

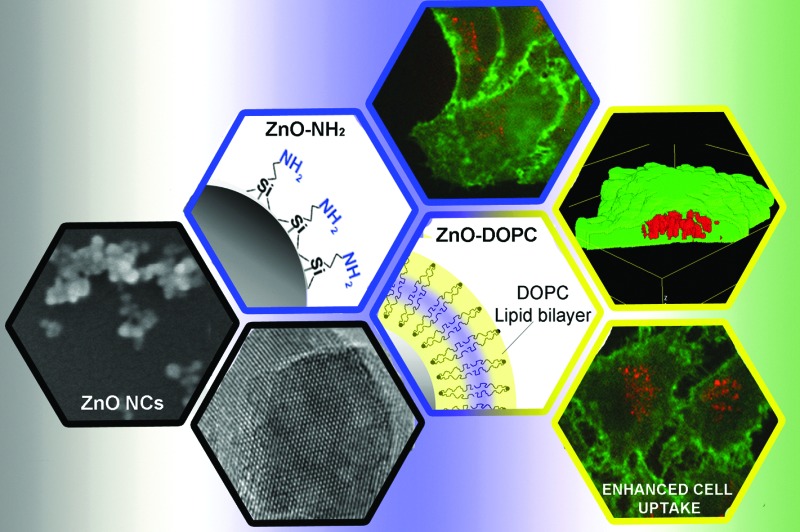
 The surface chemistry and charge of zinc oxide nanocrystals influence their behaviour in biological fluids. A novel lipid bilayer assembly is developed to shield ZnO nanocrystals improving their stability and cell internalization.
The surface chemistry and charge of zinc oxide nanocrystals influence their behaviour in biological fluids. A novel lipid bilayer assembly is developed to shield ZnO nanocrystals improving their stability and cell internalization.
Abstract
The widespread use of ZnO nanomaterials for biomedical applications, including therapeutic drug delivery or stimuli-responsive activation, as well as imaging, imposes a careful control over the colloidal stability and long-term behaviour of ZnO in biological media. Moreover, the effect of ZnO nanostructures on living cells, in particular cancer cells, is still under debate. This paper discusses the role of surface chemistry and charge of zinc oxide nanocrystals, of around 15 nm in size, which influence their behaviour in biological fluids and effect on cancer cells. In particular, we address this problem by modifying the surface of pristine ZnO nanocrystals (NCs), rich of hydroxyl groups, with positively charged amino-propyl chains or, more innovatively, by self-assembling a double-lipidic membrane, shielding the ZnO NCs. Our findings show that the prolonged immersion in simulated human plasma and in the cell culture medium leads to highly colloidally dispersed ZnO NCs only when coated by the lipidic bilayer. In contrast, the pristine and amine-functionalized NCs form huge aggregates after already one hour of immersion. Partial dissolution of these two samples into potentially cytotoxic Zn2+ cations takes place, together with the precipitation of phosphate and carbonate salts on the NCs’ surface. When exposed to living HeLa cancer cells, higher amounts of lipid-shielded ZnO NCs are internalized with respect to the other samples, thus showing a reduced cytotoxicity, based on the same amount of internalized NCs. These results pave the way for the development of novel theranostic platforms based on ZnO NCs. The new formulation of ZnO shielded with a lipid-bilayer will prevent strong aggregation and premature degradation into toxic by-products, and promote a highly efficient cell uptake for further therapeutic or diagnostic functions.
Introduction
The use of zinc oxide (ZnO) based nanoplatforms in biomedical applications is constantly increasing over time, thanks to several useful therapeutic and diagnostic functions of ZnO.1–6 Among them, photodynamic properties and rapid dissolution enhancing cytotoxicity or drug delivery were tested toward cancer cells,7 as well as green fluorescent emission strategies for bioimaging purposes.8 The current challenges for using ZnO as a theranostic tool include tailoring its nanoscale morphology, in terms of size, shape,9 aspect ratio,9–13 surface area,12 and most importantly its surface chemistry,14 to meet the desired biomedical application. For this reason, it is a prerequisite to have stable and colloidally dispersed ZnO in aqueous biological media. Actually, nano-sized and well-dispersed particles should have dimensions comparable to naturally occurring biological molecules, allowing their internalization into cells15 and enabling them to act as bio-imaging tools or to potentially affect the cellular behaviour in a therapeutic perspective.
It has been largely demonstrated from in vivo experiments that the size and surface chemistry of nanoparticles govern their site of accumulation and action in living organisms.15 In the same way, it is known that the main cytotoxicity mechanisms of ZnO are related to the capability of the nanostructure to dissolve and to be internalized by cells.16,17 Since cell uptake is significantly influenced by the surface chemistry and size of ZnO nanoparticles, it turns out that the precise control of both parameters is fundamental.
Concerning the surface chemistry, pristine ZnO nanoparticles’ surface exposes neutral hydroxyl groups, which basically govern the surface charge behaviour.13,18 In a high pH aqueous medium, the chemisorbed protons (H+) leave the NP's outer layer inducing a negatively charged surface with partially bonded oxygen atoms (ZnO–). On the other side, for low pH values, protons from the medium are most likely transferred to the particle surface, leading to positively charged ZnOH+2 groups.19 ZnO nanoparticles, characterized by an isoelectric point around pH 9–10, have a positive surface charge20 under physiological conditions. Cancer cells usually have high negative membrane potentials due to the presence of anionic phospholipids, charged proteins and carbohydrates21 on their outer plasma membrane.22,23 Therefore, electrostatic interactions with positively charged ZnO nanoparticles would be expected to enhance the uptake of ZnO nanoparticles into tumor cells, thus affecting their viability.24
Charge and surface chemistry influence also the colloidal stability of nanoparticles, a crucial property for their interaction with biological systems. The presence of capping agents or coatings enables a precise control in this regard.11 Different approaches were used to enhance biocompatibility and decrease nanoparticles’ aggregation, based on polymeric coatings, such as polyethylene glycol (PEG),25 propylene glycol,10 poly-N-vinyl-2-pyrrolidone (PVP) and others.26 However, the literature has also shown the effect of some of these stabilizers on the cytotoxicity of ZnO NPs.27
In general, several mechanisms of ZnO NPs’ cytotoxicity were proposed. Some of them include ZnO nanoparticles’ internalization inducing reactive oxygen species (ROS)28,29 and DNA damage,14,30 zinc ion release due to nanoparticles’ dissolution,31–35 and membrane dysfunction.25,36 Nevertheless, in most cases, it is unclear if ZnO dissolution occurs in the extracellular medium before cellular uptake or intracellularly after the internalization. Certain works indicate that the ZnO cytotoxicity is independent of the concentration of extracellular soluble Zn2+ in the culture medium37–39 and that a direct particle–cell contact or internalization is required for cellular toxicity.40
In agreement with other authors,41 we believe that the poor accordance between the literature results derives from a lack of control of the synthetic process, together with a large uncertainty about the surface chemistry and charge, as well as the wide size distribution of the NPs. To address these issues, in the present paper we propose a systematic approach to study the ZnO colloidal stability and cytotoxicity as a function of different and controlled surface treatments. We designed three types of ZnO nanocrystals (NCs) with different surface charge densities and chemistries: (i) bare ZnO NCs rich in hydroxyl groups, (ii) amino-propyl surface-functionalized ZnO NCs aiming at a predominant positive surface charge, and (iii) lipid-coated ZnO NCs for improving colloidal and chemical stability in biological media. We studied and tuned the colloidal stability in terms of long-lasting well-dispersed ZnO NC suspensions in different simulated body fluids for both in vitro and in vivo applications. In particular, we tested the three types of ZnO NCs in the cell culture medium and in a buffered solution mimicking the inorganic composition of the human plasma.
