

Abstract
Ischemia/reperfusion (I/R) induces leukocyte/endothelial cell adhesive interactions (LECA) in postcapillary venules and impaired endothelium-dependent, NO-mediated dilatory responses (EDD) in upstream arterioles. A large body of evidence has implicated reactive oxygen species (ROS), adherent leukocytes, and proteases in postischemic EDD dysfunction in conduit arteries. However, arterioles represent the major site for the regulation of vascular resistance, but have received less attention with regard to the mechanisms underlying their reduced responsiveness to EDD stimuli in I/R. Even though leukocytes do not roll along, adhere to, or emigrate across arteriolar endothelium in postischemic intestine, recent work indicates that I/R-induced venular LECA is causally linked to EDD in arterioles. An emerging body of evidence supports the view that I/R-induced EDD in arterioles occurs by a mechanism that is triggered by LECA in postcapillary venules and involves the formation of signals in the interstitium elicited by the proteolytic activity of emigrated leukocytes. This activity releases matricryptins from or exposes matricryptic sites in the extracellular matrix (ECM) that interact with the integrin αvβ3 to induce mast cell chymase-dependent formation of angiotensin II (Ang II). Subsequent activation of NAD(P)H oxidase by Ang II leads to the formation of oxidants which inactivate NO, resulting in arteriolar EDD dysfunction. ROS generation also promotes eNOS uncoupling in the vascular wall, exacerbating impaired EDD responses. This work establishes new links between LECA in postcapillary venules, signals generated in the interstitium by emigrated leukocytes, mast cell degranulation, and impaired EDD in upstream arterioles. Given the importance of the endothelium in regulating vascular tone, these fundamentally important findings have enormous implications for our understanding of blood flow dysregulation in conditions characterized by I/R.
Keywords: Leukocyte adhesion, leukocyte emigration, matrix metalloproteinases, calpains, matricryptins, extracellular matrix, mast cells, endothelium-dependent vasodilation, myeloperoxidase, NADPH oxidase
Introduction
Ischemia/reperfusion (I/R) is a localized pathology that often leads to multi-organ dysfunction and shock (Granger and Korthuis, 1995; Kalogeris, Baines, Krenz & Korthuis, 2016; Kalogeris, Baines, Krenz & Korthuis, 2012; Rosario, Waldo, Becker, Schmid-Schonbein, 2004; Boros, 2003). One of the earliest events in the pathogenesis of I/R is microvascular dysfunction that is accompanied by a diminution in nitric oxide (NO) production by the endothelium. This pronounced reduction in NO bioavailability has been implicated as a trigger for leukocyte/ endothelial cell adhesive interactions (LECA) and increased permeability in postcapillary venules (Boros, 2003; Granger, 2003; Granger and Korthuis, 1995; Kalogeris et al, 2012; Kalogeris 2016; Kurose, Wolf, Grisham & Granger, 1994; Rosario, Waldo, Becker, Schmid-Schonbein, 2004). Reperfusion of ischemic tissues also alters vasoconstrictor responses and produces deficits in endothelium-dependent, NO-mediated vascular reactivity (EDD) in arteries and arterioles (Banda, Lefer & Granger, 1997; Cuccocrea, Mazzon, Costantino, Seraino, De Sarro & Caputi, 2000; Faraci & Didion, 2004; Gao, Korthuis & Benoit, 1994; Gao, Korthuis & Benoit, 1996; Gao, Benoit & Korthuis, 1996; Hansen, 1998; Hayward and Lefer, 1998; Hayward & Lefer, 1999; Hayward, Campbell, Shin, Scalia & Lefer, 1999; Hein, Zhang, Wang, Chang, Thengchaisri & Kuo, 2003; Kalogeris et al, 2012; Kalogeris et al, 2016; Lefer & Ma, 1994; Lefer & Aoki, 1990; Lefer, Tsao, Lefer & Ma, 1991; Ma, Tsao & Lefer, 1991; Ma, Lefer, Lefer & Rothlein, 1992; Ma, Weyrich, Lefer, Buerke, Albertine, Kishimoto, et al, 1993; Ruh, Schmidt & Vogel, 2003; Tsao, Aoki, Lefer, Johnson & Lefer, 1990; Tsao, Ma & Lefer, 1992; Touyz, 2004; Weyrich, Ma, Lefer, Albertine & Lefer, 1993; Wolin, Gupte & Oekler, 2002; Woodman, Hart & Sobey, 1997; Yokohama, Korthuis & Benoit, 1996; Zhao, Nakamura, Wang, Wilcox, Shearer, Guyton et al, 1999).
In conduit arteries, it appears that superoxide and other reactive oxygen species (ROS) play a major role the development of EDD dysfunction after I/R (Faraci & Didion, 2004; Gourdin et al, 2009; Granger, 2006; Kalogeris et al 2012; Kalogeris et al 2017; Mikhed, Daiber & Steven, 2015; Touyz, 2004; Widlansky & Gutterman, 2011; Wolin et al, 2002). Based on studies conducted in adhesion molecule-deficient mice or in animals treated with anti-adhesion reagents it appears that adherent neutrophils also contribute to postischemic impaired vasorelaxation to acetylcholine in conduit arteries (Banda et al, 1997; Hayward & Lefer, 1999; Hayward et al, 1999; Lefer & Aoki, 1990; Ma et al, 1991, 1992, 1993; Tsao et al, 1992; Weyrich et al, 1993). In sharp contrast to conduit arteries, leukocytes do not or only very rarely roll along, adhere to, or emigrate across postischemic arteriolar endothelium in most peripheral tissues, as they do in postcapillary venules (Kalogeris et al, 2012; Kalogeris et al, 2016). Thus, a role for these inflammatory phagocytes in the postischemic impairment in EDD responses that occurs in arterioles had been largely ignored. However, an emerging body of evidence suggests that there may be a mechanistic link between postcapillary venular LECA and EDD in upstream arterioles (Banda et al, 1997; Dai & Korthuis, 2017).
Although the latter studies indicate that LECA in postcapillary venules are causally linked to EDD in upstream arterial vessels, the mechanisms coupling these manifestations of I/R-induced microvascular dysfunction are only now becoming understood. The purpose of this review is to summarize the evidence implicating roles for ROS, proteases, and neutrophils in the pathogenesis of I/R-induced EDD dysfunction in conduit arterial vessels versus arterioles.
Leukocyte-Mediated Postischemic Endothelial Dysfunction in Arterioles and Venules
Endothelial cells (EC) lining the microvasculature are particularly vulnerable to the deleterious effects of I/R (Granger & Korthuis, 1995; Granger 2003; Kalogeris et al 2012; Kalogeris 2016). Prolonged ischemia (or hypoxia in cell culture models) results in depletion of intracellular energy stores, disrupted cytoskeletal architecture, EC swelling, diminished production of certain bioactive agents (e.g., prostacyclin, NO) and accelerated formation of others (e.g., endothelin, Ang II, thromboxane A2, chemotactic mediators). Hypoxic exposure induces the expression of some endothelial genes (e.g., adhesion molecules, cytokines) while suppressing others (e.g., eNOS, thrombomodulin). Reoxygenation or reperfusion exacerbates these EC responses, which occur rapidly and often without obvious morphologic evidence of cell injury in the initial period after blood flow restitution (Granger & Korthuis, 1995; Granger 2003; Kalogeris et al 2012; Kalogeris et al 2016). Even though all EC in the microcirculation are exposed to the detrimental effects of I/R, the characteristics of EC dysfunction are manifested in a site-specific manner within the functionally distinct segments of the microvasculature, as outlined below.
Postcapillary Venular Responses to I/R
Most of the inflammatory responses elicited by I/R can be attributed to EC activation in postcapillary venules (Granger & Korthuis, 1995; Granger 2003; Kalogeris et al 2012; Kalogeris 2016). Characteristic features of postcapillary venular responses to I/R include enhanced oxidant production, leukocyte rolling and stationary adhesion, transendothelial leukocyte migration, platelet-leukocyte aggregation, platelet/endothelial cell adhesion, and protein extravasation. Mast cells and macrophages also release substances that can amplify the inflammatory response to I/R (Boros, Kaszaki & Nagy, 1991; Bortolotto, Morrison, Han & Messina, 2004; Dai & Korthuis, 2011; Granger & Korthuis, 1995; Granger 2003; Kalogeris et al 2012; Kalogeris et al, 2016; Kanwar & Kubes, 1994; Kanwar & Kubes, 2004; Kanwar, Hickey & Kubes, 1998; Steiner, Gonzalez & Wood; 1985). Neutrophil recruitment in response to I/R occurs by a complex, highly dynamic and well-coordinated series of steps, as depicted in Figure 1. Experiments using immunoneutralization approaches or adhesion molecule-deficient mice have established the selectins as dominant receptors for leukocyte rolling in postischemic tissues, while the interaction between the β2-integrins (CD11/CD18) on leukocytes and intercellular adhesion molecule-1 (ICAM-1) on EC accounts for the establishment of stationary adhesive interactions (Granger & Korthuis, 1995; Granger 2003; Kalogeris et al 2012; Kalogeris et al, 2016). Leukocyte emigration requires ICAM-1, platelet-endothelial cell adhesion molecule-1 (PECAM-1), CD99, and a number of junctional adhesion molecules (JAMs). Diapedesis occurs preferentially across regions of the endothelium where there are gaps in the underlying pericyte investment and the basement membrane matrix exhibits by low expression of its constituent molecules (Voisin & Nourshargh, 2014). P-selectin expression follows a biphasic course after I/R, with an initial rapid increase due to mobilization from pre-formed pools, followed by a slower and sustained increase that is protein synthesis-dependent. E-selectin and other adhesion molecules such as ICAM-1 require more time (>3 hr) for full expression on activated EC, again by protein synthesis-dependent mechanisms.
Figure 1.

The recruitment of leukocytes to ischemic sites involves a complex cascade of highly integrated steps. When leukocytes exit capillaries they are moved to the postcapillary venular walls (margination) by hydrodynamic dispersal forces. In the presence of activating factors, the marginated leukocyte can form weak adhesive interactions that permit rolling along the endothelial surface. The rolling leukocyte can establish stationary adhesive interactions and then crawl along the endothelium to sites along the venular wall where the underlying basement membrane exhibits low expression of constituent molecules and where gaps between pericytes exist. The adherent leukocyte sends out pseudopods to move through or between adjacent endothelial cells to gain access to the subendothelial cell space, crawls along pericytes surrounding the venular endothelium until it encounters a gap between these cells, which it can use as a route of transit into the tissue space. Once in the tissue space, the emigrated leukocyte releases its cytotoxic arsenal of reactive oxygen species and hydrolytic enzymes to cleave extracellular matrix components and direct a focused attack on parenchymal cells. Figure from Kalogeris et al, 2016.
Leukocyte recruitment to postischemic tissues is promoted by ROS generated by xanthine oxidase in venular endothelium and other enzymes (e.g., NADPH oxidases in the vascular wall and adherent leukocytes, mitochondrial sources) which participate in the formation of proinflammatory stimuli (e.g., PAF, LTB4, Ang II, and activated complement components), modify the expression of adhesion molecules on the surface of leukocytes and endothelial cells (e.g., CD11b/CD18, P-selectin and ICAM-1), and reduce tissue levels of the potent anti-adhesive agent NO (Granger & Korthuis, 1995; Granger 2003; Kalogeris et al 2012; Kalogeris et al, 2016) (Figure 1). One of the earliest signs of microvascular dysfunction induced by I/R is a reduction in the restrictive properties of the endothelial barrier in postcapillary venules, an event strongly associated with postischemic oxidant stress-induced LECA (Granger & Korthuis, 1995; Granger 2003; Kalogeris et al 2012; Kalogeris et al, 2016). Recent work has also shown that leukcoyte diapedesis invoked by LTB4 induces leakage of plasma proteins across postcapillary venules by a mechanism involving neutrophilic TNF secretion (Finsterbach, Voisin, Beyrau, Williams & Nourshargh, 2014).
Arterial and Arteriolar Responses to I/R
Impaired endothelium-dependent, NO-mediated relaxation of VSM to mechanical signals (e.g., shear stress) and receptor-dependent vasodilator agonists (e.g., acetylcholine) are the primary postischemic manifestations of endothelial cell dysfunction in arteries and arterioles (Banda et al 1997; Faraci & Didion, 2004; Gourdin, Bree & De Kock, 2009; Hayward & Lefer, 1999; Hein et al, 2003; Kalogeris et al, 2012; Kalogeris et al, 2016; Lefer & Ma, 1994; Ma et al, 1991, 1992, 1993; Ruh et al, 2003; Tsao et al, 1990; Touyz, 2004; Weyrich et al, 1993; Wolin et al, 2002). Since relaxation to endothelium-independent vasodilators (eg, nitroprusside, papaverine) is preserved in postischemic vessels, it appears that vascular smooth muscle (VSM) dysfunction does not account for the impairment in arterial and arteriolar responsiveness. Rather, overproduction of superoxide by endothelium exposed to I/R accounts for the inability of arterioles to exhibit NO-dependent vasodilation since treatment with superoxide dismutase (SOD), SOD mimetics, and other antioxidants restore this response. Oxidative stress induced by I/R also contributes to impaired vasoconstrictor responsiveness (Gao et al, 1994; Gao et al, 1996; Gao et al, 1996; Yokohama et al, 1996). Work conducted in porcine coronary arterioles suggests that I/R inhibits NO-mediated vasodilation by increasing arginase activity, which reduces the availability of L-arginine to nitric oxide synthase for NO synthesis (Hein et al, 2003). However, since neither arginase inhibition nor L-arginine supplementation completely restored NO-mediated vasodilator responses, postischemic NO quenching by ROS and/or reduction in eNOS expression likely account for a significant proportion of EDD dysfunction.
