

Abstract
Background
VSL#3 is a probiotic compound that has been used in the treatment of Inflammatory Bowel Disease (IBD). T-cell protein tyrosine phosphatase (TCPTP) is the protein product of the IBD candidate gene, PTPN2, and we have previously shown that it protects epithelial barrier function. The aim of this study was to investigate if VSL#3 improves intestinal epithelial barrier function against the effects of the IBD associated pro-inflammatory cytokine, interferon-gamma (IFN-γ) via activation of TCPTP.
Methods
Polarized monolayers of T84 intestinal epithelial cell (IEC) were treated with increasing concentrations of VSL#3 to determine effects on TCPTP expression and enzymatic activity. Therapeutic effects of VSL#3 against barrier disruption by IFN-γ were measured by transepithelial electrical resistance (TER) and FITC-dextran permeability. A novel TCPTP-deficient HT-29 IEC line was generated to study the role of TCPTP in mediating the effects of VSL#3. Tight junction protein distribution was assessed by confocal microscopy.
Results
VSL#3 increased TCPTP protein levels and enzymatic activity, correlating with a VSL#3-induced decrease in IFN-γ signaling. VSL#3 corrected the decrease in TER and the increase in epithelial permeability induced by IFN-γ. Moreover, the restorative effect of VSL#3 against IFN-γ signaling, epithelial permeability defects, altered expression and localization of the tight junction proteins claudin-2, occludin and ZO-1, were not realized in stable TCPTP/(PTPN2)-deficient HT-29 intestinal epithelial cells.
Conclusion
VSL#3 reduces IFN-γ signaling and IFN-γ-induced epithelial barrier defects in a TCPTP-dependent manner. These data point to a key role for TCPTP as a therapeutic target for restoration of barrier function by probiotics.
Keywords: Claudin-2, Inflammation, IFN-γ, PTPN2, STAT-1
INTRODUCTION
Inflammatory Bowel Disease (IBD) comprises two separate but related conditions, Crohn’s Disease (CD) and Ulcerative Colitis (UC), and is characterized by chronic inflammation of the gastrointestinal tract. Both subtypes of IBD are idiopathic in nature and pathogenesis of the disease is a multi-factorial event.(1) Disease progression has been attributed to genetic predisposition, environmental triggers, and an inappropriate immune response to normally harmless commensal flora in the gut. The goal of IBD therapy is to induce and sustain clinical remission; however, many of these therapies are not suitable for long-term maintenance of the disease due to undesirable side effects. Probiotics are non-harmful bacteria that have been shown to convey beneficial effects to host intestinal health.(2–4) One such probiotic mixture is VSL#3, a combination that consists of various strains of lyophilized bifidobacterium (Bifidobacterium breve DSM 24732, B. longum DSM 24736, B. infantis DSM 24737), lactobacilli (Lactobacillus acidophilus DSM 24735, L. plantarum DSM 24730, L. paracasei DSM 24733, L. delbrueckii subsp. bulgaricus DSM 24734), and Streptococcus thermophilus (DSM 24731).(5) This bacterial compound has shown efficacy in the treatment of ulcerative colitis, and in preventing post-operative recurrence of Crohn’s Disease, however, the mechanisms underlying these beneficial effects are still poorly understood.(4, 6–8)
In IBD, pro-inflammatory cytokines such as IFN-γ and tumor necrosis factor-α (TNF-α) are present at high concentrations, with elevated levels of IFN-γ predominating in CD. Previously, it has been shown that probiotic DNA can reduce the secretion of the pro-inflammatory cytokine IFN-γ in the large intestine.(9) Therefore, it was of interest to examine the effect of VSL#3 on other regulators of IFN-γ signaling. Specifically, we were interested in the IBD candidate gene, protein tyrosine phosphatase non-receptor type 2 (PTPN2), and its protein product, TCPTP, a negative regulator of IFN-γ signaling in intestinal epithelial cells. TCPTP is a member of the protein tyrosine phosphatase family (PTP) whose members share a highly conserved catalytic motif.(10, 11) PTPs play an integral role in regulating a variety of basic cellular processes including cell growth and differentiation.(12) Among TCPTP substrates are the phosphotyrosine residues on the receptors for epidermal growth factor and insulin.(13, 14) Additionally, TCPTP negatively regulates phosphorylation of the IFN-γ receptor (IFNγR) and the IFN-γ signaling molecules, signal transducers and activators of transcription (STAT)1 and 3.(15, 16) Dephosphorylation of STATs by TCPTP leads to their inactivation and termination of STAT-induced transcription.(15) TCPTP, therefore, plays an integral role in regulating IFN-γ signaling and maintaining cellular homeostasis.
Genome-wide-association-studies (GWAS) have revealed that individuals with single nucleotide polymorphic mutations in the locus of the PTPN2 gene that encodes TCPTP, have an increased susceptibility to developing CD and UC.(17–19) Preliminary evidence indicates that these SNPs lead to loss-of-function mutations in TCPTP.(20) While it has been shown that TCPTP expression and activity is increased in response to IFN-γ, reduced TCPTP expression leads to increased effects of IFN-γ on STAT signaling and a greater reduction in barrier function.(21–23) Therefore, agents that increase the activity of TCPTP are of general interest as modifiers of inflammatory signaling events.(24, 25) The goal of this project was to determine the effects of different VSL#3 concentrations and incubation times on TCPTP expression and activity. Additionally, we sought to examine whether VSL#3 affects TCPTP regulation of IFN-γ signaling and can correct downstream effects on barrier integrity. This information could potentially provide insights into the mechanisms by which probiotics are able to treat IBD, the timing of administration and appropriate concentration required for probiotics to exert an effect, and a mechanism to support VSL#3’s administration to counter IFN-γ driven inflammatory events.
EXPERIMENTAL PROCEDURES
Materials
VSL#3 packets with 450 billion bacteria per sachet (gift from Professor Claudio De Simone), human recombinant IFN-γ (Roche, Mannheim, Germany), cycloheximide (Sigma-Aldrich, St. Louis, MO, USA), monoclonal mouse anti-TCPTP antibody CF-4, which detects the 45-kilodalton and the 48-kilodalton isoforms (Calbiochem, San Diego, CA), mouse anti-TCPTP (Ab-1) antibody (EMD Millipore, Billerica, MA), anti-phospho-STAT1 (Tyr701), anti-STAT1, (Cell Signaling Technologies, Danvers, MA), Claudin-2, Occludin and ZO-1 (Invitrogen, Waltham, Massachusetts, USA) and monoclonal mouse anti-β-Actin (Sigma) were obtained from the sources noted. Millicell culture plate inserts were purchased from Millipore Corporation (Millipore, Bedford, MA). McCoy’s 5A and DMEM media were purchased form Corning Inc, (Corning, NY). All other reagents were of analytical grade and acquired commercially.
Tissue Culture
Human T84 colonic epithelial cells (passages 20 – 40) were grown in 1:1 Dulbecco’s modified Eagle’s/Ham’s F-12 medium with L-glutamine and 15mM HEPES (Mediatech Inc., Manassas, VA, USA), supplemented with 5% newborn calf serum (GIBCO BRL) at 37°C in 5% CO2. Cells were fed twice a week and then seeded onto 30-mm Millicell transwell polycarbonate filters (0.5×106 cells) for Western blot analysis. When grown on polycarbonate filters, T84 cells acquire the polarized phenotype of native colonic epithelia. For barrier function studies, T84 cells were seeded onto 12mm Millicell-HA culture plate inserts (filter membranes) and grown for 12-20 days before study, at which time they had stable values of transepithelial electrical resistance ≥ 1000 Ω·cm2. IFN-γ (1000U/mL) was added basolaterally, cycloheximide (35.5μM) bilaterally, and VSL#3 (102, 104, 106, 108 CFU/mL diluted in serum-free media) apically. Commensal E. coli K12 bacteria were cultured overnight in LB medium shaking at 37 °C (250 rpm). For studies with conditioned media (CM), VSL#3 was incubated in cell culture media and the bacteria then removed by filtration prior to the addition of VSL#3-CM to the apical surface of polarized T84 monolayers. HT-29 and HEK293T cells were grown in McCoy’s 5A and DMEM media respectively with 10% heat-inactivated fetal bovine serum and 1% glutamine and penicillin (100U/ml)/streptomycin (100 μg/ml). For heat-killed (HK) experiments the bacteria (K12 and VSL#3 - 106 CFU/mL) were diluted using serum free media and heat-killed at 95°C for 30 minutes as previously described.(26) The samples were set aside for 10 minutes prior to addition to the apical surface of T84 cells for 9 hrs incubation.
RNA Isolation and Real-Time Polymerase Chain Reaction
Total RNA was isolated and DNA was removed from T84 cells using the Direct-zol RNA MiniPrep kit (Zymogen, Irvine, CA) according to manufacturer’s instructions. RNA purity and concentration were assessed by absorbance at 260 and 280 nm. Complementary DNA (cDNA) synthesis was performed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative Reverse-Transcriptase Polymerase Chain Reaction was performed using MESA GREEN qPCR MasterMix Plus for SYBR Assays (Eurogentec, San Diego, CA) on a StepOnePlus Real-Time PCR system using Step One Software v2.0 (Applied Biosystems). Measurements were performed in triplicate, human GAPDH was used as an endogenous control, and results were analyzed by the ΔΔCT method.