We observed a profound effect of the ZnO NCs’ surface properties on the cellular internalization and cytotoxicity profiles in in vitro cancer cells. Therefore, the precise control of ZnO nanoparticles’ surface is crucial to customize and develop new opportunities in cancer diagnosis and therapies. In detail, we demonstrate that engineering the surface of ZnO NCs with a shielding lipidic bilayer leads to a profound enhancement of the colloidal and chemical stability in biological and simulated media. Moreover, these properties are related to a reduced cytotoxicity and to an appreciable enhancement of cellular internalization of this novel hybrid nanoconstruct.
Our results are therefore of paramount importance to further improve advanced nano-imaging solutions and to design alternative therapeutic plans through the use of ZnO NCs when administered to cancer cells.
Experimental
Synthesis of ZnO nanocrystals
Pristine zinc oxide nanocrystals (ZnO NCs) were synthetized by a wet chemical method using zinc acetate dihydrate (Zn(CH3COO)2·H2O) and sodium hydroxide (NaOH) as precursors and methanol as a solvent. In detail, 0.818 g (3.73 mmol) of Zn(CH3COO)2·H2O was dissolved in 42 mL of methanol in a 100 mL round bottom flask and heated to 60 °C under vigorous stirring. When the temperature reached 60 °C, 318 μL of bi-distilled water (from a Direct Q3 system, Millipore) and a solution of 0.289 g (7.22 mmol) of NaOH in 23 mL of methanol were dropwise added to the zinc acetate solution. The resulting synthesis mixture was maintained, under continuous stirring, at 60 °C for 2.15 h and then washed two times with fresh ethanol using a repeated centrifugation–redispersion process.
Preparation of ZnO–NH2 nanocrystals
The amino-propyl functionalized sample (ZnO–NH2 NCs) was obtained by the reaction of pristine zinc oxide nanocrystals and 3-aminopropyltrimethoxysilane (APTMS) as follows: 100 mg (1.23 mmol) of ZnO NCs, dispersed in ethanol, was heated to 78 °C in a 100 mL flask under continuous stirring and a nitrogen gas flow. After about 15 minutes, 21.4 μL of APTMS was added to the solution; this volume of the functionalizing agent corresponds to 0.123 mmol (22.05 g), equal to 10 mol% of total ZnO amount. The obtained mixture was refluxed under a nitrogen gas flow for 6 h and then washed two times to remove the unbound APTMS molecules.
Preparation of ZnO–DOPC nanocrystals
To prepare lipid-coated nanocrystals (ZnO–DOPC NCs), a solvent exchange method was used. 2.5 mg of DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) in chloroform was dried under vacuum overnight and re-dispersed in a 1 mL mixture of 40 vol% ethanol and 60 vol% bi-distilled water. Afterwards, a volume corresponding to 1 mg of ZnO NCs was centrifuged, in order to remove all the ethanol, and then the pellet was suspended in 100 μL of lipid solution. The addition of water (700 μL) results in the formation of a supported lipid bilayer on the surface of ZnO NCs. The obtained ZnO–DOPC NCs were washed with water two times to remove the unbound lipids. For the bio-stability assays, the whole procedure was scaled-up to obtain 100 mg of ZnO–DOPC NCs. For lipid-coated nanocrystals used in the fluorescence experiments, two labelling steps were added: liposomes were labeled with Bodipy-FL-DHPE by adding 0.5 μg of fluorescent dye at 2.5 mg of lipids before evaporating the chloroform. Amino-propyl functionalized nanocrystals (ZnO–NH2 NCs) were coupled with ATTO550-NHS ester dye, at a ratio of 2 μg per mg of NCs. The suspension of dye and ZnO–NH2 NCs in ethanol was kept in the dark under continuous stirring overnight and then the sample was washed two times with fresh ethanol in order to remove the unbound dye molecules.
Bio-stability assays
For the bio-stability assays, the three types of ZnO nanocrystals were tested in Simulated Body Fluid (SBF), and cell culture medium (EMEM) completed with 10% of fetal bovine serum.
To prepare the SBF, the following reagents were dissolved in 500 mL of bi-distilled water: 7.996 g NaCl, 0.350 g NaHCO3, 0.224 g KCl, 0.228 g K2HPO4·3H2O, 0.305 g MgCl2·6H2O, 0.278 g CaCl2, 0.071 g Na2SO4, 40 mL HCl 1 M, and 6.057 g NH2C(CH2OH)3 (TRIS). The pH of the solution was then regulated at 7.4 at 37 °C and the solution was made up to 1.0 L and stored at 4 °C until use.
The bio-stability tests were performed at a concentration of 2 mg mL–1, by suspending 25 mg of each ZnO NC sample (opportunely separated from ethanol or water with a centrifugation and washing step) in 12.5 mL of SBF or EMEM pre-heated at 37 °C. The samples were maintained under continuous stirring (150 rpm) at a constant temperature of 37 °C. At selected times (1, 24, 72 h and 25 days), 2.5 mL of the suspension was collected and centrifuged. The supernatant was collected and opportunely diluted for ICP-MS analysis, whereas the ZnO NCs were washed with bi-distilled water twice and characterized.
Physico-chemical characterization
The particle size and Zeta potential of the three samples were determined using dynamic light scattering (Zetasizer Nano ZS90, Malvern). DLS measurements were performed by suspending 500 μg of nanoparticles in 1 mL of various tested media (ethanol, water, SBF or EMEM). Zeta potential measurements were performed in bi-distilled water and the pH was adjusted with HCl and NaOH 1 M.
The morphology of the nanocrystals was studied by Field Emission Scanning Electron Microscopy (FESEM, Auriga and Merlin, Karl Zeiss) and Transmission Electron Microscopy (TEM, Tecnai F20ST, FEI) before and after the bio-stability assays. The samples were prepared by diluting 5 μL of nanocrystals’ suspension in bi-distilled water (final concentration 25 μg mL–1) and drying a drop of the resulting suspension on a silica wafer or on a holey carbon-coated copper grid for FESEM and TEM, respectively.
Fourier transform infrared spectra (FT-IR, recorded in transmission mode with a Bruker Equinox 55 in the region of 4000–400 cm–1) and energy dispersive spectra (EDS, Oxford Instruments, coupled to the FESEM Merlin, Karl Zeiss) were recorded in order to verify the success of functionalization procedures and to establish the precipitation of organic and inorganic compounds and their nature during bio-stability assays.
The formation of the supported lipid bilayer on the surface of ZnO–DOPC samples was also confirmed by fluorescence co-localization experiments. A fully motorized wide-field inverted microscope (Eclipse TiE from Nikon) equipped with a high resolution sCMOS camera (Zyla 4.2 Plus from Andor) was used with an immersion-oil 60× objective.
The crystalline structure of the material was analysed by X-ray diffraction using an X'Pert diffractometer with θ–2θ Bragg–Brentano configuration using Cu-Kα radiation (λ = 1.54 Å, 40 kV and 30 mA). The spectra were collected in the range of 20–60° after each bio-stability assay step, in order to verify the preservation of nanoparticles’ crystalline structure and the presence of crystalline precipitates.
Internalization experiments in HeLa cells
For cell experiments, HeLa cells (ATCC® CCL-2) were cultivated in DMEM supplemented with 10 vol% FBS and 1 vol% PenStrep. All reagents were purchased from Sigma-Merck.