LECA in Postcapillary Venules IS Required for the Development of Arteriolar EDD
Since leukocytes do not roll along, adhere to, or emigrate across arteries or arterioles in the postischemic intestine (Granger & Korthuis, 1995; Granger 2003; Kalogeris et al 2012; Kalogeris et al, 2016) (or most other tissues, the heart being a notable exception (Hayward and Lefer, 1999; Ma et al, 1991, 1992, 1993; Tsao et al, 1990; Weyrich et al, 1993), a role for venular leukocyte adhesion in arteriolar endothelium-dependent vasoregulation has been largely ignored. However, the recent demonstration that arterial tissue obtained from mice that are genetically deficient in P-selectin, CD11/CD18, or ICAM-1 do not exhibit EDD after I/R compared to wild-type animals (Banda et al, 1997; Zamboni, Stephenson, Roth, Suchy & Russell, 1997) suggests that LECA interactions in postcapillary venules may be causally linked to endothelial dysfunction in upstream conduit arteries. In support of this concept, we have recently demonstrated that both ACH and flow-dependent vasodilation are impaired in intestinal arterioles isolated from wild-type but not P-selectin deficient mice (Dai & Korthuis, 2017). An identical pattern of response was noted when arteriolar responses to ACH were observed by intravital microscopic examination in the intact mesenteric microcirculation of neutropenic rats. Similar observations have been made in hypercholesterolemia models and following stroke (Kim, Carter & Harris, 2007; Petrault, Ouk, Gautier, Laprais, Gele, Bastide, et al, 2004). Our previous work shows that sensitivity to vasoconstrictor stimuli is reduced in postischemic intestinal arteries and arterioles, effects that were mitigated by antioxidant treatment but were not abolished by neutrophil depletion (Gao et al, 1994; Gao et al, 1996). These are important observations since arterioles are primarily responsible for controlling the distribution of blood flow within the microcirculation and suggest that leukocyte adhesion is required specifically for the production of EDD dysfunction in postischemic intestinal arterioles, but not responses to vasoconstrictor stimuli. As noted above, studies conducted in coronary arteries obtained after myocardial I/R support these conclusions (Banda et al, 1997; Hayward and Lefer, 1999; Ma et al, 1991, 1992, 1993 Tsao et al, 1990; Weyrich et al, 1993; Zamboni, Stephenson, Roth, Suchy & Russell, 1997). However, a critical difference between these vascular beds is that I/R induces neutrophil adherence to coronary arteries but not arterioles in other vascular beds.
While it is important to re-emphasize that we have not observed leukocyte rolling or adhesion in arterioles of the postischemic mesentery or intestine, there are conditions wherein such adhesive interactions occur in arterioles. As noted above, leukocytes do adhere to aortic tissue and coronary arteries and arterioles after I/R and may contribute to EDD in those vessels (Banda et al, 1997; Cuccocrea et al, 2000; Hayward and Lefer, 1999; Ma et al, 1991, 1992, 1993; Tsao et al, 1990; Weyrich et al, 1993; Zamboni, Stephenson, Roth, Suchy & Russell, 1997). Monocytes clearly adhere to large artery endothelium in hypercholesterolemic animals and humans, especially in regions containing fatty streaks and disrupted flow patterns. In addition, cigarette smoke exposure induces leukocyte adhesion in arterioles in the dorsal skin-fold of hamsters (Lehr, Frei & Arfors, 1994). In the cat mesentery, reductions in shear rate, as occurs in ischemia, produce pronounced increases in the number of adherent leukocytes in postcapillary venules (Perry & Granger, 1991). On the other hand, leukocyte adhesion in arterioles was occasionally observed, but the magnitude of the reduction (by >80-90%) in shear rate required to produce this effect was much greater and the numbers of adherent cells were far lower in arterioles (∼10% of that noted in venules). We emphasize that we do not observe leukocyte adherence to arterioles during ischemia or reperfusion in rodents, perhaps because shear rate is reduced to 25% of control during ischemia in our model. Leukocyte adhesion is also occasionally observed in intestinal arterioles of diabetic animals (Lash & Bohlen, 1991) and has been noted after nitric oxide synthase inhibition or angiotensin II (Ang II) or TNF exposure (Alvarez & Sanz, 2001; Alvarez, Piqueras, Bello, Canet Moreno, Kubes, et al, 2001; Alvarez, Cerda-Nicolas, Nabah, Mata, Issekutz, Panes, et al, 2004; Nabah, Mateo, Cerda-Nicolas, Alvarez, Martinez, Issekutz, et al, 2005; Piqueras Kubes, Alvarez, O'Connor, Issekutz, Esplugues, et al, 2000; Sumagin & Sarelius, 2006). Whether leukocytes adhering to arteriolar walls in these models participate in EDD dysfunction has not been evaluated.
Interestingly, the effects of nitric oxide synthase inhibition are mediated by Ang II (Nabah et al, 2005). Ang II has also been shown to upregulate arteriolar P-selectin, E-selectin, ICAM-1 and VCAM-1 expression when assessed 4 hrs after reperfusion (Alvarez et al, 2004). While these observations have potentially important implications for our work, it is should be noted that one hour of exposure to Ang II fails to promote leukocyte adhesion in arterioles, but does induce significant adhesion in venules over this time frame (Alvarez & Sans, 2001; Alvarez et al 2001; Alvarez et al 2004; Petnehazy, Cooper, Stokes, Russell, Wood & Granger, 2006; Piqueras et al, 2000; Yusof, Kamada, Gaskin & Korthuis, 2007). Rather, leukocyte adhesion did not occur in arterioles unless they were exposed to Ang II for four hours (Alvarez et al 2004). This may explain why we fail to note arteriolar leukocyte adhesion in our model as all responses are monitored within 90 min of reperfusion. These Ang II-induced postcapillary adhesive interactions occur by a mast cell-independent mechanism (Alvarez et al, 2001; Piqueras et al, 2000), an observation consistent with our postulate that mast cell degranulation occurs secondary to leukocyte emigration (see discussion of the role of mast cells in I/R-induced EDD dysfunction below) and is a cause rather than an effect of Ang II generation. In addition, it should be noted that mononuclear cells appear to be the major adherent cell type in response to Ang II (Alvarez et al, 2004), while neutrophils are the principal infiltrating leukocyte subtype during the first 60-120 min of intestinal reperfusion (Oliver, Specian, Perry & Granger, 1991). EDD dysfunction is noted in mesenteric arterioles after hemorrhagic shock, an effect that was not abolished by neutrophil depletion (Zakaria, Garrison, Kawabe & Harris, 2004). Thus, a role for neutrophils in EDD cannot be extrapolated to all conditions characterized by I/R. Interestingly, leukocyte rolling may be more frequent in pulmonary arterioles than in venules under control conditions (Kuebler Kuhnle, Groh & Getz, 1994; Yamaguchi, Nishio, Sato, Tsumura, Ichihara, Kudo, et al 1997). It is not clear why leukocyte adhesion occurs in arterioles under the aforementioned conditions, but not in our model, but most likely relates to the different interventions used to elicit arteriolar leukocyte adhesion (diabetes, cigarette smoke exposure vs I/R), the different organs (heart, skeletal muscle vs gut) and/or species (cat vs murine or rat mesentery) studied, level of shear stress reduction required to elicit occasional arteriolar leukocyte adhesion, the presence of a much larger population of mast cells in the small intestine vs skeletal muscle, time frame over which responses occur (4 hrs continuous exposure to elevated Ang II are required to elicit leukocyte adhesion to arterioles), or dose of interventions (Ang II) or conditions (eg systemic shock vs local I/R) tested.
Whatever the explanation, an important and often overlooked fact is that the endothelium appears be protected from a misguided attack by adherent leukocytes. It has been suggested that the fluid dynamics of a leukocyte's intravascular environment may provide constant modulating signals that activate certain leukocyte functions, while simultaneously inhibiting others (Moazzam, DeLano, Zweifach & Schmid-Schonbein, 1997). As a result of these actions, it appears that proinflammatory signals activate a behavioral program in leukocytes that contributes to the development of leukocyte/endothelial interactions and promotes extravasation. However, protective mechanisms existent in the vasculature and in the leukocyte have evolved to keep leukocytes from secreting mediators at the wrong place or at an inappropriate time. For example, intracellular and plasma antioxidants or endogenous protease inhibitors neutralize the toxicity of mediators secreted by adherent leukocytes. In addition, shear stress may downregulate neutrophilic oxidant production and protease release by adherent cells but activates calcium and other ion channels important in regulating cell motility, thereby protecting the endothelium from leukocyte-derived mediators, while allowing their extravasation (Moazzam, DeLano, Zweifach & Schmid-Schonbein, 1997). However, once emigrated, the leukocyte is removed from shear stress-induced inhibition, and the extravasated cells can now mobilize their cytotoxic arsenal to direct a focused attack on extracellular matrix components. This postulate is consistent with our hypothesis that events occurring secondary to leukocyte emigration modify EDD processes in arterioles (see Figure 2 and discussion below). Since inhibition of leukocyte rolling or adhesion also prevents emigration, the potential role of postcapillary venular leukocyte emigration in the development of EDD in arterioles may be blocked by anti-adhesion interventions or is absent in mice deficient in P-selectin.
Figure 2.
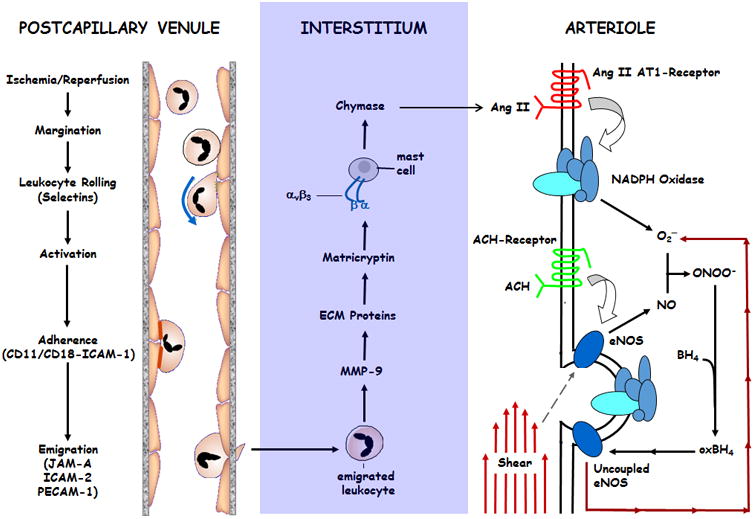
Rather than adhering directly to arterial vessels to produce arteriolar EDD dysfunction after I/R, the mechanism by which activated neutrophils disrupt NO-dependent vasoregulation requires adhesive interactions and subsequent emigration across postcapillary venular walls. Upon entry into the extravascular compartment, these inflammatory phagocytes release proteases, such as matrix metalloproteinase-9 (MMP-9), to generate signals within the interstitium that provoke the release of mast cell-derived chymase. In turn, chymase catalyzes the conversion of angiotensin I (Ang I) to angiotensin II (Ang II), which instigates NADPH oxidase-dependent production of reactive oxygen species (ROS). This oxidative stress is exacerbated by postischemic eNOS uncoupling, which results in the production of superoxide instead of NO (not shown). Postischemic ROS may also be derived from xanthine oxidase and mitochondrial sources (not shown). I/R-induced ROS production quenches NO produced in response to mechanical stimuli (e.g., shear stress) or EDD agonists (e.g., acetylcholine), resulting in impaired EDD responses in arterioles. Figure modified from Dai & Korthuis, 2011.
While the aforementioned studies provide strong support for the concept that events initiated by leukocytes adhering to postcapillary venules contribute to I/R-induced EDD in upstream arteries and arterioles, the mechanisms underlying this effect are entirely unclear. There are at least three potential explanations for ability of leukocyte/endothelial cells adhesive interactions in postcapillary venules to influence endothelial function in upstream arteries and arterioles. First, an ascending propagated signal could be initiated by leukocytes adhering to postcapillary venules in postischemic tissues via cell-cell communication between adjacent microvascular cells to upstream arterioles (Banda et al, 1997; Segal & Duling, 1986). Interestingly, after exposure to I/R, adherent leukocytes express connexin 43 that is localized to their points of contact with endothelial cells (Jara, Boric & Saez, 1995). It has been suggested that this may allow for heterotypic intercellular gap junctional communication to regulate diapedesis, but also perhaps be a source for initiating signaling to upstream arterioles. This may represent a less likely explanation because of the long distance the conducted response would have to travel, especially if a decrement in the conducted response occurs as the signal is propagated, as has been reported for conducted vasodilatory responses to acetylcholine in arterioles (Segal and Duling, 1986). Moreover, conducted responses to agonists appear to be quite sensitive to ischemia, with enhanced spatial decay (as reflected by reductions in mechanical length constants) occurring in response to arterial or venous occlusion (de With, Haug, van der Heijden & Segal, 2005; Lin & Duling, 1994). More importantly, connexin expression is very sparse or absent in venules (Tallini, Brekke, Shui, Doran, Hwang, Nakai, et al, 2007; Schmidt, Wolfle, Boettcher & de Wit, 2008), an observation consistent with our demonstration that gap junction inhibitors do not prevent the leukocyte-dependent arteriolar EDD (Dai and Korthuis, 2017). Second, transfer of a highly diffusible mediator produced by adherent leukocytes or the underlying endothelium in the postcapillary venule to adjacent arterioles with countercurrent flow could account for this response (Kim, Carter & Harris, 39). However, anatomic considerations preclude this as a likely explanation for the aforementioned observations in the aorta and superior mesenteric artery and for arterioles not arranged in parallel with venules. A third possibility is release of inflammatory mediators the produce hyperreactive vasoconstrictor responses that limit NO-dependent dilation in arterial vessels, as has been observed in atherosclerotic plaques (Sato, Shirai, Hontani, Shinooka, Hasegawa, Kichise, et al, 2017). Similar responses have been suggested to play a role in vascular smooth muscle hyperreactivity associated with variant angina (Lanza & Maseri, 2000). This explanation may not pertain to our observations because atherosclerosis is not present. Mast cell-derived mediators produce vasoconstriction of hamster cheek pouch arterioles after I/R, but fail to modify NO-dependent dilator responses to acetylcholine in skeletal muscle (Keller, 1997). In addition, our previous work indicates that mesenteric vessels become hyporesponsive to vasoconstrictor stimuli such as norepinephrine, vasopressin and Ang II (Gao et al, 1994, 1996, 1996; Yokoyama et al, 1996). On the other hand, intestinal arterioles exhibit reduced vascular caliber after I/R or intermittent hypoxia, an effect that is likely due to reduced bioavailability of NO and/or H2S (Gonzalez Bosc, Osmond, Giermakowska, Pace, Riggs, Jackson-Weaver, et al 2017; Kanagy, Szabo & Papapetropoulos, 2017; Kurose et al, 1994; Osmond & Kanagy, 2014).
