

Abstract
Cue-induced reinstatement of alcohol-seeking is a hallmark behavioral pathology of addiction. Evidence suggests that reinstatement (e.g., relapse), may be regulated by cell signaling systems that underlie neuroplasticity. A variety of plasticity events require activation of calcium calmodulin-dependent protein kinase II (CaMKII) in components of the reward pathway, such as the nucleus accumbens and amygdala. We sought to determine if cue-induced reinstatement of alcohol-seeking behavior is associated with changes in the activation state (e.g., phosphorylation) of CaMKII-T286. Male C57BL/6J mice (n = 14) were trained to lever press on a fixed-ratio-4 schedule of sweetened alcohol (2% sucrose + 9% EtOH) reinforcement. After 14-d of extinction (no cues or reinforcers), mice underwent a response-contingent reinstatement (n = 7) vs. an additional day of extinction (n = 7). Brains were removed immediately after the test and processed for evaluation of pCaMKII-T286 immunoreactivity (IR). Number of pCaMKII-T286 positive cells/mm2 was quantified from coronal brain sections using Bioquant Image Analysis software. Mice emitted significantly more responses on the alcohol vs. the inactive lever throughout the baseline phase with average alcohol intake of 1.1 ± 0.03 g/kg/1-h. During extinction, responses on the alcohol lever decreased to inactive lever levels by day 7. Significant cue-induced reinstatement of alcohol-seeking was observed during a single test with no effects on the inactive lever. Reinstatement was associated with increased pCaMKII-T286 IR specifically in amygdala (LA and BLA), nucleus accumbens (AcbSh), lateral septum, mediodorsal thalamus, and piriform cortex as compared to extinction control. Brain regions showing no change included the dorsal striatum, medial septum, cingulate cortex, habenula, paraventricular thalamus, and ventral hypothalamus. These results show response contingent cue-induced reinstatement of alcohol-seeking behavior is associated with selective increases in pCaMKII-T286 in specific reward- and memory-related brain regions of male C57BL/6J mice. Primary molecular mechanisms of associative learning and memory may regulate relapse in alcohol addiction.
Keywords: CaMKII, CaMKII-T286, Cue-induced, Reinstatement, Relapse, Addiction, Alcohol, Ethanol olfactory, Nucleus accumbens, Amygdala, Thalamus, Hypothalamus, Piriform cortex, Striatum, Habenula
1. Introduction
Relapse to alcohol-seeking during abstinence is a hallmark behavioral pathology of addiction and a major clinical problem. Associative learning and memory processes (e.g., forms of neuroplasticity) play a principal role in relapse. Clinical studies conducted with abstinent alcoholics indicate that exposure to environmental cues previously associated with alcohol promote craving and relapse (Carter and Tiffany, 1999; Kaplan et al., 1985) and activate brain regions that regulate learning, memory, and reward, such as the amygdala (Schneider et al., 2001; Wiers et al., 2015; Wiers et al., 2014). Thus, elucidating adaptations in mechanisms of plasticity has the potential to move the field forward in understanding how drug-paired cues control behavior, and may identify novel therapeutic targets for the medical management of addiction (Berke and Hyman, 2000; Grueter et al., 2012; Kalivas and Volkow, 2011; Kauer and Malenka, 2007; Kelley, 2004; Kourrich et al., 2015; Mulholland and Chandler, 2007; Niehaus et al., 2009). Cell signaling systems that subserve synaptic plasticity are primary candidate mechanisms because they transduce pleiotropic drug effects, such as changes in membrane receptor activity, into enduring modifications in neural function (Hyman, 2005). Thus, a key challenge for the field is to determine if cell signaling systems that regulate normal learning, memory, and plasticity also underlie maladaptive associative learning and memory processes such as cue-induced alcohol-seeking behavior.
Calcium calmodulin-dependent protein kinase II (CaMKII) is a family of Ca2+-activated Ser/Thr protein kinases that mediates many intracellular responses in the brain, including regulation of membrane current, neurotransmitter synthesis and release, and is widely considered to be a primary mechanism of neuroplasticity (Colbran and Brown, 2004; Hudmon and Schulman, 2002; Lisman et al., 2002). Of the four known isoforms of CaMKII (α, β, γ, and δ), CaMKIIα and CaMKIIβ are the most prevalent in the brain, where they form mixed α/ β holoenzymes. The CaMKII holoenzyme is activated via phosphorylation when neuronal depolarization leads to Ca2+ entry into the cell through multiple sources including ionotropic glutamate receptors (AMPA and NMDA) and L-type voltage-gated calcium channels. Calmodulin then binds calcium to form a Ca2+/calmodulin complex that leads to CaMKII phosphorylation at a threonine 286 site (pCaMKII-T286). Following activation, pCaMKII-286 can translocate to the membrane and/or postsynaptic density where it facilitates glutamate receptor (i.e., NMDA, AMPA) activity (Sessoms-Sikes et al., 2005). Activation of glutamate receptors is critical for learning, memory, and addictive processes.
An important feature of CaMKII signal transduction that may subserve its role in synaptic and behavioral plasticity is that once activated, the kinase enters a relatively autonomous state. That is, autophosphorylation of CaMKII-T286 renders the kinase resistant to phosphatases and allows kinase activity to continue for long periods of time after the Ca2+ signal diminishes (Soderling et al., 2001). This prolonged and independent function of CaMKII led to the widely held hypothesis that CaMKIIα may represent a molecular “switch,” or “memory molecule,” that encodes long-term memory and other enduring changes in behavior and synaptic plasticity (Hudmon and Schulman, 2002; Lisman et al., 2002; Soderling et al., 2001). These findings support the hypothesis that CaMKII activity may underlie long-term enduring pathological behaviors associated with alcoholism, such as learning that occurs when alcohol becomes a positive reinforcer, and during cue-induced relapse-like behavior, which is a form of associative memory.
Consistent with this notion, we have shown that moderate home-cage alcohol drinking or low-dose operant alcohol self-administration increases total CaMKIIα protein expression in amygdala and nucleus accumbens, and CaMKII phosphorylation (pCaMKII-T286) in C57BL/6J mouse amygdala (Salling et al., 2016). Higher levels of alcohol exposure induced by binge-like alcohol drinking also increase CaMKII protein expression in the nucleus accumbens (Lee et al., 2016). At the mechanistic level, pharmacological inhibition of CaMKII activity in the amygdala reduces operant alcohol self-administration in C57BL/6J mice (Salling et al., 2016) and alcohol-preferring P-rats (Cannady et al., 2016), indicating that the reinforcing effects of alcohol require CaMKII activation in the amygdala. Moreover, we have also shown that CaMKII activity in the amygdala is required for AMPA receptor activation-induced escalation of operant alcohol self-administration by P-rats (Cannady et al., 2016). Mice carrying a global point mutation that blocks CaMKIIα-T286 autophosphorylation (Giese et al., 1998) show blunted acquisition of voluntary home cage alcohol drinking (Easton et al., 2013). By contrast, inhibition of CaMKII in the prefrontal cortex increased the positive reinforcing effects of alcohol (Faccidomo et al., 2016), suggesting that CaMKII regulation of alcohol self-administration is brain region, or neural circuit, dependent. Overall, these findings indicate that CaMKII activity plays a vital role in the positive reinforcing effects of alcohol in mice and rats, demonstrating interspecies generality of this mechanistic effect.
It is not known if CaMKII activation is associated with cue-induced reinstatement of alcohol-seeking behavior, which is a preclinical model of relapse that relies on associative learning and memory. Here we sought to establish the feasibility of this approach in mice using a procedure adapted from our rat studies (Schroeder et al., 2008) to show: 1) extinction of alcohol reinforced responding; 2) response-contingent cue-induced reinstatement of alcohol-seeking; and 3) potential changes in pCaMKII-T286 immunoreactivity (IR) in corticolimbic brain regions known to regulate relapse-like behavior, such as the amygdala and nucleus accumbens.
2. Materials and methods
2.1. Animals
Male C57BL/6J mice (n = 16) arrived at 10 weeks of age from Jackson Laboratory (Bar Harbor, ME). Mice were group-housed in clear, polycarbonate cages lined with corncob bedding. Cages were covered by stainless steel wire lids through which Purina rodent chow and water was available ad libitum. The vivarium was maintained on a 12 h:12 h reverse light/dark cycle (lights off at 0800) with temperature at 21 ± 1 °C. Mean ± SEM body weight ranged from (25.8 ± 0.5 g) on training day 1 to (29.4 ± 0.9 g) on the last experimental day, indicating normal growth and development. All procedures were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill, and animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council (U.S.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals. et al., 2011).
2.2. Self-administration apparatus
Alcohol self-administration, extinction and reinstatement sessions were conducted in Plexiglas operant chambers designed for mice (Med Associates, Georgia, VT) measuring 15.9 × 14 × 12.7 cm with stainless steel grid floors. Each chamber was housed in a sound-attenuating cubicle equipped with a house fan that provided ventilation and helped mask external noise. Chambers were equipped with two ultra-sensitive stainless-steel response levers with a stimulus light located above each lever. An alcohol solution was delivered via a syringe mounted to a programmable pump (PHM-100, Med Associates), which delivered 0.014 ml per activation into a stainless-steel cup. The operant conditioning chambers were interfaced (Med Associates, Med-PC Software) to a Windows compatible desktop computer, which was programmed to record all lever presses, and control liquid deliveries and stimulus lights.
