

Abstract
Rationale:
Hereditary spherocytosis (HS) is an inherited disorder characterized by the presence of spherical-shaped red blood cells (RBCs) on the peripheral blood (PB) smear. To date, a number of mutations in 5 genes have been identified and the mutations in SPTB gene account for about 20% patients.
Patient concerns:
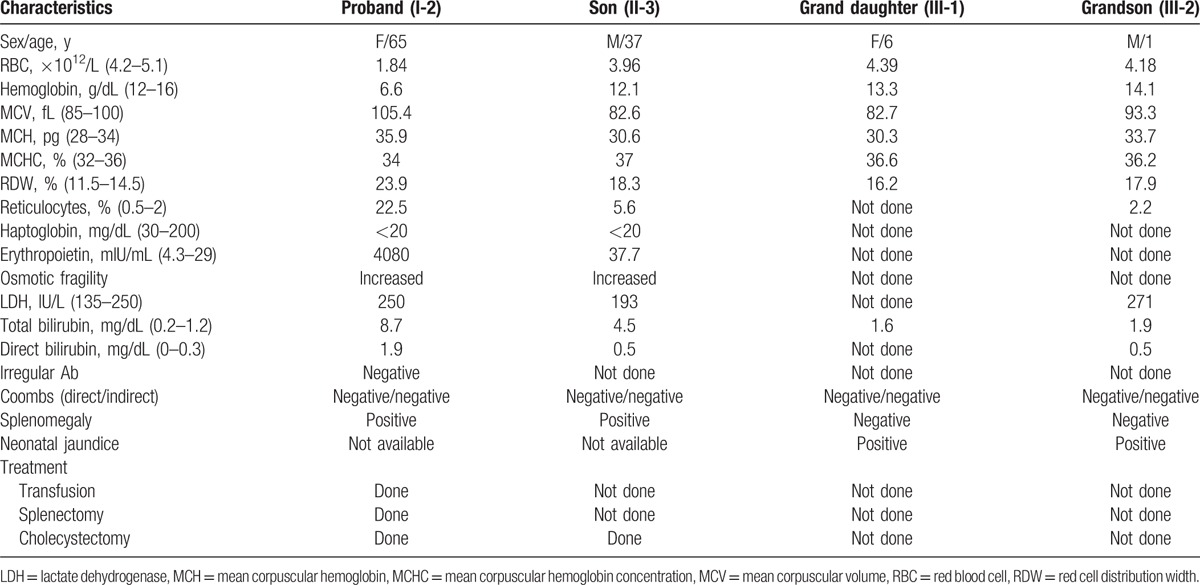
A 65-year-old female had been diagnosed as hemolytic anemia 30 years ago, based on a history of persistent anemia and hyperbilirubinemia for several years. She received RBC transfusion several times and a cholecystectomy roughly 20 years ago before. Round, densely staining spherical-shaped erythrocytes (spherocytes) were frequently found on the PB smear. Numerous spherocytes were frequently found in the PB smears of symptomatic family members, her 3rd son and his 2 grandchildren.
Diagnosis:
One heterozygous mutation of SPTB was identified by targeted next-generation sequencing (NGS). The nonsense mutation, c.1956G>A (p.Trp652∗), in exon 13 was confirmed by Sanger sequencing and thus the proband was diagnosed with HS.
Interventions:
The proband underwent a splenectomy due to transfusion-refractory anemia and splenomegaly.
Outcomes:
After the splenectomy, her hemoglobin level improved to normal range (14.1 g/dL) and her bilirubin levels decreased dramatically (total bilirubin 1.9 mg/dL; direct bilirubin 0.6 mg/dL).
Lessons:
We suggest that NGS of causative genes could be a useful diagnostic tool for the genetically heterogeneous RBC membrane disorders, especially in cases with a mild or atypical clinical manifestation.