The day before particle incubation the cells were seeded onto 8-well ibiTreat slides (Ibidi) at a concentration of 5000 cells per well containing 300 μL of medium. They were incubated with the NCs at a final concentration of 18 μg mL–1 for 24, 48 and 72 h. NCs were prepared and labelled as described above, only replacing Atto550 by Atto633 NHS ester dye. After incubation for 24–72 h, the cell membrane was stained immediately before imaging using WGA488 (Thermofisher Scientific) and washed with the medium after an incubation period of about 1 minute. Cells and particles were imaged at 37 °C under a 5% CO2 humidified atmosphere using spinning disc microscopy (Zeiss Cell Observer SD utilizing a Yokogawa spinning disk unit CSU-X1). The objective was a 1.40 NA 63× Plan apochromat oil immersion objective (Zeiss). Atto633 was imaged with 639 nm and WGA488 with 488 nm laser excitation. For two colour detection a dichroic mirror (560 nm, Semrock) and band-pass filters 525/50 and 690/60 (both Semrock) were used in the detection path. Separate images for each fluorescence channel were acquired using two separate electron multiplier charge coupled device (EMCCD) cameras (Photometrics Evolve TM).
The stacks of confocal fluorescence images of cells interacting with nanoparticles were analysed applying the Particle_in_Cell-3D plugin based on the ImageJ software and free to download at the ImageJ Documentation Portal.42 For each experimental condition, 13 cells were randomly selected from the confocal z-stacks and independently analysed. The fluorescence image of the cell membrane in each confocal plane was transformed into a 3D mask of the cell.
By applying this mask to the corresponding particle fluorescence images, intra- and extra-cellular nanoparticles were automatically discriminated. To get a semiquantitative result, for each cell the sum of all pixel intensities corresponding to the intracellular nanoparticle fluorescence signal was finally calculated and then averaged over all cells corresponding to the same experimental condition. The fluorescence intensity threshold of nanoparticles was kept constant for all the stacks.
Cytotoxicity experiments
HeLa cells were seeded at a concentration of 5000 cells per well into 96 well plates (Corning) containing a final volume of 100 μL of medium. One day after seeding they were incubated with NCs at the desired concentration. 72 h after incubation with NCs, MTT tests were performed according to the standard protocol.
Results and discussion
Zinc oxide nanocrystals
For this study, we selected a wet chemical route for obtaining small ZnO nanocrystals (ZnO NCs) with a spherical shape, as reported in detail in the Experimental section. A fast synthetic approach, modified from that already reported in the literature,43 was applied involving zinc acetate dihydrate and sodium hydroxide in a molar ratio Zn2+ : OH– of 1 : 1.94 and in polar methanol solvent at mild temperature (60 °C) according to the following chemical reactions:Zn2+ + 2OH– → Zn(OH)2Zn(OH)2 + 2OH– → Zn(OH)42–Zn(OH)42– → ZnO + H2O +2OH–We obtained spherical shaped, well-dispersed nanocrystals having a size of about 14 nm in diameter (Fig. 1a, measured from Field Emission Scanning Electron Microscopy (FESEM) with the application of a thin Pt conductive layer.). Both Transmission Electron Microscopy (TEM) and Scanning Transmission Electron Microscopy (STEM) also confirmed the round shape of the ZnO NCs and their size (Fig. 1a). The literature indicates indeed to maintain the Zn2+ : OH– molar ratio between 1 : 1.6 and 1 : 1.95 to obtain ZnO particles with a diameter below 15 nm. The X-ray diffraction pattern (Fig. S1 in the ESI†) shows that the ZnO NCs are in the single-phase wurtzite crystalline structure (according to the JCPDS 36-1451, hexagonal, space group P63mc). The Debye–Scherrer equation applied to the broad diffraction peaks confirms the average size of nanocrystallites, being around 15 nm. After functionalization with either aminopropyl groups (ZnO–NH2 in Fig. 1b) or the lipid layer (ZnO–DOPC in Fig. 1c), the morphology and the crystalline structure of the NCs are not modified (Fig. S1 in the ESI†), except in the size of the ZnO–DOPC sample, which presents larger particles of around 20 nm.
Fig. 1. The three ZnO nanocrystals used in this study: (a) pristine ZnO NCs, (b) amine-propyl functionalized ZnO–NH2 NCs, and (c) lipid-coated ZnO–DOPC NCs. From left to right: scheme of the particles, FESEM, TEM and STEM images for each NC type. For FESEM images all the NCs were coated by a thin layer of Pt. The scale bar is 50 nm in all FESEM and TEM images, whereas it is 20 nm in all the three STEM images (right column).

We can suppose the formation of a supported lipid bilayer of a few nm in thickness on the surface of ZnO NCs, as previously reported by some of us using the solvent-exchange methods applied to a lipid solution in the presence of mesoporous silica nanoparticles.44 The approach relies on the effects that a solvent has on the self-assembly behaviour of lipids. Since the interaction between lipid molecules and alcoholic solutions (ethanol or isopropanol) is favourable, the lipids are solvated and exist as monomers at low water concentrations. However, by adding water to the solution, the lipids begin to self-assemble into liposomes, constituted by a lipid bilayer.45 In this arrangement, the hydrophobic tails of the lipids are protected from water. A similar behaviour is observed in the presence of the surface of a nanoparticle, which offers a favourable support for the lipid bilayer self-assembly.44
The successful functionalization procedure of pristine ZnO, leading to ZnO–NH2 and ZnO–DOPC, is supported by both Fourier Transform-InfraRed (FT-IR) and Z-potential analyses. The FT-IR spectra (Fig. 2a) of ZnO and ZnO–NH2 nanocrystals show in general some common features among the two different ZnO samples, such as an intense mode at 440 cm–1 typical of Zn–O vibration and a broad band from 3600 to 3200 cm–1, which is due to the stretching vibration of hydroxyl groups on the ZnO surface. In addition, the two spectra show with different intensities the C O and C–O vibration modes at 1570 and 1420 cm–1, as well as the symmetric and antisymmetric stretching of –CH2 and –CH3 groups at 2860 and 2925 cm–1, respectively. These vibrations are attributed to residual acetate groups on the ZnO surface due to the precursors used or to methoxy groups derived from the reflux conditions, used in the synthetic procedure.46 However, in the case of ZnO–NH2 NCs, the 2860 and 2925 cm–1 stretching modes are more intense because of the presence of the propyl chain of the amine-functional group, thus confirming the successful functionalization of the ZnO surface. The ZnO–NH2 NCs’ spectrum also shows an absorption peak near 3200 cm–1, characteristic of primary amines, and new bands at 1020 and 1100 cm–1, corresponding to the symmetric and asymmetric stretching of the O–Si–O groups, respectively, present in the aminopropyl-trimethoxysilane (APTMS) chain. Moreover, the band at 3800 cm–1 and the one from 3600 to 3400 cm–1, due to the stretching vibrations of hydroxyl groups on the ZnO surface, are less pronounced in the ZnO–NH2 sample with respect to the pristine ZnO. Since the APTMS moiety links through hydroxyl groups to the oxide surface (Zn–OH), leading to Zn–O–SiR bonds, the above-mentioned-observation further confirms the successful amine functionalization of our ZnO NCs.
Fig. 2. (a) Fourier Transform-InfraRed (FT-IR) spectra and (b) Z-potential measurements of the pristine ZnO and functionalized ZnO–NH2 and ZnO–DOPC NCs.

The formation of a phospholipidic layer on the ZnO surface was confirmed by the presence of intense peaks at 2860, 2925 and 1750 cm–1 in the ZnO–DOPC spectrum. These peaks are assigned to the stretching vibrations of –CHx and C O groups, respectively, present in the phospholipid hydrophobic tails. Moreover, the characteristic stretching of P O and P–O appeared at 1100 and 1230 cm–1. The disappearance of the –OH broad band from 3600 to 3200 cm–1 and the attenuation of the Zn–O bond vibration peak at 440 cm–1 further confirm the success of ZnO interaction with DOPC molecules.