2.3. Alcohol self-administration, extinction, and reinstatement procedure
A summary of the behavioral procedures is shown in the time line in Fig. 1A.
Fig. 1.

Experimental procedures. (A) Timeline of experimental procedures showing each phase in weeks. (B) Diagram of the self-administration chambers showing 3 behavioral phases: baseline self-administration, extinction, and reinstatement. Cues were present during self-administration and reinstatement test. EtOH was available only during the self-administration phase.
2.3.1. Self-administration
Following one week of acclimation to housing conditions, mice were deprived of fluid for 20 h prior to initial training in the operant chamber (Fig. 1B, left). The first three days of training were 16-h sessions with sucrose (5% w/v) reinforcement initially on a fixed-ratio 1 (FR1) schedule of reinforcement that progressed to an FR4 schedule. All subsequent self-administration sessions were 1-h in duration and followed a FR4 schedule of reinforcement. Mice were then trained, during 28 daily 1-h sessions, to self-administer a sweetened alcohol solution (9% EtOH (v/v) + 2% sucrose (w/v)) by using a sucrose fading procedure as previously described (Faccidomo et al., 2009; Salling et al., 2008; Salling et al., 2016). Subsequently the sweetened alcohol solution is referred to as “alcohol.” Self-administration procedures were continued for 28 sessions (7 days per week). During all operant self-administration sessions, responses on the alcohol-paired lever resulted in the presentation of a compound stimulus consisting of a visual cue light and the auditory stimulus of pump activation from a fixed spatial location. Responses on the inactive lever produced no programmed consequences. Fluid cups were inspected at the end of each session to verify intake.
2.3.2. Extinction
Following the 28-day baseline, self-administration behavior was extinguished by removing all programmed consequences of lever press responding (i.e., absence of cue-light, pump sound, and sweetened alcohol delivery) (Fig. 1B, middle). Extinction training continued during 14 daily (2 weeks) 1-h sessions.
2.3.3. Cue-induced reinstatement
Reinstatement testing was conducted during a single session, one day following the last extinction session. For reinstatement testing and subsequent immunohistochemistry, mice were divided into two subgroups. First, an extinction control group (n = 7) continued extinction training for an additional session. A second group (n = 7) was given a response contingent cue-induced reinstatement test in which lever press responses were followed by presentation of the cue light and auditory cue (as during self-administration training on a FR4 schedule) in the absence of alcohol/sucrose reinforcement to examine cue-induced alcohol-seeking behavior, as previously reported by our group (Schroeder et al., 2008) (Fig. 1B, right). For the reinstatement group only, fluid cups were also primed with 0.14 ml of the alcohol/sucrose solution (equivalent to one reinforcer) prior to session onset to provide orogustatory stimuli in addition to the cue-light and pump sound.
2.3.4. Behavioral data analysis
The principal behavioral parameters of operant ethanol self-administration were number of active and inactive lever presses, and volume of ethanol consumed (mls). Alcohol intake (g/kg) was determined using each animal’s daily body weight and the volume of ethanol delivered per total number of reinforcers (0.14 ml of 9% v/v ethanol). Self-administration and extinction data were analyzed separately by two-way repeated measures analysis of variance (RM ANOVA) with factors for lever (active vs. inactive) and day (GraphPad Software Inc., La Jolla, CA USA). Multiple comparisons were conducted with Sidak’s test.
Total response data were analyzed from the reinstatement test via unpaired t-test comparison of two groups (extinction vs. reinstatement).
2.4. Immunohistochemistry
2.4.1. Tissue collection and fixation
Immediately after the reinstatement test, extinction and reinstatement groups (n = 7 each were deeply anesthetized with pentobarbital (150 mg/kg) and intracardially perfused with ice-cold phosphate buffered saline (PBS; 0.1 M) followed by 4% paraformaldehyde. Brains were removed and post-fixed in paraformaldehyde for 48 h, and stored in PBS at 4 °C. Coronal brain sections (40 μM) were prepared using a vibratome (Leica VT1000S, Leica Microsystems, Buffalo Grove, IL) and were stored at −20 °C in cryoprotectant until immunohistochemistry processing.
2.4.2. Immunodetection
Analysis of pCaMKII-T286 immunoreactivity (IR) was performed as previously described (Agoglia et al., 2015; Cannady et al., 2016; Faccidomo et al., 2015; Schroeder et al., 2008; Spanos et al., 2012) using a 2 day protocol. On day 1, free-floating coronal sections were washed with 0.1 M PBS before being gently bathed in 1% H2O2 to inhibit endogenous peroxidase activity. Antigen retrieval was performed by immersing tissue in Citra buffer (1×; Biogenics, Napa, CA) for 30 min at 70 °C. Sections were blocked with 5% normal goat serum (Vector Labs, Burlingame, CA) in PBS with 0.1% Triton X (PBSTx) for 1 h and incubated with a rabbit polyclonal antibody to pCamKII-T286 (Abcam, ab32678 at 1:1500 dilution) overnight at 4 °C. On day 2, sections were washed in PBS-TX and then incubated with a goat anti-rabbit secondary antibody for 1 h at room temperature. Finally, antibody bound pCaMKII-T286 was visualized using a DAKO-DAB chromogen (Agilent Technologies, Carpinteria, CA), sections were counterstained with Toluidine Blue and were mounted on slides for analysis.
2.4.3. Antibody validation
This pCaMKII-T286 antibody (Abcam, ab32678) was validated by the vendor to be specific for the ~50 kDa alpha CaM Kinase II subunit and the ~60 kDa beta CaM Kinase II subunit phosphorylated at Thr286 in Western blots. Immunolabeling was blocked by the phosphopeptide used as the antigen but not by the corresponding dephosphopeptide. We also validated the antibody using Western blots in untreated C57BL/6J mouse frontal cortex and nucleus accumbens tissue and verified labeling of two bands at ~50 and ~60 kDa. No other bands were detected (data not shown). Thus, this antibody labels activation (phosphorylation) of the CaMKII α/β holoenzyme at the T286.
2.4.4. Visualization and analysis
Quantification of pCaMKII-T286 IR was conducted at 10× magnification using a light microscope (Olympus CX41, Olympus, Tokyo) connected to a computer with Bioquant Image Anaysis software (Bioquant Life Science Software v.8.40.20; Bioquant Corp, Nashville, TN). A digital camera (Regita OEM fast, QImaging, Burnaby, BC) attached to the microscope was used to acquire all images. The microscope, camera, and software were background corrected and normalized to preset light levels to ensure fidelity of data acquisition. The number of pCaMKIIα-T286 immunopositive cells from extinction and reinstatement test conditions was obtained from a circumscribed field encompassing the brain region or sub-nucleus of interest, divided by the area, and analyzed as cells/mm2. Data represent the average of bilateral measurements from 2 or 3 coronal sections through each brain region. Tissue was coded throughout the experiment so the experimenter was blind to the treatment conditions during analysis. Immunohistochemistry data were analyzed by unpaired t-test after verifying lack of statistical outliers (Grubb’s test, GraphPad Software). Differences in degrees of freedom among brain regions reflects data exclusion due to tissue loss (n = 1 from each EXT and REINST conditions, all brain regions), irregular staining (n = 1–2 from both EXT and REINST; lateral septum and mediodorsal thalamus) and improper anatomical coordinates (n = 1 from each EXT and REINS; piriform cortex). A list of all brain regions analyzed and cell count data are shown in Table 1. Data from brain regions that showed significant changes are reproduced graphically accompanied by photomicrographs.
Table 1.
pCaMKII-T286 IR in specific brain regions following cue-induced reinstatement of alcohol-seeking behavior vs. extinction control.
| Brain region | Extinction | Reinstatement | |
|---|---|---|---|
| Nucleus accumbens | Core | 167.0 ± 42.9 | 207.2 ± 34.8 |
| Shell | 153.8 ± 28.5 | 284.5 ± 23.9a | |
| Dorsal striatum | Medial | 128.6 ± 48.6 | 116.6 ± 25.2 |
| Lateral | 87.9 ± 45.2 | 51.9 ± 12.7 | |
| Septum | Medial | 247.7 ± 24.1 | 233.8 ± 31.7 |
| Lateral | 221.9 ± 37.5 | 324.7 ± 25.8a | |
| Piriform cortex | 116.3 ± 19.5 | 165.1 ± 14.9a | |
| Cingulate cortex | CG1 | 441.6 ± 111.2 | 396.9 ± 131.4 |
| CG2 | 287.8 ± 52.2 | 323.3 ± 16.1 | |
| Septohippocampal nucleus | 171.6 ± 27.7 | 191.2 ± 33.3 | |
| Amygdala | Lateral | 149.6 ± 25.6 | 299.3 ± 58.9a |
| Basolateral | 112.3 ± 26.6 | 262.6 ± 61.7a | |
| Central | 231.1 ± 22.1 | 263.6 ± 33.8 | |
| Habenula | Lateral | 182.2 ± 34.9 | 211.8 ± 38.2 |
| Thalamus | Paraventricular | 219.1 ± 53.5 | 272.8 ± 39.0 |
| Intermediodorsal | 234.0 ± 27.0 | 271.9 ± 47.3 | |
| Mediodorsal | 166.9 ± 43.5 | 229.6 ± 41.8a | |
| Reuniens | 195.4 ± 58.5 | 168.6 ± 42.1 | |
| Hypothalamus | Ventromedial | 195.8 ± 37.9 | 211.6 ± 20.0 |
| Dorsomedial | 193.4 ± 30.4 | 195.5 ± 41.9 |
Data represent MEAN ± SEM cells/mm2.