Two key observations led us to propose a fourth possibility (Figure 2) which we believe provides the most likely explanation for the ability of LECA in postcapillary venules to induce EDD in upstream arterioles. First, we and others have demonstrated that prevention of I/R-induced leukocyte rolling or stationary adhesion completely prevents leukocyte emigration across postcapillary venules (Kalogeris et al, 2012; 2016). Second, tantalizing evidence suggests that proteolytic fragments of extracellular matrix proteins alter vasoreactivity in arterioles (Gloe, Sohn, Meininger & Pohl, 2002; Hein, Platts, Waitkus-Edwards, Mousa & Meininger, 2001; Hill & Meininger, 2012; Martinez-Lemus, Wu, Wilson, Hill, Davis, Davis & Meininger, 2003; Martinez-Lemus, Sun, Trache, Trzciakowski & Meininger, 2005; Mogford, Davis, Platts & Meininger, 1996; Wu, Mogford, Platts, Davis, Meininger & Davis, 1998). These observations led us to hypothesize that the oxidative/proteolytic activity of emigrating leukocytes may lead to the formation of nascent mediators or exposure of previously cryptic effectors in the interstitium that directly, or through activation of downstream mediators, produce impaired arteriolar responses to endothelium-dependent agonists and shear stress, which will be discussed next (Figure 2).
Mediators Generated by the Proteolytic Activity of Emigrated Leukocytes are Essential for the Development of EDD Dysfunction in I/R
In order to produce tissue injury, adherent leukocytes must extravasate into the tissues (Granger & Korthuis, 1995; Kalogeris et al, 2012, 2016). This removes leukocytes from the inhibiting effects of shear stress imposed by the flowing blood (Moazzam et al, 1997), thereby allowing the infiltrating leukocyte to disrupt subendothelial basement membrane components, hydrolyze extracellular matrix (ECM) proteins, and injure parenchymal cells by releasing cytotoxic oxidants and/or hydrolytic enzymes (Granger & Korthuis, 1995; Kalogeris et al, 2012, 2016; Welbourne, Goldman, Paterson, Valeri, Shepro & Hechtman, 1987). This postulate is supported by our observation that treatment with phalloidin, which we have previously shown completely prevents postischemic leukocyte emigration without influencing I/R-induced leukocyte rolling or adhesion, oxidant production or protease release by activated leukocytes, CD18 clustering, leukocyte adherence to plastic, or leukocyte emigration across inert membranes (Asako, Wolf, Granger & Korthuis, 2002; Korthuis, Carden, Kvietys, Shepro & Fuseler, 1991; Shigematsu, Ishida, Gute & Korthuis, 1999), prevented EDD in postischemic rat mesentery and in vessels isolated from murine intestine after I/R. These observations suggest that adherent leukocytes must emigrate into the tissues to influence vasodilator responses to ACH in arterioles. Since ACH responses were similar in arterioles of control rats (no-ischemia) in the presence and absence of phalloidin, it does not appear that this agent directly influences the responses to acetylcholine. However, it is possible that flow-dependent vasodilation in isolated arterioles may be influenced by this agent, which acts to prevent leukocyte emigration by stabilizing F-actin in endothelial cells, since cytoskeletal elements play an important role in transmitting mechanical signals in response increased shear stress.
It is important to note that we have not observed a single leukocyte to adhere to arteriolar endothelium in our model of intestinal I/R. However, because leukocytes do adhere to the endothelium in other conditions, we determined whether perfusion of isolated arterioles with neutrophils and subsequently exposed to PAF plus LTB4 plus Ang II for 60-90 min would induce adhesion. These agents failed to induce arteriolar leukocyte adhesion, even when pseudo-shear rates were concomitantly reduced to 25% of control, and did not impair ACH responses. Similarly, PAF plus LTB4 alone (ie, in the absence of neutrophil perfusion) failed to alter arteriolar ACH responses. Our latter observations are particularly relevant to I/R, because PAF, LTB4, and Ang II have been shown to play a major role in postischemic leukocyte adhesion in venules (Granger & Korthuis, 1995; Kalogeris et al, 2012, 2016; Petnehazy et al, 2007; Yusof et al, 2007). These data indicate that leukocytes do not adhere to arteriolar endothelium in our model and also indicate that neutrophils flowing down arterioles do not modify ACH responses, even in the presence of activating factors and reduced shear rates.
Although much attention has focused on the role of reactive oxygen metabolites, less emphasis has been placed on elucidating the role of leukocyte proteases in the pathogenesis of I/R injury. However, it is clear that hydrolytic enzymes released by leukocytes, such as collagenase, elastase and matrix metalloproteinase-9, degrade important structural components of the endothelial glycocalyx, basement membrane, and ECM, thereby contributing to leukocyte-dependent microvascular barrier dysfunction in postischemic tissues (Altshuler, Kistler & Schmid-Schonbein, 2016; Carden & Korthuis, 1996; Granger & Korthuis, 1995; Kalogeris et al, 2012, 2016; Schmid-Schonbein, 2016).
Among the proteases involved in the pathogenesis of I/R-induced microvascular dysfunction and tissue injury, matrix metalloproteinase-9 (MMP-9; 92 Kd gelatinase, gelatinase B) appears to play a prominent role. The MMP family consists of at least 20 zinc-containing serine proteases that participate in tissue and vascular remodeling, tumor metastasis, angiogenesis, and contribute to several phases of inflammation (Fujimura, Gasche, Morita-Juimura, Massengale, Kawase & Chan, 1999; Gasche, Jujimura, Morita-Fujimura, Copin, Kawase & Chang, 1999; Rosario et al, 2004; Yang and Rosenberg, 2015). These structurally complex endoproteases are also known as matrixins or metzincins and are found in many cell types, including fibroblasts, osteoclasts, keratinocytes, leukocytes, and endothelial cells. MMPs are secreted by cells in an inactive form (zymogen) that can be activated by proteolytic cleavage. Recent evidence indicates that MMPs can also be activated by hypochlorous acid (HOCl), a potent oxidant formed by neutrophilic myeloperoxidase (MPO) (Fu, Kassim, Parks & Heinecke, 2001; Yang & Rosenberg, 2015). MPO also inactivates tissue inhibitor of matrix metalloproteinases-1, allowing this neutrophilic enzyme to disinhibit MMP activity (Wang, Rosen, Madtes, Shao, Martin & Heineke, 2007). On the other hand, HOCl can also restrain the activity of some MMPs, providing a mechanism to fine tune proteolysis during inflammatory states (Fu, Kao, Bergt, Kassim, Huq, d'Avignon, et al, 2004). MMP-9 is expressed by endothelial cells during reperfusion (Gasche et al, 1999; Fujimura et al, 1999). Neutrophils infiltrating postishemic intestine also release MMP-9, which leads to extracellular matrix turnover (Rosario et al, 2004). MMP-9 has been implicated in parenchymal cell injury induced by myocardial infarction, hemorrhagic stroke, sepsis, and in liver and intestinal I/R (Camp, Tyagi, Aru, Hayden, Mehta & Tyagi, 2004; Fujimura et al, 1999; Gasche et al, 1999; Heo, Lucero, Abumiya, Koziol, Copeland & del Zoppo, 1999; Kuyvenhoven, Molenaar, Verspaget, Veldman, Palareti, Legnani, et al, 2004, Rosario et al, 2004; Smith and Gabler, 1995). Indeed, Rosario et al (2004) demonstrated that MMP-9 is highly expressed in the wall of the postischemic small intestine and appears to be derived from infiltrating neutrophils. Furthermore, these investigators demonstrated that pancreatic trypsin promotes rapid proteolytic activation of MMP-9 in the wall of the small intestine after I/R, allowing activated monomers to dimerize to the active form of the protease in postischemic gut, which in turn, amplifies the inflammatory response to I/R (ie, exacerbated postischemic leukocyte rolling, adhesion, and infiltration) and was associated with extensive morphologic mucosal damage.
Proteolytic digestion of ECM proteins has been shown to reveal hidden (latent) bioactive epitopes (e.g., the conserved amino acid sequence Arg-Gly-Asp or RGD) or release of proinflammatory mediators, collectively referred to as matricryptic effectors (Davis, 2010; Davis, Bayless, Davis & Meininger, 2000). With regard to vascular function, Meininger, Davis and coworkers (Gloe, Sohn, Meininger & Pohl, 2002; Hein, Platts, Waitkus-Edwards, Mousa & Meininger, 2001; Hill & Meininger, 2012; Martinez-Lemus, Wu, Wilson, Hill, Davis, Davis & Meininger, 2003; Martinez-Lemus, Sun, Trache, Trzciakowski & Meininger, 2005; Mogford, Davis, Platts & Meininger, 1996; Wu, Mogford, Platts, Davis, Meininger & Davis, 1998) were the first to demonstrate that synthetic RGD-containing peptides and proteolytic degradation products of extracellular matrix proteins such as collagen type I alter arteriolar tone. Furthermore, they demonstrated that synthetic RGDs interact with the integrin αvβ3 (and other integrins) to alter myogenic tone. These studies led us to postulate that proteolytic digestion of ECM proteins by leukocytes infiltrating the postischemic intestine may modify arteriolar vasoregulatory responses in postischemic tissues or in other inflammatory states by an integrin-dependent mechanism. Support for this concept is provided by the observations that treatment with broad-spectrum anti-proteases, MMP-selective inhibitors or agents targeting αvβ3 prevented postischemic arteriolar EDD dysfunction (Dai and Korthuis, 2017). Moreover, ACH responses were preserved in MMP-9-deficient mice, but not MMP-2-/- mice, subjected to I/R compared to wild-type animals. Importantly, arterioles isolated from non-ischemic rat mesenteries demonstrated EDD dysfunction when incubated with postischemic intestinal lymph (as a surrogate for interstitial fluid) draining reperfused intestines of donor rats, but not when exposed to lymph obtained from rats treated with protease inhibitors during I/R. The latter studies support the concept that signals generated in the interstitium after I/R act to disrupt EDD function. In addition to these effects, MMP inhibition with doxycycline has been shown to improve EDD function by inhibiting oxidative stress and improving NO bioavailability (Castro, Rizzi, Ceron, Guimaraes, Rodrigues, Bendhack et al, 2012). Studies conducted with MMP inhibitors or in MMP-9 deficient mice also show that endothelial MMPs influence vascular reactivity (Cena, Lalu, Rosenfeldt & Schulz, 2008; Su, Palen, Lucchesi & Matrougui, 2006).
Mast Cells Contribute to I/R-induced Arteriolar EDD
Because mast cell density is very high in gastrointestinal tissues (Nilsson, Costa, Metcalfe, 1999), it is not surprising that these perivascular cells play a much more prominent role in the inflammatory response to I/R in the small bowel (Bortolotto et al, 2004; Kanwar et al, 1998). Intestinal mast cells degranulate during reperfusion (Kanwar & Kubes, 1994, 1994; Kanwar et al, 1998; Miller & Pemberton; 2003), but mast cell degranulation does not influence postischemic P-selectin expression in the first 10-30 min of reperfusion (Eppihimer, Russell, Anderson, Epstein, Laroux & Granger, 1997). This time frame is consistent with our contention that leukocyte extravasation must occur first to produce mediators that contribute to mast cell degranulation and subsequent induction of P-selectin expression. However, treatment with mast cell stabilizers is effective in preventing I/R-induced mucosal barrier disruption, increased vascular permeability, and leukocyte infiltration in the small intestine of mice and rats after reperfusion for 60 min (Kanwar & Kubes, 1994, 1994, Kanwar et al, 1998), a time frame that allows for neutrophil diapedesis-dependent mast cell degranulation to occur first.