The Z-potential (Fig. 2b) of the three samples was evaluated in water at different pH values, obtaining different behaviour and isoelectric points (IEP) depending on the surface properties of the ZnO NCs and thus on the surface functionalization. For pristine ZnO, the hydroxyl groups at the surface can protonate at acidic pH, forming ZnOH2+ species, whereas at highly basic pH, ZnO– species are formed.20 The measured isoelectric point (IEP) for pristine ZnO is at pH 9.85, in good agreement with the literature values.20 Therefore, at lower pH, and in particular at neutral pH, interesting for biological applications, the pristine ZnO NCs show a positive Z-potential.
Higher positive values of Z-potential are even observed for the ZnO–NH2 NCs: actually, at acidic pH the amine groups at the ZnO surface can protonate, forming –NH3+ species and the IEP of these amine-functionalized NCs is shifted to pH 11. In contrast, an important shift to lower Z-potential values is obtained for the ZnO–DOPC sample, having an IEP at pH 7.8. The DOPC phospholipid, showing both a negative phosphate and a positive amine head group (see the molecule scheme in Fig. S2 of the ESI†), neutralizes the surface charge of ZnO and flattens its Z-potential behaviour, with slightly positive values at acidic pH and slightly negative values at basic pH. The comparison of the Z-potential behaviour of ZnO–NH2 and ZnO–DOPC with that of pristine ZnO gives a clear confirmation that both chemical functionalization and coverage by phospholipids of ZnO NCs worked successfully.
It is worth mentioning that, since ZnO is prone to degradation at acidic pH, here every measurement at each pH value was followed by a DLS check, still showing a good count rate and detecting the presence of the nanocrystals.
Further evidence of the lipid bilayer formation of the ZnO NCs is obtained by fluorescence microscopy co-localization experiments in wide-field configuration (Fig. 3). The DOPC lipid formulation was conjugated with 1% Bodipy-DHPE lipid, showing fluorescence excitation at around 488 nm (Fig. 3a). Additionally, ZnO NCs functionalized with amino-propyl groups can be efficiently coupled with Atto550-NHS ester dye, having fluorescence excitation at 550 nm (Fig. 3b). By merging the two distinct images, it is worth noting that all the bright spots in both green and red channels co-localize in the same position (Fig. 3c and Fig. S3 in the ESI†). Different experiments were carried out, showing a high percentage of red-labelled NCs co-localizing with the green-labelled lipids. The conjugation yield was evaluated to be 95.8%. In addition, since ZnO NCs can emit in the green channel when excited by UV-light, we have also imaged the ZnO NCs by exciting them at 340 nm and collecting their fluorescence in the blue channel (Fig. S3c in the ESI†). We can actually co-localize in the same position the ZnO NCs’ fluorescent emission together with the Atto550 dye in the red channel attached to the ZnO surface and the lipid shell in the green one, thus shielding the nanocrystals.
Fig. 3. Wide-field fluorescence image of the (a) lipid shell labeled by 1% Bodipy-DHPE; (b) ZnO–NH2 NCs conjugated with Atto550-NHS ester; and (c) the merged channels showing complete co-localization. Scale bar: 10 μm.

The EDS analysis for the three samples (reported in Table S3 of the ESI†) shows that the samples are of ZnO material. In the case of ZnO–DOPC NCs, the presence of an increased amount of carbon and a small amount of phosphorus confirms the presence of lipids.
The colloidal stability of the three ZnO NCs was proven by DLS (Fig. 4), monitoring their behaviour in various media, primarily in ethanol and water solutions, then in the physiological simulated media (SBF) and in the cell culture medium (EMEM) completed with 10% of fetal bovine serum. The aim of using the last two solutions is to unravel the behaviour of ZnO NCs and the influence of surface charge and chemistry on the promotion of colloidal stability in both simulated and biological media. The SBF is an inorganic buffered salt solution simulating the composition of human plasma. The DLS behaviours versus time (as further shown in Fig. 5) would roughly simulate what could happen when the above mentioned nanocrystals are in contact with plasma fluids, i.e. for a hypothetical injection into living systems for further therapy or diagnostic purposes. It is indeed of prominent importance to avoid aggregation in the bloodstream of nanoparticles conceived to have therapeutic or diagnostic effects on a pathological site of interest in the organisms. Moreover, an efficient cell uptake and thus therapeutic/diagnostic treatments can be achieved solely by having well-dispersed nanoparticles, i.e. in the cell culture medium (EMEM) for in vitro experiments.
Fig. 4. Dynamic Light Scattering (DLS) measurements in number % of the ZnO samples, pristine ZnO NCs (dotted curves), ZnO–NH2 NCs (dashed curves), and ZnO–DOPC NCs (solid curves) in different media (ethanol, water, SBF, and EMEM).

Fig. 5. DLS analysis comparing the behaviour of pristine ZnO NCs (dotted curve) and ZnO–DOPC NCs (solid curve) in terms of mean hydrodynamic size (z-average) in (a) SBF and (b) EMEM for both short (1 hour) and (c) long (25 days) time periods.

Concerning the ZnO–DOPC NCs, the self-assembled lipid bilayers on the ZnO surface can be disassembled by the presence of alcoholic solutions;45 thus the DLS measurements were only carried out in water-based solutions (Fig. 4 from the second to the fourth panel). We observed that ZnO NCs are well dispersed in both EtOH (ZnO hydrodynamic diameter: 35 nm) and water media (ZnO: 44 nm). In contrast, the ZnO–NH2 NCs show aggregation in ethanol (ZnO–NH2: 59 and 106 nm) and good dispersion in water solution (ZnO–NH2: 88 nm). It is also worth noting that the functional layers, both lipidic and amine-moieties, contribute to a larger hydrodynamic diameter of ZnO NCs in water solution (with ZnO–DOPC NCs’ mean diameter of 122 nm). In SBF, ZnO and ZnO–NH2 NCs form large aggregates of micrometer size, whereas ZnO–DOPC NCs remain highly colloidally dispersed (with a hydrodynamic diameter of 66 nm). The improved ZnO–DOPC colloidal stability is further confirmed in EMEM (a narrow hydrodynamic peak at 104 nm). We attribute this colloidally stable behaviour to the lipid shell stabilization, shielding the ZnO NCs. In particular, the lipid coating limits the contact with the ZnO surface preventing possible interactions with solution's components. It is also worth noting that in EMEM also the ZnO–NH2 NCs show a less extent of aggregation (215 nm) compared to pristine ZnO NCs (1255 nm)
Biostability assays in simulated body fluid and cell culture media
The previous DLS results showed that the lipid-coating provides a significant enhancement of colloidal stability in both SBF and EMEM fluids. Thus, to monitor this behaviour over time, we recorded in real-time the z-average (mean hydrodynamic diameter in nm weighed on the scattered light intensity from the ensemble collection of particles) of ZnO–DOPC NCs and their uncoated counterparts (ZnO NCs and ZnO–NH2 NCs) in both SBF and EMEM over time (Fig. 5). It is worth mentioning that we also tried to record DLS analysis longer than 60 minutes in SBF; however further measurements did not meet the quality criteria due to the high instability of the ZnO NC and ZnO–NH2 NC samples in SBF for t > 60 min. For consistency, the ZnO–DOPC NCs were not further measured. In contrast, we can report the data in EMEM also for long time periods for the three samples.
