDL‐C. In addition to identifying potential therapeutic targets, broader profiling of the lipid/lipoprotein changes in response to statin therapy could identify independent markers of benefit from statin therapy, and could support more precision‐medicine, individualized approaches to cardiovascular patient care.
Methods
Study Population
The study population is derived from a primary prevention randomized, controlled clinical trial of rosuvastatin versus placebo (JUPITER [Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin]; NCT00239681).20 The JUPITER trial investigators randomized 17 802 individuals (women ≥60 years, men ≥50 years) without past history of coronary disease, stroke, or diabetes mellitus, all of whom had low or normal LDL‐C (<130 mg/dL), but elevated high‐sensitivity C‐reactive protein (≥2.0 mg/L), to rosuvastatin 20 mg daily versus placebo. Individuals with triglycerides >500 mg/dL were excluded from trial entry. The study enrolled a multiethnic population of women and men. The relative risk reduction in the primary composite incident ASCVD end point was 44% with rosuvastatin. After trial completion, in a subset of randomly selected JUPITER participants with sufficient plasma available, we performed 400‐MHz proton nuclear magnetic resonance (1H‐NMR) spectroscopy‐based lipoprotein analysis of fasting blood samples collected at baseline (pretreatment) and at 12 months (on study drug). Data were analyzed from 9423 participants with all measures of interest at both time points. The design of this secondary analysis is observational in nature. Institutional approval was granted and subjects provided informed consent.
Laboratory Analysis
Lipid measurements were performed on fasting samples by a central laboratory. LDL‐C was calculated by the Friedewald equation (total cholesterol minus high‐density lipoprotein cholesterol [HDL‐C] minus triglycerides/5) when triglycerides were <400 mg/dL and measured by ultracentrifugation when triglycerides were ≥400 mg/dL.21 Chemically measured triglycerides were quantified using a colorimetric assay. HDL‐C was assayed in supernatant after heparin‐manganese precipitation of apolipoprotein B–containing proteins. 1H‐NMR spectroscopy (400‐MHz) LipoProfile III measurements were performed by LipoScience (now LabCorp). 1H‐NMR was used to quantify the total concentration of VLDL‐Chylomicron particles (VLDL‐p) and size‐based VLDL subclasses (large VLDL‐p/chylomicrons [>60 nm in diameter], medium VLDL‐p [42–60 nm], and small VLDL‐p [29–42 nm]). Total VLDL‐C was measured by NMR as the sum of cholesterol contained in all VLDL subclasses. Concentrations of intermediate‐density lipoprotein were also examined. We also examined a formulaic estimation of remnant cholesterol (RC)22 which was calculated from standard lipid measures as total cholesterol minus HDL‐C minus LDL‐C. (This was near equivalent to calculating RC as triglycerides divided by 5, because LDL‐C was calculated from the Friedewald equation in most JUPITER participants.)
Outcomes
The primary outcome in this study was the JUPITER trial primary composite incident ASCVD end point, defined as the occurrence of either myocardial infarction, stroke, hospitalization for unstable angina, arterial revascularization, or cardiovascular death.20 As in the primary trial, we counted only end points confirmed upon medical record review by an end points committee masked to treatment assignment.
Statistical Analyses
For baseline and follow‐up continuous variables, including clinical data and lipoprotein measures, we tabulated medians and interquartile ranges, and percentages for categorical variables. Demographic and biochemical differences between groups were compared with the Kruskal–Wallis test or χ2 test. All P values were 2‐tailed using α=0.05.
An overview of the study is shown in Figure 1. In the primary analysis, using risk‐factor–adjusted Cox proportional hazards models, we evaluated residual risk of incident ASCVD among statin‐allocated participants who achieved a minimal (<median) or larger (≥median) absolute change (always a reduction) in each cholesterol/lipoprotein of interest: LDL‐C, triglycerides, chylomicron/large VLDL‐p, medium VLDL‐p, small VLDL‐p, and VLDL‐C. Changes in non‐HDL‐C, apolipoprotein B, IDL‐p, and formulaic RC were also examined. Risk estimates were examined in the 2 statin response groups, in relation to risk in placebo‐allocated participants (reference). Differences between response group relative hazards were tested for significance (Wald test). In addition to examining risk by absolute marker change, risk based on percent marker change was examined in sensitivity analyses. The exposure time was calculated as the time from randomization to end point occurrence.
Figure 1.