Keywords: hereditary spherocytosis, nonsense mutation, SPTB gene, targeted next-generation sequencing
1. Introduction
Hereditary spherocytosis (HS) is a common inherited red cell membrane disorders characterized by nonautoimmune hemolytic anemia, jaundice, splenomegaly, and gallstone.[1] HS has a heterogeneous spectrum of clinical severity, and its prevalence is 1.39 cases per 100,000 people of Chinese population.[2] The main lesion in HS is loss of red blood cell (RBC) membrane surface, leading to reduced deformability due to defects in the RBC membrane proteins such as ankyrin, spectrin, band 3, or protein 4.2.[1]
Erythrocytic beta spectrin (SPTB) plays a role in RBC membrane organization and stability, along with ankyrin. The protein encoded by this locus functions in stability of RBC membranes, and mutations in this gene have been associated with HS type 2, hereditary elliptocytosis, and neonatal hemolytic anemia.[3] Beta spectrins are typically composed of 4 structural domains: actin binding domain, dimerization domain, spectrin repeats, and ankyrin binding domain. Spectrin and ankyrin interact to tether the spectrin cytoskeleton to the RBC membrane as major components of the RBC membrane skeleton. The structure of the spectrin binding domain of ankyrin and the ankyrin binding domain of spectrin have been solved to elucidate the structural basis for ankyrin–spectrin recognition.[4,5] The structure of spectrin repeats shows that these repeats are similar to all other spectrin repeats. SPTB mutations have been detected in about 20% of all affected individuals with HS who usually exhibit an autosomal dominant (AD) inheritance. Clinical manifestation ranges from mild to severe depending on the degree of the RBC membrane defect.[6]
Recent advances in next-generation sequencing (NGS) technology have led to a paradigm shift, leading the laboratory field of genetic testing away from Sanger sequencing.[7] Cost-effective, high-throughput NGS has led to the clinical implementation of targeted NGS or whole exome sequencing (WES).[8] WES has contributed greatly to the discovery of novel mutations responsible for Mendelian diseases.[9] It is widely employed as a diagnostic method, as it allows researchers to screen the entire coding regions of genes.[10,11] In this report, we identified a novel SPTB mutation responsible for HS in a Korean family using targeted NGS. This demonstrates that targeted NGS is an effective method both for identifying novel causal mutations and for diagnosing additional disease cases.
2. Case presentation
A 65-year-old female (Fig. 1A, individual I-2) was referred to the Department of Internal Medicine, Daejeon St. Mary's Hospital (Daejeon, Republic of Korea) for a further evaluation. She had been diagnosed as hemolytic anemia 30 years ago, based on a medical history of persistent anemia and hyperbilirubinemia for several years. She received RBC transfusion several times and a cholecystectomy roughly 20 years ago before without knowing the reason for the operations. The laboratory findings at the time of admission to our hospital were as follows: hemoglobin, 6.6 g/dL; red cell distribution width, 23.9%; reticulocytes, 22.5%; haptoglobin, <20 mg/dL; erythropoietin, 4080 mIU/mL; total bilirubin, 8.7 mg/dL; direct bilirubin, 1.9 mg/dL; and lactate dehydrogenase, 250 IU/L. Autoimmune hemolytic anemia was ruled out because irregular antibody screening, Coombs test, and cold agglutinin test were negative, even though increased osmotic fragility. On the peripheral blood (PB) smear, round, densely staining spherical-shaped RBCs (spherocytes) were frequently found about 10 to 20 cells per high power field, that lack central pallor and have a smaller than normal diameter (Fig. 1B). Pedigree analysis of the proband's family members revealed that her 3rd son (Fig. 1A, individual II-3) showed similar symptoms such as anemia and jaundice, 2 grandchildren (Fig. 1A, individuals III-1 and III-2) experienced neonatal jaundice after birth (Table 1). Numerous spherocytes were frequently found in the PB smears of symptomatic family members. Based on these laboratory findings and clinical manifestations, the proband and symptomatic family members were provisionally diagnosed as HS. At 2 years later, the proband underwent a splenectomy due to transfusion-refractory anemia and splenomegaly. After the splenectomy, her hemoglobin level improved to normal range (14.1 g/dL) and her bilirubin levels decreased dramatically (total bilirubin 1.9 mg/dL; direct bilirubin 0.6 mg/dL).