In Fig. 5a, the mean hydrodynamic size (z-average) of the pristine ZnO NCs is relatively high, ranging from 3529 nm to 4870 nm, meaning that this sample immediately forms huge aggregates when exposed to the SBF solution. A visual check was also performed, noting that pristine ZnO NCs form a white, fluffy precipitate at the bottom of the measurement cuvette almost immediately after contact with SBF. A similar behaviour was observed for the pristine ZnO within the first hour in EMEM, with z-average ranging from a minimum of 1600 to a maximum of 1850 nm. Aggregation in both SBF and EMEM was recorded also for the ZnO–NH2 NCs. In contrast, the ZnO–DOPC sample remained with a constant and small z-average (from a minimum value of 194 nm to a maximum of 314 nm) in both SBF and EMEM solutions for the first hour of the experiments (Fig. 5a and b), accounting for a well-dispersed and stable sample. When analysing the data in EMEM for long periods (Fig. 5c) the z-average of ZnO–DOPC maintains a constant value of about 200–300 nm for the first 72 hours, increasing up to 500 nm after 25 days. In contrast, the pristine ZnO NCs account for a decreasing z-average (starting from 1800 nm down to 360 nm) accompanied by a decrease of the signal count rate, which corresponds to particle precipitation. The ZnO–NH2 NCs show a constant value at around 1100 nm, consisting of aggregated but still suspended particles.
FESEM analyses were performed on the three different NCs after selected time steps (1 h, 72 h, and 25 days) of the biostability assays in both SBF and EMEM (Fig. 6 and Fig. S4, S5 in the ESI†).
Fig. 6. FESEM images of the biostability behaviour of the three ZnO nanocrystals after 25 days assay times in SBF (left column) and EMEM (right column): (a–d) pristine ZnO, (b–e) ZnO–NH2, and (c–f) lipid-shell coated ZnO–DOPC.

Both ZnO and ZnO–NH2 NCs show strong aggregation already from the first hour, as confirmed by the previous DLS analysis. Both kinds of NCs tend to form spherical aggregates, whose dimension increases with time. The images of pristine ZnO NCs in both SBF and EMEM solutions after 1 and 72 hours still reveal the morphology of the single NCs (see the inset of Fig. S4a and S5a at the corresponding times in the ESI†). In contrast, after 25 days in SBF (Fig. 6a and Fig. S4a bottom and related inset), the sample appears strongly aggregated and the single NCs are almost indistinguishable. This behaviour is also evident for ZnO NCs after 25 days in EMEM solution (Fig. 6d) in which the aggregates are constituted by a smooth matrix that seems to incorporate the nanocrystals.
Similar considerations can be applied for the ZnO–NH2 samples, forming spherical micrometer-sized aggregates in SBF after 25 days (Fig. 6b). Large and compact clusters are formed in EMEM with the presence of precipitates, possibly derived from the solution (Fig. S5b in the ESI† after 72 h), and a smooth matrix encapsulating the ZnO–NH2 NCs is visible for the long time step of 25 days (Fig. 6e).
In contrast with the previous results, the ZnO–DOPC NC sample shows well distinguishable round-shaped particles even after biodegradation tests. This behaviour is observed for all time points in both SBF (Fig. S4 in the ESI†) and EMEM (Fig. S5 in the ESI†). In particular, tiny and well-dispersed nanoparticles can be observed after 25 days in SBF (Fig. 6c) and until 72 hours in EMEM (Fig. S5c in the ESI†). Formation of some aggregates for a longer time period, i.e. 25 days in EMEM (Fig. 6f), can be observed together with the presence of a soft matrix possibly derived from the medium. Strikingly, the well distinguishable ZnO–DOPC NCs observed in the FESEM analysis after 72 h and 25 days in both SBF and EMEM media are in fair agreement with the DLS data. In contrast, it is important to note that in both ZnO and ZnO–NH2 NC samples after 25 days, the nanocrystals are no longer distinguishable. In both cases, particle aggregation, fusion and dissolution occur, as demonstrated below with further characterization techniques.
The interaction between SBF and EMEM solutions and nanocrystal surfaces, already supposed when observing FESEM images, is confirmed by both FT-IR and EDS analyses which highlight the presence of more elements in addition to the forecasted Zn and O.
EDS results (reported in Tables S3 and S4 along with SBF and EMEM compositions in Tables S1 and S2 of the ESI†) show the presence of carbon, phosphorus, sodium, calcium and chlorine for all the three samples, i.e. ZnO, ZnO–NH2 and ZnO–DOPC NCs, at different biostability times in both SBF and EMEM. For samples in contact with EMEM, the presence of sulphur, potassium, magnesium and a higher content of carbon is detected, due to the high amount of proteins, amino acids and other biomolecules in the cell culture medium. These data confirm that the matrix incorporating the nanocrystals, in particular visible for the ZnO and ZnO–NH2 samples in EMEM, is made of such biomolecules.
Further confirmations are given from FT-IR spectra (Fig. 7), which show increasing vibrational peaks in terms of frequency and intensity, with respect to the untreated NCs, at increasing times of biostability assays. It is, in particular, worth mentioning an increasing peak, in all the three kind of samples, at 1045 cm–1, ascribed to the stretching vibration of the P–O bond. As also evidenced by EDS analysis, we assume that phosphate derivatives (PO43–) precipitate on these sample surfaces due to contact with either SBF or EMEM, both containing phosphate compounds. In particular, at long biostability times (i.e. 72 hours and 25 days), peaks at 1220 cm–1 and 1150 cm–1 appear, related to the stretching of P O bonds, again attributed to the phosphate groups. Stretching typical of carbonate groups (C–O) also appears, in accordance with the increase of carbon content revealed by EDS analysis.
Fig. 7. Fourier-Transform InfraRed (FT-IR) spectra of the (a) pristine ZnO NCs, (b) ZnO–NH2 NCs, and (c) ZnO–DOPC NCs after different time steps of biostability assays in SBF (left) and EMEM (right).

Concerning the ZnO–DOPC, the presence of a phospholipid layer on the ZnO surface was already confirmed by the presence of peaks at 2860, 2925 cm–1 (stretching vibrations of –CHx) and 1750 cm–1 (C O groups) that maintain a constant intensity at different biostability times in both SBF and EMEM solutions. Also in both ZnO and ZnO–NH2 samples, the vibrations at 2955, 2915, and 2855 cm–1 related to the stretching of –CH2 and –CH3 groups are already present in the spectra of the untreated materials, as observed in Fig. 3a. However, longer biostability times in both SBF and EMEM produce an increase of these stretching peaks in both ZnO and ZnO–NH2 NCs accompanied by a constant decrease of the hydroxyl stretching vibration (–OH) between 3600 and 3000 cm–1. Here two different hypotheses could be made, depending on the solution composition. In SBF medium, we assume that the oxide surface of the NCs reacts with some components of the solution, in particular with TRIS (Tris(hydroxymethyl)aminomethane chlorohydrate), leading to partial hydrolysis and reaction with methyl groups of the buffer component, responsible for the increase of the –CHx stretching vibrations observed in both ZnO and ZnO–NH2 samples. A consequence of this reactive behaviour could also be partial dissolution of the zinc oxide surfaces, i.e. for both ZnO and ZnO–NH2 NCs, as further reported below.
In EMEM, we hypothesize that the ZnO and ZnO–NH2 NC samples aggregate within a soft matrix of proteins and amino acids and all other biomolecules present in the serum, as already observed by FESEM. The gradual reduction of –OH group stretching could account for the interaction of these groups at the ZnO surface with the bound molecules. These behaviours can also be associated with the slight dissolution.
In contrast to the case of the pristine ZnO NCs and the ZnO–NH2 NCs, a strong increase of the –OH stretching band from 3600 to 3200 cm–1 in the ZnO–DOPC NCs is observed (Fig. 7c). We attribute these variations to the physical adsorption and coordination with water molecules from both SBF and EMEM media on the phospholipidic layer, positively influencing the high colloidal stability of the ZnO–DOPC sample in water-based media, in agreement with the DLS results of Fig. 5.