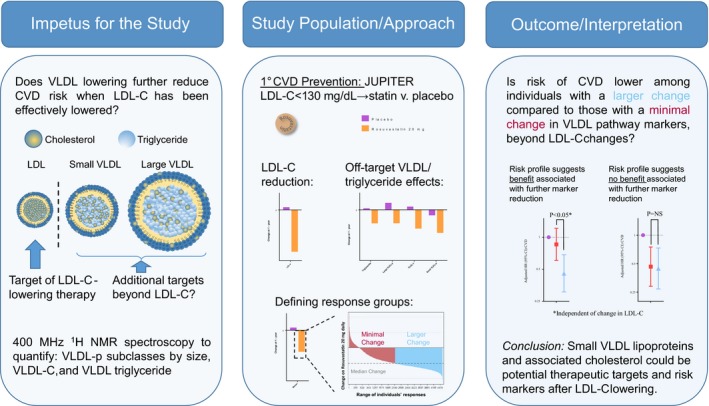
Illustrative schematic overview of study hypothesis, approach, and interpretation (hypothetical result shown). CVD indicates cardiovascular disease; HR, hazard ratio; JUPITER, Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin trial; LDL‐C, low‐density lipoprotein cholesterol; max., maximum; med., median; mo., months; VLDL, very‐low‐density lipoprotein; VLDL‐C, VLDL cholesterol; VLDL‐p, VLDL lipoprotein particle concentration.
Adjusted Cox proportional survival models were examined, adjusting for baseline marker level, age, sex, race, smoking, body mass index, systolic blood pressure, fasting glucose, baseline HDL‐C, and baseline high‐sensitivity C‐reactive protein. To determine whether potential reductions in risk associated with reductions in VLDLs and their contents were independent of reductions in LDL‐C, we incrementally adjusted for each individual's statin‐related change in LDL‐C. We also examined Spearman correlations between change in LDL‐C and changes in the VLDL‐related markers. To determine whether patient compliance with statin therapy could explain reductions in risk, we performed a sensitivity analysis including only those individuals confirmed to be taking statin at 1 year, beginning the follow‐up at that time. Analyses were performed using SAS software (version 9.3; SAS Institute Inc., Cary, NC).
Results
Study Population
Included study participants had a median (interquartile range) age of 66 (60, 71) years and were 36% female. Except for race/ethnicity, patients in the current study were generally representative of those in the parent JUPITER trial (Table 1). Baseline and on‐statin, respectively, median (25th, 75th percentile) marker levels were: LDL‐C, 109 (96, 119) and 54 (43, 70) mg/dL; triglycerides, 119 (88, 169) and 101 (76, 138) mg/dL; and HDL‐C, 49 (41, 60) and 53 (43, 64) mg/dL. There were a total of 211 primary events observed over a mean 2.0‐year (maximum 5.0 years) follow‐up, representing 21 442 person‐years of follow‐up.
Table 1.
Baseline Characteristics in the Study Sample vs the Original JUPITER Cohort
| Characteristic | Current JUPITER Substudy (N=9423) | Overall JUPITER Trial (N=17 802) |
|---|---|---|
| Age, y | 66 (60, 71) | 66 (60, 71) |
| Women | 36 | 38 |
| Rosuvastatin | 49 | 50 |
| Race/ethnicitya | ||
| White | 85.1 | 71.2 |
| Black | 5.2 | 12.5 |
| Asian | 1.5 | 1.6 |
| Hispanic | 7.5 | 12.7 |
| Other/unknown | 0.7 | 1.96 |
| Body mass index, kg/m2 | 28.5 (25.6, 32.0) | 28.3 (25.3, 32.0) |
| Hypertension | 56 | 57 |
| Systolic blood pressure, mm Hg | 134 (124, 146) | 134 (124, 145) |
| Diastolic blood pressure, mm Hg | 80 (75, 86) | 80 (75, 87) |
| Current smoker | 14 | 16 |
| Family history of premature coronary disease | 13 | 11 |
| Glucose, mg/dL | 95 (89, 102) | 94 (88, 102) |
| hsCRP, mg/L | 4.05 (2.75, 6.65) | 4.25 (2.85, 7.10) |
| LDL‐C, mg/dL | 109 (96, 119) | 108 (94, 119) |
| Apolipoprotein B, mg/dL | 109 (97, 122) | 109 (95 122) |
| Triglycerides, mg/dL | 119 (88, 169) | 118 (85, 169) |
| HDL‐C, mg/dL | 49 (41, 60) | 49 (40, 60) |
Values shown are median (25th, 75th percentile) or proportion (%). HDL‐C indicates high‐density lipoprotein cholesterol; hsCRP high‐sensitivity C‐reactive protein; JUPITER, Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin trial; LDL‐C, low‐density lipoprotein cholesterol.
Proportions do not sum to 100% given rounding.