Figure 1.
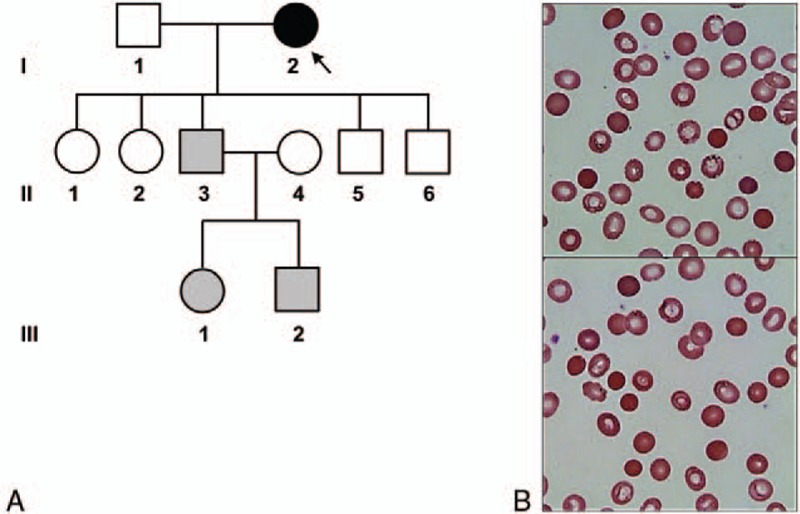
Pedigree analysis and peripheral blood smears of the proband. (A) Pedigree analysis of a Korean hereditary spherocytosis with a novel nonsense mutation in SPTB. Proband (indicated by the arrow) revealed the c.1956G>A; p.Trp652∗ in the heterozygous state. Gray symbols indicate clinically affected individuals not tested for the mutation. (B) Peripheral blood smears demonstrate moderate spherocytosis, about 10 to 20 cells per high power field (Wright–Giemsa stain, ×1000 magnification).
Table 1.
Laboratory findings and clinical characteristics in a Korean family with hereditary spherocytosis.
To confirm the genetic cause of HS, genetic testing using targeted NGS that consists of genes related anemia and bone marrow failure syndromes was performed in the proband. The gene panel included 5 HS-associated genes such as ANK1 (HS type 1, OMIM 182900), SPTB (HS type 2, OMIM 616649), SPTA1 (HS type 3, OMIM 270970), SLC4A1 (HS type 4, OMIM 612653), and EPB42 (HS type 5, OMIM 612690). All subjects provided written informed consent for clinical and molecular analyses, and the study protocol was approved by the Institute Review Board of the Catholic University of Korea.
Briefly, genomic DNA was extracted from the PB. Capture of the target regions was performed with reagents from a custom design HaloPlex Target Enrichment kit (Agilent Technologies, Inc., Santa Clara, CA). Massively parallel sequencing was performed on the Illumina HiSeq 2000 platform (Illumina, Inc., San Diego, CA). Average coverage of depth of the entire panel was 101×, and 98.3% of targeted bases were covered by 10× sequence reads. Sequence reads were aligned to hg19 with Burrow–Wheeler Aligner (version 0.7.12, MEM algorithm). Duplicate reads were removed by using Picard-tools1.96. Local realignment and base quality recalibration were done by the Genome Analysis Toolkit (GATK ver 3.5). Variant calling was performed by GATK HaplotypeCaller. Variants were annotated by SnpEff ver 4.2. As a result, 1 heterozygous nonsense mutation, c.1956G>A; p.Trp652∗ (reference sequence: NM_001024858.2), was identified in the exon 13 of SPTB gene (Fig. 2). This novel mutation was not found in public population sequence databases such as 1000 Genomes Project Database, ESP6500, and ExAC, as well as in Korean population sequence databases (KRGDB, http://152.99.75.168/KRGDB/menuPages/intro.jsp). Any (likely) pathogenic variants were not identified in other 4 HS-associated genes according to the American College of Medicine and Genetics guidelines for the interpretation of sequence variation. Thus, this nonsense mutation of the SPTB was confirmed to cause HS in the proband and may affect symptomatic family members who could not be tested for the mutation.
