

Abstract
Background
Carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG132), a peptide aldehyde proteasome inhibitor, can inhibit tumor progression by inactivating nuclear factor (NF)-κB signaling. Paclitaxel (PTX) is part of a routine regimen for the treatment of breast cancer. However, activation of the NF-κB pathway after treatment with PTX confers insensitivity to this drug. This study investigated the potential effect of MG132 as a co-treatment with PTX against breast cancer, and clarifies the underlying molecular mechanisms.
Material/Methods
Breast cancer cells were treated with PTX, MG132, or PTX plus MG132, and the therapeutic effects were evaluated phenotypically. A mouse model of breast cancer was used to determine the combined effect of PTX plus MG132 in vivo.
Results
Treatment with PTX plus MG132 suppressed aggressive phenotypes of breast cancer cells more effectively than PTX alone. Consistently, MG132 also enhanced the suppressive effect of PTX on tumor growth in C57BL/6 mice. Significantly, activation of the NF-κB pathway by PTX was attenuated by MG132.
Conclusions
Based on our findings, we suggest the application of MG132 in clinical practice in combination with PTX for the treatment of breast cancer.
MeSH Keywords: Breast Neoplasms, NF-kappa B, Paclitaxel, Proteasome Inhibitors
Background
Breast cancer is one of the most common cancers worldwide [1]. To date, despite advances in surgery and radiotherapy, paclitaxel (PTX)-based combinational adjuvant chemotherapy is still considered standard treatment for patients with breast cancer. Clinically, some patients respond very positively to PTX-based combinational chemotherapies and tolerate the treatment well. In contrast, others exhibit no initial benefit or the treatment must be stopped because of severe adverse effects [2].
PTX reportedly exerts an anti-tumor effect by inhibiting cancer cell division. However, PTX can also up-regulate nuclear factor (NF)-κB activity, which might lead to cancer cell resistance to chemotherapeutic agents [3]. In addition, targeted degradation of inhibitor of κB (IκB) has been reported in cancer cells during PTX-based treatment, followed by decreased therapeutic responses [3].
Destruction of IκB activates NF-κB, which is rapidly translocated into the nucleus [4–6], where NF-κB unites homologous sites in the promoter areas of genes encoding pro-inflammatory cytokines, cell adhesion molecules, and anti-apoptotic proteins [7–10]. Moreover, NF-κB has a role in regulating cell viability and apoptosis [11–13]. NF-κB is up-regulated in many human cancers, including breast cancer, and is involved in the development and progression of various cancers [14,15]. Activation of the NF-κB-mediated transcriptional response is an important feature of certain malignancies [16,17], and may promote tumor cell survival and reduce the effectiveness of drug or radiation therapies [18,19]. Importantly, NF-κB also activates the transcription of IκB and itself, effectively serving as a molecular augmenter to sustain its own transcriptional activity.
Inhibiting the degradation of IκB can effectively block NF-κB signaling. Furthermore, phosphorylation of specific serine residues in the N-terminus of IκB can cause ubiquitination of internal lysine residues [20–22], marking the protein for proteasomal degradation. Recently, participation of the proteasome in the functional regulation and degradation of proteins, such as oncoproteins, cell cycle proteins, and tumor suppressors, has been extensively investigated [23]. In addition, proteasome inhibitors can inhibit NF-κB signaling by causing degradation of IκB [24].
Carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG132) is a peptide aldehyde proteasome inhibitor capable of also inhibiting other proteases such as calpains and cathepsins. MG132 exhibits an anti-tumor effect in multiple tumor models and cell lines. For example, MG132 inhibits invasion and induces apoptosis in human malignant pleural mesothelioma [25] and glioblastoma [26] cells. As an adjuvant agent, MG132 can promote curative effects in a wide variety of tumors [26]. However, the role of MG132 administration in breast cancers has not been studied extensively. Furthermore, its potential role in enhancing the efficacy of PTX by degrading IκB has not been investigated. Based on its role in other cancers, we hypothesized that MG132 enhances the therapeutic effect of PTX on breast cancer cells, and that NF-κB signaling is involved in this process. To test this hypothesis, the present study investigated whether a combination of MG132 and PTX would increase the efficacy of treatment compared with PTX alone. In addition, we sought to clarify the underlying molecular mechanisms.