Preparation of Cytoplasmic Lysates
On the day of the experiment, cells from inserts containing T84 monolayers were suspended in ice cold lysis buffer (50mM Tris, 150mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 20μM NaF, 1mM EDTA, 1μg/ml antipain, 1μg/ml pepstatin, 1μg/ml leupeptin, 1mM NaVO3, 100μg/ml phenylmethylsulfonyl fluoride), vortexed thoroughly, and subjected to lysis using a .22 gauge needle. Cells were centrifuged at 10,000 rpm for 10 min to remove insoluble material, and an aliquot was removed from each sample to determine protein content (Bio-Rad protein assay according to manufacturer’s instructions). Samples were resuspended in loading buffer (50mM Tris, pH 6.8, 2% SDS, 100 mM dithiothreitol, 0.2% bromophenol blue, 20% glycerol) and boiled for 5 min.
Western Blotting
Samples suspended in loading buffer were loaded onto a 4-15% gradient polyacrylamide gel in order to resolve proteins. The proteins were transferred onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA). The membrane was incubated in blocking buffer (5% BSA in 0.1% TBST) for 1 hr followed by overnight incubation of the membrane in blocking buffer containing primary antibody diluted 1:1000. This was followed by four 15-min washes with 0.1% TBST. After washing, secondary antibody (goat-anti-rabbit, Cell Signaling, Danvers, MA, or goat-anti-mouse IgG conjugated to horseradish perodixase, BD Pharmingen, San Diego, CA) diluted 1:2000 was added to the membrane and incubated for 30 min. This was followed by four more 15-min washes with wash buffer. The membrane was then treated with chemiluminescent solution according to the manufacturer’s directions (Thermo Scientific, Rockford, IL) and exposed to film. Densitometric analysis of Western blots was performed using Image J software (NIH).
Immunoprecipitation
6 × 106 T84 and HT29 cells were cultured in 75 cm2 flasks. Following stimulation, cells were scraped and suspended in 1 mL of ice-cold PBS. Cells were centrifuged for 5 min at 5,000 rpm. The pellet was fully resuspended and lysed in 500 μl of ice-cold lysis buffer (5% IGEPAL CA-360, 750 mM NaCl, 250 mM Tris-Cl, pH 8.0, 1μg/ml antipain, 1μg/ml pepstatin, 1μg/ml leupeptin, 100μg/ml phenylmethylsulfonyl fluoride). Lysed cells were centrifuged for 10 minutes at 10,000 rpm to pellet DNA and small nonsoluble particles. 1 μg of monoclonal mouse anti-TCPTP CF-4 antibody was added per sample and placed on a rotating platform at 4°C for 1 hr. This was followed by the addition of 30 μl of the 50% (v/v) protein A-sepharose beads to the cell lysate/antibody mix and allowed to incubate on a rotating platform at 4°C for 1 hr. Samples were then centrifuged briefly to pellet the protein A-Sepharose-antibody-antigen complexes, and the complex was washed three times with cold lysis buffer followed by three more washes with phosphatase reaction buffer (25 mM Hepes, 50 mM NaCl, 1 mM ditheothreitol). Beads were resuspended in phosphatase reaction buffer and allowed to warm to room temperature before the initiation of the phosphatase assay.
Phosphatase Activity Assay
Phosphatase activity was assessed using the EnzChek Phosphatase Assay Kit (Molecular Probes, Eugene OR) according to manufacturer’s instructions using the fluorescent phosphatase substrate, 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP). Upon dephosphorylation, DiFMUP fluoresces at an excitation/emission wavelength of 360/460. TCPTP was first immunoprecipitated from whole cell lysates using anti-TCPTP antibody (see above). Immunoprecipitates were incubated for 15 minutes with DiFMUP after which fluorescence was detected with a SpectraMax M2 Fluorescence Microplate reader using SoftMax Pro v5 Software (Molecular Devices, Sunnyvale, CA). Fluorescence was measured every 15 minutes for the first hour and every 30 minutes thereafter. Measurements were performed in triplicate. A sample from each immunoprecipitation was run on SDS-PAGE and probed for TCPTP to confirm equal protein loading. To account for any differences in overall phosphatase amounts, fluorescence activity units gathered from each assay were compared to TCPTP densitometric values obtained from Western blotting. Thus, values represent the specific activity of TCPTP rather than total quantities of the enzyme.
Small Interfering RNA Transfection
2 × 106 T84 cells were seeded 3 days before transfection. For TCPTP, 100 pmol of 3 different annealed Silencer pre-designed small interfering RNA (siRNA) oligonucleotides (Applied Biosystems, Foster City, CA) were transfected into T84 cells using the Amaxa nucleofector system (Lonza, Walkersville, MD). After transfection, cells were cultured on filter membranes for 72 hours before treatment. Control siRNA SMARTpool (Upstate Biotechnology/Dharmacon, Lake Placid, NY) (100 pmol/transfection) was used as a negative control.
Stable shRNA PTPN2-deficient Cell Line Generation
The shRNA targeting human PTPN2 and scrambled control shRNA were purchased from Sigma-Aldrich (St. Louis, MO). HEK293T cells (1×106) were seeded in a 100 mm dish and allowed to grow overnight. The control shRNA and shRNA against PTPN2 were then transfected into HEK293T cells along with packaging vector (pR8.2) and ENV plasmid (pMDG.2) (gifts from Dr. R. Daniel Beauchamp, Vanderbilt University) using Effectene reagent (Qiagen, Valencia, CA). After 18 hrs of transfection, the HEK293T cells were washed and fresh media was added to the cells. After 48 hrs, the cell supernatant was collected and filtered through a 0.45 μM filter (Millipore, Billerica, MA). The filtered media was used to infect HT-29 cells by mixing equal amounts of fresh media with polybrene (5 mg/ml). After 72–96 hrs of infection, cells were stably selected using puromycin (1 μg/ml). The stable cell lines were maintained with 500ng/ml puromycin. PTPN2 knockdown was confirmed by both RT-PCR as well as by immunoblotting methods.
Transepithelial Electrical Resistance
Transepithelial electrical resistance (TER) across T84 and HT-29 cell monolayers was assessed by voltohmeter (WPI, Sarasota, FL) and companion electrodes (Milipore, Bedford, MA). Measurements were calculated in Ω.cm2 and expressed as a percentage of the baseline measurement.
Transepithelial Permeability Studies
Transepithelial permeability was measured as 4-kilodalton fluorescein isothiocyanate-dextran (FITC-dextran) (Sigma Chemical Co, St. Louis, MO) flux across IEC monolayers. Following VSL#3 and IFN-γ treatment, cells were washed (3X) and incubated for 30 minutes at 37°C with Ringer’s solution to equilibrate. FITC-Dextran (1 mg/mL) was added to the apical compartment of the monolayer. Two hours later, 100 μL of the basolateral solution was removed, and fluorescence was detected using a microplate reader (Molecular Devices, Sunnyvale, CA).
Immunofluorescence Staining and Quantification
HT-29-Control and PTPN2 KD cell lines were plated on coverslips (12-well) and cultured for 3 days. After treatment, the cells were washed (x2) with PBS and fixed with 100% methanol for 5 minutes at −20°C. Cells were washed (x2) with PBS and permeabilized with 0.3% Triton X-100 in PBS for 30 min at room temperature. Cells washed (x2) with PBS and blocked with 10% normal donkey serum (Jackson ImmunoResearch) for 30 min at room temperature. Cells were incubated overnight at 4°C with primary antibodies diluted in 1% normal donkey serum and 0.01% Tween-20 in PBS. Next day, cells were washed (x5) times for 5 min at room temperature with 0.01% Tween-20 in PBS (PBS-T). The cells were incubated with appropriate secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 594, for 1 h at room temperature. At the end of the incubation, cells were washed (x5) with PBS-T and then washed in water and mounted with ProLong Gold plus DAPI (Invitrogen). All images were captured with a Leica DM5500 microscope attached with DFC365 FX camera using 63X oil immersion objective with an additional 2x digital zoom. The individual images were converted to tiff files with the LAS-AF lite software and Photoshop (Adobe) was used to create the final figures. The total fluorescence measurements were performed with Image J software and densitometric analysis was performed on 9-10 images from 4 different fields of view.(27)
Statistical Analysis
All data are means ± SEM for a series of experiments. Statistical analysis was performed by Student’s unpaired t-test or analysis of variance (ANOVA) and Student-Newman-Keuls post-test using Graph Pad Instat 3 software (Graph Pad Software, La Jolla, CA). P values < 0.05 were considered significant.