Figure 2.
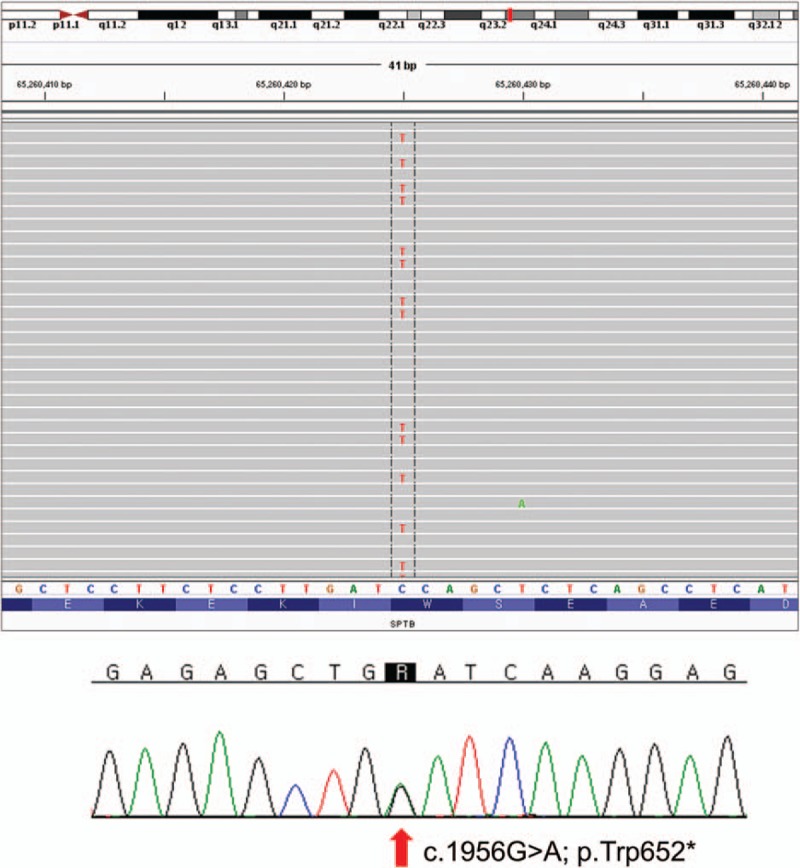
One heterozygous mutation of SPTB was identified by targeted next-generation sequencing and confirmed by Sanger sequencing. A novel nonsense mutation annotated as c.1956G>A; p.Trp652∗ in exon 13.
3. Discussion
Here, we describe a Korean family with 4 members affected clinically by HS. To the best of our knowledge, this is the first report describing HS with the novel SPTB mutation detected by targeted NGS in a Korean family. Among them, novel nonsense mutation (p.Trp652∗) of SPTB was identified by targeted NGS and confirmed by Sanger sequencing in the proband. Unfortunately, genetic testing was not available in symptomatic family members. The proband's son received a cholecystectomy and her grandchildren experienced neonatal hyperbilirubinemia, indicating a high possibility of spherocytosis, in spite of his incomplete medical history. On the other hand, abnormal spherocytes are trapped and destroyed in the spleen and this is the main cause of hemolysis in HS. Common complications are hemolytic episodes, cholelithiasis, and aplastic crises. Splenectomy is curative but should be undertaken only after careful assessment of the risks and benefits.[1] In our proband, spleen removal was performed as an effective therapeutic option, thus, eliminated anemia, hyperbilirubinemia, and lowers the high reticulocyte count to nearly normal levels, effectively.