Lipoprotein Response to Statin Therapy
Rosuvastatin therapy generally produced large absolute reductions in LDL‐C (median [25th, 75th percentile]=−51.0 [−65.0, −31.0] mg/dL; Figure 2). Conversely, statin therapy resulted in smaller, more variable absolute changes in triglycerides (−17.0 [−48.0, 5.0] mg/dL; Figure 3), large VLDL‐p (−0.3 [−1.7, 0.7] mmol/L; Figure 3), medium VLDL‐p (−0.6 [−6.0, 4.4] mmol/L), small VLDL‐p (−6.9 [−17.4, 2.9] mmol/L; Figure 3), VLDL‐C (−2.6 [−5.9, 0.6] mg/dL; Figure 3), and calculated RC (−3.0 [−10.0, 1.0] mg/dL). Correlation (Spearman r) between absolute change in LDL‐C and VLDL‐related markers was: triglycerides (r=0.15), large VLDL‐p (r=0.16), medium VLDL‐p (r=0.18), small VLDL‐p (r=0.20), VLDL‐C (r=0.29), and formualic RC (r=0.15). Baseline small VLDL‐p and VLDL‐C levels were higher among those with larger reductions in these markers compared with those with more minimal changes (Tables 2 and 3).
Figure 2.
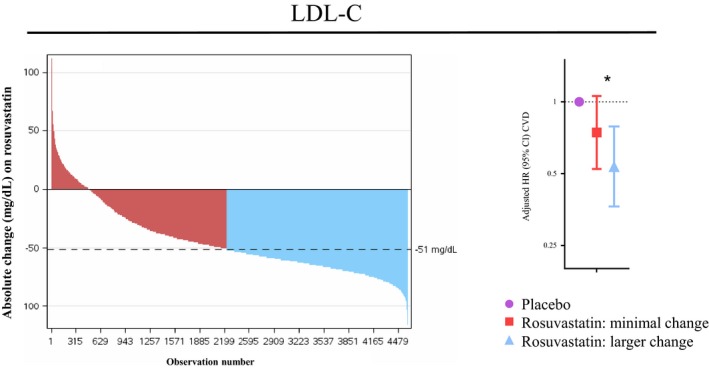
Waterfall plot23 demonstrating the range of individuals’ changes in LDL‐C with rosuvastatin 20 mg daily. Subjects were divided into Minimal Response (red) and Larger Response (blue) groups based on whether they had greater than or less than the median change (a reduction) in LDL‐C. Cox models are adjusted for age, sex, race, smoking, body mass index, systolic blood pressure, fasting glucose, baseline HDL‐C, baseline hsCRP, and baseline LDL‐C and used placebo‐allocated individuals as a reference. *Between‐response group comparison among participants in this substudy (N=9423) P=0.17 and among all JUPITER participants with LDL‐C measure (N=15 546) P=0.01.
Figure 3.
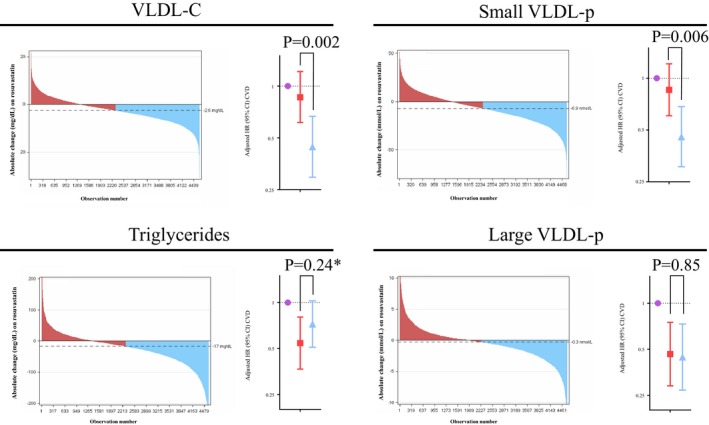
Waterfall plot demonstrating the range of individuals’ absolute changes in VLDL‐C, small and large VLDL‐p, and triglycerides with rosuvastatin 20 mg daily. Subjects were divided into Minimal Response (red) and Larger Response (blue) groups based on whether they had greater than or less than the median reduction in the given marker. Cox models (comparing risk with those in the current substudy allocated to placebo as the reference population) are adjusted for age, sex, race, smoking, body mass index, systolic blood pressure, fasting glucose, baseline HDL‐C, baseline hsCRP, baseline VLDL‐related marker level, and change in LDL‐C. Among those on rosuvastatin, the median (IQR) on‐treatment LDL‐C levels in the Minimal Response and Larger Response groups were 57 (45, 78) and 52 (42.0, 66.0) mg/dL, respectively, in the small VLDL‐p change group and 58 (45, 82) and 51 (45, 65) mg/dL, respectively, in the VLDL‐C change group. *Between‐response group comparison among participants in this stubstudy (N=9423) P=0.24 and among all JUPITER participants with triglyceride measure (N=15 546) P=0.28.