Two types of mast cells are present in small intestine: connective tissue mast cells, which are located in the submucosa and muscularis layers of the bowel wall and in the mesentery, and mucosal mast cells, which as the name implies reside in the mucosa (Nilsson et al, 1999). These two types of mast cells release different mediators and may respond to distinct stimuli (Nilsson et al, 1999). For example, chymase, a chymotrypsin-like serine protease that catalyzes the formation of Ang II, is mainly contained in secretory granules of connective tissue mast cells and is released only by these mast cells, and not mucosal mast cells, in rats and humans (Miller & Pemberton, 2003; Nilsson et al 1999). In normal tissues, mast cell chymase is stored in an inactive form, but acquires the ability to form Ang II following secretion from degranulating mast cells (Dell'Italia & Husain, 2004; Gaboury, Johnston, Niu & Kubes, 1995; Jin, Takai, Okamoto, Muramatsu & Miyazaki, 2004; Miller & Pemberton, 2003; Miyazaki and Takai, 2001; Nilsson et al 1999; Richard, Hurel-Merle, Scalbert, Ferry, Lallemand, Bessou, et al, 2001). Importantly, this enzyme can account for as much as 80-90% of Ang II formation in the heart and is a significant source of the peptide in damaged arterial vessels, indicating that chymase can become the chief source of Ang II in injured tissues. Thus, mast cell chymase is an important source Ang II in injured vessels but because Ang II derived from this enzymatic source does not influence blood pressure, it appears that that the effects of chymase-derived Ang II are restricted to the local environment (Miyazaki & Takai, 2001; Richard et al, 2001; Dell'Italia & Husain, 2004; Suzuki et al, 1998).
Given this background and the facts that: 1) mast cells can be activated to release chymase and other proinflammatory mediators on exposure to RGDs, a secretory process that appears to be mediated by αvβ3 (Nilsson et al, 1999; Shimuzu, Irani, Brown, Ashman & Schwartz, 2001), 2) mast cell chymase has been implicated lipopolysaccharide-induced vasomotor dysfunction in arterioles, an effect that is mediated by Ang II-dependent production of reactive oxygen species (Suzuki, Coughey Gao & Rubinstein, 1998), and 3) mast cell chymase also plays an important role in activating pro-MMP-2 and -9, which may serve to amplify the effect of MMPs in I/R (Ishida, Takai, Murano, Nishikawa, Inoue, Murano, et al, 2008; Tchougounova, Lundequist, Fajardo, Winberg, Abrink & Pejler, 2005), we postulated that leukocyte-derived MMP-9 acts to expose latent matricryptic effectors in the extracellular matrix which interact with β3 integrin binding sites on mast cells, thereby stimulating chymase secretion by mast cells (Figure 2). Chymase-dependent formation of Ang II in turn activates NAD(P)H oxidase to produce superoxide, quenching NO and leading to impaired vascular responses to endothelium-dependent vasodilators (Figure 2). This hypothesis is supported by the observations that treatment with a mast cell stabilizer prevented the I/R-induced impairment in ACH-induced vasodilation in mesenteric arterioles, as did treatment with a chymase inhibitor. Importantly, postischemic mast cell degranulation in the rat mesentery was attenuated in animals treated with phalloidin (which prevents leukocyte emigration), protease inhibitors, or β3 immunoneutralization (Dai & Korthuis, 2017). In contrast, superfusing the mesentery with compound 48/80 promoted mast cell degranulation that was not inhibited by phalloidin and was associated with EDD. These latter observations suggest that mast cell degranulation per se can promote EDD in arterioles and that phalloidin does not directly influence mast cell degranulation. Taken together, our results suggest that the proteolytic activity of emigrating leukocytes may generate effectors that promote mast cell degranulation via the integrin αvβ, and that chymase release by mast cells modifies postischemic arteriolar responses to ACH. This integrin is also upregulated on cerebral microvessels after stroke (Abumiya, Lucero, Heo, Tagaya, Kosiol, Copeland et al, 1999), suggesting the possibility of a more direct effect of αvβ on EDD function since ligation of this integrin promotes vasodilation (Mogford et al, 1996).
Ang II-dependent, NAD(P)H Oxidase-mediated Production of Reactive Oxygen Species (ROS) Contribute to eNOS Uncoupling and EDD in Arterioles after I/R
Overproduction of superoxide by postischemic endothelium largely accounts for the inability of arterioles to exhibit endothelium-dependent, NO-mediated relaxation since treatment with superoxide dismutase and other antioxidants restore this response in arterial tissue (Kalogeris et al, 2012, 2016). Although superoxide generation appears to be pivotal in producing postischemic EDD dysfunction, it also participates in the generation of other ROS that may influence arteriolar function (Cifuentes & Pagano, 2006; Gourdin, Bree & De Kock, 2009; Lyle & Griendling, 2005; Mehta & Griendling, 2007; Pagano and Gutterman, 2007; Rey, Cifuentes, Kiarash, Quinn & Pagano, 2001; Rey Li, Carretero, Garvin & Pagano, 2002; Touyz, 2004; Wolin et al, 2002). Reaction of superoxide with NO generates peroxynitrite (ONOO-) and other reactive nitrogen oxide species which are potentially deleterious (see next paragraph). Dismutation of superoxide spontaneously or by enzymatic action of SOD produces hydrogen peroxide, which can react with reduced transition metals to form the highly reactive hydroxyl radical. Hydrogen peroxide can also be metabolized by neutrophilic myeloperoxidase (MPO) to form hypochlorous acid and other oxidizing species. MPO can also act as an NO oxidase, thereby reducing the bioactivity of NO and increasing oxidative stress, thereby contributing to EDD dysfunction (Baldus, Heitzer, Eiserich, Lau, Mollnau, Ortak et al, 2004; Tian, Ding, Peng & Lu, 2017).
Despite the wealth of information supporting a role for oxidants in arteriolar EDD dysfunction, the source of superoxide that inactivates NO remains unclear. In addition to mitochondrial sources, superoxide and/or hydrogen peroxide can be formed by many enzymes. Two of the most important sources in normal vessels are cytochrome P450 and the NAD(P)H oxidases, while xanthine oxidase, uncoupled eNOS, and mitochondrial respiratory chain appear to play an important role in postischemic tissues (Kalogeris et al, 2012, 2016). It is well-known that xanthine oxidase-derived oxidants play an important role in the genesis of I/R injury by participating in the formation of pro-inflammatory mediators such as PAF and LTB4 and by inactivating NO, which normally produces powerful anti-adhesive effects. The net effects of these actions are to promote leukocyte adhesion and emigration in postcapillary venules. Superoxide formed by the NAD(P)H oxidases appears to play a more dominant role in impaired EDD noted in arterial vessels in a variety of pathologic states. An interesting and important feature of eNOS is its ability to produce superoxide under certain conditions, such as L-arginine or tetrahydrobiopterin (BH4) deficiency, dephosphorylation of threonine 495, and formation of eNOS monomers (Forstermann, 2006; Moens & Kass, 2006; Munzel, Daiber, Ullrich & Mulsch, 2005). Functional eNOS converts L-arginine and O2 to NO and L-citrulline, an enzymatic process that consumes NADPH and requires Ca2+/calmodulin, flavin adenine dinucleotide, flavin monocleotide, and BH4. When uncoupled by absence of L-arginine or BH4, electrons normally flowing from the reductase domain of one subunit to the oxygenase domain of the other subunit are diverted to molecular oxygen rather than L-arginine or the cofactor BH4, resulting in superoxide production. Thus, oxygen reduction and NO formation are uncoupled and superoxide is generated from the eNOS oxygenase domain by dissociation of the ferrous-dioxygen complex.
The NAD(P)H oxidases (Nox) of the cardiovascular system are expressed in endothelial cells, vascular smooth muscle cells, and fibroblasts in the vascular wall, as well as in cardiac myocytes (Lyle & Griendling, 2005; Mehta & Griendling, 2007; Touyz, 2004; Wolin et al, 2002). While the cardiovascular oxidases are similar to the neutrophil NADPH oxidase, they represent a unique family of enzymes (Nox1-5). The vascular Nox's are low output, slow-release enzymes relative to the phagocytic enzyme and exhibit moderate constitutive activity that is absent in neutrophils. Phagocytic NADPH oxidase consists of 4 major subunits: a membrane associated complex of 2 subunits, gp91phox and p22phox, and a cytosolic complex with two components, p67phox and p47phox. Under baseline conditions, these components are kept apart, rendering the enzyme inactive. On stimulation, p47phox becomes heavily phosphorylated and, along with p67phox, moves to the cell membrane and binds the membrane complex, allowing for activation. The membrane portion then transfers an electron from NADPH to molecular oxygen, forming superoxide. The vascular forms use NADH and NADPH to drive superoxide production and vary in their subunit composition and cellular location (Lyle & Griendling, 2005; Mehta & Griendling, 2007; Touyz, 2004; Wolin et al, 2002). One of the most important attributes of the vascular Nox's is its responsiveness to hormones (eg, Ang II, thrombin, lactosylceramide, PAF, IL-1, TNF, etc), shear forces, and local metabolic factors (eg, lactate), with the oxidase of each cell type exhibiting differential responsiveness to such stimuli (Lyle & Griendling, 2005; Mehta & Griendling, 2007; Touyz, 2004; Wolin et al, 2002).
Of particular interest to our work is the fact that Ang II increases Nox activity in vascular smooth muscle, an effect mediated by lipoxygenase metabolites of arachidonic acid (Lyle & Griendling, 2005; Mehta & Griendling, 2007; Touyz, 2004; Wolin et al, 2002). Moreover, the intestinal vasculature exhibits extraordinarily high concentrations of Ang II receptors (Riaz, Wang, Schramm, Sato, Menger, Jeppsson et al, 2004; Gunther, Gimbrone & Alexander, 2004). Work from our laboratory also implicates Ang II in postischemic inflammatory responses in the small intestine (Yusof et al, 2007). In this study, I/R increased Ang II levels while AT1 receptor blockade, chymase inhibition, and a CGRP receptor antagonist attenuated postischemic leukocyte adhesion. Superfusion of control intestine (no ischemia) with Ang II for 2 hr induced leukocyte adhesion, an effect prevented by concomitant Nox inhibition. Ang II may also influence I/R-induced fluid accumulation via its effect to induce αvβ3 integrin-mediated dependent collagen contraction by fibroblasts (Nunohiro, Ashizawa, Graf, Hsueh & Yanoh, 1999), an action that has been suggested to create highly negative interstitial fluid pressure, thereby facilitating edema formation (Reed, Berg, Gjerde & Rubin, 2001). Applying a force to integrins has been shown to activate Nox by an Ang II-dependent mechanism (Browe & Baumgaarten, 2004, 2006). Of high significance to our central hypothesis (Figure 2) is the growing body of evidence supporting the notion that the products of the Nox's and eNOS (superoxide and NO, respectively) rapidly interact to form ONOO-, which can oxidize the essential NOS co-factor, BH4, to biologically inactive products (Forstermann, 2006; Moens & Kass, 2006). As a consequence, oxygen reduction uncouples from NO synthesis, thereby transforming functional, NO-producing eNOS into a dysfunctional superoxide-producing enzyme. Indeed, Nox-driven eNOS uncoupling by oxidation of BH4 has been implicated in the vascular dysfunction associated with hypertension, hypercholesterolemia, and diabetes, which can be attenuated by administration of exogenous BH4 (Elrod, Duranski, Langston, Greer, Tao, Dugas, et al 2006; Forstermann, 2006; Moens & Kass, 2006; Munzel Daiber, Ullrich & Mulsch, 2005; Stokes, Dugas, Tang, Garg, Guidry, Bryan, 2009). Moreover, myocardial infarct size, cardiac superoxide generation, and coronary artery endothelial dysfunction are attenuated by provision of exogenous BH4, findings which support the concept that eNOS uncoupling contributes to I/R injury in the heart (Dumitrescu, Biondi, Xia, Cardounel, Druhan, Ambrosio et al, 2007; Forstermann, 2006; Moens & Kass, 2006). Taken together, these studies suggest the possibility that Nox-derived superoxide reduces the bioavailability of NO and thus EDD during I/R by at least two mechanisms related to the formation of peroxynitrite: 1) superoxide combines with NO formed by a still functional eNOS, producing ONOO-, an effect that effectively scavenges NO as it is produced, thereby quenching the direct effects of NO to produce vasodilation, and 2) ONOO- produces eNOS uncoupling secondary to BH4 oxidation, transforming a functional eNOS into a dysfunctional, superoxide producing enzyme. This fuels a futile cycle wherein uncoupled eNOS-derived superoxide contributes to BH4 oxidation and leads to further eNOS uncoupling.
I/R-induced superoxide production has been implicated in EDD dysfunction in coronary and skeletal muscle arteries, and we have obtained evidence extending this concept to intestinal arterioles. Superfusing the mesentery with the cell-permeant SOD mimetic (Mn-TBAP), BH4, or the Nox inhibitor apocynin attenuated impaired arteriolar responses to acetylcholine that are induced by I/R. The apocynin data not only provides a second line of evidence supporting a role for oxidants in the development of impaired responses to acetylcholine but also suggest that Nox may represent an important source of reactive oxygen species in vessels subjected to I/R, while the eNOS cofactor studies support a role for uncoupled eNOS. Interestingly, topical application of candesartan, an Ang II AT1 receptor antagonist was as effective as Mn-TBAP and apocynin in preventing I/R-induced arteriolar dysfunction. It is difficult to determine from this data whether the treatments directly exert their effects on arteriolar responses or to their ability to prevent I/R-induced leukocyte adhesion. However, arteriolar ROS production was attenuated by neutrophil depletion as well as by addition of Mn-TBAP, candesartan, or apocyin to the solution bathing mesenteric arterioles after I/R. More importantly, ROS production by naïve arterioles incubated with postischemic lymph was attenuated by addition of Mn-TBAP, candesartan, or apocynin to the lymph bathing the vessels. Taken together, these studies strongly support the notion that there is a link between effectors produced in the interstitium by the proteolytic activity of emigrated leukocytes and impaired responses of arterioles to endothelium-dependent, NO-mediated vasodilator responses (Figure 2). The fact that treatment with Mn-TBAP, BH4, apocynin, or candesartan restores ACH-dependent responses in postischemic mesentery and prevents oxidant production in arteriolar vessels exposed to postischemic lymph supports our hypothesis that emigrating leukocytes generate effectors that induce oxidant production in arterioles by an Ang II-dependent, Nox-mediated mechanism that leads to eNOS uncoupling. Taken together, these data suggest that I/R-induced, Ang II-dependent Nox oxidase activation may be a source of superoxide that promotes eNOS uncoupling, inactivates NO and induces EDD dysfunction. The fact that L-NIO partially reduced, but SOD mimetic and apocynin treatment, largely abolished postischemic superoxide generation in arterioles bathed in postischemic lymph supports this view. We also measured Ang II levels in plasma draining the ischemic region and demonstrated that I/R induced a 3-fold increase that was prevented by prior neutrophil depletion.