RESULTS
VSL#3 increased TCPTP protein but not mRNA expression
To study whether VSL#3 had any effect on TCPTP expression in human IECs, T84 cells grown as monolayers on permeable supports were treated with increasing concentrations of VSL#3 or the control commensal E. coli K12 (102, 104, 106 and 108 CFU/mL) for 3, 9, and 24 hours. VSL#3 and K12 were applied only to the apical compartment as VSL#3 is administered via the oral route in vivo. Western blotting indicated that 102, 104 CFU/mL (p<0.01, Fig. 1A,B) and 106 CFU/mL significantly increased TCPTP protein levels over untreated cells and cells treated with K12 following a 3, 9 and 24 hr incubation (p<0.01-p<0.001; n=3 in triplicate). 106 CFU/mL of VSL#3 at 9 hr induced the largest increase in TCPTP levels of all the conditions tested over the untreated and K12 control conditions (p<0.001 vs. media alone or K12 106 CFU/mL, 9 hr; ; n=3 in triplicate; Fig. 1B). At the 24 hr time point, the lower dose of 104 CFU/mL of VSL#3 significantly increased TCPTP expression to (210 ± 15 % over the untreated condition; p<0.001; Fig. 1B) and over K12 (p<0.001; n=3; Fig 1B). VSL#3 was also administered to T84 monolayers at a concentration of 108 CFU/mL; however, this concentration caused a significant decrease in TCPTP and actin expression, indicating a damaging effect on epithelial cell viability. Therefore, this concentration of VSL#3 was not pursued in further studies. K12 at 108 CFU/mL did not appear to compromise cell viability and induced a slight, but statistically significant, increase in TCPTP levels at 9 and 24 hrs (p<0.05 vs. untreated control; n=3 in triplicate; Fig 1B). Interestingly, RT-PCR analysis revealed no increase in TCPTP messenger RNA (mRNA) levels in cells treated with VSL#3 compared to untreated T84 cells (Fig. 1C). Therefore, in an effort to further elucidate the mechanism by which VSL#3 increased TCPTP protein levels, the ribosomal inhibitor, cycloheximide, was used to investigate whether VSL#3 induced TCPTP protein synthesis. Previous studies employing the T84 cell line used cycloheximide at a final concentration of 35.5 μM to inhibit protein synthesis.(25, 28) Therefore, T84 cells grown as monolayers on permeable supports were treated with cycloheximide (35.5 μM) for 30 minutes followed by the addition of VSL#3 (106 CFU/mL) for 9 hours as this concentration and time point induced the greatest increase in TCPTP protein (c.f. Fig 1B). Western blotting indicated a significant increase in TCPTP protein levels in cells treated with 106 CFU/mL of VSL#3 (p < 0.05, Fig. 1D). Pre-administration of cycloheximide completely blocked VSL#3 induction of TCPTP protein to levels comparable with that in cells treated with DMSO vehicle control alone (Fig. 1D). These data suggest that the effect of VSL#3 on TCPTP protein involves protein synthesis although we cannot rule out a possible impact of VSL#3 in limiting degradation of TCPTP protein. The exact nature of regulation of TCPTP protein levels will be the focus of future studies.
FIGURE 1. VSL#3 increased TCPTP protein expression in T84 intestinal epithelial cells.
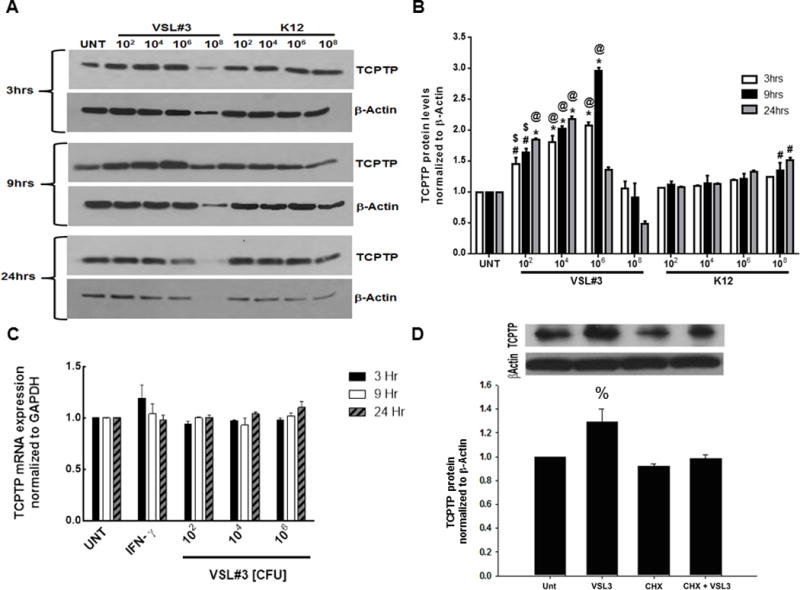
(A & B) Western blotting indicated that cytoplasmic TCPTP protein levels were significantly increased by VSL#3 at 102, 104 and 106 CFU/mL (n = 3 in triplicate for each condition) concentrations over 3, 9 and 24 hrs compared to untreated cells, or cells treated with equivalent concentrations of the non-invading commensal E. coli, K12. Maximal TCPTP protein levels were observed at 9hr treatment with 106 CFU/mL (p<0.01 vs. control). (C) RT-PCR analysis was performed to measure TCPTP mRNA levels in untreated T84 cells and cells treated with 102, 104, and 106 CFU/ml VSL#3 over 3, 9 and 24 hours (n = 3). Experiments were performed in triplicate. There was no statistical significance between the various concentration and incubation time conditions of probiotic (VSL#3) with respect to TCPTP mRNA levels. (D) Pre-treatment with 35.5 μM cycloheximide prior to addition of VSL#3 (106 CFU/mL) for 9 hours significantly blocked VSL#3 induction of TCPTP protein (p<0.05; n=4). %,p<0.05; #,p<0.01; *,p< 0.001 compared to untreated control. $,p<0.01, @,p<0.001 vs. corresponding K12 condition. Statistical analysis was performed by ANOVA and Student-Newman-Keuls post-hoc test.
VSL#3 increased TCPTP enzymatic activity in intestinal epithelial cells
Next, we investigated whether VSL#3 altered TCPTP enzymatic activity. T84 cell monolayers were treated with VSL#3 at varying concentrations (102, 104, 106CFU/mL) for 9 hrs, as this time point exhibited peak TCPTP expression in response to VSL#3 (c.f. Fig 1B). Following incubation for 9 hrs with or without VSL#3, TCPTP was immunoprecipitated from whole T84 cell lysates. The phosphatase assay was initiated by the addition of the substrate (DiFMUP) to TCPTP immunoprecipitates and enzymatic activity of TCPTP was measured over 120 minutes. After 30 minutes of treatment, 102, 104 and 106 CFU/mL of VSL#3 significantly increased TCPTP activity (p<0.001, n=5; Fig. 2A). This was maintained through 90 and 120 minutes with 106 CFU/mL of VSL#3 inducing a maximal increase in activity of 55 ± 9% by 120 minutes (p<0.001, n=5, Fig. 2A). A time course response of TCPTP activity after incubation of T84 cells with 106 CFU/mL VSL#3 was also performed. Incubation for 3, 9 and 24 hrs significantly increased activity above untreated controls with 24 hrs incubation having the greatest effect on TCPTP activity vs. untreated when corrected for protein level (p<0.001; n=4; Fig 2B).
FIGURE 2. VSL#3 increased TCPTP enzymatic activity in T84 intestinal epithelial cells.
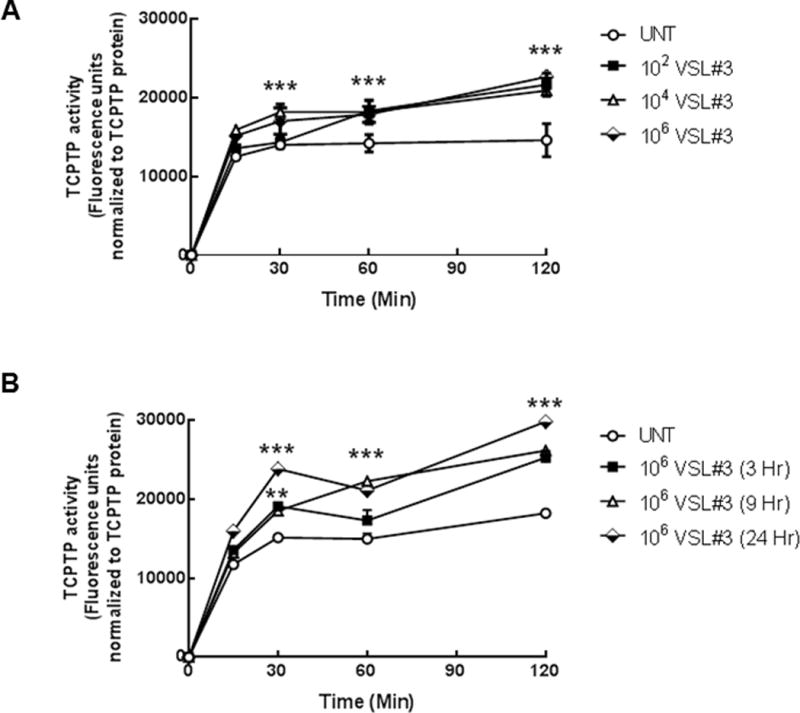
(A) T84 cell monolayers were treated with VSL#3 at varying concentrations (102, 104, 106 CFU/mL) for 9 hrs. TCPTP was immunoprecipitated from whole cell lysates and TCPTP activity was assessed over 120 minutes (n = 5). A sample from each immunoprecipitation was run on an SDS-PAGE gel and following transfer to PVDF membrane, probed for TCPTP to confirm equal protein loading. Fluorescence activity units were expressed relative to TCPTP protein levels to account for any differences in overall phosphatase amounts. 106 CFU/mL induced the greatest increase in TCPTP enzymatic activity (p<0.001, n=5). (B) A time course of incubation of T84 cells with 106 CFU/mL was performed for 3, 9 and 24 hrs. VSL#3 maximally increased activity above untreated controls after 9 hr incubation (p<0.001; n=4). All data are expressed as a percentage of the control ± SEM and analyzed by ANOVA and Student-Newman-Keuls post-hoc test. Asterisks denote significant differences vs. the respective untreated control. (***,p< 0.001 compared to the respective untreated time point).