Indicates statistically different from Extinction, t-test.
2.4.5. Brain regions
Based on prior results (Schroeder et al., 2008), analysis of pCaMKII-T286 was initially focused on subnuclei of the nucleus accumbens (AcbC and AcbSh; Bregma 1.0 ± 0.25 mm) and amygdala (LA, BLA, and CeA; Bregma 1.6 ± 0.5 mm). However, prominent pCaMKII-T286 IR was observed in structures adjacent to the nucleus accumbens (dorsal striatum, frontal and anterior piriform cortices, and lateral and medial septum) and amygdala (habenula, midline thalamus, and hypothalamus) and was also analyzed. The arcuate nucleus, medial habenula, and hippocampus were not analyzed due to excessively dark background and/or irregular staining.
3. Results
3.1. Alcohol self-administration
Alcohol self-administration increased as a function of time and stabilized during the 28-day exposure period. Two-way RM ANOVA showed that C57BL/6J mice exhibited significantly more responses on the alcohol lever as compared to the inactive lever [F(1,26) = 38.6, p < 0.0001] throughout the baseline phase (Fig. 2A, left). There was also a significant effect of Time [F(27,702) = 6.6, p < 0.0001] and a significant Lever x Time interaction [F(27,702) = 3.3, p < 0.0001] indicating that the reinforcing effects of alcohol increased, or escalated, over time (Fig. 2A, left). Sidak’s multiple comparison tests showed that responses on the active lever were significantly greater than on the inactive lever in all cases except on days 1 and 3 (Fig. 2A, left, p < 0.05). Sidak’s multiple comparisons within the active lever condition also showed that there were no significant differences in response totals after day 17 (p > 0.05) indicating that self-administration behavior was stable during the last 10 days of baseline. As a result, dosage of self-administered alcohol per 1-h session (MEAN ± SEM) increased from (0.4 ± 0.07 g/kg) on day 1 to (1.02 ± 0.08 g/kg) on day 17 and remained stable during the last 10 days of baseline (range: 0.98 ± 0.08–1.25 ± 0.18 g/kg; mean: 1.09 ± 0.10 g/kg).
Fig. 2.

(A) Acquisition and extinction alcohol-seeking behavior. Acquisition of alcohol self-administration (Alcohol Self-Admin) is shown on the left and extinction on the right. Data are plotted as mean ( ± SEM) total responses per session on the active and inactive levers. * - significantly different from inactive lever control on the same day. (B) Cue-induced reinstatement of alcohol-seeking behavior. Data are shown as mean ( ± SEM) total responses during the single reinstatement test. Bars show performance of sub-groups that continued on extinction training (Ext) or underwent a reinstatement (Reinst) test. Data represent performance of n = 7 mice per group. * - significantly different from Ext, t-test.
3.2. Extinction
During the 14 days of extinction, mice emitted significantly more responses on the lever that previously produced alcohol reinforcement [F(1,26) = 6.28, p < 0.05] (Fig. 2A, right). Although there was a main effect of time [F(13,388) = 23.3, p < 0.0001] indicating that responding on both the active (alcohol) and inactive levers decreased as a function of time, the magnitude of change was greater for responses on the alcohol lever as indicated by the significant statistical interaction between lever (active vs. inactive) and time [F(13,388) = 10.1, p < 0.0001]. Sidak’s multiple comparisons showed that responses on the alcohol-associated lever decreased as a function of time and were greater than inactive lever responding only on the first 6 days of (Fig. 2A, right). Overall, these data show extinction of alcohol reinforced responding (i.e., equivalent responding on levers that previously produced alcohol or no consequences) when all consequences of lever pressing were removed for 14 days. In addition, subsequent group differences on reinstatement tests were not a function of differential between-group rates of extinction.
3.3. Cue-induced reinstatement
3.3.1. Sub-group assignment
Prior to the reinstatement test, mice were divided into two subgroups for subsequent extinction or reinstatement testing. Behaviorally matched sub-groups were derived by averaging active lever presses over the last 5 self-administration sessions (MEAN ± SEM Reinstatement:144 ± 19.6 vs. Extinction: 138 ± 43.7) and the last 5 extinction sessions (MEAN ± SEM Reinstatement: 40.3 ± 5.9 vs Extinction 37.4 ± 4.6) followed by t-test comparisons to ensure lack of group differences. Two mice were excluded from analyses due to low response totals during self-administration, which resulted in final group sizes of n = 7 for the extinction and reinstatement conditions. Grubb’s test was used to verify lack of statistical outliers following sub-group formation.
3.3.2. Reinstatement test
Mice underwent a single reinstatement test in which responses on the previously active (alcohol-associated) lever produced either: no consequence (Ext group) or presentation of discrete response-contingent cues (Reinst group) that were previously paired with reinforcer delivery during self-administration baseline (i.e., stimulus light above the lever and pump sound). Mice in the reinstatement group emitted significantly more responses on the active lever, which produced cues (light and pump sound), as compared to the group that continued extinction, t(12) = 2.4, p = 0.03 (Fig. 2B). These behavioral data demonstrate cue-induced reinstatement of alcohol-seeking behavior on the previously active lever that was associated with alcohol and cue presentation. Cue-induced reinstatement of approximately 50% of baseline alcohol reinforced responding is generally consistent with prior studies in alcohol preferring P-rats (Cannady et al., 2013; Schroeder et al., 2008) and C57BL/6J mice (Tsiang and Janak, 2006; Loos et al., 2013 #777).
3.4. Effect of cue-induced reinstatement on pCaMKII-T286 IR
Response-contingent cue-induced reinstatement of alcohol-seeking behavior was associated with increased pCaMKII-T286 IR in specific corticolimbic nuclei: nucleus accumbens (AcbSh), cortex (anterior PIR), lateral septum, amygdala (LA and BLA), and mediodorsal thalamus. No changes were observed in other nuclei that showed prominent pCaMKII-T286 IR including central amygdala, dorsal striatum (medial and lateral), paraventricular thalamus, lateral habenula, and hypothalamus (dorsomedial and ventromedial) (Table 1).
3.4.1. Nucleus accumbens (core and shell)
Cue-induced reinstatement of alcohol-seeking behavior produced sub-regional effects on CaMKII-T286 phosphorylation in the nucleus accumbens. There was no change in MEAN ± SEM pCaMKII-T286 cells/mm2 in the nucleus accumbens core, t(10) = 0.73, p = 0.48. However, pCaMKII-T286 IR in the nucleus accumbens shell (AcbSh) was increased approximately 1.85 fold, t(10) = 3.5, p = 0.005 (Fig. 3A). The cytological pattern of pCaMKII-T286 IR in the nucleus accumbens following extinction and reinstatement are shown in Fig. 3B.
Fig. 3.
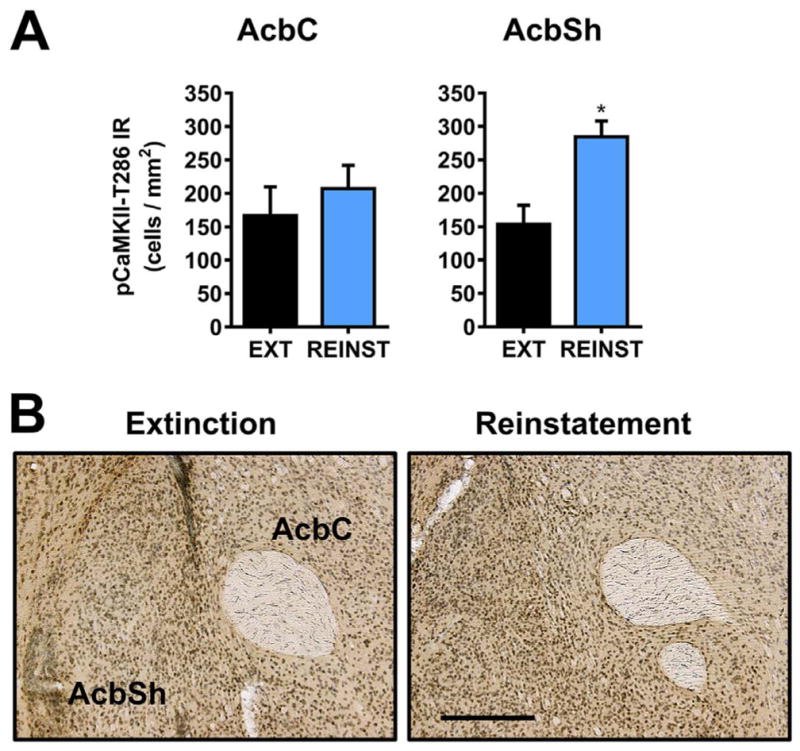
Effect of cue-induced reinstatement of alcohol-seeking behavior on CaMKII-T286 activation in the nucleus accumbens. (A) pCaMKII-T286 immunoreactivity (IR) was selectively increased in the nucleus accumbens shell (AcbSh), but not the core (AcbC) following cue-induced reinstatement (REINST) as compared to extinction (EXT). (B). Representative photomicrographs of the cytological pattern of pCaMKII-T286 IR in the nucleus accumbens. Data are plotted as MEAN ± SEM positive cells/mm2. * - significantly different from EXT, p < 0.05, t-test. Scale bar represents 250 μm.