Summary and Future Directions
A large body of evidence indicates that neutrophils and ROS derived from Nox and uncoupled eNOS contribute to I/R-induced EDD dysfunction in conduit arteries and arterioles. However, until very recently, it was not clear whether these contributing factors were mechanistically linked in arterioles, where LECA is not observed after I/R. More recent work has implicated a role for mast cells and MMPs in postischemic arteriolar dysfunction and has tied these disparate findings into a cohesive mechanism (Figure 2). Neutrophils participate in the development of postischemic arteriolar EDD dysfunction by a mechanism that is initiated by adhesive interactions between these inflammatory phagocytes and postcapillary venular endothelial cells. As neutrophils migrate across postcapillary venular walls and gain access to the interstitial space, they become activated and release a variety of hydrolytic enzymes, including matrix metalloproteinase-9. The enzymatic activity of this and other proteases appears to cleave components of the extracellular matrix, thereby exposing matricryptic sites or releasing matricryptins that activate mast cells by an αvβ3 integrin-dependent mechanism. Ensuing mast cell degranulation releases chymase, an enzyme that catalyzes the conversion of Ang I to Ang II. Ang II-dependent Nox activation results in ROS production, which interacts with NO produced by eNOS in response to shear stress or endothelium-dependent vasodilator agonists, producing peroxynitrite (and other reactive nitrogen oxide species). Peroxynitrite may exacerbate EDD dysfunction by producing eNOS uncoupling secondary to BH4 oxidation, transforming a functional eNOS into a dysfunctional, superoxide-producing enzyme. This fuels a futile cycle wherein uncoupled eNOS-derived superoxide contributes to BH4 oxidation and leads to further eNOS uncoupling. Understanding this pathway provides important insights into the pathogenesis of arteriolar dysfunction in I/R that may extrapolate to other conditions characterized by enhanced leukocyte infiltration into the tissues.
While this is an appealing hypothesis that links together adhesive events occurring in postcapillary venules to signals generated in the interstitium that ultimately act to impair responses to endothelium-dependent stimuli in upstream arterioles, it also provokes a number of questions for future studies. For example, it is not clear what role protease-dependent receptor cleavage (Schmid-Schonbein, 2016; Mazor & Schmid-Schonbein, 2015) might play in postischemic EDD dysfunction. Moreover, interesting recent work has demonstrated that pentraxin 3, a soluble pattern recognition receptor that plays an important role in innate immune responses, induces EDD dysfunction via a P-selectin/MMP-dependent pathway that may feed into our hypothesized mechanism (Carrizzo, Lenzi, Procaccini, Damato, Biagioni, Ambrosio et al, 2015). The role of neutrophil extracellular traps (NETs) in I/R-induced microvascular dysfunction and tissue injury has been established (Albadawi, Oklu, Raacke Malley, O'Keefe, Uong, Cormier et al, 2016; Ge, Zhou, Ji, Lu, Zhang, Zhang et al, 2015; Jansen, Emal, Teske Dessing, Florquin & Roelofs, 2017; Nakazawa, Kumar, Marschner, Desai, Holderied, Rath et al, 2017; Pallet, 2017), but their potential contribution to EDD dysfunction in arterial tissues is unknown. Interestingly, pentraxin 3 is a key component of NETs (Liu, Chuang, Tsai & Yu, 2014), suggesting a possible linkage to our hypothesized mechanism for neutrophil-initiated EDD dysfunction in I/R.
In addition to MMPs, another group of proteases, the calpains, have been shown to participate in agonist-induced EDD (Hein, Rosa, Ren XU & Kuo, 2015), improve collateral-dependent perfusion and reduce infarct size after myocardial ischemia (Potz, Sabe, Abid & Sellke, 2015; Sabe, Potz, Elmadhun, Liu, Feng, Abid, et al, 2016), and contribute to EDD dysfunction associated with risk factors for cardiovascular disease (Chen, Zhao, Ni, Tang, Shan, Cepinskas, et al, 2014; Cheng, Jiang, Pansuria, Fang, Mai, Mallilankaraman, et al, 2015; Kishore, Benedict & Cheng, 2015; Nangle, Cotter & Cameron, 2006; Stalker, Gong & Scalia, 2005; Randriamboavonjy, Kyseolova, Elgheznawy, Zukunft, Wittig & Fleming, 2017; Stalker, Skvarka & Scalia, 2003, Wang, Peng, Zhang & Liu, 2008) or activation of Ang II type 1 receptors (Scalia, Gong, Berzins, Freund, Feather, Landesberg et al, 2011). However, the role of these proteases in I/R-induced EDD dysfunction has not been evaluated. Similarly, while inhibition of MMP activity, neutrophil depletion, prevention of NETosis, and ROS scavenging have been shown to protect against vascular hyporeactivity in endotoxemia in vivo or in arterial tissue exposed to bacterial toxins (Buerke, Sibelius, Grandel, Buerke, Grimminger, Seeger, et al, 2002; Castro, Cena, Cho, Walsh & Schulz, 2012; Cena, Lalu, Rosenfelt & Schulz, 2008; Cena, Lalu, Cho, Chow, Bagdan, Daniel, et al, 2009; Gardiner & Andrews, 2012; Nin, El-Assar, Sanchez, Ferruelo, Sahchez-Derrer, Martinez-Caro, et al 2011; Wenceslau, McCarthy, Szasz, Goulopoulou & Webb, 2015), it is not clear whether these pathologic events are mechanistically linked as described for I/R.
Acknowledgments
This work was supported by grants the National Institutes of Health (HL095486, GM115553 and AA022108).
Abbreviations
- ACH
acetylcholine
- Ang II
angiotensin II
- AT1-R
Ang II type 1 receptor
- BH4
tetrahydrobiopterin
- EC
endothelial cell
- ECAM
endothelial cell adhesion molecule
- ECM
extracellular matrix
- EDD
endothelium-dependent, NO-mediated vasodilator
- eNOS
endothelial nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- H2O2
hydrogen peroxide
- I/R
ischemia/reperfusion
- JAMs
junctional adhesion molecules A, B, & C
- Knockout
-/-
- LECA
leukocyte/endothelial cell adhesive interactions
- LTB4
leukotriene B4
- MAb
monoclonal antibody
- MMP
matrix metalloproteinase
- MPO
myeloperoxidase
- NO
nitric oxide
- Nox
NAD(P)H oxidase
- O2-
superoxide anion
- ONOO-
peroxynitrite
- PAF
platelet-activating factor
- RGD
Arg-Gly-Asp peptide
- RNOS
reactive nitrogen oxide species
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TNF
tumor necrosis factor
Footnotes
Conflict of Interest Statement: The authors have no conflicts of interest to declare.
References
- Abumiya T, Lucero J, Heo JH, Tagaya M, Koziol JA, Copeland BR, del Zoppo GJ. Activated microvessels express vascular endothelial growth factor and integrin alpha(v)beta3 during focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:1038–1050. doi: 10.1097/00004647-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Albadawi H, Oklu R, Raacke Malley RE, O'Keefe RM, Uong TP, Cormier NR, Watkins MT. Effect of DNase I treatment and neutrophil depletion on acute limb ischemia-reperfusion injury in mice. J Vasc Surg. 2016;64:484–493. doi: 10.1016/j.jvs.2015.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler AE, Kistler EB, Schmid-Schonbein GW. Autodigestion: Proteolytic Degradation and Multiple Organ Failure in Shock. Shock. 2016;45:483–489. doi: 10.1097/SHK.0000000000000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez A, Cerda-Nicolas M, Naim Abu Nabah Y, Mata M, Issekutz AC, Panes J, Lobb RR, Sanz MJ. Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood. 2004;104:402–408. doi: 10.1182/blood-2003-08-2974. [DOI] [PubMed] [Google Scholar]
- Alvarez A, Sanz MJ. Reactive oxygen species mediate angiotensin II-induced leukocyte-endothelial cell interactions in vivo. J Leukoc Biol. 2001;70:199–206. [PubMed] [Google Scholar]
- Alvarez A, Piqueras L, Bello R, Canet A, Moreno L, Kubes P, Sanz MJ. Angiotensin II is involved in nitric oxide synthase and cyclo-oxygenase inhibition-induced leukocyte-endothelial cell interactions in vivo. Br J Pharmacol. 2001;132:677–684. doi: 10.1038/sj.bjp.0703867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asako H, Wolf RE, Granger DN, Korthuis RJ. Phalloidin prevents leukocyte emigration induced by proinflammatory stimuli in rat mesentery. Am J Physiol. 1992;263:H1637–1642. doi: 10.1152/ajpheart.1992.263.6.H1637. [DOI] [PubMed] [Google Scholar]
- Banda MA, Lefer DJ, Granger DN. Postischemic endothelium-dependent vascular reactivity is preserved in adhesion molecule-deficient mice. Am J Physiol. 1997;273:H2721–2725. doi: 10.1152/ajpheart.1997.273.6.H2721. [DOI] [PubMed] [Google Scholar]
- Baldus S, Heitzer T, Eiserich JP, Lau D, Mollnau H, Ortak M, Petri S, Goldmann B, Duchstein HJ, Berger J, Helmchen U, Freeman BA, Meinertz T, Munzel T. Myeloperoxidase enhances nitric oxide catabolism during myocardial ischemia and reperfusion. Free Radic Biol Med. 2004;37:902–911. doi: 10.1016/j.freeradbiomed.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Boros M. Microcirculatory dysfunction during intestinal ischemia-reperfusion. Acta Physiol Hung. 2003;90:263–279. doi: 10.1556/APhysiol.90.2003.4.1. [DOI] [PubMed] [Google Scholar]
- Boros M, Kaszaki J, Nagy S. Histamine release during intestinal ischemia-reperfusion: role of iron ions and hydrogen peroxide. Circ Shock. 1991;35:174–180. [PubMed] [Google Scholar]
- Bortolotto SK, Morrison WA, Han X, Messina A. Mast cells play a pivotal role in ischaemia reperfusion injury to skeletal muscles. Lab Invest. 2004;84:1103–1111. doi: 10.1038/labinvest.3700126. [DOI] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl- current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol. 2004;124:273–287. doi: 10.1085/jgp.200409040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM. EGFR kinase regulates volume-sensitive chloride current elicited by integrin stretch via PI-3K and NADPH oxidase in ventricular myocytes. J Gen Physiol. 2006;127:237–251. doi: 10.1085/jgp.200509366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerke M, Sibelius U, Grandel U, Buerke U, Grimminger F, Seeger W, Meyer J, Darius H. Staphylococcus aureus alpha toxin mediates polymorphonuclear leukocyte-induced vasocontraction and endothelial dysfunction. Shock. 2002;17:30–35. doi: 10.1097/00024382-200201000-00006. [DOI] [PubMed] [Google Scholar]
- Camp TM, Tyagi SC, Aru GM, Hayden MR, Mehta JL, Tyagi SC. Doxycycline ameliorates ischemic and border-zone remodeling and endothelial dysfunction after myocardial infarction in rats. J Heart Lung Transplant. 2004;23:729–736. doi: 10.1016/j.healun.2003.06.005. [DOI] [PubMed] [Google Scholar]
- Carden DL, Korthuis RJ. Protease inhibition attenuates microvascular dysfunction in postischemic skeletal muscle. Am J Physiol. 1996;271:H1947–1952. doi: 10.1152/ajpheart.1996.271.5.H1947. [DOI] [PubMed] [Google Scholar]
- Carrizzo A, Lenzi P, Procaccini C, Damato A, Biagioni F, Ambrosio M, Amodio G, Remondelli P, Del Giudice C, Izzo R, Malovini A, Formisano L, Gigantino V, Madonna M, Puca AA, Trimarco B, Matarese G, Fornai F, Vecchione C. Pentraxin 3 Induces Vascular Endothelial Dysfunction Through a P-selectin/Matrix Metalloproteinase-1 Pathway. Circulation. 2015;131:1495–1505. doi: 10.1161/CIRCULATIONAHA.114.014822. discussion 1505. [DOI] [PubMed] [Google Scholar]
- Castro MM, Cena J, Cho WJ, Walsh MP, Schulz R. Matrix metalloproteinase-2 proteolysis of calponin-1 contributes to vascular hypocontractility in endotoxemic rats. Arterioscler Thromb Vasc Biol. 2012;32:662–668. doi: 10.1161/ATVBAHA.111.242685. [DOI] [PubMed] [Google Scholar]
- Castro MM, Rizzi E, Ceron CS, Guimaraes DA, Rodrigues GJ, Bendhack LM, Gerlach RF, Tanus-Santos JE. Doxycycline ameliorates 2K-1C hypertension-induced vascular dysfunction in rats by attenuating oxidative stress and improving nitric oxide bioavailability. Nitric Oxide. 2012;26:162–168. doi: 10.1016/j.niox.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Cena J, Lalu MM, Rosenfelt C, Schulz R. Endothelial dependence of matrix metalloproteinase-mediated vascular hyporeactivity caused by lipopolysaccharide. Eur J Pharmacol. 2008;582:116–122. doi: 10.1016/j.ejphar.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Cena JJ, Lalu MM, Cho WJ, Chow AK, Bagdan ML, Daniel EE, Castro MM, Schulz R. Inhibition of matrix metalloproteinase activity in vivo protects against vascular hyporeactivity in endotoxemia. Am J Physiol Heart Circ Physiol. 2010;298:H45–51. doi: 10.1152/ajpheart.00273.2009. [DOI] [PubMed] [Google Scholar]