By comparing the X-ray diffraction patterns of the three ZnO nanocrystals before and after the biostability assays in both SBF and EMEM at different time steps (Fig. S6 in the ESI†), one can notice a slight reduction of the diffraction intensities of the wurtzite reflections, i.e. (100), (002) and (101) peaks for both ZnO and ZnO–NH2 NCs. Therefore, we can further support the previous FT-IR results assuming that partial, but not complete, hydrolysis and dissolution of the ZnO and ZnO–NH2 surfaces takes place when they are in contact with SBF and EMEM. In some samples, we observed the appearance of sharp peaks at around 32° attributed to NaCl crystals.
In contrast, the diffraction peaks of the ZnO–DOPC NCs after the different assay times did not decrease, owing to the high chemical stability of the sample and the maintenance of the initial crystalline structure.
As a further check of the crystalline quality of ZnO, High Resolution Transmission Electron Microscopy (HR-TEM) was performed. In Fig. 9 and 10 we show a comparison of each NC structure before and after prolonged immersion (25 days) in either SBF or EMEM. All the pictures show the presence of single-crystalline mono-domains, as confirmed by the Fast Fourier Transform (FFT) in the inset of the pictures, which refers to the white squared area. These data therefore suggest that the crystalline integrity of the three different nanocrystals is still preserved after prolonged immersion in different simulated and biological media. This result is not in contradiction with the previous FT-IR and XRD data, where only slight and partial dissolution was hypothesized.
Fig. 9. HR-TEM, SAED and STEM imaging of the amine-functionalized ZnO–NH2 nanocrystals: (a) before biostability treatments and in comparison after (b) 25 days in SBF solution and (c) 25 days in EMEM. The scale bar is 5 nm.

Fig. 10. HR-TEM, SAED and STEM imaging of the lipid-shielded ZnO–DOPC nanocrystals: (a) before biostability treatments and in comparison after (b) 25 days in SBF solution and (c) 25 days in EMEM. The scale bar is 5 nm.

In particular, the HR-TEM images of the ZnO–DOPC sample (especially in Fig. 10c, left panel) show the NCs immersed in an amorphous matrix. Considering that the sample was neither stained nor fixed for this imaging, this amorphous structure could be attributed to the phospholipidic shell.
The STEM images help to clarify the nanostructures’ aggregation before and after the biostability treatments. Distinct nanocrystals are observed for all the three samples before biostability tests (Fig. 8a, 9a and 10a), whereas strong aggregation is visible in particular for the ZnO NC samples (Fig. 8b and c), and to a less extent for the ZnO–NH2 samples (Fig. 9b and c), after 25 days in both SBF and EMEM. The as-prepared ZnO–DOPC is characterized by nanocrystals (Fig. 10a) that, still after 25 days of contact with simulated buffer and biological fluids, are embedded by a smooth matrix, attributed to the presence of phospholipids completely immersing the inorganic crystalline structure (see also Fig. S7 in the ESI† for other magnified STEM images). All the STEM measurements are in complete agreement with DLS and FESEM characterization.
Fig. 8. HR-TEM, SAED and STEM imaging of the pristine ZnO nanocrystals: (a) before biostability treatments and in comparison after (b) 25 days in SBF solution and (c) 25 days in EMEM. The scale bar is 5 nm.

ICP-MS analyses were also carried out on the SBF and EMEM solutions at each biostability time point, as reported in Fig. 11, monitoring the zinc, phosphorus and calcium elements, with the last two elements being highly reactive towards the ZnO surface. In SBF and EMEM solutions, for both ZnO and ZnO–NH2 NCs, an increase of the Zn2+ cations is clearly observed in the first hour from 0 to a maximum of 180 ppm for ZnO NCs and 121 ppm for ZnO–NH2 NCs in SBF and up to 165 ppm for ZnO NCs and 188 ppm for ZnO–NH2 NCs in EMEM. These amounts roughly correspond to a range of dissolution between 7.5 and 11 mol% of zinc with respect to the initial amount of zinc from the NCs. These trends therefore underline the slight dissolution of these nanocrystalline structures in both media, confirming the FT-IR and XRD data reported above. In addition, a strong reduction of the phosphorus content is assessed from SBF (from 40 ppm to 0.5 ppm) and a bit less in EMEM (from 40 ppm to 10 ppm), confirming the precipitation of phosphate species on both ZnO and ZnO–NH2 sample surfaces already observed with EDS and FT-IR analyses. In contrast, the level of calcium cations does not change in SBF for the whole duration of the assays, which is also confirmed by the almost negligible presence of calcium-containing species on the ZnO and ZnO–NH2 surfaces in SBF, found in the previous FT-IR and EDS characterization. Literature studies report a high degree of interaction between the phosphate anions and ZnO nanoparticles.47 In silica or bioglass-based nanomaterial assays in SBF, co-precipitation of calcium cations with phosphate anions was observed leading to the formation of the hydroxyapatite compound on the material surfaces.48–50 However, we believe that in the present case, the strong interaction between zinc and phosphate is detrimental for the combination of such anions with Ca2+, and the sole precipitation of carbonate and phosphate groups on ZnO and ZnO–NH2 NCs takes place. In addition, no evidence of hydroxyapatite can be observed (as reported in the X-ray diffractograms of Fig. S5, evidencing the only presence of ZnO-related wurtzite phase) in any of the three samples.
Fig. 11. ICP-MS analysis of (a) zinc, (b) calcium, and (c) phosphorus elements present in SBF and EMEM at different time steps of the biostability assays from the three ZnO-based NCs.

Differently, in EMEM the amount of calcium cations slowly decreases (from 15 ppm to around 5 ppm) when in contact with the ZnO and ZnO–NH2 NCs, in accordance with EDS analysis (Table S4, ESI†) detecting the presence of calcium in all samples.
In remarkable contrast with the previous results, the composition of SBF and EMEM solutions in terms of phosphorus and calcium content does not vary significantly when in contact with the ZnO–DOPC NCs. In particular, the presence of Zn2+ ions is almost not detectable in the used media for the whole assay time, i.e. until 25 days. This evidence confirms the previous results, showing no hydrolysis nor dissolution of the lipid-coated ZnO samples. An oscillating behaviour, with a tendency for reduction, can be just observed for the phosphorus content in EMEM for the ZnO–DOPC NCs. This behaviour is a clear proof of the precipitation of phosphate groups on this sample surface, as also previously confirmed by the FT-IR and EDS analysis.
All these findings support the idea that a phospholipidic shielding on the ZnO surface is very effective: (i) it enables the stabilization of the ZnO NCs as a colloidal suspension in different water and physiological media; (ii) it prevents their partial degradation, preserving the physico-chemical properties of the ZnO NCs; and (iii) most importantly, it avoids the release of potentially cytotoxic Zn2+ cations. In our opinion, these findings can be possible solely by the formation of a complete and dense coverage of the phospholipidic bilayer, self-assembled on the ZnO surface and able to prevent ZnO NCs’ interactions with the surrounding medium.
Internalization and cytotoxicity experiments in HeLa cells
Several studies demonstrated that ZnO NCs’ cytotoxicity and uptake efficacy are strictly influenced by nanocrystals’ surface chemistry.19,51,52 To evaluate the effects of different surface functionalizations, the toxicity and cellular uptake of the pristine ZnO NCs and the functionalized ones were assessed on human epithelial carcinoma cells (HeLa). It is worth mentioning here that the methods used for these cellular studies impose the use of dye-labelled nanocrystals. As also reported in the Experimental section, we used the amine-functional group at the ZnO NC surface to covalently bind an ATTO-550 NHS ester dye. Therefore, any further comparison among the functionalized nanocrystals is performed between the ZnO–NH2 and ZnO–NH2–DOPC.