3.4.2. Striatum (dorsomedial and dorsolateral)
No changes in were observed in MEAN ± SEM pCaMKII-T286 cells/mm2 in the dorsomedial striatum, t(10) = 0.17, p = 0.86, or dorsolateral striatum, t(10) = 0.77, p = 0.46, following cue-induced reinstatement (Table 1).
3.4.3. Septum (medial and lateral) and septohippocampal nucleus
Analysis of septal nuclei following cue-induced reinstatement identified a 1.46 fold increase in pCaMKII-T286 IR specifically in the lateral septum, t(7) = 2.13, p < 0.05 (Fig. 4A). Representative photomicrographs illustrating the cytological pattern of pCaMKII-T286 IR in septal nuclei following extinction and reinstatement are shown in Fig. 4B. No changes in pCaMKII-T286 IR were observed in the medial septum, t(10) = 0.39, p = 0.70), or septohippocampal nucleus, t(10) = 0.45, p = 0.66 (Table 1).
Fig. 4.

Effect of cue-induced reinstatement of alcohol-seeking behavior on CaMKII-T286 activation in the septum and piriform cortex. (A, C) pCaMKII-T286 immunoreactivity (IR) was increased in the lateral septum (A) and piriform cortex (C) following cue-induced reinstatement as compared to extinction (EXT). (B, D). Representative photomicrographs of the cytological pattern of pCaMKII-T286 IR. Data are plotted as MEAN ± SEM positive cells/mm2. * - significantly different from EXT, p < 0.05, t-test. Scale bar represents 500 μm.
3.4.4. Piriform and cingulate cortex
Analysis of the anterior piriform cortex, lateral to the nucleus accumbens, showed that reinstatement was associated with a significant increase in pCaMKII-T286 IR [t(8) = 1.99, p < 0.05] (Fig. 4C). Representative photomicrographs illustrating the cytological pattern of pCaMKII-T286 IR in the anterior piriform are shown in Fig. 4D.
No changes were observed in in pCaMKII-T286 IR in the cingulate cortex subregions CG1, t(10) = 0.26, p = 0.80, or CG2, t(10) = 0.65, p = 0.53 (Table 1).
3.4.5. Amygdala (lateral, basolateral, and central)
Cue-induced reinstatement of alcohol-seeking behavior was associated with a significant increase in CaMKII-T286 phosphorylation (i.e., activation) specifically in the lateral nuclei of the amygdala (Fig. 5A). Analysis showed that pCaMKII-T286 IR was increased approximately 2-fold in both the lateral amygdala (LA) [t(11) = 2.5, p = 0.03] and basolateral amygdala (BLA) [t(11) = 2.4, p = 0.04] following response-contingent exposure to cues previously associated with alcohol delivery. There was no significant change in pCaMKII-T286 IR in the central amygdala (CeA), t(11) = 0.82, p = 0.42. Representative photomicrographs illustrating the cytological pattern of pCaMKII-T286 IR in subnuclei of the amygdala following extinction and reinstatement are shown in Fig. 5B.
Fig. 5.
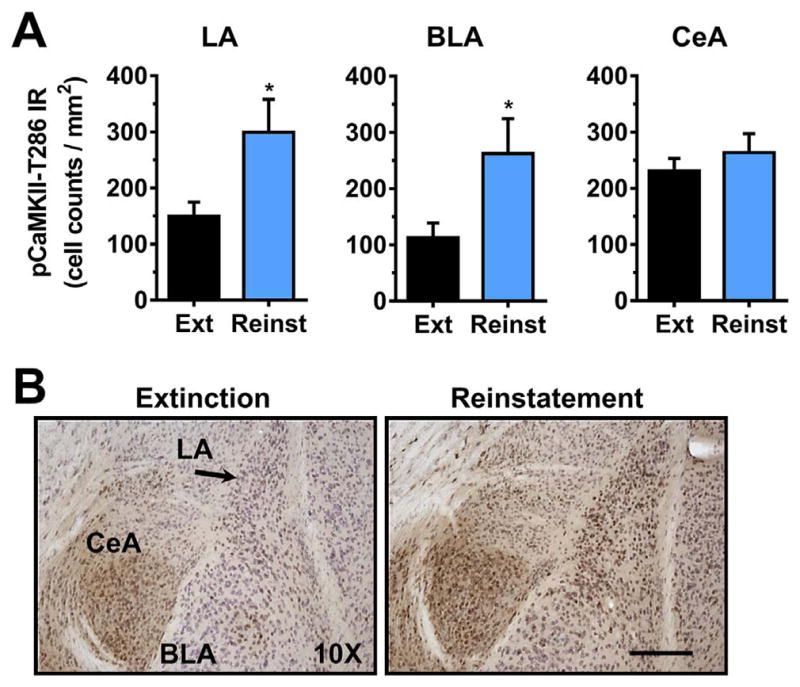
Effect of cue-induced reinstatement of alcohol-seeking behavior on CaMKII-T286 activation in the amygdala. (A) pCaMKII-T286 immunoreactivity (IR) was increased selectively in the lateral (LA) and basolateral (BLA) following reinstatement (REINST) as compared to extinction (EXT). No change was observed in the central amygdala (CeA). (B). Representative photomicrographs of the cytological pattern of pCaMKII-T286 IR. Data are plotted as MEAN ± SEM positive cells/mm2. * - significantly different from EXT, p < 0.05, t-test. Scale bar represents 250 μm.
3.4.6. Thalamus (paraventricular, intermediodorsal, mediodorsal and nucleus reuniens) and lateral habenula
Cue-induced reinstatement of alcohol-seeking behavior was associated with a site-specific increase in pCaMKII-T286 IR in the mediodorsal thalamus [t(8) = 1.9, p = 0.04] (Fig. 6A). No changes were observed in paraventricular or intermediodorsal thalamus, or the more ventral nucleus reuniens (Fig. 6A, Table 1). No changes in pCaMKII-T286 IR were observed in the lateral habenula (Table 1). pCaMKII-T286 IR in the medial habenula was consistently intense regardless of experimental condition, which precluded differentiation of specific cells for analysis. Representative photomicrographs illustrating the cytological pattern of pCaMKII-T286 IR in subnuclei of the dorsal thalamus following extinction and reinstatement are shown in Fig. 6B. The nucleus reuniens is not pictured.
Fig. 6.

Effect of cue-induced reinstatement of alcohol-seeking behavior on CaMKII-T286 activation in habenula and midline dorsal thalamic nuclei. (A) pCaMKII-T286 immunoreactivity (IR) was increased selectively in the mediodorsal thalamus (MD) following reinstatement (REINST) as compared to extinction (EXT). (B). Representative photomicrographs of the cytological pattern of pCaMKII-T286 IR. Data are plotted as MEAN ± SEM positive cells/mm2. * - significantly different from EXT, p < 0.05, t-test. Scale bar represents 250 μm.
3.4.7. Hypothalamus
There were no changes in pCaMKII-T286 in the ventromedial or ventrolateral hypothalamus associated with cue-induced reinstatement of alcohol-seeking behavior (Table 1).
4. Discussion
Addiction can be conceptualized as a disorder of neuroplasticity (Chandler, 2003; Kalivas, 2009; Kauer and Malenka, 2007; Koob and Volkow, 2010; McCool, 2011; Nestler, 2001; Winder et al., 2002) involving maladaptive associative learning and memory (Berke and Hyman, 2000; Hyman, 2005; Kelley, 2004). Consequently, exposure to environmental stimuli that have been paired with a drug of abuse, such as alcohol, can induce craving and relapse in humans (Carter and Tiffany, 1999; Papachristou et al., 2014; Schneider et al., 2001; Seo and Sinha, 2014) and relapse-like behavior in animal models (Chaudhri et al., 2008; Le and Shaham, 2002; Maccioni et al., 2007). CaMKII-T286 phosphorylation is a primary molecular mechanism of neuroplasticity at the cellular level (Colbran and Brown, 2004; Malinow et al., 1989; Silva et al., 1992b) and is required for learning, memory and associative conditioning (Mayford et al., 1996; Rodrigues et al., 2004; Silva et al., 1992a). Here we show that response-contingent exposure to environmental stimuli that were previously paired with the positive reinforcing effects of alcohol (associative conditioning) reinstates alcohol-seeking behavior and increases pCaMKII-T286 IR in specific reward-related brain regions in C57BL/6J mice. Thus, alcohol-paired environmental stimuli may express motivational significance, in part, by activation of this fundamental molecular mechanism of neuroplasticity.