VSL#3 attenuated IFN-γ signaling in a TCPTP-dependent manner
Having determined that VSL#3 stimulated TCPTP activity, and given the importance of TCPTP as a negative regulator of IFN-γ signaling, it was of interest to establish whether VSL#3 had any effect on IFN-γ signaling and the phosphorylation state of the IFN-γ downstream signaling molecule, and TCPTP substrate, STAT1. A pathophysiologically relevant concentration of IFN-γ (1000 U/mL) was added basolaterally to T84 monolayers.(29) IFN-γ (24 hrs) increased STAT1 phosphorylation but increasing concentrations of VSL#3 alone had no effect on STAT1 phosphorylation as revealed by Western blotting analysis (Fig 3A). In subsequent studies, and following 24 hrs treatment with cytokine, VSL#3 (102, 104, 106 CFU/mL) was added apically for 9 hrs as determined by data in Fig 1B. IFN-γ increased total STAT-1 expression at 24 hr, but as expected and consistent with our previous observations, IFN-γ increased STAT-1 phosphorylation relative to total STAT1 compared to untreated cells (p<0.001, Fig. 3B,C).(21, 25) Administration of VSL#3 for 9 hrs following 24 hrs of IFN-γ treatment significantly reduced STAT-1 phosphorylation in a dose-dependent manner compared to IFN-γ-treated cells alone (p<0.05, n=3; Fig. 3B,C). K12 (106 CFU/mL) had no effect on IFN-γ-induced phosphorylation of STAT1 normalized to total STAT1 and β-actin even though total STAT1 was reduced in the IFN-γ + K12 condition (Fig 3B,C). These data indicate that administration of VSL#3, even after IFN-γ has activated downstream signaling pathways, can alleviate IFN-γ signaling in a therapeutic manner. Next, we investigated whether the attenuation of STAT1 phosphorylation observed in T84 cells co-administered VSL#3 with IFN-γ was actually mediated by TCPTP. T84 cells were transfected with either TCPTP-specific siRNA or nonspecific control siRNA, seeded on permeable supports, and allowed to grow for 72 hrs. Nonspecific effects of siRNA transfections on overall protein levels were not observed, as shown by equivalent levels of the loading control, β-actin, in each experiment. Following the addition of IFN-γ (1000 U/mL) to cell monolayers, Western blotting revealed that IFN-γ induced STAT1 phosphorylation following 24 hrs treatment and that this level was significantly increased in both control and TCPTP siRNA transfected cells compared with untreated conditions. (p < 0.001; n=4-5; Fig. 3D,E). As expected, a decrease in STAT1 phosphorylation was observed in control siRNA transfected cells administered VSL#3 (106 CFU/mL, 9hrs) after IFN-γ vs. IFN-γ alone (p < 0.05, n=5; Fig. 3E). Interestingly, the decrease in STAT1 phosphorylation caused by VSL#3 was not observed in TCPTP knockdown cells compared to control siRNA-treated cells (p < 0.05, n=4; Fig. 3E). Together, these data indicate that TCPTP is the likely mediator through which VSL#3 reduces IFN-γ-induced STAT1 phosphorylation.
FIGURE 3. VSL#3 attenuated STAT1 signaling in a TCPTP-dependent manner.
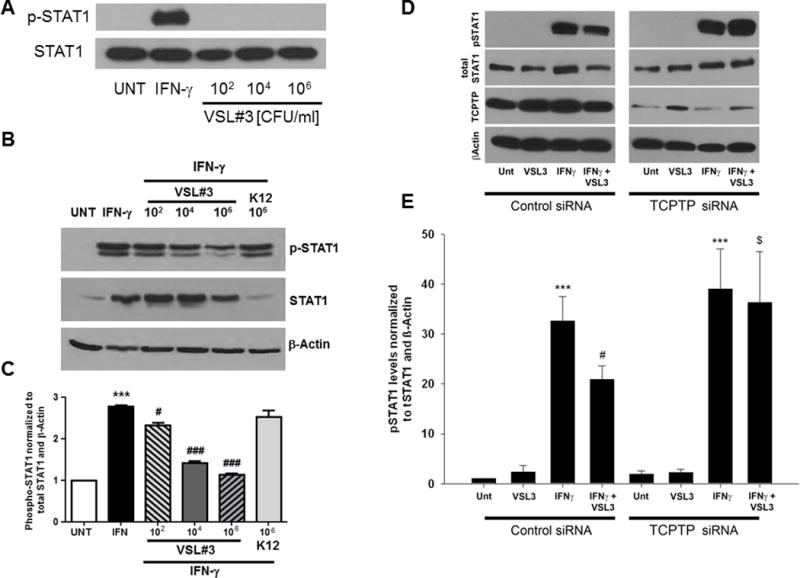
(A) T84 monolayers were basolaterally treated with IFN-γ (1000 U/ml) for 24 hrs or VSL#3 (102, 104, 106, 108 CFU/mL) apically for 9 hrs and STAT1 phosphorylation and total STAT1 levels were determined by subsequent Western blotting of cell lysates. While IFN-γ as expected increased phospho-STAT1, VSL#3 alone was without effect (representative blot of 3 experiments. (B,C) Densitometric analysis of Western blots from T84 monolayers treated basolaterally with IFN-γ (1000 U/ml) for 24 hrs showed a significant increase in STAT1 phosphorylation relative to total STAT1 and β-actin (p<0.01; n=3). Treatment of IFN-γ-incubated T84 monolayers at 24 hrs with VSL#3 (104, 106 CFU/ml) for 9 hours caused a significant reduction in p-STAT1 levels (p<0.05-p<0.001; n=3). K12 (106 CFU/ml) had no effect on IFN-γ induced STAT1 phosphorylation. (D) T84 cells were transfected with either TCPTP-specific siRNA or nonspecific control siRNA. Cells were treated with IFN-γ (1000 U/ml) or vehicle control for 24 hr followed by VSL#3 (106 CFU/ml) or serum-free media for 9 hours. Whole cell lysates were processed by SDS-PAGE electrophoresis and STAT1 phosphorylation and total STAT1 levels were determined by Western blotting. β-Actin was used throughout as a loading control and the relative protein levels were assessed by densitometry. (E) Densitometric analysis showed that IFN-γ (1000 U/ml) significantly increased STAT1 phosphorylation relative to total STAT1 levels (p<0.001; n=3). A decrease in STAT1 phosphorylation was observed in control siRNA transfected cells administered VSL#3 vs. IFN-γ alone (p<0.05; n=3). This effect of VSL#3 was lost in TCPTP-siRNA transfected T84 cells (p<0.05 vs. IFN-γ + VSL#3 in control siRNA cells). (n = 3). β-Actin was used throughout as a loading control and the relative protein levels were assessed by densitometry. All data are expressed as a percentage of the control ± SEM and analyzed by ANOVA and Student-Newman-Keuls post-hoc test. Asterisks denote significant differences vs. the respective control. (**,p<0.01, ***,p<0.001 compared to untreated control, #,p<0.05, compared to IFN-γ-treated cells; $,p<0.05 compared to IFNγ+VSL#3 treatment of control siRNA-transfected cells).
VSL#3 increased intestinal epithelial barrier integrity and restored barrier function following inflammatory cytokine exposure
To determine how VSL#3 affects the integrity of T84 epithelial-cell monolayers, concentration-response experiments with VSL#3 were performed and resulting TER changes were recorded over 24 hours. VSL#3 (102, 104, 106 CFU/mL, n = 3) increased TER compared to untreated cells following 9 and 24 hr treatment. The greatest increase in TER over untreated was observed with 106 CFU/mL of VSL#3 with incubation times of 9 hours and 24 hours. At 9 hours, cells treated with 106 CFU/mL of VSL#3 had a TER that was significantly greater than untreated samples (p < 0.05, n=3; Fig. 4A). At 24 hours, the condition with 106 CFU/mL of VSL#3 had a TER that was 38 ± 6 % greater than untreated samples (p < 0.05; n=3; Fig. 4A). Given the strong effect of VSL#3 in increasing TER, it was of interest to determine whether VSL#3 could enhance intestinal epithelial barrier function when administered following challenge with IFN-γ. IFN-γ has been reported to decrease TER in T84 cells by as much as 40% as early as 24 hours after treatment.(30, 31) To investigate a possible therapeutic effect of VSL#3 on a compromised barrier, IFN-γ was added basolaterally (1000 U/mL) once T84 cells had established a stable monolayer in which the baseline TER before treatment was greater than 1000 Ohms/cm2. Following IFN-γ treatment for 24 hrs, cells were then untreated or incubated with VSL#3 or K12 at 106 CFU/mL for 9 hrs. After a combined 33 hrs of treatment, IFN-γ-exposed cells treated with VSL#3 had a final TER that was significantly greater than IFN-γ-treated cells alone indicating partial recovery of TER (p < 0.001; n=3; Fig. 4B). The IFN-γ-induced decrease in TER (p<0.001; Fig 4B) was not rescued by K12 treatment. To determine if the restorative effect of VSL#3 on TER at 33 hrs (p<0.001 vs. IFN-γ; n=3 in triplicate; Fig 4C) required viable bacteria, we performed the same rescue experiment using heat-killed VSL#3 and K12 (Fig 4C). Data with regular VSL#3 or K12 (REG) reflect the 33 hr condition in panel B. Heat-killed VSL#3 failed to rescue the reduction in TER caused by IFN-γ (Fig 4C). IFN-γ also significantly increased epithelial cell macromolecule permeability following 33 hrs of treatment as measured by 4 kDa FITC-dextran flux (p < 0.001, Fig. 4D). Administration of VSL#3 (10 μM) for 9 hrs, but not K12, following 24 hrs of IFN-γ treatment, significantly reduced the flux of FITC-dextran across T84 cell monolayers (p < 0.001; n=4 in triplicate; Fig. 4C). Heat-killed VSL#3 were completely unable to rescue FD4 permeability (p<0.001 vs. VSL#3 [REG] + IFN-γ; n=3 in triplicate; Fig 4D) while heat-killed K12 were without effect (Fig 4D). These studies revealed a therapeutic role for VSL#3 in the regulation of epithelial barrier function with partial recovery of TER and full recovery from FD4 permeability in IFN-γ-exposed IEC monolayers. To determine if the therapeutic effect of VSL#3 was due to the physical presence of bacteria or possibly to secreted factors, we repeated these experiments using conditioned media from VSL#3 cultures (106 CFU/mL). Following 24 hr basolateral incubation with IFN-γ, or vehicle (control), cells were incubated apically with filtered conditioned media or serum-free media for 9 hrs and TER and FITC-dextran permeability were assessed. VSL#3 conditioned media (VSL#3-CM) partially reversed the drop in TER caused by IFN-γ treatment (p<0.01; n=3; Fig 4E). VSL#3-CM also caused a decrease in FD4 permeability vs. IFN-γ alone although this did not reach statistical significance (Fig 4F). Therefore while partially effective, VSL#3-CM did not reverse the influence of IFN-γ on parameters of barrier function indicating a need for whole bacteria to fully manifest the recuperative effect on barrier function.