Table 2.
Baseline and Select On‐Treatment Demographic and Biochemical Variables Among Those With Minimal or Larger Change in Small VLDL‐p
| ∆ Small VLDL‐p | ||
|---|---|---|
| Minimal Changea | Larger Changea | |
| Baseline clinical and biochemical variables | ||
| Age, y | 66 (60, 71) | 66 (60, 71) |
| Women | 38.0 | 35.6 |
| Race (white) | 84.7 | 85.5 |
| BMI, kg/m2 | 28.6 (25.6, 32.3) | 28.4 (25.7, 31.8) |
| Hypertension | 59.1 | 54.3 |
| Current smoker | 13.9 | 13.8 |
| Family CAD history | 12.6 | 13.5 |
| Glucose, mg/dL | 95.0 (88.0, 103.0) | 95.0 (88.0, 102.0) |
| hsCRP, mg/L | 4.15 (2.85, 6.85) | 3.95 (2.70, 6.45) |
| LDL‐C, mg/dL | 108.0 (94.0, 119.0) | 110.0 (96.0, 120.0) |
| HDL‐C, mg/dL | 50.0 (41.0, 61.1) | 48.0 (41.0, 59.0) |
| Triglycerides, mg/dL | 117.0 (83.0, 168.0) | 122.0 (91.0, 174.0) |
| On‐statin lipid variables | ||
| LDL‐C, mg/dL | 57.0 (45.0, 78.0) | 52.0 (42.0, 66.0) |
| HDL‐C, mg/dL | 53.0 (43.5, 65.0) | 52.0 (43.0, 64.0) |
| Triglycerides, mg/dL | 106.0 (77.0, 145.0) | 97.0 (75.0, 132.0) |
| Small VLDL‐p (mmol/L)—baseline, change, and on‐statin levels | ||
| Baseline | 17.9 (11.5, 26.3) | 35.8 (26.8, 46.0) |
| Change | 2.8 (−2.3, 9.8) | −17.4 (−25.0, −11.5) |
| 12‐mo | 22.6 (15.3, 32.1) | 16.0 (9.9, 23.6) |
BMI indicates body mass index; CAD, coronary artery disease; HDL‐C indicates high‐density lipoprotein cholesterol; hsCRP high‐sensitivity C‐reactive protein; LDL‐C, low‐density lipoprotein cholesterol; VLDL‐p, VLDL lipoprotein particle concentration.
Median (25th, 75th percentile) or proportion (%).
Table 3.
Baseline and Select On‐Treatment Demographic and Biochemical Variables Among Those With Minimal or Larger Change in VLDL‐C
| ∆ VLDL‐C | ||
|---|---|---|
| Minimal Changea | Larger changea | |
| Baseline clinical and biochemical variables | ||
| Age, y | 66.0 (61.0, 71.0) | 66.0 (60.0, 71.0) |
| Women | 38.6 | 35.0 |
| Race (white) | 84.2 | 86.0 |
| BMI, kg/m2 | 28.5 (25.5, 32.1) | 28.5 (25.8, 31.9) |
| Hypertension | 57.8 | 55.6 |
| Current smoker | 13.7 | 14.0 |
| Family CAD history | 12.6 | 13.6 |
| Glucose, mg/dL | 95.0 (88.0, 102.0) | 95.0 (89.0, 102.0) |
| hsCRP, mg/L | 4.25 (2.90, 7.05) | 3.80 (2.70, 6.15) |
| LDL‐C, mg/dL | 107.0 (92.0119.0) | 111.0 (98.0, 121.0) |
| HDL‐C, mg/dL | 51.0 (41.0, 62.0) | 48.0 (40.0, 58.0) |
| Triglycerides, mg/dL | 113.0 (82.0, 160.0) | 127.0 (94.0, 181.0) |
| On‐statin lipid variables | ||
| LDL‐C, mg/dL | 58.0 (45.0, 82.0) | 51.0 (42.0, 63.0) |
| HDL‐C, mg/dL | 53.0 (43.0, 65.0) | 53.0 (44.0, 63.0) |
| Triglycerides, mg/dL | 108.0 (79.0, 150.0) | 95.0 (74.0, 126.0) |
| VLDL‐C (mg/dL)—baseline, change, and on‐statin levels | ||
| Baseline | 9.2 (6.6, 12.7) | 15.2 (12, 18.9) |
| Change | 0.6 (−1.1, 3.0) | −5.9 (−8.3, −4.1) |
| 12‐mo | 10.6 (7.3, 14.5) | 8.3 (6.0, 11.6) |
BMI indicates body mass index; CAD, coronary artery disease; HDL‐C indicates high‐density lipoprotein cholesterol; hsCRP high‐sensitivity C‐reactive protein; LDL‐C, low‐density lipoprotein cholesterol; VLDL‐p, VLDL lipoprotein particle concentration.