This premise is supported by growing evidence indicating that alcohol alters CaMKII expression and phosphorylation in brain regions that regulate the reinforcing effects of alcohol, such as the cortex, nucleus accumbens, and amygdala. Alcohol transiently increases CaMKII activity in mouse oocytes (Winston and Maro, 1995). Similarly, chronic alcohol produces a 25% increase in kinase activity in rat cortical astrocytes (Smith and Navratilova, 1999). Results from in vivo studies show that forced exposure to alcohol in the drinking water from conception through postnatal day 8, 30, or 90 upregulates CaMKIIα protein in rat cortex (Mahadev et al., 2001) and that binge-like drinking (~6 g/kg/day) increases expression of CaMKII in the nucleus accumbens of adult mice (Lee et al., 2016). We have shown that voluntary alcohol drinking (24-h home-cage) increases total CaMKIIα protein expression in mouse amygdala and nucleus accumbens, and that low-dose operant self-administration (~1 g/kg/day) increases pCaMKII-T286 IR in the amygdala immediately after intake (Salling et al., 2016). In the present study, exposure to stimuli previously paired with operant sweetened alcohol self-administration (positive reinforcement) increased pCaMKII-T286 in lateral amygdala structures (LA and BLA) and nucleus accumbens shell. Together these findings suggest that stimuli that are paired alcohol self-administration become associated with the pharmacological effect of alcohol on CaMKII-T286 phosphorylation in reward-related brain regions; thus, acquiring conditioned reinforcing properties that can induce CaMKII-T286 activation (phosphorylation) and support relapse-like behavior. Undoing or redirecting the maladaptive appropriation of CaMKII-T286 activation may be useful in the medical management of relapse and other behavioral pathologies associated with alcohol addiction.
A key finding from this study is that cue-induced reinstatement of alcohol-seeking behavior was associated with increased CaMKII-T286 IR in the nucleus accumbens shell (AcbSh), with no effect in the accumbens core (AcbC) or dorsal striatum (medial or lateral). The nucleus accumbens is a major component of the mesolimbic reward pathway and a primary site of alcohol’s positive reinforcing effects (Hodge et al., 1995; Hodge et al., 1997; Hodge et al., 1992; Olive et al., 2000; Samson and Hodge, 1993; Samson et al., 1992), and evidence suggests that stress- and cue-induced reinstatement of alcohol-seeking behavior is associated with increased neural activity specifically in the AcbSh (Funk et al., 2006; Schroeder et al., 2008). These findings are consistent with other evidence indicating that CaMKII-T286 activation in the AcbSh is associated with reinstatement of opiate- and stimulant-seeking behavior. For instance, priming-induced reinstatement of morphine-seeking is associated with increased pCaMKII-T286 in the AcbSh, but not AcbC, as compared to extinction controls (Liu et al., 2012). Short-term abstinence from cocaine self-administration is associated with increased CaMKII-T286 expression in the nucleus accumbens and medial prefrontal cortex, but not dorsolateral striatum (Caffino et al., 2014). Reinstatement of cocaine-seeking behavior is also associated with increased pCaMKII-T286 expression specifically in the AcbSh but not AcbC (Anderson et al., 2008). Together, these findings suggest that reinstatement of alcohol-, opiate-, and cocaine-seeking are all associated with upregulated CaMKII-T286 activation in the AcbSh. This shared molecular adaptation across drug classes underscores the significance of delineating the fundamental processes by which all drugs of abuse gain control over neural function to produce behavioral pathologies, such as relapse.
Sub-regional specificity was also observed in the amygdala where cue-induced reinstatement of alcohol-seeking behavior was accompanied by a significant increase in pCamKII-T286 IR in the lateral and basolateral amygdala, but not in the central nucleus. An abundance of evidence indicates that cue-induced relapse to alcohol-seeking behavior is associated with adaptations in the amygdala. Exposure to alcohol-related visual cues (Wiers et al., 2015; Wiers et al., 2014) or alcohol odor (Schneider et al., 2001) increases craving and neural activity in the amygdala of abstinent alcohol-dependent patients as measured by fMRI. Rodent studies show that exposure to cues paired with alcohol increases markers of neural activation in the basolateral amygdala including ERK1/2 MAP kinase phosphorylation (Radwanska et al., 2008; Schroeder et al., 2008) and Fos expression (Jupp et al., 2011; Radwanska et al., 2008). Further, cue-induced reinstatement of alcohol-seeking is associated with increased glutamate transmission in the basolateral amygdala (Gass et al., 2011), which should increase intracellular Ca2+ that binds calmodulin and promotes activation of CaMKII via phosphorylation at T286. Following activation, CaMKII can translocate to the neuronal membrane where it drives insertion of glutamate AMPA receptors into the synapse and enhances their activity via phosphorylation of the GluA1-S831 subunit (Hayashi et al., 2000; Lu et al., 2010; Opazo et al., 2010). We have shown that operant alcohol self-administration upregulates pCaMKII-T286 and pGluA1-S831 in the amygdala (Salling et al., 2016) and that CaMKII-dependent activation of AMPARs promotes cue-induced reinstatement of alcohol-seeking behavior (Cannady et al., 2013). Thus, the increased pCaMKII-T286 IR observed in the LA and BLA may reflect enhanced release of glutamate and enhanced AMPAR activity in the lateral nuclei of the amygdala during reinstatement. Thus, these results support future assessment of GluA1-S831 phosphorylation in reinstatement, and regulation of relapse by a CaMKII-AMPAR mechanism in specific brain regions.
Selective activation of CaMKII in specific subnuclei of the amygdala is consistent with differential functions of these anatomically distinct sub-regions. The amygdala is a heterogeneous set of nuclei consisting of several anatomically and functionally distinct nuclei. The lateral structures (i.e., LA and BLA) appear cortex-like and consist of mainly of glutamatergic projections neurons, whereas neurons in central structures (i.e., CeA) appear striatum-like and are mostly GABAergic (reviewed by (Ehrlich et al., 2009)). Recent evidence supports the idea that processing of associative learning information occurs primarily in the LA, whereas instrumental, or operant responses, are directed via the CeA (Balleine and Killcross, 2006; Maren and Quirk, 2004). Accordingly, CaMKII activity in the CeA regulates the primary positive reinforcing effects of alcohol (Salling et al., 2016). By contrast, exposure to pairings of conditioned stimuli (CS or cues) and unconditioned stimuli (US, shock, food, etc.) is processed via sensory modality-specific thalamocortical inputs to the LA (Pare et al., 2004), supporting the view that the LA is the “gatekeeper” of the amygdala and a key substrate of associative learning and memory (LeDoux, 2007). Importantly, conditioned responses (i.e., response to a specific cue) require CaMKII activity in the lateral amygdala (Rodrigues et al., 2004) and pharmacological inhibition of CaMKII in the basolateral amygdala reduced cue-induced reinstatement of cocaine-seeking behavior (Rich et al., 2016). CaMKII activity in lateral amygdala nuclei represents a novel and untapped potential mechanism of cue-induced relapse.
A significant finding from this study is that reinstatement of alcohol-seeking behavior was associated with an increase in pCaMKII-T286 in the anterior piriform cortex. The piriform is part of the olfactory cortex and plays a critical role in odor memory (Wilson and Stevenson, 2003) and odor cue-reward processing (Truchet et al., 2002). The piriform functions as an association cortex and has strong connections with the amygdala, which play a key role in linking odors with emotional or motivational value (Sadrian and Wilson, 2015). Prior research has shown that odor cue-induced reinstatement of alcohol-seeking behavior was associated with increased Fos expression in the piriform cortex of rats (Jupp et al., 2011). In the present study, reinstatement was induced by response-contingent exposure to a stimulus complex that included a single non-contingent alcohol reinforcer available only at the start of the session. No other alcohol was present during reinstatement testing. Under these conditions, exposure to the small volume of alcohol may have provided sufficient olfactory stimulation to increase pCaMKII-T286 IR in the piriform directly or through odor-based memory processing. Alternatively, the BLA sends CaMKII-positive glutamatergic projections to the piriform, allowing the amygdala to modulate odor processing (Luna and Morozov, 2012; Majak et al., 2004; Sadrian and Wilson, 2015). Thus, the concomitant increase in pCaMKII-T286 IR in the BLA and piriform may indicate circuit activation induced by interactive processing of the compound cue and CaMKII-dependent processing of alcohol reinforcement in the amygdala (e.g., (Salling et al., 2016)). Indeed, exposure to alcohol odor increases measures of craving, and neural activity in the amygdala, in recently abstinent alcoholic patients (Schneider et al., 2001) suggesting potential interaction of motivational and odor memory systems in relapse. Thus, CaMKII-dependent piriform activity may play a vital role in cue-induced relapse where alcohol odor is associated with the rewarding pharmacological effects of the drug, such as when individuals enjoy the smell of wine prior to consumption.