- Cena J, Lalu MM, Rosenfelt C, Schulz R. Endothelial dependence of matrix metalloproteinase-mediated vascular hyporeactivity caused by lipopolysaccharide. Eur J Pharmacol. 2008;582:116–122. doi: 10.1016/j.ejphar.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Chen AY, Ha JN, Delano FA, Schmid-Schonbein GW. Receptor cleavage and P-selectin-dependent reduction of leukocyte adhesion in the spontaneously hypertensive rat. J Leukoc Biol. 2012;92:183–194. doi: 10.1189/jlb.0112010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Zhao Q, Ni R, Tang F, Shan L, Cepinskas I, Cepinskas G, Wang W, Schiller PW, Peng T. Inhibition of calpain reduces oxidative stress and attenuates endothelial dysfunction in diabetes. Cardiovasc Diabetol. 2014;13:88. doi: 10.1186/1475-2840-13-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Jiang X, Pansuria M, Fang P, Mai J, Mallilankaraman K, Gandhirajan RK, Eguchi S, Scalia R, Madesh M, Yang X, Wang H. Hyperhomocysteinemia and hyperglycemia induce and potentiate endothelial dysfunction via mu-calpain activation. Diabetes. 2015;64:947–959. doi: 10.2337/db14-0784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes ME, Pagano PJ. Targeting reactive oxygen species in hypertension. Curr Opin Nephrol Hypertens. 2006;15:179–186. doi: 10.1097/01.mnh.0000214776.19233.68. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Costantino G, Serraino I, De Sarro A, Caputi AP. Effects of n-acetylcysteine in a rat model of ischemia and reperfusion injury. Cardiovasc Res. 2000;47:537–548. doi: 10.1016/s0008-6363(00)00018-3. [DOI] [PubMed] [Google Scholar]
- Dai H, Korthuis RJ. Mast cell proteases and inflammation. Drug Discov Today Dis Models. 2011;8:47–55. doi: 10.1016/j.ddmod.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Korthuis RJ. Mechanisms of arteriolar endothelium-dependent vasodilatory dysfunction in ischemia/reperfusion: role of proteases and mast cells 2017 [Google Scholar]
- Dai H, Korthuis RJ. Role of reactive oxygen species and chymase in postischemic arteriolar endothelial dysfunction 2017 [Google Scholar]
- Davis GE. Matricryptic sites control tissue injury responses in the cardiovascular system: relationships to pattern recognition receptor regulated events. J Mol Cell Cardiol. 2010;48:454–460.c. doi: 10.1016/j.yjmcc.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol. 2000;156:1489–1498. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano FA, Chen AY, Wu KI, Tran ED, Rodrigues SF, Schmid-Schonbein GW. The Autodigestion Hypothesis and Receptor Cleavage in Diabetes and Hypertension. Drug Discov Today Dis Models. 2011;8:37–46. doi: 10.1016/j.ddmod.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano FA, Schmid-Schonbein GW. Proteinase activity and receptor cleavage: mechanism for insulin resistance in the spontaneously hypertensive rat. Hypertension. 2008;52:415–423. doi: 10.1161/HYPERTENSIONAHA.107.104356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano FA, Zhang H, Tran EE, Zhang C, Schmid-Schonbein GW. A New Hypothesis for Insulin Resistance in Hypertension Due to Receptor Cleavage. Expert Rev Endocrinol Metab. 2010;5:149–158. doi: 10.1586/eem.09.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Italia LJ, Husain A. Dissecting the role of chymase in angiotensin II formation and heart and blood vessel diseases. Curr Opin Cardiol. 2002;17:374–379. doi: 10.1097/00001573-200207000-00009. [DOI] [PubMed] [Google Scholar]
- de With MC, Haug SJ, Brigitte van der Heijden EP, Segal SS. Ischemia-reperfusion impairs ascending vasodilation in feed arteries of hamster skeletal muscle. Microcirculation. 2005;12:551–561. doi: 10.1080/10739680500253451. [DOI] [PubMed] [Google Scholar]
- Dumitrescu C, Biondi R, Xia Y, Cardounel AJ, Druhan LJ, Ambrosio G, Zweier JL. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci U S A. 2007;104:15081–15086. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod JW, Duranski MR, Langston W, Greer JJ, Tao L, Dugas TR, Kevil CG, Champion HC, Lefer DJ. eNOS gene therapy exacerbates hepatic ischemia-reperfusion injury in diabetes: a role for eNOS uncoupling. Circ Res. 2006;99:78–85. doi: 10.1161/01.RES.0000231306.03510.77. [DOI] [PubMed] [Google Scholar]
- Eppihimer MJ, Russell J, Anderson DC, Epstein CJ, Laroux S, Granger DN. Modulation of P-selectin expression in the postischemic intestinal microvasculature. Am J Physiol. 1997;273:G1326–1332. doi: 10.1152/ajpgi.1997.273.6.G1326. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- Finsterbusch M, Voisin MB, Beyrau M, Williams TJ, Nourshargh S. Neutrophils recruited by chemoattractants in vivo induce microvascular protein leakage through secretion of TNF. J Exp Med. 2014;211:1307–1314. doi: 10.1084/jem.20132413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U. Janus-faced role of endothelial NO synthase in vascular disease: uncoupling of oxygen reduction from NO synthesis and its pharmacological reversal. Biol Chem. 2006;387:1521–1533. doi: 10.1515/BC.2006.190. [DOI] [PubMed] [Google Scholar]
- Fu X, Kao JL, Bergt C, Kassim SY, Huq NP, d'Avignon A, Parks WC, Mecham RP, Heinecke JW. Oxidative cross-linking of tryptophan to glycine restrains matrix metalloproteinase activity: specific structural motifs control protein oxidation. J Biol Chem. 2004;279:6209–6212. doi: 10.1074/jbc.C300506200. [DOI] [PubMed] [Google Scholar]
- Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- Gaboury JP, Johnston B, Niu XF, Kubes P. Mechanisms underlying acute mast cell-induced leukocyte rolling and adhesion in vivo. J Immunol. 1995;154:804–813. [PubMed] [Google Scholar]
- Gao H, Korthuis RJ, Benoit JN. Effects of reactive oxygen metabolites on norepinephrine-induced vasoconstriction. Free Radic Biol Med. 1994;16:839–843. doi: 10.1016/0891-5849(94)90201-1. [DOI] [PubMed] [Google Scholar]
- Gao H, Korthuis RJ, Benoit JN. Effects of hypoxia/reoxygenation on aortic vasoconstrictor responsiveness. Free Radic Biol Med. 1996;21:591–600. doi: 10.1016/0891-5849(96)00157-8. [DOI] [PubMed] [Google Scholar]
- Gao H, Korthuis RJ, Benoit JN. Hypoxia/reoxygenation selectively impairs alpha 1b-adrenoceptor function in small mesenteric arteries. Am J Physiol. 1996;271:G820–823. doi: 10.1152/ajpgi.1996.271.5.G820. [DOI] [PubMed] [Google Scholar]
- Gardiner EE, Andrews RK. Neutrophil extracellular traps (NETs) and infection-related vascular dysfunction. Blood Rev. 2012;26:255–259. doi: 10.1016/j.blre.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Fujimura M, Morita-Fujimura Y, Copin JC, Kawase M, Massengale J, Chan PH. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999;19:1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- Gaskin FS, Dai H, Korthuis RJ. Postischemic endothelium-dependent, but not vasoconstrictor agonist-induced, arteriolar reactivity is preserved in neutropenic and P-selectin-deficient animals 2017 [Google Scholar]
- Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, Ma YQ, Zhao JH, Li YM. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol. 2015;308:H500–509. doi: 10.1152/ajpheart.00381.2014. [DOI] [PubMed] [Google Scholar]
- Gloe T, Sohn HY, Meininger GA, Pohl U. Shear stress-induced release of basic fibroblast growth factor from endothelial cells is mediated by matrix interaction via integrin alpha(v)beta3. J Biol Chem. 2002;277:23453–23458. doi: 10.1074/jbc.M203889200. [DOI] [PubMed] [Google Scholar]
- Gonzalez Bosc LV, Osmond JM, Giermakowska WK, Pace CE, Riggs JL, Jackson-Weaver O, Kanagy NL. NFAT regulation of cystathionine gamma-lyase expression in endothelial cells is impaired in rats exposed to intermittent hypoxia. Am J Physiol Heart Circ Physiol. 2017;312:H791–H799. doi: 10.1152/ajpheart.00952.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdin MJ, Bree B, De Kock M. The impact of ischaemia-reperfusion on the blood vessel. Eur J Anaesthesiol. 2009;26:537–547. doi: 10.1097/EJA.0b013e328324b7c2. [DOI] [PubMed] [Google Scholar]
- Granger DN, Korthuis RJ. Physiologic mechanisms of postischemic tissue injury. Annu Rev Physiol. 1995;57:311–332. doi: 10.1146/annurev.ph.57.030195.001523. [DOI] [PubMed] [Google Scholar]
- Gunther S, Gimbrone MA, Jr, Alexander RW. Identification and characterization of the high affinity vascular angiotensin II receptor in rat mesenteric artery. Circ Res. 1980;47:278–286. doi: 10.1161/01.res.47.2.278. [DOI] [PubMed] [Google Scholar]
- Hansen PR. Inflammatory alterations in the myocardial microcirculation. J Mol Cell Cardiol. 1998;30:2555–2559. doi: 10.1006/jmcc.1998.0827. [DOI] [PubMed] [Google Scholar]
- Hayward R, Campbell B, Shin YK, Scalia R, Lefer AM. Recombinant soluble P-selectin glycoprotein ligand-1 protects against myocardial ischemic reperfusion injury in cats. Cardiovasc Res. 1999;41:65–76. doi: 10.1016/s0008-6363(98)00266-1. [DOI] [PubMed] [Google Scholar]
- Hayward R, Lefer AM. Time course of endothelial-neutrophil interaction in splanchnic artery ischemia-reperfusion. Am J Physiol. 1998;275:H2080–2086. doi: 10.1152/ajpheart.1998.275.6.H2080. [DOI] [PubMed] [Google Scholar]
- Hayward R, Lefer AM. Acute mesenteric ischemia and reperfusion: protective effects of recombinant soluble P-selectin glycoprotein ligand-1. Shock. 1999;12:201–207. [PubMed] [Google Scholar]
- Hein TW, Platts SH, Waitkus-Edwards KR, Kuo L, Mousa SA, Meininger GA. Integrin-binding peptides containing RGD produce coronary arteriolar dilation via cyclooxygenase activation. Am J Physiol Heart Circ Physiol. 2001;281:H2378–2384. doi: 10.1152/ajpheart.2001.281.6.H2378. [DOI] [PubMed] [Google Scholar]
- Hein TW, Rosa RH, Jr, Ren Y, Xu W, Kuo L. VEGF Receptor-2-Linked PI3K/Calpain/SIRT1 Activation Mediates Retinal Arteriolar Dilations to VEGF and Shear Stress. Invest Ophthalmol Vis Sci. 2015;56:5381–5389. doi: 10.1167/iovs15-16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein TW, Zhang C, Wang W, Chang CI, Thengchaisri N, Kuo L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. FASEB J. 2003;17:2328–2330. doi: 10.1096/fj.03-0115fje. [DOI] [PubMed] [Google Scholar]
- Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- Hill MA, Meininger GA. Arteriolar vascular smooth muscle cells: mechanotransducers in a complex environment. Int J Biochem Cell Biol. 2012;44:1505–1510. doi: 10.1016/j.biocel.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida K, Takai S, Murano M, Nishikawa T, Inoue T, Murano N, Inoue N, Jin D, Umegaki E, Higuchi K, Miyazaki M. Role of chymase-dependent matrix metalloproteinase-9 activation in mice with dextran sodium sulfate-induced colitis. J Pharmacol Exp Ther. 2008;324:422–426. doi: 10.1124/jpet.107.131946. [DOI] [PubMed] [Google Scholar]
- Jansen MP, Emal D, Teske GJ, Dessing MC, Florquin S, Roelofs JJ. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. 2017;91:352–364. doi: 10.1016/j.kint.2016.08.006. [DOI] [PubMed] [Google Scholar]
- Jara PI, Boric MP, Saez JC. Leukocytes express connexin 43 after activation with lipopolysaccharide and appear to form gap junctions with endothelial cells after ischemia-reperfusion. Proc Natl Acad Sci U S A. 1995;92:7011–7015. doi: 10.1073/pnas.92.15.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D, Takai S, Okamoto Y, Muramatsu M, Miyazaki M. Chymase-derived angiotensin II and arrhythmias after myocardial infarction. Nihon Yakurigaku Zasshi. 2004;124:77–82. doi: 10.1254/fpj.124.77. [DOI] [PubMed] [Google Scholar]
- Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/Reperfusion. Compr Physiol. 2016;7:113–170. doi: 10.1002/cphy.c160006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanagy NL, Szabo C, Papapetropoulos A. Vascular biology of hydrogen sulfide. Am J Physiol Cell Physiol. 2017;312:C537–C549. doi: 10.1152/ajpcell.00329.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwar S, Hickey MJ, Kubes P. Postischemic inflammation: a role for mast cells in intestine but not in skeletal muscle. Am J Physiol. 1998;275:G212–218. doi: 10.1152/ajpgi.1998.275.2.G212. [DOI] [PubMed] [Google Scholar]
- Kanwar S, Kubes P. Ischemia/reperfusion-induced granulocyte influx is a multistep process mediated by mast cells. Microcirculation. 1994;1:175–182. doi: 10.3109/10739689409148272. [DOI] [PubMed] [Google Scholar]
- Kanwar S, Kubes P. Mast cells contribute to ischemia-reperfusion-induced granulocyte infiltration and intestinal dysfunction. Am J Physiol. 1994;267:G316–321. doi: 10.1152/ajpgi.1994.267.2.G316. [DOI] [PubMed] [Google Scholar]
- Keller MW. Arteriolar constriction in skeletal muscle during vascular stunning: role of mast cells. Am J Physiol. 1997;272:H2154–2163. doi: 10.1152/ajpheart.1997.272.5.H2154. [DOI] [PubMed] [Google Scholar]
- Kim MH, Carter PR, Harris NR. P-selectin-mediated adhesion impairs endothelium-dependent arteriolar dilation in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2007;292:H632–638. doi: 10.1152/ajpheart.00780.2006. [DOI] [PubMed] [Google Scholar]
- Kishore R, Benedict C, Cheng Z. mu-Calpain as a Novel Target for Impairment of Nitric Oxide-Mediated Vascular Relaxation in Diabetes: A Mini Review. J Mol Genet Med. 2015;9 doi: 10.4172/1747-0862.1000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korthuis RJ, Carden DL, Kvietys PR, Shepro D, Fuseler J. Phalloidin attenuates postischemic neutrophil infiltration and increased microvascular permeability. J Appl Physiol (1985) 1991;71:1261–1269. doi: 10.1152/jappl.1991.71.4.1261. [DOI] [PubMed] [Google Scholar]
- Kuebler WM, Kuhnle GE, Groh J, Goetz AE. Leukocyte kinetics in pulmonary microcirculation: intravital fluorescence microscopic study. J Appl Physiol (1985) 1994;76:65–71. doi: 10.1152/jappl.1994.76.1.65. [DOI] [PubMed] [Google Scholar]
- Kurose I, Anderson DC, Miyasaka M, Tamatani T, Paulson JC, Todd RF, Rusche JR, Granger DN. Molecular determinants of reperfusion-induced leukocyte adhesion and vascular protein leakage. Circ Res. 1994;74:336–343. doi: 10.1161/01.res.74.2.336. [DOI] [PubMed] [Google Scholar]
- Kurose I, Wolf R, Grisham MB, Granger DN. Modulation of ischemia/reperfusion-induced microvascular dysfunction by nitric oxide. Circ Res. 1994;74:376–382. doi: 10.1161/01.res.74.3.376. [DOI] [PubMed] [Google Scholar]
- Kuyvenhoven JP, Molenaar IQ, Verspaget HW, Veldman MG, Palareti G, Legnani C, Moolenburgh SE, Terpstra OT, Lamers CB, van Hoek B, Porte RJ. Plasma MMP-2 and MMP-9 and their inhibitors TIMP-1 and TIMP-2 during human orthotopic liver transplantation. The effect of aprotinin and the relation to ischemia/reperfusion injury. Thromb Haemost. 2004;91:506–513. doi: 10.1160/TH03-05-0272. [DOI] [PubMed] [Google Scholar]
- Lanza GA, Maseri A. Coronary Artery Spasm. Curr Treat Options Cardiovasc Med. 2000;2:83–90. doi: 10.1007/s11936-000-0031-0. [DOI] [PubMed] [Google Scholar]
- Lash JM, Bohlen HG. Structural and functional origins of suppressed acetylcholine vasodilation in diabetic rat intestinal arterioles. Circ Res. 1991;69:1259–1268. doi: 10.1161/01.res.69.5.1259. [DOI] [PubMed] [Google Scholar]
- Lefer AM, Aoki N. Leukocyte-dependent and leukocyte-independent mechanisms of impairment of endothelium-mediated vasodilation. Blood Vessels. 1990;27:162–168. doi: 10.1159/000158807. [DOI] [PubMed] [Google Scholar]
- Lefer AM, Ma XL. Endothelial dysfunction in the splanchnic circulation following ischemia and reperfusion. J Cardiovas Pharmacol. 1994;17(Suppl 3):S186–S190. [Google Scholar]
- Lefer AM, Tsao PS, Lefer DJ, Ma XL. Role of endothelial dysfunction in the pathogenesis of reperfusion injury after myocardial ischemia. FASEB J. 1991;5:2029–2034. doi: 10.1096/fasebj.5.7.2010056. [DOI] [PubMed] [Google Scholar]
- Lehr HA, Frei B, Arfors KE. Vitamin C prevents cigarette smoke-induced leukocyte aggregation and adhesion to endothelium in vivo. Proc Natl Acad Sci U S A. 1994;91:7688–7692. doi: 10.1073/pnas.91.16.7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Duling BR. Vulnerability of conducted vasomotor response to ischemia. Am J Physiol. 1994;267:H2363–2370. doi: 10.1152/ajpheart.1994.267.6.H2363. [DOI] [PubMed] [Google Scholar]
- Liu FC, Chuang YH, Tsai YF, Yu HP. Role of neutrophil extracellular traps following injury. Shock. 2014;41:491–498. doi: 10.1097/SHK.0000000000000146. [DOI] [PubMed] [Google Scholar]
- Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda) 2006;21:269–280. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- Ma XL, Lefer DJ, Lefer AM, Rothlein R. Coronary endothelial and cardiac protective effects of a monoclonal antibody to intercellular adhesion molecule-1 in myocardial ischemia and reperfusion. Circulation. 1992;86:937–946. doi: 10.1161/01.cir.86.3.937. [DOI] [PubMed] [Google Scholar]
- Ma XL, Tsao PS, Lefer AM. Antibody to CD-18 exerts endothelial and cardiac protective effects in myocardial ischemia and reperfusion. J Clin Invest. 1991;88:1237–1243. doi: 10.1172/JCI115427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XL, Weyrich AS, Lefer DJ, Buerke M, Albertine KH, Kishimoto TK, Lefer AM. Monoclonal antibody to L-selectin attenuates neutrophil accumulation and protects ischemic reperfused cat myocardium. Circulation. 1993;88:649–658. doi: 10.1161/01.cir.88.2.649. [DOI] [PubMed] [Google Scholar]
- Martinez-Lemus LA, Sun Z, Trache A, Trzciakowski JP, Meininger GA. Integrins and regulation of the microcirculation: from arterioles to molecular studies using atomic force microscopy. Microcirculation. 2005;12:99–112. doi: 10.1080/10739680590896054. [DOI] [PubMed] [Google Scholar]
- Martinez-Lemus LA, Wu X, Wilson E, Hill MA, Davis GE, Davis MJ, Meininger GA. Integrins as unique receptors for vascular control. J Vasc Res. 2003;40:211–233. doi: 10.1159/000071886. [DOI] [PubMed] [Google Scholar]
- Mazor R, Schmid-Schonbein GW. Proteolytic receptor cleavage in the pathogenesis of blood rheology and co-morbidities in metabolic syndrome. Early forms of autodigestion. Biorheology. 2015;52:337–352. doi: 10.3233/BIR-15045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- Mikhed Y, Daiber A, Steven S. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int J Mol Sci. 2015;16:15918–15953. doi: 10.3390/ijms160715918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller HR, Pemberton AD. Tissue-specific expression of mast cell granule serine proteinases and their role in inflammation in the lung and gut. Immunology. 2002;105:375–390. doi: 10.1046/j.1365-2567.2002.01375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Takai S. Local angiotensin II-generating system in vascular tissues: the roles of chymase. Hypertens Res. 2001;24:189–193. doi: 10.1291/hypres.24.189. [DOI] [PubMed] [Google Scholar]
- Moazzam F, DeLano FA, Zweifach BW, Schmid-Schonbein GW. The leukocyte response to fluid stress. Proc Natl Acad Sci U S A. 1997;94:5338–5343. doi: 10.1073/pnas.94.10.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2006;26:2439–2444. doi: 10.1161/01.ATV.0000243924.00970.cb. [DOI] [PubMed] [Google Scholar]
- Mogford JE, Davis GE, Platts SH, Meininger GA. Vascular smooth muscle alpha v beta 3 integrin mediates arteriolar vasodilation in response to RGD peptides. Circ Res. 1996;79:821–826. doi: 10.1161/01.res.79.4.821. [DOI] [PubMed] [Google Scholar]
- Munzel T, Daiber A, Ullrich V, Mulsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol. 2005;25:1551–1557. doi: 10.1161/01.ATV.0000168896.64927.bb. [DOI] [PubMed] [Google Scholar]
- Nabah YN, Mateo T, Cerda-Nicolas M, Alvarez A, Martinez M, Issekutz AC, Sanz MJ. L-NAME induces direct arteriolar leukocyte adhesion, which is mainly mediated by angiotensin-II. Microcirculation. 2005;12:443–453. doi: 10.1080/10739680590960962. [DOI] [PubMed] [Google Scholar]
- Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, Kraft F, Lei Y, Fukasawa Y, Moeckel GW, Angelotti ML, Liapis H, Anders HJ. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J Am Soc Nephrol. 2017;28:1753–1768. doi: 10.1681/ASN.2016080925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangle MR, Cotter MA, Cameron NE. The calpain inhibitor, A-705253, corrects penile nitrergic nerve dysfunction in diabetic mice. Eur J Pharmacol. 2006;538:148–153. doi: 10.1016/j.ejphar.2006.03.068. [DOI] [PubMed] [Google Scholar]
- Nilsson G, Costa JJ, Metcalfe DD. Mast cells and basophils. In: Gallin John I, Snyderman Ralph., editors. Inflammation: Basic Principles and Clinical Correlates. 3rd. 1999. [Google Scholar]
- Nin N, El-Assar M, Sanchez C, Ferruelo A, Sanchez-Ferrer A, Martinez-Caro L, Rojas Y, Paula M, Hurtado J, Esteban A, Lorente JA. Vascular dysfunction in sepsis: effects of the peroxynitrite decomposition catalyst MnTMPyP. Shock. 2011;36:156–161. doi: 10.1097/SHK.0b013e31821e50de. [DOI] [PubMed] [Google Scholar]
- Nunohiro T, Ashizawa N, Graf K, Hsueh WA, Yano K. Angiotensin II promotes integrin-mediated collagen gel contraction by adult rat cardiac fibroblasts. Jpn Heart J. 1999;40:461–469. doi: 10.1536/jhj.40.461. [DOI] [PubMed] [Google Scholar]
- Oliver MG, Specian RD, Perry MA, Granger DN. Morphologic assessment of leukocyte-endothelial cell interactions in mesenteric venules subjected to ischemia and reperfusion. Inflammation. 1991;15:331–346. doi: 10.1007/BF00917350. [DOI] [PubMed] [Google Scholar]
- Osmond JM, Kanagy NL. Modulation of hydrogen sulfide by vascular hypoxia. Hypoxia (Auckl) 2014;2:117–126. doi: 10.2147/HP.S51589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano PJ, Gutterman DD. The adventitia: the outs and ins of vascular disease. Cardiovasc Res. 2007;75:636–639. doi: 10.1016/j.cardiores.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallet N. Neutrophil Extracellular Traps Orchestrate Necroinflammation. J Am Soc Nephrol. 2017;28:1670–1672. doi: 10.1681/ASN.2017010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MA, Granger DN. Role of CD11/CD18 in shear rate-dependent leukocyte-endothelial cell interactions in cat mesenteric venules. J Clin Invest. 1991;87:1798–1804. doi: 10.1172/JCI115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petnehazy T, Cooper D, Stokes KY, Russell J, Wood KC, Granger DN. Angiotensin II type 1 receptors and the intestinal microvascular dysfunction induced by ischemia and reperfusion. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1203–1210. doi: 10.1152/ajpgi.00578.2005. [DOI] [PubMed] [Google Scholar]
- Petrault O, Ouk T, Gautier S, Laprais M, Gele P, Bastide M, Bordet R. Pharmacological neutropenia prevents endothelial dysfunction but not smooth muscle functions impairment induced by middle cerebral artery occlusion. Br J Pharmacol. 2005;144:1051–1058. doi: 10.1038/sj.bjp.0706124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piqueras L, Kubes P, Alvarez A, O'Connor E, Issekutz AC, Esplugues JV, Sanz MJ. Angiotensin II induces leukocyte-endothelial cell interactions in vivo via AT(1) and AT(2) receptor-mediated P-selectin upregulation. Circulation. 2000;102:2118–2123. doi: 10.1161/01.cir.102.17.2118. [DOI] [PubMed] [Google Scholar]
- Potz BA, Sabe AA, Abid MR, Sellke FW. Calpains and Coronary Vascular Disease. Circ J. 2016;80:4–10. doi: 10.1253/circj.CJ-15-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randriamboavonjy V, Kyselova A, Elgheznawy A, Zukunft S, Wittig I, Fleming I. Calpain 1 cleaves and inactivates prostacyclin synthase in mesenteric arteries from diabetic mice. Basic Res Cardiol. 2017;112:10. doi: 10.1007/s00395-016-0596-8. [DOI] [PubMed] [Google Scholar]