FIGURE 4. VSL#3 increases intestinal epithelial barrier integrity and restores barrier function following inflammatory cytokine exposure.
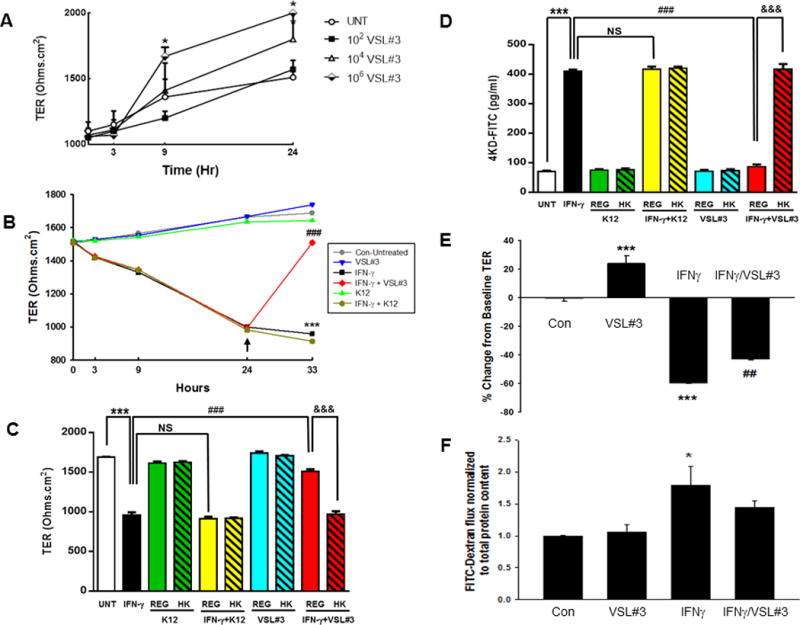
T84 cells were plated on transwells and grown for 10 days. (A) Media was changed on alternate days and the TER was measured at 3, 9 and 24hrs after adding increasing concentrations of VSL#3. 106 CFU/mL alone significantly increased TER above untreated (p < 0.05; n=3). (B) IFN-γ was added basolaterally and reduced TER by 24 hrs. Untreated or IFN-γ treated cells were incubated apically at the 24 hr time point with K12 control bacteria or VSL#3 at 106 CFU/mL for 9 hours (total 33hrs). The IFN-γ-induced decrease in TER at 33 hrs (***,p<0.001; n=3) was significantly reversed by VSL#3 (###,p<0.001; n=3 in triplicate) while K12 had no effect. (C) Viable (Regular [REG]; solid bars) vs. heat-killed (HK) bacteria (hatched bars) were added apically to the untreated or IFN-γ (24 hr) conditions and the effect on TER at 33 hrs was determined. (D) At 33 hrs, cells from the same experiment as (C) were washed with PBS to remove bacteria and FITC-Dextran (4 kD (FD4); 1 mg/mL) was added to the apical compartment to measure macromolecule permeability. Two hours later, 100 μL of the basolateral solution was removed, and fluorescence was measured. IFN-γ alone significantly increased FD4 permeability vs. control cells (***,p<0.001; n=3 in triplicate). Treatment with VSL#3 but not K12 (106 CFU/mL) significantly increased TER and reduced permeability vs. IFN-γ alone (###,p<0.001; n=3). The rescue of TER and reversal of IFN-γ induction of FD4 permeability by VSL#3 (REG) was lost when VSL#3 were heat-killed (&&&,p<0.001 vs. IFN-γ + REG VSL#3). (E) Filtered conditioned media from VSL#3 (106 CFU/mL) was added apically to polarized T84 cells at 24 hrs after IFN-γ or vehicle treatment (Control). VSL#3 conditioned media (CM) alone increased TER vs. control (p<0.001; n=3). VSL#3 CM partially reversed the IFN-γ-induced drop in TER although TER was still significantly lower than control (p<0.01; n=3). (F) FD4 permeability in the same cells was assessed at the end of the treatment period (33 hrs after initial treatment with IFN-γ). IFN-γ significantly increased FD4 permeability (p<0.05) and this effect was partially lost following treatment with VSL#3 CM (n=3).
VSL#3 recovery of IFN-γ-induced barrier defects is TCPTP-dependent
We next investigated whether the recuperative effects of VSL#3 on barrier function changes caused by IFN-γ were mediated by TCPTP. These studies were conducted using a novel PTPN2 (TCPTP) shRNA stable knockdown HT-29 cell line (PTPN2-KD) generated in our lab, along with a complementary control shRNA transfected HT-29 cell line (Con-shRNA)(Fig 5A,B). Resting TER in PTPN2-KD cells was significantly reduced compared with Con-shRNA HT-29 monolayers indicating a role for TCPTP in basal regulation of TER (p<0.001; n=3; Fig 5C). In Con-shRNA cells, administration of VSL#3 (106 CFU/ml) 24 hrs after IFN-γ promoted a significant recovery of TER vs. IFN-γ alone (p<0.001; n=3; Fig 5D). This restorative effect was significantly reduced in PTPN2-KD cells (p<0.001; n=3; Fig 5C). Interestingly, the drop in TER caused by IFN-γ occurred more rapidly in PTPN2-KD cells vs. control cells as a significantly greater relative decrease in TER occurred in PTPN2-KD cells at the 9 hr time point (p<0.05; n=3; Fig 5D). Further analysis of the extent of the restorative effect of VSL#3 against the drop in TER caused by IFN-γ indicated that in control cells, VSL#3 caused a recovery in TER of 30 ± 1.2 % and this was reduced to 12.7 ± 3.2% in PTPN2-KD cells (p<0.05; n=3; Fig 5E). These data indicate that TCPTP is required for recovery of TER by VSL#3 in IFN-γ pre-treated IEC monolayers. Following VSL#3 (106 CFU/mL) and IFN-γ treatment, FITC-Dextran (4 kD; 1 mg/mL) permeability was determined. (F) Untreated KD cells showed increased FD4 permeability vs. untreated control cells (p<0.001; n=3; Fig 5F) while IFN-γ treatment significantly increased permeability in both cell types (p<0.05; n=3 respectively). VSL#3 significantly reduced permeability vs. IFN-γ alone in C-shRNA cells (p<0.01; n=3), but this effect was completely inhibited in KD cells. These data indicate that TCPTP is required to mediate VSL#3 reversal of IFN-γ-induced macromolecule permeability.
FIGURE 5. Restorative effects of VSL#3 on barrier function are TCPTP-dependent.