Confirmation of hereditary RBC membrane disorders at a molecular level is important for not only clinical management of the patient but also genetic counseling. Understanding of the genotype–phenotype correlation is valuable to prepare genetic-based practice in HS. Ankyrin gene (up to 50%) is most commonly responsible for HS, followed by mutations in spectrin gene (SPTB, up to 20%; SPTA1, up to 5%), SLC4A1 (up to 15%), and EPB42 (up to 10%).[12] In the Korean population, ANK1 and SPTB are the major HS which corresponded to AD inheritance.[13] In other ethnic population, ANK1 or SPTB mutations were frequently detected: ANK1 mutations in 31% (15/49) Japanese HS,[14]SPTB mutations in 25% (10/40) the United States and Europe HS.[15]
To date, 16 missense/nonsense mutations in SPTB have been reported among patients with HS.[13] Like the novel p.Trp652∗ mutation reported in this report, 7 have been nonsense mutations sparsely located in the parts of beta spectrin repeats. Deficient beta spectrin protein levels due to nonsense mutations sited on beta spectrin repeats have been described as a cause of HS.[13] Beta spectrin proteins containing spectrin di-repeat structures may serve a controlled flexibility to membrane-associated scaffolds, as well as an intrinsic mechanosensing switch designed to control the disposition of ligands and signal-transducing molecules in response to cellular deformation or stretch.[5] Our proband shows increased osmotic fragility, which is well-known pathological feature of HS type 2 caused by an SPTB mutation.
RBC membrane disorders can be readily screened by various laboratory approaches such as PB smear, osmotic fragility test, eosin-5′-maleimide flow cytometry,[16] 2-dimensional gel electrophoresis,[17] and sodium dodecyl sulfate polyacrylamide gel electrophoresis.[18] However, the confirmatory diagnosis of HS is based on mutational analysis of gene encoding RBC membrane proteins.[19] An attempt to diagnose the process was described using Sanger sequencing of multiple genes in a sequential manner, which is, however, labor-intensive and expensive. Because RBC membrane disorders are clinically and genetically heterogeneous diseases with broadly overlapping clinical symptoms.[20] Defects in various membrane proteins involved in linking the lipid bilayer to membrane skeleton result in loss in membrane cohesion leading to surface area loss.[20] Recently, the advent NGS is a time and cost-effective method of detecting causal mutations associate to conventional genetic tests, especially when clinical manifestations of various conditions and a large number of candidate genes are involved.[21] In addition, the phenotypic variance with the mutation detected by NGS could be verified, comparing clinical characteristics of family members with and without the mutation.[22]
4. Conclusion
In summary, a novel nonsense mutation (p.Trp652∗) in SPTB was identified using NGS in a Korean family affected by HS. Five known causative genes involving in RBC cytoskeleton formation are considered in HS. Moreover, more than 20 genes are associated with hyperbilirubinemia and bilirubin metabolism.[22] Thus, to exactly discover the mutation causing the patient's clinical manifestations, both pedigree analysis and genetic testing are required simultaneously. We suggest that NGS of causative genes could be a useful diagnostic tool for the genetically heterogeneous RBC membrane disorders, especially in cases with a mild or atypical clinical manifestation.
Acknowledgment
The authors would like to acknowledge the financial support of the Catholic Medical Center Research Foundation during the 2017 program year.
Footnotes
Abbreviations: AD = autosomal dominant, HS = hereditary spherocytosis, NGS = next-generation sequencing, PB = peripheral blood, RBC = red blood cell, WES = whole exome sequencing.
The authors have no conflicts of interest to disclose.