The cellular uptake of HeLa cells treated with 18 μg mL–1 of ZnO–NH2 and ZnO–NH2–DOPC nanocrystals was qualitatively analysed based on fluorescence images after 24, 48 and 72 hours by Spinning Disk microscopy (Fig. 12). After 24 hours of incubation, the two different types of nanocrystals showed a comparable uptake rate. After longer incubation times (48 and 72 hours), the DOPC-coated NCs showed a statistically significant (P < 0.02) higher intensity of fluorescence possibly related to their higher internalization (Fig. 12d).
Fig. 12. (a) Fluorescence quantification of ZnO–NH2 and ZnO–NH2–DOPC nanocrystals in HeLa cells after 24, 48 and 72 hours incubation. Data are expressed as mean ± SD of three experiments. P = two-way ANOVA test. (b) 3D representation of ZnO–NH2 nanocrystals uptaken into an HeLa cell. Intracellular NCs (in red) can be observed through the window opened in the cell membrane region (green). The z-stack images were processed with Particle_in_Cell-3D Macro. 3D scale bars = 3 μm. Representative fluorescent images of HeLa cell membranes (in green) incubated for 72 hours with fluorescent (c) ZnO–NH2 and (d) ZnO–NH2–DOPC nanocrystals (in red). Scale bars: 5 μm.

The effects of different concentrations of ZnO, ZnO–NH2 and ZnO–NH2–DOPC nanocrystals on HeLa cell culture for 72 hours are shown in Fig. 13. From a quantitative point of view, these cytotoxicity data confirmed ZnO nanocrystals’ toxic effect against HeLa cells only in the case of high NC concentration.53 Actually, the three kinds of ZnO NCs had no visible cytotoxic effect on HeLa cells after 72 hours treatment up to a concentration of about 18 μg mL–1. On the contrary, all of them showed a significant cytotoxic effect when used at concentrations as high as 90 μg mL–1. The data trend of ZnO–NH2 and ZnO–NH2–DOPC NCs shows a significantly higher toxicity compared with the ZnO NCs’ treatment, respectively at the concentrations of 36 and 18 μg mL–1. This enhanced cytotoxicity could be partially explained by their better dispersion in the biological medium compared to their unfunctionalized counterpart (as reported in the DLS results of Fig. 4). In this scenario, both ZnO–NH2 and ZnO–NH2–DOPC NCs would be more easily uptaken by cancer cells.
Fig. 13. Cell viability of HeLa cells after 72 hours exposure to ZnO, ZnO–NH2 and ZnO–NH2–DOPC nanocrystals. The mitochondrial activity was measured with MTT assay and data were expressed as mean ± SD of three experiments. *p < 0.05, **p < 0.01.

In addition, our results suggest that the extent of internalization is not the only parameter affecting cell toxicity. After 72 hours of treatment, ZnO–NH2–DOPC showed a greater degree of intracellular fluorescence compared to that of the amino-propyl functionalized ZnO–NH2 NCs although the increased uptake was not associated to an enhanced cytotoxic effect.26
By considering all the previous results, these data confirm that intracellular release of zinc ions plays a key role in the definition of the cytotoxic effect, as already reported in the literature.40 Our results suggest that the presence of the lipid-coating limits the access of the ZnO NCs’ surface to the lysosomal pH, preventing metal ion dissolution. Consequently, the lipid-shell significantly reduces the cytotoxic effect on the cultured cell lines, despite the high internalization levels of the ZnO–DOPC NCs.
Conclusions
We have reported the biostability behaviour in simulated and biological media, as well as the cell internalization and cytotoxicity, of three different kinds of ZnO nanomaterials. In particular, pristine nanocrystals (ZnO NCs), amino-propyl functionalized ZnO–NH2 NCs and phospholipid-shielded ZnO–DOPC NCs were studied. The results have shown an important effect of the surface chemistry and charge on the biostability behaviour at different time steps.
In particular, we have observed the strong aggregation and slight dissolution behaviour of both ZnO and ZnO–NH2 NCs already at short time steps in water-based media, i.e. SBF, simulating the inorganic composition of human blood, and EMEM, the cell culture media.
In contrast, we have verified that the successful formation of a self-assembled phospholipid bilayer on the ZnO nanocrystals’ surface leads to a more biostable nanoconstruct. Actually, the lipid-shell prevents NCs’ aggregation in water-based media, improves the colloidal stability over time until 25 days and avoids NCs’ dissolution into cytotoxic Zn2+ cations. This new formulation also improves the internalization of the nanocrystals into HeLa cancer cells. Strikingly, the higher level of internalization of ZnO–NH2–DOPC NCs compared to the uncoated ZnO–NH2 counterpart is not reflected by a higher cytotoxic effect.
The above reported results give a clear insight into the behaviour of ZnO nanomaterials in biological media, indicating that an appropriate surface shielding, i.e. by a phospholipid bilayer, is fundamental to develop multifunctional ZnO NCs for therapeutic and bio-imaging applications in cancer cells.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
This work has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 678151 – Project Acronym “TROJANANOHORSE” – ERC starting Grant) and by a Regional program entitled “Attrarre Docenti di Qualità con Starting Grant” from the Compagnia di Sanpaolo, D. R. 349-2016. The help of Mr Mauro Raimondo in the acquisition of FESEM images and Ms Carla Celozzi and Mr Vanni Martignoni in ICP-MS analysis is gratefully acknowledged.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb02229h
References
- Stassi S., Chiadò A., Cauda V., Palmara G., Canavese G., Laurenti M., Ricciardi C. Anal. Bioanal. Chem. 2017;409:2615–2625. doi: 10.1007/s00216-017-0204-2. [DOI] [PubMed] [Google Scholar]
- Laurenti M., Stassi S., Canavese G., Cauda V. Adv. Mater. Interfaces. 2017;4:1600758. [Google Scholar]
- Laurenti M., Verna A., Fontana M., Stassi S., Canavese G., Marasso S. L., Cauda V. Adv. Mater. Interfaces. 2016;3:1600110. [Google Scholar]
- Cauda V., Motto P., Perrone D., Piccinini G., Demarchi D. Nanoscale Res. Lett. 2014;9:53. doi: 10.1186/1556-276X-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X., Cao X., Guo H., Li T., Jie Y., Wang N., Wang Z. L. ACS Nano. 2016;10:8038–8044. doi: 10.1021/acsnano.6b04121. [DOI] [PubMed] [Google Scholar]
- Yeh P.-H., Li Z., Wang Z. L. Adv. Mater. 2009;21:4975–4978. doi: 10.1002/adma.200902172. [DOI] [PubMed] [Google Scholar]
- Xiong H.-M. Adv. Mater. 2013;25:5329–5335. doi: 10.1002/adma.201301732. [DOI] [PubMed] [Google Scholar]