Median (25th, 75th percentile) or proportion (%).
Residual Risk
With on‐statin marker changes, 2 patterns of risk reduction were observed. First, similar to LDL‐C (Figure 2),23 incrementally greater reductions in VLDL‐C and small VLDL‐p (Figure 3) were associated with incrementally greater ASCVD risk reductions. Compared with placebo, the hazard ratio (95% confidence interval) for ASCVD among those with minimal and larger on‐statin changes in VLDL‐C was 0.86 (0.61, 1.21) and 0.44 (0.30, 0.67), respectively, and for small VLDL‐p was 0.86 (0.61, 1.21) and 0.46 (0.31, 0.68), respectively. Differences in risk between the statin response groups remained significant after incrementally adjusting for each individual's on‐statin absolute change in LDL‐C (P=0.002 for VLDL‐C and P=0.006 for small VLDL‐p; Figure 3). Conversely, although statin‐allocated individuals were at lower risk of ASCVD events compared with placebo, there was no incremental risk reduction associated with incrementally greater reductions in triglycerides or large VLDL‐p (P≥0.24; Figure 3), nor for medium VLDL‐p, formulaic RC, nor intermediate‐density lipoprotein (P≥0.35 for all).
In sensitivity analyses, the results were similar when the analysis was performed using percent marker change with randomized statin allocation. Results for apolipoprotein B and non‐HDL‐C resembled those for LDL‐C. Additional adjustment of the main absolute change‐based analysis for baseline LDLc level did not appreciably affect the results. In sensitivity analyses restricted to those confirmed on study drug at 1 year without events preceding to this determination, the patterns of association persisted (Table S1), although, with less power, between‐group differences were no longer statistically significant. Finally, a sensitivity analysis examining changes in LDL‐C and triglycerides in the broader JUPITER population in whom these standard measures were available (N=15 546) revealed significant differences in response groups for LDL‐C (P=0.01), but not triglycerides (P=0.28).
Discussion
Our study was designed to provide insight into whether reductions in VLDLs and their contents are associated with incremental reductions in residual ASCVD risk, beyond reductions in LDL‐C. Similar to LDL‐C,23, 24 we observed a dose‐response reduction in incident ASCVD risk with greater reductions in NMR‐measured VLDL‐C and small VLDL‐p. Change in these lipoproteins was not strongly correlated with change in LDL‐C on‐statin, and the incremental risk reductions associated with reductions in VLDL‐C and small VLDL‐p remained significant after accounting for each participant's change in LDL‐C. Conversely, although allocation to statin was associated with reduced ASCVD risk compared with placebo, there was no further reduction in risk with greater reductions in chemically measured triglycerides nor in NMR‐measured large VLDL‐p/chylomicrons or medium VLDL‐p. Overall, these results support a potentially important role for small VLDL lipoproteins and their associated cholesterol in the development of ASCVD events, and suggest that, beyond reductions in LDL‐C, greater reductions in these particular lipoproteins may possibly confer incremental ASCVD risk reduction. Importantly, risk associated with these smaller atherogenic triglyceride‐rich lipoproteins was not captured by the chemically measured triglyceride level.
The implementation of broad pharmacological‐ and lifestyle‐based preventive strategies targeting LDL‐C has resulted in appreciable reductions in LDL‐C on the population level.1, 2, 3 At the same time, there is histopathological evidence that the composition and morphology of human atherosclerotic plaque has changed in recent years in parallel with these secular trends in LDL‐C reduction,25 compelling a shifting focus toward residual, non‐LDL‐C mediators of ASCVD,26 including VLDL.5, 11 To investigate this potential risk pathway, we performed detailed 1H‐NMR spectroscopy to profile changes in VLDL lipoprotein species and their associated cholesterol and triglycerides in relation to changes in risk. Our results support the hypothesis that VLDL species are important contributors to ASCVD risk, and raise hope that their targeted reduction may possibly further reduce event rates beyond reductions in LDL‐C.
Experimental evidence suggests that VLDLs and their contents could contribute to the pathogenesis of ASCVD, potentially through increased expression of proinflammatory cytokines, adhesion molecules, and coagulation factors, as well as enhanced recruitment and attachment of monocytes to endothelium, induction of endothelial cell apoptosis, and impairment of the anti‐inflammatory properties of HDL.5, 10, 12, 13, 14, 15, 16 Differences in VLDL size and composition may determine their predilection for uptake into the neointima of atheroma.27 In our study, reductions in the smaller VLDL lipoproteins and VLDL‐C, but not in larger VLDL lipoproteins/chylomicrons (nor in plasma triglyceride), were associated with lower risk, consistent with these experimental observations. Given that triglyceride relative to cholesterol content decreases progressively as VLDL size decreases, our findings appear concordant with these past experimental observations.