References
- [1].Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet 2008;372:1411–26. [DOI] [PubMed] [Google Scholar]
- [2].Wang C, Cui Y, Li Y, et al. A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1978 to 2013 and estimation of the prevalence of the disease using a disease model. Intractable Rare Dis Res 2015;4:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Delaunay J. Molecular basis of red cell membrane disorders. Acta Haematol 2002;108:210–8. [DOI] [PubMed] [Google Scholar]
- [4].Ipsaro JJ, Huang L, Mondragon A. Structures of the spectrin-ankyrin interaction binding domains. Blood 2009;113:5385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Stabach PR, Simonovic I, Ranieri MA, et al. The structure of the ankyrin-binding site of beta-spectrin reveals how tandem spectrin-repeats generate unique ligand-binding properties. Blood 2009;113:5377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kim Y, Park J, Kim M. Diagnostic approaches for inherited hemolytic anemia in the genetic era. Blood Res 2017;52:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Koboldt DC, Steinberg KM, Larson DE, et al. The next-generation sequencing revolution and its impact on genomics. Cell 2013;155:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med 2014;370:2418–25. [DOI] [PubMed] [Google Scholar]
- [9].Rabbani B, Mahdieh N, Hosomichi K, et al. Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet 2012;57:621–32. [DOI] [PubMed] [Google Scholar]
- [10].Levenson D. Whole-exome sequencing emerges as clinical diagnostic tool: testing method proves useful for diagnosing wide range of genetic disorders. Am J Med Genet A 2014;164A:ix–x. [DOI] [PubMed] [Google Scholar]
- [11].Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. Jama 2014;312:1870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].An X, Mohandas N. Disorders of red cell membrane. Br J Haematol 2008;141:367–75. [DOI] [PubMed] [Google Scholar]
- [13].Park J, Jeong DC, Yoo J, et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin Genet 2016;90:69–78. [DOI] [PubMed] [Google Scholar]
- [14].Nakanishi H, Kanzaki A, Yawata A, et al. Ankyrin gene mutations in Japanese patients with hereditary spherocytosis. Int J Hematol 2001;73:54–63. [DOI] [PubMed] [Google Scholar]
- [15].Hassoun H, Vassiliadis JN, Murray J, et al. Characterization of the underlying molecular defect in hereditary spherocytosis associated with spectrin deficiency. Blood 1997;90:398–406. [PubMed] [Google Scholar]
- [16].Joshi P, Aggarwal A, Jamwal M, et al. A comparative evaluation of Eosin-5′-maleimide flow cytometry reveals a high diagnostic efficacy for hereditary spherocytosis. Int J Lab Hematol 2016;38:520–6. [DOI] [PubMed] [Google Scholar]
- [17].Demiralp DO, Peker S, Turgut B, et al. Comprehensive identification of erythrocyte membrane protein deficiency by 2D gel electrophoresis based proteomic analysis in hereditary elliptocytosis and spherocytosis. Proteomics Clin Appl 2012;6:403–11. [DOI] [PubMed] [Google Scholar]
- [18].Crisp RL, Solari L, Vota D, et al. A prospective study to assess the predictive value for hereditary spherocytosis using five laboratory tests (cryohemolysis test, eosin-5′-maleimide flow cytometry, osmotic fragility test, autohemolysis test, and SDS-PAGE) on 50 hereditary spherocytosis families in Argentina. Ann Hematol 2011;90:625–34. [DOI] [PubMed] [Google Scholar]
- [19].Chen J, Zhou Y, Gao Y, et al. A genetic features and gene interaction study for identifying the genes that cause hereditary spherocytosis. Hematology 2017;22:240–7. [DOI] [PubMed] [Google Scholar]
- [20].Da Costa L, Galimand J, Fenneteau O, et al. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev 2013;27:167–78. [DOI] [PubMed] [Google Scholar]
- [21].He Y, Jia S, Dewan RK, et al. Novel mutations in patients with hereditary red blood cell membrane disorders using next-generation sequencing. Gene 2017;627:556–62. [DOI] [PubMed] [Google Scholar]
- [22].Han JH, Kim S, Jang H, et al. Identification of a novel p.Q1772X ANK1 mutation in a Korean family with hereditary spherocytosis. PLoS ONE 2015;10:e0131251. [DOI] [PMC free article] [PubMed] [Google Scholar]