Material and Methods
Reagents
Penicillin, streptomycin, and fetal bovine serum (FBS) were purchased from Life Technologies (Shanghai, China). Secondary and primary antibodies were purchased from Proteintech (Rosemont, IL, USA), except for the matrix metalloproteinase (MMP)2 antibody, which was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell lines and cell culture
MCF-7 cells (a human breast cancer cell line) and human umbilical vein endothelial cells (HUVECs) (a normal cell line) were acquired from the Institute of Biochemistry and Cell Biology, CAS (Shanghai, China). The mouse breast cancer cell line EO771 was provided by Professor Helen Arthur, Institute of Genetic Medicine, Newcastle University, UK. EO771 cells, MCF-7 cells, and HUVECs were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS (Gibco, Gaithersburg, MD, USA).
Cell viability assay
Cells (2×103) were added to 96-well plates with 100 μL of DMEM in each well and were treated with MG132 (0, 0.25, 0.5, 1, and 2 μM) or PTX (0, 0.1, 0.5, 1, and 2 μM) for 24, 48, and 72 h. Combination treatments included MG132 plus PTX at concentrations of 0.25 and 0.1 μM, or 0.5 and 0.5 μM, respectively, for 24 h. Then, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (5 mg/mL in phosphate-buffered saline (PBS) was added at 10 μL per well and the plates were incubated in 5% CO2 at 37°C for 4 h. To solubilize the formazan crystals, 150 μL of dimethyl sulfoxide was added. Absorbance of each well was measured at 490 nm on a microplate reader (M200PRO; Tecan, Männedorf, Switzerland). To assess the synergistic inhibitory effect of the combined drugs, we used the coefficient of drug interaction (CDI) as described previously [27,28]. The CDI was calculated using the following formula: CDI=AB/(A×B) where A and B are the single drug groups compared to the control group, and AB is the combination group compared to the control group. When the CDI is less than, greater than, or equal to 1, the drug interaction is deemed synergistic, antagonistic, or additive, respectively. A CDI of less than 0.7 indicates a significant synergistic effect [27,28].
Flow cytometric analysis of apoptosis
Fluorescein isothiocyante (FITC)-labeled Annexin V and 7-aminoactinomycin D (7AAD) were used to determine the extent of apoptosis, following the manufacturer’s instructions. Briefly, ice-cold PBS was used to wash the cells twice. The cells (1×106 cells/mL) were then resuspended in 1 mL 1× binding buffer, followed by addition of 100 μL 1× binding buffer containing 5 μL FITC-labeled Annexin V. The samples were vortexed lightly, incubated in a dark chamber for 10 min, and then incubated with 5 μL 7AAD. Finally, 400 μL 1× binding buffer was added to each tube. Flow cytometry was performed on a BD Calibur instrument (BD Biosciences, San Jose, CA, USA) within 1 h of incubation and FlowJo software (FlowJo, Ashland, OR, USA) was used for analysis.
Cell cycle analysis
After treatments as described in the cell viability assay section, cells were washed twice with PBS and then trypsinized. The detached cells were collected and added to 1 mL pre-cooled 70% ethanol for over 2 h. The cells were then incubated at 37°C for 1 h with 1 mL RNase (0.25 mg/mL). After incubation, the cells were resuspended in a propidium iodide solution (50 μg/mL) and incubated for 30 min in the dark. Flow cytometry was used to analyze the cell cycle.