Results from this study also show that cue-induced reinstatement of alcohol-seeking behavior is associated with increased CaMKII-T286 IR in the lateral septum (dorsal, intermediate, and ventral sub-nuclei combined) with no change in the medial septum. The lateral septum regulates neural and behavioral aspects of motivation and addiction (Sheehan et al., 2004), and this region has long been hypothesized to regulate alcohol drinking (Myers and Privette, 1989; Ryabinin et al., 2003). Moreover, recent evidence indicates that GABAergic outputs of the lateral septum regulate alcohol-induced dopamine release in the nucleus accumbens (Jonsson et al., 2017), suggesting a mechanism by which the lateral septum may regulate alcohol reinforcement. Although, prior data linking the lateral septum to reinstatement of alcohol-seeking behavior is lacking, it appears to play a key mechanistic role in cocaine reinstatement. For example, both cue- and stress-induced reinstatement of cocaine-seeking behavior is associated with increased Fos expression in lateral septum (Briand et al., 2010; Mahler and Aston-Jones, 2012). Moreover, a circuit from hippocampal area CA3 to the ventral tegmentum utilizes the lateral septum as a relay point in the regulation of context-induced cocaine reinstatement (Luo et al., 2011). Since the hippocampal projections to lateral septum are glutamatergic (Luo et al., 2011), upregulation of pCAMKII-T286 observed in the present may reflect increased excitatory drive via hippocampal projections.
pCaMKII-T286 IR was observed in a variety of thalamic nuclei (Table 1). However, reinstatement of alcohol-seeking behavior was associated with a selective increase in pCaMKII-T286 in the mediodorsal thalamus (MD, central, medial, and lateral combined). The MD influences numerous cognitive processes including memory, decision making, and reward processing (Chakraborty et al., 2016; Mitchell and Chakraborty, 2013). Lesions of the MD disrupt cue-induced lever selection in a food or sucrose operant behavioral task but have no effect on behavioral control by the reinforcer (Ostlund and Balleine, 2008). Similarly, contextual fear conditioning is disrupted by MD lesions but had no effect on post-shock freezing (Li et al., 2004), suggesting a specific role for the MD in response to conditioned stimuli as opposed to primary appetitive or aversive stimuli. Specificity of MD response to cue-based stimulus control has been confirmed in cocaine reinstatement studies. Cue-induced reinstatement of cocaine-seeking behavior in rats was associated with increased Fos expression in the MD (central, medial, and lateral nuclei) (James et al., 2011) but GABAergic inactivation of the MD had no effect on shock-induced reinstatement of cocaine responding (McFarland et al., 2004). Although thiamine-deficient alcoholics with Wernicke-Korsakoff syndrome exhibit mediodorsal thalamic lesions (Halliday et al., 1994) and show high propensity for relapse (Trevisan et al., 1998), potential mechanistic involvement of this brain region in alcohol relapse has not been investigated. Given the general role of this this midline thalamic structure in cue-related memory processing, our finding that a primary molecular mechanism of memory, pCaMKII-T286, is upregulated by exposure to alcohol-paired cues strongly suggests that the MD may play a critical role in alcohol relapse-like behavior.
Although this study shows an association between CaMKII-T286 activation and cue-induced reinstatement, it is important to consider several methodological caveats when interpreting the data. First, the results from this study do not address potential mechanistic regulation by CaMKII. Additional work inhibiting CaMKII-T286 phosphorylation in specific brain regions, such as the amygdala or nucleus accumbens, prior to reinstatement testing could evaluate brain site-specific mechanistic regulation of relapse-like behavior. Second, the two-group design used here (Extinction vs. Reinstatement) leaves open several questions, including the potential effect of extinction training on CaMKII-T286 phosphorylation, which could be addressed by comparison to mice that are actively self-administering alcohol, or to an extinction-naïve group. These additional controls could be added to future studies to more fully address the potential role of neuroplasticity in cue-induced reinstatement. Third, self-administration sessions were conducted with an alcohol solution containing sucrose (2% w/v). Sweetened solutions have been used in preclinical alcohol research for many years and offer a variety of advantages over unsweetened alcohol, including increased intake by rodents and face validity to the human condition where sweeteners are commonly added to alcohol during the initial binge-intoxication stage of addiction (reviewed by (Crabbe et al., 2011)). Sweetened alcohol was chosen as the reinforcer for the present study for three primary reasons. First, in our laboratory, mouse operant behavior under control of sweetened alcohol is highly stable during baseline and testing phases of mechanistic studies (Salling et al., 2008) and generates meaningful blood alcohol levels in a 1-h session (Faccidomo et al., 2009). Second, we have shown that CaMKII-T286 phosphorylation is increased in amygdala subregions of mice that lever-pressed for sweetened alcohol versus sucrose-only reinforcement (Salling et al., 2016). Third, the positive reinforcing effects of sweetened alcohol are specifically regulated by CaMKII activity, as compared to sucrose alone (Faccidomo et al., 2016). These findings indicate that using sweetened alcohol is a viable strategy for both assessing specific alcohol-induced neurobiological adaptations in CaMKII-T286 phosphorylation and for evaluating mechanistic regulation of drug taking behavior. Thus, interpretations of the present findings as alcohol-specific are relatively parsimonious considering prior results, but definitive conclusions require further research with a sucrose-only control or unsweetened alcohol reinstatement model.
5. Conclusion
Results from the present study indicate that cue-induced reinstatement of sweetened alcohol-seeking behavior is associated with an increase in CaMKII-T286 phosphorylation in specific subnuclei of corticolimbic brain regions that are known to regulate various aspects of learning, memory, and reward. CaMKII is enriched at excitatory glutamatergic synapses and represents one of the most abundant proteins in the postsynaptic density (PSD). CaMKII activity is both necessary and sufficient to induce plasticity at many excitatory synapses, and is required for learning and memory behavior (Lisman et al., 2002). Autophosphorylation of CaMKII-T286 appears to function as a molecular switch that promotes self-perpetuating biochemical reactions, which are required for persistent changes in behavior (Sweatt, 2016). In this regard, pCaMKII-T286 is an ideal molecular mechanism by which alcohol and other drugs of abuse may subvert adaptive neural mechanisms to produce long-term maladaptive changes in behavior. Future studies that manipulate CaMKII activity in the brain regions identified in the present study will shed new light on the molecular determinants of relapse.
Footnotes
Support: This work was supported by NIH grants R37 AA014983 and P60 AA011605 to CWH and by institutional support from the Bowles Center for Alcohol Studies at UNC Chapel Hill.