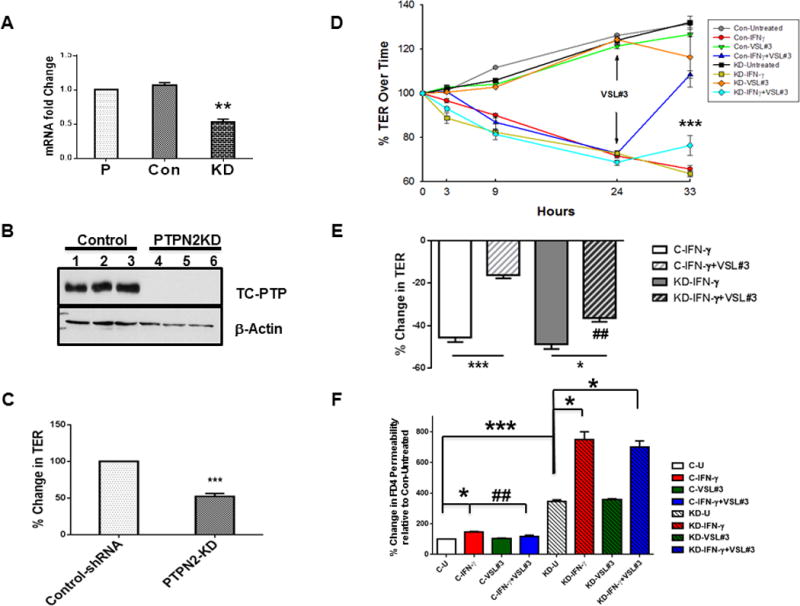
PTPN2-shRNA stable knockdown of PTPN2 in HT-29 IEC was achieved using a lentiviral transfected shRNA (KD). Control (scrambled) shRNA transfected cells and parental HT-29 cells were also used to quantify mRNA levels of PTPN2. (A) PTPN2-KD cells showed a significant reduction in TCPTP mRNA relative to parental cell line (p<0.01; n=3; data expressed as fold change from parental cell line following normalization to GAPDH levels). Con-shRNA transfected cells showed no difference vs. parental HT-29 cells. (B) Control shRNA (Control clones 1-3), and shRNA directed against PTPN2 (KD clones 4-6) were lysed with RIPA buffer and analyzed by Western blot for TCPTP to confirm knockdown of TCPTP protein in PTPN2-KD cells (n=3). β-actin was probed as a loading control. (C) PTPN2-KD significantly reduced the resting TER value by 50±4% compared with Con-shRNA transfected cells (p<0.001; n=3). (D-F) Established monolayers of C-shRNA and TCPTP-shRNA cells were treated with IFN-γ (basolaterally) for 24 hrs followed by apical incubation with VSL#3 (106 CFU/mL) for an additional 9 hrs. (D) The percent change in TER from respective untreated conditions (control shRNA untreated or PTPN2-KD untreated) over the course of treatment was calculated. VSL#3 rescue of TER was partially reversed by TCPTP KD (p<0.001 vs. C-IFN-γ + VSL#3; n=3 in triplicate). (E) The contribution of TCPTP to the restorative effect of VSL#3 on TER modified by IFN-γ in control (C-IFN-γ) and PTPN2-KD (KD-IFN-γ) cells was calculated. Loss of TCPTP significantly, but not completely, reversed the effect of VSL#3 on TER (p<0.01 between KD-IFN-γ + VSL#3 vs. Con IFN-γ + VSL#3; n=3 in triplicate). Following VSL#3 (106 CFU/mL) and IFN-γ treatment, cells were washed with PBS and FITC-Dextran (4 kD; 1 mg/mL) was added to the apical compartment. After two hours 100 μL of the basolateral solution was removed and fluorescence was measured. (F) Untreated KD cells showed increased FD4 permeability vs. untreated control cells (p<0.001; n=3 in triplicate) while IFN-γ treatment significantly increased permeability in both cell types (p<0.05; n=3 respectively). VSL#3 significantly reduced permeability vs. IFN-γ alone in C-shRNA cells (p<0.01; n=3 in triplicate), but this effect was completely inhibited in KD cells (KD-IFN-γ+VSL#3 bar). These data indicate that VSL#3 rescue of IFN-γ effects on TER and permeability is TCPTP-dependent. [*, p<0.05 vs. untreated condition (n=3; triplicates); ***,p<0.001 vs. control untreated; ##, p<0.001 vs. Control-IFN-γ].
TCPTP is required for VSL#3 correction of tight junction protein localization
IFN-γ has previously been shown to compromise barrier function by promoting removal and internalization of key tight junction proteins away from the tight junction resulting in their subsequent degradation.(25, 30, 32, 33) To identify if the VSL#3-induced recovery of barrier function was associated with recovery of tight junction proteins, we performed immunofluorescence staining for the transmembrane protein occludin, the regulatory molecule ZO-1 and the cation-pore forming transmembrane protein claudin-2. IFN-γ treatment for 24 hrs was followed by 9 hrs treatment with VSL#3 as previously. IFN-γ alone reduced both ZO-1 and occludin levels in control and PTPN2 shRNA transfected HT-29 cells while VSL#3 treatment rescued this effect (p<0.001; n=3; Fig 6 A,B). The VSL#3 promoted recovery of occludin and ZO-1 was restricted in PTPN2-KD cells indicating an integral role for TCPTP. In addition, IFN-γ increased claudin-2 expression in control and PTPN2-KD HT-29 cells and this was largely reversed by VSL#3 in control cells (p<0.01; n=3; Fig 6A, B). However, the corrective effect of VSL#3 on claudin-2 was almost completely lost in PTPN2-KD cells. These data indicate that loss of TCPTP severely mitigated the capacity of VSL#3 to correct IFN-γ-induced alterations of tight junction composition and are consistent with the effects on barrier function identified in Fig 5.
FIGURE 6. TCPTP mediates VSL#3 normalization of tight junction proteins in IFN-γ-treated IEC.
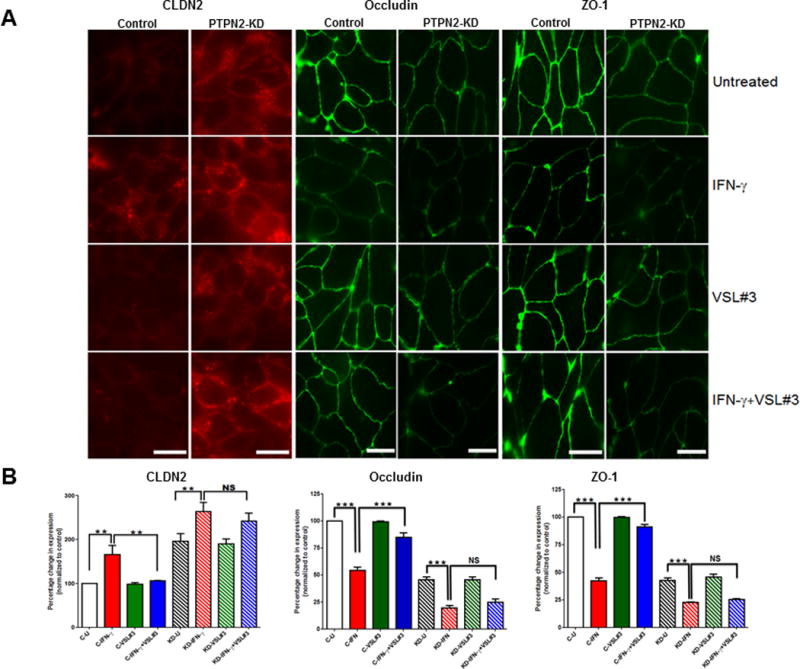
Control shRNA or PTPN2-KD HT-29 IEC were grown on coverslips for 3 days. Cells were treated with IFN-γ or control media for 24 hrs prior to incubation with VSL#3 (106 CFU/mL) or serum-free media for a further 9 hrs. (A) At the end of treatment, cells were fixed with methanol and stained for Claudin-2 (left 2 panels); Occludin (middle 2 panels); or ZO-1 (right 2 panels) (n = 3 experiments, scale bar = 25μm). (B) The total fluorescence intensity of claudin-2, occludin and ZO-1 antibody was measured by Image J software (see Methods section) in unaltered images (9–10 images from 4 fields of view) and data were expressed as % change in expression compared with untreated control cells (C-U). VSL#3 significantly reversed the IFN-γ-induced increase in claudin-2 and decrease in occludin and ZO-1 respectively in control shRNA cells. This effect was lost in PTPN2-KD cells. Statistical significance was calculated using one-way analysis of variance followed by Student-Newman-Keuls post-test. (*,p<0.01 and **,p<0.001, ns = non-significant).