The finding that greater triglyceride reduction was not associated with incremental risk reduction is of interest, given that this marker has been taken as a surrogate for this lipoprotein family in clinical practice and research. However, although in this substudy greater triglyceride reduction with statin was not associated with improved outcomes, the median on‐statin triglyceride level was only 101 mg/dL. Meta‐analysis of clinical trials of triglyceride‐lowering, including ACCORD (Action to Control Cardiovascular Risk in Diabetes), FIELD (Fenofibrate Intervention and Event Lowering in Diabetes), and others, demonstrated that patients with low levels of triglyceride (<200 mg/dL) may not derive benefit from further reduction; however, individuals with elevated baseline levels (>200 mg/dL) might experience reductions in ASCVD risk with triglyceride lowering.28 Thus, our findings are consistent with past analyses and likely do not carry negative implications for ongoing and planned triglyceride‐reduction–based clinical trials, such as REDUCE‐IT (Reduction of Cardiovascular Events Outcomes), STRENGTH (Study to Assess Statin Residual Risk Reduction With Epanova in High CV Risk Patients With Hypertriglyceridemia), and PROMINENT (Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides IN Patients With Diabetes), in which patient inclusion is limited to those with triglyceride levels 200 to 499 mg/dL. Rather, the findings related to small VLDL species suggest that even among individuals in whom targeted triglyceride reduction has historically failed to reduce events (those without elevated triglyceride), some members of this lipoprotein family might remain as important residual risk targets for alternative approaches. Overall, these results suggest that a more granular understanding of an individual's circulating lipid/lipoprotein milieu could be required to successfully identify and treat the spectrum of relevant triglyceride‐related mediators in this residual ASCVD risk pathway. The results also support profiling of broader lipid/lipoprotein changes in response to statin therapy as independent markers of individual benefit from statin therapy, and could support more individualized approaches to statin administration.
The results should be viewed in the context of the study design. To our knowledge, this is the first study to examine whether pharmacological reductions in 1H‐NMR‐profiled VLDL lipoprotein particles may reduce residual risk. However, given the relatively low event rates in this substudy, our results should be viewed as hypothesis‐generating and it is possible that power to detect smaller differences between treatment response groups may have been limited. It is possible that larger studies could enable between‐group comparisons defined by greater separation of treatment responses (focusing on more “extreme” biomarker groups29) and could detect significant between‐group differences. Furthermore, we used the variable effects of statins to “model” the effects that dedicated reductions in VLDL lipoproteins might have on ASCVD events. Given that the mechanism of statin‐mediated VLDL and triglyceride change is incompletely understood, it remains uncertain whether dedicated pharmacological therapies to reduce small remnant VLDLs would confer incremental benefit. Selection criteria utilized in JUPITER (LDL‐C <130 mg/dL, high‐sensitivity C‐reactive protein ≥2.0 mg/L, and triglyceride <500 mg/dL) might limit external generalizability, particularly the exclusion of individuals with medication‐treated diabetes mellitus or high triglycerides, given that VLDLs have been hypothesized to be particularly relevant disease mediators in this latter population. Additionally, although multiple statistical tests are performed herein, many are performed on correlated biomarkers as supportive sensitivity analyses, the results of which aligned with those of the primary analysis. Furthermore, the findings herein are supported by previous biological and epidemiological studies from multiple cohorts.30 Finally, although the JUPITER trial was initially randomized, the current findings represent a secondary analysis and therefore should be considered observational in nature. Overall, these hypothesis‐generating findings warrant further study.
In conclusion, pharmacological reductions in VLDL and their associated cholesterol may potentially provide incremental ASCVD residual risk reduction among individuals with adequate LDL‐C lowering on statin. Some of these atherogenic lipoproteins may lie outside the scope of what is reflected by chemically measured triglyceride levels.
Sources of Funding
During the study period, Lawler received support from NIH T32 (HL007575), NIH LRP, Brigham and Women's Hospital, the Peter Munk Cardiac Centre, and the Heart and Stroke/Richard Lewar Centre of Excellence. Akinkoulie receives support from NIH T32 (HL007575). The research for this article was supported by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health under award numbers R01HL117861 and R01HL134811 to Mora, as well as by the Molino Family Trust. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The JUPITER trial was funded by AstraZeneca, who had no role in the funding, design, and execution of this secondary study. LipoScience (now LabCorp; Raleigh, NC) performed the NMR lipoprotein analysis for this study at no additional cost.