References
- Agoglia AE, Sharko AC, Psilos KE, Holstein SE, Reid GT, Hodge CW. Alcohol alters the activation of ERK1/2, a functional regulator of binge alcohol drinking in adult C57BL/6J mice. Alcohol Clin Exp Res. 2015;39:463–475. doi: 10.1111/acer.12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, et al. CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci. 2008;11:344–353. doi: 10.1038/nn2054. [DOI] [PubMed] [Google Scholar]
- Balleine BW, Killcross S. Parallel incentive processing: an integrated view of amygdala function. Trends Neurosci. 2006;29:272–279. doi: 10.1016/j.tins.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Briand LA, Vassoler FM, Pierce RC, Valentino RJ, Blendy JA. Ventral tegmental afferents in stress-induced reinstatement: the role of cAMP response element-binding protein. J Neurosci. 2010;30:16149–16159. doi: 10.1523/JNEUROSCI.2827-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffino L, Cassina C, Giannotti G, Orru A, Moro F, Di Clemente A, et al. Short-term abstinence from cocaine self-administration, but not passive cocaine infusion, elevates alphaCaMKII autophosphorylation in the rat nucleus accumbens and medial prefrontal cortex. Int J Neuropsychopharmacol. 2014;17:323–329. doi: 10.1017/S1461145713000916. [DOI] [PubMed] [Google Scholar]
- Cannady R, Fisher KR, Durant B, Besheer J, Hodge CW. Enhanced AMPA receptor activity increases operant alcohol self-administration and cue-induced reinstatement. Addict Biol. 2013;18:54–65. doi: 10.1111/adb.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannady R, Fisher KR, Graham C, Crayle J, Besheer J, Hodge CW. Potentiation of amygdala AMPA receptor activity selectively promotes escalated alcohol self-administration in a CaMKII-dependent manner. Addict Biol. 2016;22:652–664. doi: 10.1111/adb.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BL, Tiffany ST. Meta-analysis of cue-reactivity in addiction research. Addiction. 1999;94:327–340. [PubMed] [Google Scholar]
- Chakraborty S, Kolling N, Walton ME, Mitchell AS. Critical role for the mediodorsal thalamus in permitting rapid reward-guided updating in stochastic reward environments. elife. 2016:5. doi: 10.7554/eLife.13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler LJ. Ethanol and brain plasticity: receptors and molecular networks of the postsynaptic density as targets of ethanol. Pharmacol Ther. 2003;99:311–326. doi: 10.1016/s0163-7258(03)00096-2. [DOI] [PubMed] [Google Scholar]
- Chaudhri N, Sahuque LL, Cone JJ, Janak PH. Reinstated ethanol-seeking in rats is modulated by environmental context and requires the nucleus accumbens core. Eur J Neurosci. 2008;28:2288–2298. doi: 10.1111/j.1460-9568.2008.06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14:318–327. doi: 10.1016/j.conb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Harris RA, Koob GF. Preclinical studies of alcohol binge drinking. Ann N Y Acad Sci. 2011;1216:24–40. doi: 10.1111/j.1749-6632.2010.05895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton AC, Lucchesi W, Lourdusamy A, Lenz B, Solati J, Golub Y, et al. alphaCaMKII autophosphorylation controls the establishment of alcohol drinking behavior. Neuropsychopharmacology. 2013;38:1636–1647. doi: 10.1038/npp.2013.60. official publication of the American College of Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Faccidomo S, Besheer J, Stanford PC, Hodge CW. Increased operant responding for ethanol in male C57BL/6J mice: specific regulation by the ERK1/2, but not JNK, MAP kinase pathway. Psychopharmacology. 2009;204:135–147. doi: 10.1007/s00213-008-1444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccidomo S, Salling MC, Galunas C, Hodge CW. Operant ethanol self-administration increases extracellular-signal regulated protein kinase (ERK) phosphorylation in reward-related brain regions: selective regulation of positive reinforcement in the prefrontal cortex of C57BL/6J mice. Psychopharmacology. 2015;232:3417–3430. doi: 10.1007/s00213-015-3993-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccidomo S, Reid GT, Agoglia AE, Ademola SA, Hodge CW. CaMKII inhibition in the prefrontal cortex specifically increases the positive reinforcing effects of sweetened alcohol in C57BL/6J mice. Behav Brain Res. 2016;298:286–290. doi: 10.1016/j.bbr.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk D, Li Z, Le AD. Effects of environmental and pharmacological stressors on c-fos and corticotropin-releasing factor mRNA in rat brain: relationship to the reinstatement of alcohol seeking. Neuroscience. 2006;138:235–243. doi: 10.1016/j.neuroscience.2005.10.062. [DOI] [PubMed] [Google Scholar]
- Gass JT, Sinclair CM, Cleva RM, Widholm JJ, Olive MF. Alcohol-seeking behavior is associated with increased glutamate transmission in basolateral amygdala and nucleus accumbens as measured by glutamateoxidase-coated biosensors. Addict Biol. 2011;16:215–228. doi: 10.1111/j.1369-1600.2010.00262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- Grueter BA, Rothwell PE, Malenka RC. Integrating synaptic plasticity and striatal circuit function in addiction. Curr Opin Neurobiol. 2012;22:545–551. doi: 10.1016/j.conb.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday G, Cullen K, Harding A. Neuropathological correlates of memory dysfunction in the Wernicke-Korsakoff syndrome. Alcohol Alcohol Suppl. 1994;2:245–251. [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Samson HH, Haraguchi M. Microinjections of dopamine agonists in the nucleus accumbens increase ethanol-reinforced responding. Pharmacol Biochem Behav. 1992;43:249–254. doi: 10.1016/0091-3057(92)90665-3. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Chappelle AM, Samson HH. GABAergic transmission in the nucleus accumbens is involved in the termination of ethanol self-administration in rats. Alcohol Clin Exp Res. 1995;19:1486–1493. doi: 10.1111/j.1530-0277.1995.tb01012.x. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Samson HH, Chappelle AM. Alcohol self-administration: further examination of the role of dopamine receptors in the nucleus accumbens. Alcohol Clin Exp Res. 1997;21:1083–1091. doi: 10.1111/j.1530-0277.1997.tb04257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–1422. doi: 10.1176/appi.ajp.162.8.1414. [DOI] [PubMed] [Google Scholar]
- James MH, Charnley JL, Flynn JR, Smith DW, Dayas CV. Propensity to ‘relapse’ following exposure to cocaine cues is associated with the recruitment of specific thalamic and epithalamic nuclei. Neuroscience. 2011;199:235–242. doi: 10.1016/j.neuroscience.2011.09.047. [DOI] [PubMed] [Google Scholar]
- Jonsson S, Morud J, Stomberg R, Ericson M, Soderpalm B. Involvement of lateral septum in alcohol’s dopamine-elevating effect in the rat. Addict Biol. 2017;22:93–102. doi: 10.1111/adb.12297. [DOI] [PubMed] [Google Scholar]
- Jupp B, Krstew E, Dezsi G, Lawrence AJ. Discrete cue-conditioned alcohol-seeking after protracted abstinence: pattern of neural activation and involvement of orexin(1) receptors. Br J Pharmacol. 2011;162:880–889. doi: 10.1111/j.1476-5381.2010.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry. 2011;16:974–986. doi: 10.1038/mp.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan RF, Cooney NL, Baker LH, Gillespie RA, Meyer RE, Pomerleau OF. Reactivity to alcohol-related cues: physiological and subjective responses in alcoholics and nonproblem drinkers. J Stud Alcohol. 1985;46:267–272. doi: 10.15288/jsa.1985.46.267. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. official publication of the American College of Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Calu DJ, Bonci A. Intrinsic plasticity: an emerging player in addiction. Nat Rev Neurosci. 2015;16:173–184. doi: 10.1038/nrn3877. [DOI] [PubMed] [Google Scholar]
- Le A, Shaham Y. Neurobiology of relapse to alcohol in rats. Pharmacol Ther. 2002;94:137–156. doi: 10.1016/s0163-7258(02)00200-0. [DOI] [PubMed] [Google Scholar]
- LeDoux J. The amygdala. Curr Biol. 2007;17:R868–74. doi: 10.1016/j.cub.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Lee KM, Coelho MA, McGregor HA, Solton NR, Cohen M, Szumlinski KK. Adolescent mice are resilient to alcohol withdrawal-induced anxiety and changes in indices of glutamate function within the nucleus accumbens. Front Cell Neurosci. 2016;10:265. doi: 10.3389/fncel.2016.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XB, Inoue T, Nakagawa S, Koyama T. Effect of mediodorsal thalamic nucleus lesion on contextual fear conditioning in rats. Brain Res. 2004;1008:261–272. doi: 10.1016/j.brainres.2004.02.038. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Liu Z, Liu XD, Zhang JJ, Yu LC. Increases in alphaCaMKII phosphorylated on Thr286 in the nucleus accumbens shell but not the core during priming-induced reinstatement of morphine-seeking in rats. Neurosci Lett. 2012;526:39–44. doi: 10.1016/j.neulet.2012.07.042. [DOI] [PubMed] [Google Scholar]
- Loos M, Staal J, Smit AB, De Vries TJ, Spijker S. Enhanced alcohol self-administration and reinstatement in a highly impulsive, inattentive recombinant inbred mouse strain. Front Behav Neurosci. 2013;7:151. doi: 10.3389/fnbeh.2013.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Isozaki K, Roche KW, Nicoll RA. Synaptic targeting of AMPA receptors is regulated by a CaMKII site in the first intracellular loop of GluA1. Proc Natl Acad Sci U S A. 2010;107:22266–22271. doi: 10.1073/pnas.1016289107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna VM, Morozov A. Input-specific excitation of olfactory cortex microcircuits. Front Neural Circuits. 2012;6:69. doi: 10.3389/fncir.2012.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo AH, Tahsili-Fahadan P, Wise RA, Lupica CR, Aston-Jones G. Linking context with reward: a functional circuit from hippocampal CA3 to ventral tegmental area. Science. 2011;333:353–357. doi: 10.1126/science.1204622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccioni P, Orru A, Korkosz A, Gessa GL, Carai MA, Colombo G, et al. Cue-induced reinstatement of ethanol seeking in Sardinian alcohol-preferring rats. Alcohol. 2007;41:31–39. doi: 10.1016/j.alcohol.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Chetty CS, Vemuri MC. Effect of prenatal and postnatal ethanol exposure on Ca2+/calmodulin-dependent protein kinase II in rat cerebral cortex. Alcohol. 2001;23:183–188. doi: 10.1016/s0741-8329(01)00133-1. [DOI] [PubMed] [Google Scholar]