- Hong H., Shi J., Yang Y., Zhang Y., Engle J. W., Nickles R. J., Wang X., Cai W. Nano Lett. 2011;11:3744–3750. doi: 10.1021/nl201782m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Zhang J., Guo J., Zhang J., Ding F., Li L., Sun Z. Toxicol. Lett. 2010;199:389–397. doi: 10.1016/j.toxlet.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Applerot G., Lipovsky A., Dror R., Perkas N., Nitzan Y., Lubart R., Gedanken A. Adv. Funct. Mater. 2009;19:842–852. [Google Scholar]
- Nair S., Sasidharan A., Divya Rani V. V., Menon D., Nair S., Manzoor K., Raina S. J. Mater. Sci.: Mater. Med. 2009;20:235. doi: 10.1007/s10856-008-3548-5. [DOI] [PubMed] [Google Scholar]
- Yin H., Casey P. S. Chemosphere. 2015;124:116–121. doi: 10.1016/j.chemosphere.2014.11.076. [DOI] [PubMed] [Google Scholar]
- Stassi S., Cauda V., Ottone C., Chiodoni A., Pirri C. F., Canavese G. Nano Energy. 2015;13:474–481. [Google Scholar]
- Yin H., Casey P. S., McCall M. J., Fenech M. Langmuir. 2010;26:15399–15408. doi: 10.1021/la101033n. [DOI] [PubMed] [Google Scholar]
- Albanese A., Tang P. S., Chan W. C. W. Annu. Rev. Biomed. Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- Xia T., Kovochich M., Liong M., Madler L., Gilbert B., Shi H., Yeh J. I., Zink J. I., Nel A. E. ACS Nano. 2008;2:2121–2134. doi: 10.1021/nn800511k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller K. H., Kulkarni J., Motskin M., Goode A., Winship P., Skepper J. N., Ryan M. P., Porter A. E. ACS Nano. 2010;4:6767–6779. doi: 10.1021/nn101192z. [DOI] [PubMed] [Google Scholar]
- Qu F. Y., Morais P. C. J. Chem. Phys. 1999;111:8588–8594. [Google Scholar]
- Punnoose A., Dodge K., Rasmussen J. W., Chess J., Wingett D., Anders C. ACS Sustainable Chem. Eng. 2014;2:1666–1673. doi: 10.1021/sc500140x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degen A., Kosec M. J. Eur. Ceram. Soc. 2000;20:667–673. [Google Scholar]
- Panchal R. G. Biochem. Pharmacol. 1998;55:247–252. doi: 10.1016/s0006-2952(97)00240-2. [DOI] [PubMed] [Google Scholar]
- Abercrombie M., Ambrose E. J. Cancer Res. 1962;22:525–548. [PubMed] [Google Scholar]
- Papo N., Shahar M., Eisenbach L., Shai Y. J. Biol. Chem. 2003;278:21018–21023. doi: 10.1074/jbc.M211204200. [DOI] [PubMed] [Google Scholar]
- Leroueil P. R., Hong S., Mecke A., Baker J. R., Orr B. G., Banaszak Holl M. M. Acc. Chem. Res. 2007;40:335–342. doi: 10.1021/ar600012y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Jiang Y., Ding Y., Povey M., York D. J. Nanopart. Res. 2007;9:479–489. [Google Scholar]
- Zeng K., Li J., Zhang Z., Yan M., Liao Y., Zhang X., Zhao C. J. Mater. Chem. B. 2015;3:5249–5260. doi: 10.1039/c5tb00486a. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Newton B., Lewis E., Fu P. P., Kafoury R., Ray P. C., Yu H. Toxicol. In Vitro. 2015;29:762–768. doi: 10.1016/j.tiv.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai J., Shoji S., Igarashi H., Hashimoto A., Kokugan T., Shimizu M., Kojima H. J. Ferment. Bioeng. 1998;86:521–522. [Google Scholar]
- Sawai J., Kawada E., Kanou F., Igarashi H., Hashimoto A., Kokugan T., Shimizu M. J. Chem. Eng. Jpn. 1996;29:627–633. [Google Scholar]
- Sharma V., Shukla R. K., Saxena N., Parmar D., Das M., Dhawan A. Toxicol. Lett. 2009;185:211–218. doi: 10.1016/j.toxlet.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Wong S. W. Y., Leung P. T. Y., Djurišić A. B., Leung K. M. Y. Anal. Bioanal. Chem. 2010;396:609–618. doi: 10.1007/s00216-009-3249-z. [DOI] [PubMed] [Google Scholar]
- Yang H., Liu C., Yang D., Zhang H., Xi Z. J. Appl. Toxicol. 2009;29:69–78. doi: 10.1002/jat.1385. [DOI] [PubMed] [Google Scholar]
- Xiaoyong D., Qixia L., Wenting C., Yanli W., Minghong W., Haijiao Z., Zheng J. Nanotechnology. 2009;20:115101. [Google Scholar]
- Xia T., Kovochich M., Liong M., Mädler L., Gilbert B., Shi H., Yeh J. I., Zink J. I., Nel A. E. ACS Nano. 2008;2:2121–2134. doi: 10.1021/nn800511k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S., Pokhrel S., Xia T., Gilbert B., Ji Z., Schowalter M., Rosenauer A., Damoiseaux R., Bradley K. A., Mädler L., Nel A. E. ACS Nano. 2010;4:15–29. doi: 10.1021/nn901503q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Xie C. Colloids Surf., B. 2006;47:140–145. doi: 10.1016/j.colsurfb.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Moos P. J., Chung K., Woessner D., Honeggar M., Cutler N. S., Veranth J. M. Chem. Res. Toxicol. 2010;23:733–739. doi: 10.1021/tx900203v. [DOI] [PubMed] [Google Scholar]
- Feltis B. N., O'Keefe S. J., Harford A. J., Piva T. J., Turney T. W., Wright P. F. A. Nanotoxicology. 2012;6:757–765. doi: 10.3109/17435390.2011.620718. [DOI] [PubMed] [Google Scholar]
- Xu M., Li J., Hanagata N., Su H., Chen H., Fujita D. Nanoscale. 2013;5:4763–4769. doi: 10.1039/c3nr34251d. [DOI] [PubMed] [Google Scholar]
- Shen C., James S. A., de Jonge M. D., Turney T. W., Wright P. F. A., Feltis B. N. Toxicol. Sci. 2013;136:120–130. doi: 10.1093/toxsci/kft187. [DOI] [PubMed] [Google Scholar]
- Rasmussen J. W., Martinez E., Louka P., Wingett D. G. Expert Opin. Drug Delivery. 2010;7:1063–1077. doi: 10.1517/17425247.2010.502560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrano A. A., Blechinger J., Osseforth C., Argyo C., Reller A., Bein T., Michaelis J., Bräuchle C. Nanomedicine. 2013;8:1815–1828. doi: 10.2217/nnm.12.178. [DOI] [PubMed] [Google Scholar]
- Pacholski C., Kornowski A., Weller H. Angew. Chem., Int. Ed. 2002;41:1188–1191. doi: 10.1002/1521-3773(20020402)41:7<1188::aid-anie1188>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Cauda V., Engelke H., Sauer A., Arcizet D., Bräuchle C., Rädler J., Bein T. Nano Lett. 2010;10:2484–2492. doi: 10.1021/nl100991w. [DOI] [PubMed] [Google Scholar]
- Hohner A. O., David M. P. C., Rädlera J. O. Biointerphases. 2010;5:1–8. doi: 10.1116/1.3319326. [DOI] [PubMed] [Google Scholar]
- Cauda V., Argyo C., Piercey D. G., Bein T. J. Am. Chem. Soc. 2011;133:6484–6486. doi: 10.1021/ja1067492. [DOI] [PubMed] [Google Scholar]
- Herrmann R., García-García F. J., Reller A. Beilstein J. Nanotechnol. 2014;5:2007–2015. doi: 10.3762/bjnano.5.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauda V., Schlossbauer A., Bein T. Microporous Mesoporous Mater. 2010;132:60–71. [Google Scholar]
- Cauda V., Onida B., Platschek B., Mühlstein L., Bein T. J. Mater. Chem. 2008;18:5888–5899. [Google Scholar]
- Cauda V., Mühlstein L., Onida B., Bein T. Microporous Mesoporous Mater. 2009;118:435–442. [Google Scholar]
- Altunbek M., Baysal A., Çulha M. Colloids Surf., B. 2014;121:106–113. doi: 10.1016/j.colsurfb.2014.05.034. [DOI] [PubMed] [Google Scholar]
- Liu J., Feng X., Wei L., Chen L., Song B., Shao L. Crit. Rev. Toxicol. 2016;46:348–384. doi: 10.3109/10408444.2015.1137864. [DOI] [PubMed] [Google Scholar]
- Pandurangan M., Enkhtaivan G., Kim D. H. J. Photochem. Photobiol., B. 2016;158:206–211. doi: 10.1016/j.jphotobiol.2016.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.