Disclosures
Mora received research grant support from Atherotech Diagnostics for research outside the current work and served as a consultant to Amgen, Lilly, Pfizer, and Quest Diagnostics and has a pending patent for the use of an NMR‐measured nonlipoprotein biomarker for colorectal cancer prediction. Glynn received research grant support from AstraZeneca. Ridker received research grant support from AstraZeneca, Novartis, Amgen, Kowa, Pfizer, and NHLBI and is listed as a co‐inventor on patents held by the Brigham and Women's Hospital related to the use of inflammatory biomarkers in cardiovascular disease (licensed to AstraZeneca and Siemens). All other authors report no disclosures.
Supporting information
Table S1. Adjusted* HR (95% CI) for the primary CVD endpoint among those achieving an absolute reduction greater than or less than the median on rosuvastatin 20 mg daily relative to placebo (reference), examining event rates after 1 year among those confirmed to be on statin (N=8420; 130 events) compared with the overall cohort.
(J Am Heart Assoc. 2017;6:e007402 DOI: 10.1161/JAHA.117.007402.)29223956
References
- 1. Carroll MD, Kit BK, Lacher DA, Shero ST, Mussolino ME. Trends in lipids and lipoproteins in US adults, 1988–2010. JAMA. 2012;308:1545–1554. [DOI] [PubMed] [Google Scholar]
- 2. Kuklina EV, Yoon PW, Keenan NL. Trends in high levels of low‐density lipoprotein cholesterol in the United States, 1999–2006. JAMA. 2009;302:2104–2110. [DOI] [PubMed] [Google Scholar]
- 3. Rosinger A, Carroll MD, Lacher D, Ogden C. Trends in total cholesterol, triglycerides, and low‐density lipoprotein in US adults, 1999–2014. JAMA Cardiol. 2017;2:339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R; Cholesterol Treatment Trialists’ (CTT) Collaborators . Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90 056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. [DOI] [PubMed] [Google Scholar]
- 5. Rosenson RS, Davidson MH, Hirsh BJ, Kathiresan S, Gaudet D. Genetics and causality of triglyceride‐rich lipoproteins in atherosclerotic cardiovascular disease. J Am Coll Cardiol. 2014;64:2525–2540. [DOI] [PubMed] [Google Scholar]
- 6. Nordestgaard BG. Triglyceride‐rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res. 2016;118:547–563. [DOI] [PubMed] [Google Scholar]
- 7. Varbo A, Nordestgaard BG. Remnant cholesterol and triglyceride‐rich lipoproteins in atherosclerosis progression and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2016;36:2133–2135. [DOI] [PubMed] [Google Scholar]
- 8. Musunuru K, Kathiresan S. Surprises from genetic analyses of lipid risk factors for atherosclerosis. Circ Res. 2016;118:579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626–635. [DOI] [PubMed] [Google Scholar]
- 10. Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, Goldberg AC, Howard WJ, Jacobson MS, Kris‐Etherton PM, Lennie TA, Levi M, Mazzone T, Pennathur S; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease . Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–2333. [DOI] [PubMed] [Google Scholar]
- 11. Khetarpal SA, Rader DJ. Triglyceride‐rich lipoproteins and coronary artery disease risk: new insights from human genetics. Arterioscler Thromb Vasc Biol. 2015;35:e3–e9. [DOI] [PubMed] [Google Scholar]
- 12. Liu L, Wen T, Zheng XY, Yang DG, Zhao SP, Xu DY, Lu GH. Remnant‐like particles accelerate endothelial progenitor cells senescence and induce cellular dysfunction via an oxidative mechanism. Atherosclerosis. 2009;202:405–414. [DOI] [PubMed] [Google Scholar]
- 13. Norata GD, Grigore L, Raselli S, Redaelli L, Hamsten A, Maggi F, Eriksson P, Catapano AL. Post‐prandial endothelial dysfunction in hypertriglyceridemic subjects: molecular mechanisms and gene expression studies. Atherosclerosis. 2007;193:321–327. [DOI] [PubMed] [Google Scholar]
- 14. Shin HK, Kim YK, Kim KY, Lee JH, Hong KW. Remnant lipoprotein particles induce apoptosis in endothelial cells by NAD(P)H oxidase‐mediated production of superoxide and cytokines via lectin‐like oxidized low‐density lipoprotein receptor‐1 activation: prevention by cilostazol. Circulation. 2004;109:1022–1028. [DOI] [PubMed] [Google Scholar]
- 15. Ting HJ, Stice JP, Schaff UY, Hui DY, Rutledge JC, Knowlton AA, Passerini AG, Simon SI. Triglyceride‐rich lipoproteins prime aortic endothelium for an enhanced inflammatory response to tumor necrosis factor‐alpha. Circ Res. 2007;100:381–390. [DOI] [PubMed] [Google Scholar]