- Reed RK, Berg A, Gjerde EA, Rubin K. Control of interstitial fluid pressure: role of beta1-integrins. Semin Nephrol. 2001;21:222–230. doi: 10.1053/snep.2001.21646. [DOI] [PubMed] [Google Scholar]
- Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- Rey FE, Li XC, Carretero OA, Garvin JL, Pagano PJ. Perivascular superoxide anion contributes to impairment of endothelium-dependent relaxation: role of gp91(phox) Circulation. 2002;106:2497–2502. doi: 10.1161/01.cir.0000038108.71560.70. [DOI] [PubMed] [Google Scholar]
- Riaz AA, Wang Y, Schramm R, Sato T, Menger MD, Jeppsson B, Thorlacius H. Role of angiotensin II in ischemia/reperfusion-induced leukocyte-endothelium interactions in the colon. FASEB J. 2004;18:881–883. doi: 10.1096/fj.03-0502fje. [DOI] [PubMed] [Google Scholar]
- Richard V, Hurel-Merle S, Scalbert E, Ferry G, Lallemand F, Bessou JP, Thuillez C. Functional evidence for a role of vascular chymase in the production of angiotensin II in isolated human arteries. Circulation. 2001;104:750–752. doi: 10.1161/hc3201.094971. [DOI] [PubMed] [Google Scholar]
- Rosario HS, Waldo SW, Becker SA, Schmid-Schonbein GW. Pancreatic trypsin increases matrix metalloproteinase-9 accumulation and activation during acute intestinal ischemia-reperfusion in the rat. Am J Pathol. 2004;164:1707–1716. doi: 10.1016/S0002-9440(10)63729-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA. Extracellular matrix inflammation in vascular cognitive impairment and dementia. Clin Sci (Lond) 2017;131:425–437. doi: 10.1042/CS20160604. [DOI] [PubMed] [Google Scholar]
- Ruh J, Schmidt E, Vogel F. Effect of N-acetylcysteine on microcirculation of mucosa in rat ileum in a model of intestinal inflammation. Dig Dis Sci. 2003;48:882–889. doi: 10.1023/a:1023087226504. [DOI] [PubMed] [Google Scholar]
- Sabe AA, Potz BA, Elmadhun NY, Liu Y, Feng J, Abid MR, Abbott JD, Senger DR, Sellke FW. Calpain inhibition improves collateral-dependent perfusion in a hypercholesterolemic swine model of chronic myocardial ischemia. J Thorac Cardiovasc Surg. 2016;151:245–252. doi: 10.1016/j.jtcvs.2015.08.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Shirai R, Hontani M, Shinooka R, Hasegawa A, Kichise T, Yamashita T, Yoshizawa H, Watanabe R, Matsuyama TA, Ishibashi-Ueda H, Koba S, Kobayashi Y, Hirano T, Watanabe T. Potent Vasoconstrictor Kisspeptin-10 Induces Atherosclerotic Plaque Progression and Instability: Reversal by its Receptor GPR54 Antagonist. J Am Heart Assoc. 2017;6 doi: 10.1161/JAHA.117.005790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalia R, Gong Y, Berzins B, Freund B, Feather D, Landesberg G, Mishra G. A novel role for calpain in the endothelial dysfunction induced by activation of angiotensin II type 1 receptor signaling. Circ Res. 2011;108:1102–1111. doi: 10.1161/CIRCRESAHA.110.229393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Schonbein GW. The autodigestion hypothesis: Proteolytic receptor cleavage in rheological and cardiovascular cell dysfunction1. Biorheology. 2016;53:179–191. doi: 10.3233/BIR-17131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt VJ, Wolfle SE, Boettcher M, de Wit C. Gap junctions synchronize vascular tone within the microcirculation. Pharmacol Rep. 2008;60:68–74. [PubMed] [Google Scholar]
- Segal SS, Duling BR. Flow control among microvessels coordinated by intercellular conduction. Science. 1986;234:868–870. doi: 10.1126/science.3775368. [DOI] [PubMed] [Google Scholar]
- Settergren M, Bohm F, Malmstrom RE, Channon KM, Pernow J. L-arginine and tetrahydrobiopterin protects against ischemia/reperfusion-induced endothelial dysfunction in patients with type 2 diabetes mellitus and coronary artery disease. Atherosclerosis. 2009;204:73–78. doi: 10.1016/j.atherosclerosis.2008.08.034. [DOI] [PubMed] [Google Scholar]
- Shigematsu S, Ishida S, Gute DC, Korthuis RJ. Bradykinin prevents postischemic leukocyte adhesion and emigration and attenuates microvascular barrier disruption. Am J Physiol. 1999;277:H161–171. doi: 10.1152/ajpheart.1999.277.1.H161. [DOI] [PubMed] [Google Scholar]
- Shimizu Y, Irani AM, Brown EJ, Ashman LK, Schwartz LB. Human mast cells derived from fetal liver cells cultured with stem cell factor express a functional CD51/CD61 (alpha v beta 3) integrin. Blood. 1995;86:930–939. [PubMed] [Google Scholar]
- Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis. 2017;8:e2812. doi: 10.1038/cddis.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalker TJ, Gong Y, Scalia R. The calcium-dependent protease calpain causes endothelial dysfunction in type 2 diabetes. Diabetes. 2005;54:1132–1140. doi: 10.2337/diabetes.54.4.1132. [DOI] [PubMed] [Google Scholar]
- Stalker TJ, Skvarka CB, Scalia R. A novel role for calpains in the endothelial dysfunction of hyperglycemia. FASEB J. 2003;17:1511–1513. doi: 10.1096/fj.02-1213fje. [DOI] [PubMed] [Google Scholar]
- Steiner DR, Gonzalez NC, Wood JG. Mast cells mediate the microvascular inflammatory response to systemic hypoxia. J Appl Physiol (1985) 2003;94:325–334. doi: 10.1152/japplphysiol.00637.2002. [DOI] [PubMed] [Google Scholar]
- Stokes KY, Calahan L, Hamric CM, Russell JM, Granger DN. CD40/CD40L contributes to hypercholesterolemia-induced microvascular inflammation. Am J Physiol Heart Circ Physiol. 2009;296:H689–697. doi: 10.1152/ajpheart.00962.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Palen DI, Lucchesi PA, Matrougui K. Mice lacking the gene encoding for MMP-9 and resistance artery reactivity. Biochem Biophys Res Commun. 2006;349:1177–1181. doi: 10.1016/j.bbrc.2006.08.189. [DOI] [PubMed] [Google Scholar]
- Sumagin R, Sarelius IH. TNF-alpha activation of arterioles and venules alters distribution and levels of ICAM-1 and affects leukocyte-endothelial cell interactions. Am J Physiol Heart Circ Physiol. 2006;291:H2116–2125. doi: 10.1152/ajpheart.00248.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Caughey GH, Gao XP, Rubinstein I. Mast cell chymase-like protease(s) modulates Escherichia coli lipopolysaccharide-induced vasomotor dysfunction in skeletal muscle in vivo. J Pharmacol Exp Ther. 1998;284:1156–1164. [PubMed] [Google Scholar]
- Tallini YN, Brekke JF, Shui B, Doran R, Hwang SM, Nakai J, Salama G, Segal SS, Kotlikoff MI. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC GCaMP2 transgenic mice. Circ Res. 2007;101:1300–1309. doi: 10.1161/CIRCRESAHA.107.149484. [DOI] [PubMed] [Google Scholar]
- Tian R, Ding Y, Peng YY, Lu N. Myeloperoxidase amplified high glucose-induced endothelial dysfunction in vasculature: Role of NADPH oxidase and hypochlorous acid. Biochem Biophys Res Commun. 2017;484:572–578. doi: 10.1016/j.bbrc.2017.01.132. [DOI] [PubMed] [Google Scholar]
- Touyz RM. Reactive oxygen species and angiotensin II signaling in vascular cells -- implications in cardiovascular disease. Braz J Med Biol Res. 2004;37:1263–1273. doi: 10.1590/s0100-879x2004000800018. [DOI] [PubMed] [Google Scholar]
- Tran ED, DeLano FA, Schmid-Schonbein GW. Enhanced matrix metalloproteinase activity in the spontaneously hypertensive rat: VEGFR-2 cleavage, endothelial apoptosis, and capillary rarefaction. J Vasc Res. 2010;47:423–431. doi: 10.1159/000281582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran ED, Yang M, Chen A, Delano FA, Murfee WL, Schmid-Schonbein GW. Matrix metalloproteinase activity causes VEGFR-2 cleavage and microvascular rarefaction in rat mesentery. Microcirculation. 2011;18:228–237. doi: 10.1111/j.1549-8719.2011.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao PS, Aoki N, Lefer DJ, Johnson G, 3rd, Lefer AM. Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation. 1990;82:1402–1412. doi: 10.1161/01.cir.82.4.1402. [DOI] [PubMed] [Google Scholar]
- Tsao PS, Ma XL, Lefer AM. Activated neutrophils aggravate endothelial dysfunction after reperfusion of the ischemic feline myocardium. Am Heart J. 1992;123:1464–1471. doi: 10.1016/0002-8703(92)90796-x. [DOI] [PubMed] [Google Scholar]
- Voisin MB, Nourshargh S. Neutrophil transmigration: emergence of an adhesive cascade within venular walls. J Innate Immun. 2013;5:336–347. doi: 10.1159/000346659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Peng Q, Zhang J, Liu L. Na+/H+ exchanger is required for hyperglycaemia-induced endothelial dysfunction via calcium-dependent calpain. Cardiovasc Res. 2008;80:255–262. doi: 10.1093/cvr/cvn179. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rosen H, Madtes DK, Shao B, Martin TR, Heinecke JW, Fu X. Myeloperoxidase inactivates TIMP-1 by oxidizing its N-terminal cysteine residue: an oxidative mechanism for regulating proteolysis during inflammation. J Biol Chem. 2007;282:31826–31834. doi: 10.1074/jbc.M704894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welbourn CR, Goldman G, Paterson IS, Valeri CR, Shepro D, Hechtman HB. Neutrophil elastase and oxygen radicals: synergism in lung injury after hindlimb ischemia. Am J Physiol. 1991;260:H1852–1856. doi: 10.1152/ajpheart.1991.260.6.H1852. [DOI] [PubMed] [Google Scholar]
- Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol. 2015;308:H768–777. doi: 10.1152/ajpheart.00779.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich AS, Ma XY, Lefer DJ, Albertine KH, Lefer AM. In vivo neutralization of P-selectin protects feline heart and endothelium in myocardial ischemia and reperfusion injury. J Clin Invest. 1993;91:2620–2629. doi: 10.1172/JCI116501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antiox Redox Signal. 2011;15:1517–1530. doi: 10.1089/ars.2010.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin MS, Gupte SA, Oeckler RA. Superoxide in the vascular system. J Vasc Res. 2002;39:191–207. doi: 10.1159/000063685. [DOI] [PubMed] [Google Scholar]
- Woodman OL, Hart JL, Sobey CG. Prevention of ischaemia-induced coronary vascular dysfunction. Int J Cardiol. 1997;62 Suppl 2:S91–99. doi: 10.1016/s0167-5273(97)00246-5. [DOI] [PubMed] [Google Scholar]
- Wu X, Mogford JE, Platts SH, Davis GE, Meininger GA, Davis MJ. Modulation of calcium current in arteriolar smooth muscle by alphav beta3 and alpha5 beta1 integrin ligands. J Cell Biol. 1998;143:241–252. doi: 10.1083/jcb.143.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Nishio K, Sato N, Tsumura H, Ichihara A, Kudo H, Aoki T, Naoki K, Suzuki K, Miyata A, Suzuki Y, Morooka S. Leukocyte kinetics in the pulmonary microcirculation: observations using real-time confocal luminescence microscopy coupled with high-speed video analysis. Lab Invest. 1997;76:809–822. [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 2015;1623:30–38. doi: 10.1016/j.brainres.2015.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Korthuis RJ, Benoit JN. Hypoxia-reoxygenation impairs endothelium-dependent relaxation in isolated rat aorta. Am J Physiol. 1996;270:R1126–1131. doi: 10.1152/ajpregu.1996.270.5.R1126. [DOI] [PubMed] [Google Scholar]
- Yusof M, Kamada K, Gaskin FS, Korthuis RJ. Angiotensin II mediates postischemic leukocyte-endothelial interactions: role of calcitonin gene-related peptide. Am J Physiol Heart Circ Physiol. 2007;292:H3032–3037. doi: 10.1152/ajpheart.01210.2006. [DOI] [PubMed] [Google Scholar]
- Zakaria el R, Garrison RN, Kawabe T, Harris PD. Role of neutrophils on shock/resuscitation-mediated intestinal arteriolar derangements. Shock. 2004;21:248–253. doi: 10.1097/01.shk.0000111824.07309.19. [DOI] [PubMed] [Google Scholar]
- Zamboni WA, Stephenson LL, Roth AC, Suchy H, Russell RC. Ischemia-reperfusion injury in skeletal muscle: CD 18-dependent neutrophil-endothelial adhesion and arteriolar vasoconstriction. Plast Reconstr Surg. 1997;99:2002–2007. doi: 10.1097/00006534-199706000-00028. discussion 2008-2009. [DOI] [PubMed] [Google Scholar]
- Zimmerman BJ, Granger DN. Reperfusion-induced leukocyte infiltration: role of elastase. Am J Physiol. 1990;259:H390–394. doi: 10.1152/ajpheart.1990.259.2.H390. [DOI] [PubMed] [Google Scholar]