DISCUSSION
In this study we have identified that the VSL#3 probiotic is capable of increasing enzymatic activity and protein expression of the protein tyrosine phosphatase TCPTP. This phosphatase protects intestinal epithelial barrier function likely through its roles in dephosphorylating signaling targets downstream of the IFN-γ receptor. Furthermore, we have shown that VSL#3, but not the control commensal E. coli K12, can promote recovery of barrier properties in IFN-γ pre-treated IEC monolayers and these effects are mediated, at least in part, via TCPTP. The protective effects of VSL#3 on intestinal homeostasis have been demonstrated in limiting both initial as well as recurrent colitis in mouse models of intestinal inflammation including IL-10−/−, TNFΔARE, SAMP and TNBS.(34–39) Moreover, VSL#3 has proven efficacious in treating human ulcerative colitis.(6, 7) Determining the active component of VSL#3 has been pursued by a number of investigators with studies identifying biological activity of VSL#3 DNA, cell debris generated by sonication and conditioned medium from VSL#3 as well as from several bacterial strains found in VSL#3, in addition to the effects of whole VSL#3.(9, 40, 41) In the present study, we observed that viable VSL#3 was absolutely required for the restorative effect on barrier function suggesting possible interactions with innate immune signaling components on intestinal epithelial cells. Moreover, VSL#3 conditioned media partially restored TER and FD4 permeability changes in IFN-γ pre-treated IEC monolayers but not to the same extent as whole bacteria. The efficacy of both whole viable bacteria and conditioned media, at least in part, may provide a clue as to the mechanism by which VSL#3 increases TCPTP protein levels without increasing mRNA transcription. Petrof et al. demonstrated that VSL#3, including conditioned medium, can inhibit proteasomal degradation of target proteins in IEC monolayers and thus inhibit TNF-α-induced NF-κB activation. Furthermore, this inhibitory effect appeared to be mediated by a soluble factor(s).(42) One intriguing finding that we are unable to fully explain is the observation that the enzymatic activity of TCPTP was maximally elevated by VSL#3 at 106 CFU/ml at 24 hrs whereas peak VSL#3-induced expression of TCPTP occured at 9 hrs. Since the phosphatase activity assay used equal amounts of immunoprecipitated TCPTP protein, there may be some as yet undetermined post-translational modification of TCPTP that renders it more active after 24 hrs vs. 9 hrs exposure of intestinal epithelial cells to VSL#3. With respect to appropriate dosing of VSL#3 in relation to in vivo studies, Tursi et al. used a dose of 3.6 × 109 CFU twice daily in UC patients while Mennigen et al. used a dose of 13.5 × 109 CFU/ml to treat DSS colitis in mice and stated that they considered that previous studies had used insufficient concentrations of VSL#3.(7, 43) Therefore, while our concentration range understandably falls below the range of in vivo studies, it does approximate concentrations used in in vitro studies on IEC lines.(26, 44)
A number of anti-inflammatory effects of VSL#3, or VSL#3 components, have been identified through in vitro and in vivo studies that are consistent with VSL#3 efficacy in reducing inflammation in human ulcerative colitis. These include reducing the secretion of inflammatory mediators such as IL-8 and IFN-γ; increasing production of anti-inflammatory cytokines such as IL-10 and TGF-β; inhibition of NF-κB; and promotion of barrier function.(9, 26, 36, 42–46) Two of the bacteria in VSL#3, S. thermophilus and L. acidophilus, when given in combination were able to partially inhibit IFN-γ-induced disruption of colonic epithelial cell barrier properties as well as reduce phosphorylation of STAT-1 and -3.(47) Furthermore, a recent study identified that VSL#3 treatment of dendritic cells reduced lipopolysaccharide (LPS)-induced STAT1 phosphorylation and transcription of STAT-1 gene targets including various inflammatory chemokines.(48) This is in broad agreement with our observations that VSL#3 reduces STAT-1 phosphorylation by IFN-γ in IEC and our data are consistent with a role for TCPTP in mediating VSL#3 inhibition of STAT-1 activation in multiple cell types.
While several studies have demonstrated a beneficial role for VSL#3 with respect to protection of barrier function, the mechanism through which this occurs has only been partially elucidated. Otte & Podolsky identified that VSL#3 and conditioned medium had a stabilizing effect on TER by modifying localization of the tight junction protein ZO-1 and blocking the disruptive effects of Salmonella on ZO-1 localization.(44) Secreted factors from bacterial strains found in VSL#3 were also shown to protect barrier properties when co-administered with inflammatory cytokines by restricting cytokine-induced reductions in ZO-1 and occludin levels while preventing increased claudin-2.(41) Corridoni et al. observed that VSL#3 increased occludin but decreased claudin-2 expression in the SAMP-1 mouse that develops spontaneous ileitis.(38) Interestingly, they also observed changes in localization of claudin-2 in VSL#3 treated mice including decreased claudin-2 in the villus tips with more pronounced expression in crypt epithelium where claudin-2 is normally observed. In addition, claudin-2 levels were reduced in the membranes of isolated ileal epithelium but increased in the cytoplasm indicating that the beneficial effect of VSL#3 may be due in part to internalization of claudin-2 away from the tight junction.(38) IFN-γ is a well-established modulator of ZO-1 and occludin localization at the tight junction and is also capable of reducing expression of these proteins.(25, 30, 32, 33) We have previously reported that siRNA knockdown of TCPTP did not exacerbate IFN-γ-induced reductions in expression of these proteins in T84 cells.(21) Here we observed that in TCPTP stable knockdown HT-29 cells, IFN-γ treatment did in fact cause a further reduction in levels of ZO-1 and occludin suggesting context-dependent differences in the response of cells to cytokine challenge on a background of acute siRNA vs. stable knockdown of TCPTP, notwithstanding that different IEC lines were also used in these separate studies (p<0.001; n=3; Fig 6A, B). Moreover, resting levels of occludin and ZO-1 in untreated PTPN2-KD (KD-U) cells were significantly lower than untreated control shRNA transfected cells (C-U), while claudin-2 levels in untreated PTPN2-KD cells were increased (p<0.01; n=3; Fig 6A, B). These data indicate that a background of deficient TCPTP expression in the absence of further treatment can modify TJ composition. VSL#3 did largely restore occludin and ZO-1 levels and reduce claudin-2 expression following IFN-γ treatment indicating the likely molecular mechanisms for the reduction in permeability and recovery of TER. Strikingly, this effect was largely lost in PTPN2-KD cells suggesting that TCPTP is a critical mediator of VSL#3 effects on epithelial barrier integrity. This is also consistent with our observations that VSL#3 not only increased protein levels of TCPTP but also its enzymatic activity (c.f. Fig 1, 2). In addition to preserving the molecular composition of tight junctions, VSL#3 has also been shown to decrease the number of apoptotic epithelial cells in a DSS murine colitis model.(43) We did not observe an increase in IEC apoptosis in response to IFN-γ alone, as determined by caspase-3 cleavage, and therefore did not assess this parameter in the presence of VSL#3 (data not shown).
In conclusion, these studies identify a novel molecular target for the VSL#3 probiotic and suggest that TCPTP, the protein product of the IBD candidate gene, PTPN2, contributes to protection and recovery of epithelial barrier function by VSL#3. These studies also have relevance to celiac disease given that VSL#3 probiotic preparation has the capacity to hydrolyze gliadin polypeptides responsible for celiac disease inflammatory events, while SNPs in the PTPN2 gene encoding TCPTP are also associated with celiac disease. Moreover, increased intestinal permeability is also a feature of this condition.(49–51) While a functional interaction between TCPTP, VSL#3 and tight junction proteins has yet to be demonstrated in vivo, our studies do identify a functional link between these three parameters and recovery of intestinal epithelial barrier properties.
Acknowledgments
This research was supported by NIH 5R01-DK091281-03 (DFM); CCFA Career Development Award (RRM); and support from VSL Pharmaceuticals Inc.
The abbreviations used
- IBD
inflammatory bowel disease
- CD
Crohn’s Disease
- UC
Ulcerative colitis
- IEC
intestinal epithelial cell
- PTP
protein tyrosine phosphatase
- PTPN2
protein tyrosine phosphatase non-receptor type 2
- TCPTP
T-cell protein tyrosine phosphatase
- IFN-γ
interferon gamma
- TNF-α
tumor necrosis factor-alpha
- STAT
signal transducer and activator of transcription
- TER
transepithelial electrical resistance
- FITC-dextran
fluorescent isothiocyanate-dextran
References
- 1.Plevy S, Silverberg MS, Lockton S, et al. Combined serological, genetic, and inflammatory markers differentiate non-IBD, Crohn’s disease, and ulcerative colitis patients. Inflamm Bowel Dis. 2013;19:1139–1148. doi: 10.1097/MIB.0b013e318280b19e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodrigues DM, Sousa AJ, Johnson-Henry KC, et al. Probiotics are effective for the prevention and treatment of Citrobacter rodentium-induced colitis in mice. J Infect Dis. 2012;206:99–109. doi: 10.1093/infdis/jis177. [DOI] [PubMed] [Google Scholar]
- 3.Dai C, Zheng CQ, Meng FJ, et al. VSL#3 probiotics exerts the anti-inflammatory activity via PI3k/Akt and NF-kappaB pathway in rat model of DSS-induced colitis. Mol Cell Biochem. 2013;374:1–11. doi: 10.1007/s11010-012-1488-3. [DOI] [PubMed] [Google Scholar]
- 4.Miele E, Pascarella F, Giannetti E, et al. Effect of a probiotic preparation (VSL#3) on induction and maintenance of remission in children with ulcerative colitis. Am J Gastroenterol. 2009;104:437–443. doi: 10.1038/ajg.2008.118. [DOI] [PubMed] [Google Scholar]