- 16. Patel S, Puranik R, Nakhla S, Lundman P, Stocker R, Wang XS, Lambert G, Rye KA, Barter PJ, Nicholls SJ, Celermajer DS. Acute hypertriglyceridaemia in humans increases the triglyceride content and decreases the anti‐inflammatory capacity of high density lipoproteins. Atherosclerosis. 2009;204:424–428. [DOI] [PubMed] [Google Scholar]
- 17. Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Borén J, Catapano AL, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Nordestgaard BG, Ray KK, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg‐Hansen A, Watts GF; European Atherosclerosis Society Consensus Panel . Triglyceride‐rich lipoproteins and high‐density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Isley WL, Miles JM, Patterson BW, Harris WS. The effect of high‐dose simvastatin on triglyceride‐rich lipoprotein metabolism in patients with type 2 diabetes mellitus. J Lipid Res. 2006;47:193–200. [DOI] [PubMed] [Google Scholar]
- 19. Verseyden C, Meijssen S, Cabezas MC. Effects of atorvastatin on fasting plasma and marginated apolipoproteins B48 and B100 in large, triglyceride‐rich lipoproteins in familial combined hyperlipidemia. J Clin Endocrinol Metab. 2004;89:5021–5029. [DOI] [PubMed] [Google Scholar]
- 20. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ; JUPITER Study Group . Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 21. Mora S, Glynn RJ, Boekholdt SM, Nordestgaard BG, Kastelein JJ, Ridker PM. On‐treatment non‐high‐density lipoprotein cholesterol, apolipoprotein B, triglycerides, and lipid ratios in relation to residual vascular risk after treatment with potent statin therapy: JUPITER (justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin). J Am Coll Cardiol. 2012;59:1521–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Varbo A, Nordestgaard BG. Remnant cholesterol and ischemic heart disease. Curr Opin Lipidol. 2014;25:266–273. [DOI] [PubMed] [Google Scholar]
- 23. Ridker PM, Mora S, Rose L; Group JTS . Percent reduction in LDL cholesterol following high‐intensity statin therapy: potential implications for guidelines and for the prescription of emerging lipid‐lowering agents. Eur Heart J. 2016;37:1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, Macfadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ; JUPITER Trial Study Group . Reduction in C‐reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–1182. [DOI] [PubMed] [Google Scholar]
- 25. van Lammeren GW, den Ruijter HM, Vrijenhoek JE, van der Laan SW, Velema E, de Vries JP, de Kleijn DP, Vink A, de Borst GJ, Moll FL, Bots ML, Pasterkamp G. Time‐dependent changes in atherosclerotic plaque composition in patients undergoing carotid surgery. Circulation. 2014;129:2269–2276. [DOI] [PubMed] [Google Scholar]
- 26. Libby P, Pasterkamp G. Requiem for the ‘vulnerable plaque’. Eur Heart J. 2015;36:2984–2987. [DOI] [PubMed] [Google Scholar]
- 27. Brecher P, Chobanian AV, Small DM, Van Sickle W, Tercyak A, Lazzari A, Baler J. Relationship of an abnormal plasma lipoprotein to protection from atherosclerosis in the cholesterol‐fed diabetic rabbit. J Clin Invest. 1983;72:1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sacks FM, Carey VJ, Fruchart JC. Combination lipid therapy in type 2 diabetes. N Engl J Med. 2010;363:692–694; author reply, 694–695. [DOI] [PubMed] [Google Scholar]
- 29. Lawler PR, Akinkuolie AO, Ridker PM, Sniderman AD, Buring JE, Glynn RJ, Chasman DI, Mora S. Discordance between circulating atherogenic cholesterol mass and lipoprotein particle concentration in relation to future coronary events in women. Clin Chem. 2017;63:870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lawler PR, Akinkoulie AO, Chu AY, Shah SV, Krauss W, Craig D, Glynn R, Chasman DI, Ridker PM, Mora S. Atherogenic lipoprotein determinants of incident cardiovascular disease and residual risk among individuals with low LDL‐C. J Am Heart Assoc. 2017;6:e005549 DOI: 10.1161/JAHA.117.005549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Adjusted* HR (95% CI) for the primary CVD endpoint among those achieving an absolute reduction greater than or less than the median on rosuvastatin 20 mg daily relative to placebo (reference), examining event rates after 1 year among those confirmed to be on statin (N=8420; 130 events) compared with the overall cohort.