- Mahler SV, Aston-Jones GS. Fos activation of selective afferents to ventral tegmental area during cue-induced reinstatement of cocaine seeking in rats. J Neurosci. 2012;32:13309–13326. doi: 10.1523/JNEUROSCI.2277-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majak K, Ronkko S, Kemppainen S, Pitkanen A. Projections from the amygdaloid complex to the piriform cortex: a PHA-L study in the rat. J Comp Neurol. 2004;476:414–428. doi: 10.1002/cne.20233. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McCool BA. Ethanol modulation of synaptic plasticity. Neuropharmacology. 2011;61:1097–1108. doi: 10.1016/j.neuropharm.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci. 2004;24:1551–1560. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AS, Chakraborty S. What does the mediodorsal thalamus do? Front Syst Neurosci. 2013;7:37. doi: 10.3389/fnsys.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland PJ, Chandler LJ. The thorny side of addiction: adaptive plasticity and dendritic spines. ScientificWorldJournal. 2007;7:9–21. doi: 10.1100/tsw.2007.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RD, Privette TH. A neuroanatomical substrate for alcohol drinking: identification of tetrahydropapaveroline (THP)-reactive sites in the rat brain. Brain Res Bull. 1989;22:899–911. doi: 10.1016/0361-9230(89)90035-x. [DOI] [PubMed] [Google Scholar]
- National Research Council (U.S.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals., Institute for Laboratory Animal Research (U.S.), National Academies Press (U.S.) Guide for the Care and Use of Laboratory Animals. 8. National Academies Press; Washington, D.C: 2011. [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Niehaus JL, Cruz-Bermudez ND, Kauer JA. Plasticity of addiction: a mesolimbic dopamine short-circuit? Am J Addict. 2009;18:259–271. doi: 10.1080/10550490902925946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive MF, Mehmert KK, Messing RO, Hodge CW. Reduced operant ethanol self-administration and in vivo mesolimbic dopamine responses to ethanol in PKCepsilon-deficient mice. Eur J Neurosci. 2000;12:4131–4140. doi: 10.1046/j.1460-9568.2000.00297.x. [DOI] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, et al. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Ostlund SB, Balleine BW. Differential involvement of the basolateral amygdala and mediodorsal thalamus in instrumental action selection. J Neurosci. 2008;28:4398–4405. doi: 10.1523/JNEUROSCI.5472-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papachristou H, Nederkoorn C, Giesen JC, Jansen A. Cue reactivity during treatment, and not impulsivity, predicts an initial lapse after treatment in alcohol use disorders. Addict Behav. 2014;39:737–739. doi: 10.1016/j.addbeh.2013.11.027. [DOI] [PubMed] [Google Scholar]
- Pare D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- Radwanska K, Wrobel E, Korkosz A, Rogowski A, Kostowski W, Bienkowski P, et al. Alcohol relapse induced by discrete cues activates components of AP-1 transcription factor and ERK pathway in the rat basolateral and central amygdala. Neuropsychopharmacology. 2008;33:1835–1846. doi: 10.1038/sj.npp.1301567. [DOI] [PubMed] [Google Scholar]
- Rich MT, Abbott TB, Chung L, Gulcicek EE, Stone KL, Colangelo CM, et al. Phosphoproteomic analysis reveals a novel mechanism of CaMKIIalpha regulation inversely induced by cocaine memory extinction versus reconsolidation. J Neurosci. 2016;36:7613–7627. doi: 10.1523/JNEUROSCI.1108-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE. Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci. 2004;24:3281–3288. doi: 10.1523/JNEUROSCI.5303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryabinin AE, Galvan-Rosas A, Bachtell RK, Risinger FO. High alcohol/sucrose consumption during dark circadian phase in C57BL/6J mice: involvement of hippocampus, lateral septum and urocortin-positive cells of the Edinger-Westphal nucleus. Psychopharmacology. 2003;165:296–305. doi: 10.1007/s00213-002-1284-y. [DOI] [PubMed] [Google Scholar]
- Sadrian B, Wilson DA. Optogenetic stimulation of lateral amygdala input to posterior piriform cortex modulates single-unit and ensemble odor processing. Front Neural Circuits. 2015;9:81. doi: 10.3389/fncir.2015.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salling MC, Faccidomo S, Hodge CW. Nonselective suppression of operant ethanol and sucrose self-administration by the mGluR7 positive allosteric modulator AMN082. Pharmacol Biochem Behav. 2008;91:14–20. doi: 10.1016/j.pbb.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salling MC, Faccidomo SP, Li C, Psilos K, Galunas C, Spanos M, et al. Moderate alcohol drinking and the amygdala proteome: identification and validation of calcium/calmodulin dependent kinase II and AMPA receptor activity as novel molecular mechanisms of the positive reinforcing effects of alcohol. Biol Psychiatry. 2016;79:430–442. doi: 10.1016/j.biopsych.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson HH, Hodge CW. The role of the mesoaccumbens dopamine system in ethanol reinforcement: studies using the techniques of microinjection and voltammetry. Alcohol Alcohol Suppl. 1993;2:469–474. [PubMed] [Google Scholar]
- Samson HH, Tolliver GA, Haraguchi M, Hodge CW. Alcohol self-administration: role of mesolimbic dopamine. Ann N Y Acad Sci. 1992;654:242–253. doi: 10.1111/j.1749-6632.1992.tb25971.x. [DOI] [PubMed] [Google Scholar]
- Schneider F, Habel U, Wagner M, Franke P, Salloum JB, Shah NJ, et al. Subcortical correlates of craving in recently abstinent alcoholic patients. Am J Psychiatry. 2001;158:1075–1083. doi: 10.1176/appi.ajp.158.7.1075. [DOI] [PubMed] [Google Scholar]
- Schroeder JP, Spanos M, Stevenson JR, Besheer J, Salling M, Hodge CW. Cue-induced reinstatement of alcohol-seeking behavior is associated with increased ERK1/2 phosphorylation in specific limbic brain regions: blockade by the mGluR5 antagonist MPEP. Neuropharmacology. 2008;55:546–554. doi: 10.1016/j.neuropharm.2008.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo D, Sinha R. The neurobiology of alcohol craving and relapse. Handb Clin Neurol. 2014;125:355–368. doi: 10.1016/B978-0-444-62619-6.00021-5. [DOI] [PubMed] [Google Scholar]
- Sessoms-Sikes S, Honse Y, Lovinger DM, Colbran RJ. CaMKIIalpha enhances the desensitization of NR2B-containing NMDA receptors by an autophosphorylation-dependent mechanism. Mol Cell Neurosci. 2005;29:139–147. doi: 10.1016/j.mcn.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Sheehan TP, Chambers RA, Russell DS. Regulation of affect by the lateral septum: implications for neuropsychiatry. Brain Res Brain Res Rev. 2004;46:71–117. doi: 10.1016/j.brainresrev.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992a;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992b;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Smith TL, Navratilova E. Increased calcium/calmodulin protein kinase activity in astrocytes chronically exposed to ethanol: influences on glutamate transport. Neurosci Lett. 1999;269:145–148. doi: 10.1016/s0304-3940(99)00438-3. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Chang B, Brickey D. Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2001;276:3719–3722. doi: 10.1074/jbc.R000013200. [DOI] [PubMed] [Google Scholar]
- Spanos M, Besheer J, Hodge CW. Increased sensitivity to alcohol induced changes in ERK Map kinase phosphorylation and memory disruption in adolescent as compared to adult C57BL/6J mice. Behav Brain Res. 2012;230:158–166. doi: 10.1016/j.bbr.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD. Neural plasticity and behavior - sixty years of conceptual advances. J Neurochem. 2016;139(Suppl 2):179–199. doi: 10.1111/jnc.13580. [DOI] [PubMed] [Google Scholar]
- Trevisan LA, Boutros N, Petrakis IL, Krystal JH. Complications of alcohol withdrawal: pathophysiological insights. Alcohol Health Res World. 1998;22:61–66. [PMC free article] [PubMed] [Google Scholar]
- Truchet B, Chaillan FA, Soumireu-Mourat B, Roman FS. Learning and memory of cue-reward association meaning by modifications of synaptic efficacy in dentate gyrus and piriform cortex. Hippocampus. 2002;12:600–608. doi: 10.1002/hipo.10097. [DOI] [PubMed] [Google Scholar]
- Tsiang MT, Janak PH. Alcohol seeking in C57BL/6 mice induced by conditioned cues and contexts in the extinction-reinstatement model. Alcohol. 2006;38:81–88. doi: 10.1016/j.alcohol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Wiers CE, Stelzel C, Park SQ, Gawron CK, Ludwig VU, Gutwinski S, et al. Neural correlates of alcohol-approach bias in alcohol addiction: the spirit is willing but the flesh is weak for spirits. Neuropsychopharmacology. 2014;39:688–697. doi: 10.1038/npp.2013.252. official publication of the American College of Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiers CE, Stelzel C, Gladwin TE, Park SQ, Pawelczack S, Gawron CK, et al. Effects of cognitive bias modification training on neural alcohol cue reactivity in alcohol dependence. Am J Psychiatry. 2015;172:335–343. doi: 10.1176/appi.ajp.2014.13111495. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Stevenson RJ. The fundamental role of memory in olfactory perception. Trends Neurosci. 2003;26:243–247. doi: 10.1016/S0166-2236(03)00076-6. [DOI] [PubMed] [Google Scholar]
- Winder DG, Egli RE, Schramm NL, Matthews RT. Synaptic plasticity in drug reward circuitry. Curr Mol Med. 2002;2:667–676. doi: 10.2174/1566524023361961. [DOI] [PubMed] [Google Scholar]
- Winston NJ, Maro B. Calmodulin-dependent protein kinase II is activated transiently in ethanol-stimulated mouse oocytes. Dev Biol. 1995;170:350–352. doi: 10.1006/dbio.1995.1220. [DOI] [PubMed] [Google Scholar]