- 5.Bassaganya-Riera J, Viladomiu M, Pedragosa M, et al. Probiotic bacteria produce conjugated linoleic acid locally in the gut that targets macrophage PPAR gamma to suppress colitis. PLoS One. 2012;7:e31238. doi: 10.1371/journal.pone.0031238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bibiloni R, Fedorak RN, Tannock GW, et al. VSL#3 probiotic-mixture induces remission in patients with active ulcerative colitis. Am J Gastroenterol. 2005;100:1539–1546. doi: 10.1111/j.1572-0241.2005.41794.x. [DOI] [PubMed] [Google Scholar]
- 7.Tursi A, Brandimarte G, Papa A, et al. Treatment of relapsing mild-to-moderate ulcerative colitis with the probiotic VSL#3 as adjunctive to a standard pharmaceutical treatment: a double-blind, randomized, placebo-controlled study. Am J Gastroenterol. 2010;105:2218–2227. doi: 10.1038/ajg.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pronio A, Montesani C, Butteroni C, et al. Probiotic administration in patients with ileal pouch-anal anastomosis for ulcerative colitis is associated with expansion of mucosal regulatory cells. Inflamm Bowel Dis. 2008;14:662–668. doi: 10.1002/ibd.20369. [DOI] [PubMed] [Google Scholar]
- 9.Jijon H, Backer J, Diaz H, et al. DNA from probiotic bacteria modulates murine and human epithelial and immune function. Gastroenterology. 2004;126:1358–1373. doi: 10.1053/j.gastro.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 11.Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402:1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCole DF. Phosphatase regulation of intercellular junctions. Tissue Barriers. 2013;1:e26713. doi: 10.4161/tisb.26713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mattila E, Pellinen T, Nevo J, et al. Negative regulation of EGFR signalling through integrin-alpha1beta1-mediated activation of protein tyrosine phosphatase TCPTP. Nat Cell Biol. 2005;7:78–85. doi: 10.1038/ncb1209. [DOI] [PubMed] [Google Scholar]
- 14.Galic S, Hauser C, Kahn BB, et al. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol Cell Biol. 2005;25:819–829. doi: 10.1128/MCB.25.2.819-829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shields BJ, Court NW, Hauser C, et al. Cell cycle-dependent regulation of SFK, JAK1 and STAT3 signalling by the protein tyrosine phosphatase TCPTP. Cell Cycle. 2008;7:3405–3416. doi: 10.4161/cc.7.21.6950. [DOI] [PubMed] [Google Scholar]
- 17.Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franke A, Balschun T, Karlsen TH, et al. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nature genetics. 2008;40:713–715. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 19.Glas J, Wagner J, Seiderer J, et al. PTPN2 gene variants are associated with susceptibility to both Crohn’s disease and ulcerative colitis supporting a common genetic disease background. PLoS One. 2012;7:e33682. doi: 10.1371/journal.pone.0033682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scharl M, Mwinyi J, Fischbeck A, et al. Crohn’s disease-associated polymorphism within the PTPN2 gene affects muramyl-dipeptide-induced cytokine secretion and autophagy. Inflamm Bowel Dis. 2012;18:900–912. doi: 10.1002/ibd.21913. [DOI] [PubMed] [Google Scholar]
- 21.Scharl M, Paul G, Weber A, et al. Protection of epithelial barrier function by the Crohn’s disease associated gene protein tyrosine phosphatase n2. Gastroenterology. 2009;137:2030–2040. doi: 10.1053/j.gastro.2009.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCole DF. Regulation of epithelial barrier function by the inflammatory bowel disease candidate gene, PTPN2. Ann N Y Acad Sci. 2012;1257:108–114. doi: 10.1111/j.1749-6632.2012.06522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCole DF. IBD candidate genes and intestinal barrier regulation. Inflamm Bowel Dis. 2014;20:1829–1849. doi: 10.1097/MIB.0000000000000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mattila E, Marttila H, Sahlberg N, et al. Inhibition of receptor tyrosine kinase signalling by small molecule agonist of T-cell protein tyrosine phosphatase. BMC Cancer. 2010;10:7. doi: 10.1186/1471-2407-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Penrose HM, Marchelletta RR, Krishnan M, et al. Spermidine stimulates T cell protein-tyrosine phosphatase-mediated protection of intestinal epithelial barrier function. J Biol Chem. 2013;288:32651–32662. doi: 10.1074/jbc.M113.475962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madsen K, Cornish A, Soper P, et al. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology. 2001;121:580–591. doi: 10.1053/gast.2001.27224. [DOI] [PubMed] [Google Scholar]
- 27.Burgess A, Vigneron S, Brioudes E, et al. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A. 2010;107:12564–12569. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cafferata EG, Gonzalez-Guerrico AM, Giordano L, et al. Interleukin-1beta regulates CFTR expression in human intestinal T84 cells. Biochim Biophys Acta. 2000;1500:241–248. doi: 10.1016/s0925-4439(99)00105-2. [DOI] [PubMed] [Google Scholar]
- 29.Sasaki T, Hiwatashi N, Yamazaki H, et al. The role of interferon gamma in the pathogenesis of Crohn’s disease. Gastroenterol Jpn. 1992;27:29–36. [PubMed] [Google Scholar]
- 30.Watson CJ, Hoare CJ, Garrod DR, et al. Interferon-gamma selectively increases epithelial permeability to large molecules by activating different populations of paracellular pores. J Cell Sci. 2005;118:5221–5230. doi: 10.1242/jcs.02630. [DOI] [PubMed] [Google Scholar]
- 31.Beaurepaire C, Smyth D, McKay DM. Interferon-gamma regulation of intestinal epithelial permeability. J Interferon Cytokine Res. 2009;29:133–144. doi: 10.1089/jir.2008.0057. [DOI] [PubMed] [Google Scholar]
- 32.Bruewer M, Luegering A, Kucharzik T, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–6172. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 33.Bruewer M, Utech M, Ivanov AI, et al. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005;19:923–933. doi: 10.1096/fj.04-3260com. [DOI] [PubMed] [Google Scholar]
- 34.Hoermannsperger G, Clavel T, Hoffmann M, et al. Post-translational inhibition of IP-10 secretion in IEC by probiotic bacteria: impact on chronic inflammation. PLoS One. 2009;4:e4365. doi: 10.1371/journal.pone.0004365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hormannsperger G, Clavel T, Hoffmann M, et al. Posttranslational inhibition of proinflammatory chemokine secretion in intestinal epithelial cells: implications for specific IBD indications. J Clin Gastroenterol. 2010;44(Suppl 1):S10–15. doi: 10.1097/MCG.0b013e3181e102c1. [DOI] [PubMed] [Google Scholar]
- 36.Di Giacinto C, Marinaro M, Sanchez M, et al. Probiotics ameliorate recurrent Th1-mediated murine colitis by inducing IL-10 and IL-10-dependent TGF-beta-bearing regulatory cells. J Immunol. 2005;174:3237–3246. doi: 10.4049/jimmunol.174.6.3237. [DOI] [PubMed] [Google Scholar]
- 37.Pagnini C, Saeed R, Bamias G, et al. Probiotics promote gut health through stimulation of epithelial innate immunity. Proc Natl Acad Sci U S A. 2010;107:454–459. doi: 10.1073/pnas.0910307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corridoni D, Pastorelli L, Mattioli B, et al. Probiotic bacteria regulate intestinal epithelial permeability in experimental ileitis by a TNF-dependent mechanism. PLoS One. 2012;7:e42067. doi: 10.1371/journal.pone.0042067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mennigen R, Bruewer M. Effect of probiotics on intestinal barrier function. Ann N Y Acad Sci. 2009;1165:183–189. doi: 10.1111/j.1749-6632.2009.04059.x. [DOI] [PubMed] [Google Scholar]
- 40.Schlee M, Harder J, Koten B, et al. Probiotic lactobacilli and VSL#3 induce enterocyte beta-defensin 2. Clin Exp Immunol. 2008;151:528–535. doi: 10.1111/j.1365-2249.2007.03587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ewaschuk JB, Diaz H, Meddings L, et al. Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1025–1034. doi: 10.1152/ajpgi.90227.2008. [DOI] [PubMed] [Google Scholar]
- 42.Petrof EO, Kojima K, Ropeleski MJ, et al. Probiotics inhibit nuclear factor-kappaB and induce heat shock proteins in colonic epithelial cells through proteasome inhibition. Gastroenterology. 2004;127:1474–1487. doi: 10.1053/j.gastro.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Mennigen R, Nolte K, Rijcken E, et al. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1140–1149. doi: 10.1152/ajpgi.90534.2008. [DOI] [PubMed] [Google Scholar]
- 44.Otte JM, Podolsky DK. Functional modulation of enterocytes by gram-positive and gram-negative microorganisms. Am J Physiol Gastrointest Liver Physiol. 2004;286:G613–626. doi: 10.1152/ajpgi.00341.2003. [DOI] [PubMed] [Google Scholar]
- 45.Hart AL, Lammers K, Brigidi P, et al. Modulation of human dendritic cell phenotype and function by probiotic bacteria. Gut. 2004;53:1602–1609. doi: 10.1136/gut.2003.037325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohland CL, Macnaughton WK. Probiotic bacteria and intestinal epithelial barrier function. Am J Physiol Gastrointest Liver Physiol. 2010;298:G807–819. doi: 10.1152/ajpgi.00243.2009. [DOI] [PubMed] [Google Scholar]
- 47.Resta-Lenert S, Barrett KE. Probiotics and commensals reverse TNF-alpha- and IFN-gamma-induced dysfunction in human intestinal epithelial cells. Gastroenterology. 2006;130:731–746. doi: 10.1053/j.gastro.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 48.Mariman R, Tielen F, Koning F, et al. The probiotic mixture VSL#3 dampens LPS-induced chemokine expression in human dendritic cells by inhibition of STAT-1 phosphorylation. PLoS One. 2014;9:e115676. doi: 10.1371/journal.pone.0115676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Angelis M, Rizzello CG, Fasano A, et al. VSL#3 probiotic preparation has the capacity to hydrolyze gliadin polypeptides responsible for Celiac Sprue. Biochim Biophys Acta. 2006;1762:80–93. doi: 10.1016/j.bbadis.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 50.Smyth DJ, Plagnol V, Walker NM, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767–2777. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arrieta MC, Bistritz L, Meddings JB. Alterations in intestinal permeability. Gut. 2006;55:1512–1520. doi: 10.1136/gut.2005.085373. [DOI] [PMC free article] [PubMed] [Google Scholar]