Wound healing assay
A wound healing assay was used to assess cell migration as described previously [29]. Briefly, 1×106 EO771 cells were added to each well of a 6-well plate and incubated overnight. A 200-μL pipette tip was used to make a scratch wound in the cell monolayer. Cells were gently washed 3 times with PBS, and cultivated in medium without FBS. Cell migration was determined after 24 h by analyzing a photograph of the wounded site with Image J software (National Institutes of Health, Bethesda, MD, USA).
Cell invasion assay
Matrigel-precoated Transwell inserts were used to conduct the cell invasion assay. Cells (1×105) were resuspended in 200 μL DMEM without serum and added to the top chamber. Medium (600 μL) containing 10% FBS as a chemoattractant was added to the lower compartment. After incubation at 37°C under 5% CO2 for 24 h, cells on the upper surface of the Transwell membrane were removed with a cotton swab. Cells on the lower surface of the membrane were fixed for 20 min with 4% paraformaldehyde and stained with 0.1% crystal violet for counting.
Western blotting
Total proteins were extracted using RIPA lysis buffer (Beyotime Biotechnology, Jiangsu, China). Equal amounts of protein (80 μg) were separated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and then transferred to a polyvinylidene difluoride membrane. Blocking was performed for 1 h with 5% skim milk in Tris-buffered saline containing 1% Tween 20. The following primary antibodies were incubated with the membranes at 4°C overnight: NF-κB (10745-1-AP), pNF-κB (phospho-Ser536, #AB11014), IκBα (51066-1-AP), cyclin B1 (55004-1-AP), Cdc2 (10762-1-AP), Bax (50599-2-Ig), Bcl-2 (26593-1-AP) (all 1: 1000), and MMP2 (1: 1000, sc-13595). Subsequently, membranes were incubated with anti-mouse or anti-rabbit secondary antibodies (1: 2000). A LAS4010 reader (GE Healthcare. Pittsburgh, PA, USA) was used to visualize bands stained with the EasySee Western Blot Kit (TransGen Biotech, Beijing, China). Densitometric analysis of protein bands was performed using ImageJ software with β-actin (1: 5000, Proteintech) as the control.
Nuclear/cytoplasmic extraction
Cytoplasmic proteins were separated in a buffer mixture containing 2 mM dithiothreitol, 1 mM Na3VO4, 1 mM phenylmethyl sulfonylfluoride, 1 mM NaF, and Protease Inhibitors Cocktail (Sigma-Aldrich). This was added to L1 buffer (2 mM EDTA, 50 mM Tris-HCl, pH 8.0, 10% glycerol, and 0.1% NP-40). Nuclear pellets were washed twice with the buffer mixture and then lysed in NP-40 lysis buffer (1 mM EDTA, 250 mM NaCl, 0.1% NP-40, 10% glycerol, 50 mM Tris-HCl, pH 8.0) to which proteasome inhibitors cocktail was added. A bicinchoninic acid protein assay kit (Boster, Wuhan, China) was used to quantify protein concentrations.
Immunofluorescent cell staining
Cells (3×103) were grown on chamber slides and fixed with paraformaldehyde for 10 min. Triton X-100 (0.5% in PBS) was added for another 10 min to permeabilize the cells. After washing with PBS, cells were blocked with 1% bovine serum albumin and 5% normal goat serum for 1 h. Cells were then incubated with rabbit polyclonal antibodies specific for pNF-κB (1: 100) at 4°C overnight. After another washing in PBS, the cells were incubated for 1 h in the dark at 37°C with secondary antibodies (1: 2000). Finally, the cells were stained for 5 min using 4′,6-diamidino-2-phenylindole (Solarbio, Beijing, China) and photographed under a fluorescence microscope.
Animal models of breast cancer
A total of 24 C57BL/6 female mice aged 8–12 weeks were used. All experimental procedures were approved by the First Affiliated Hospital of Jinzhou Medical University Ethics Committee. Trypsinization was used to detach EO771 cells. Orthotopic allografts were performed by injecting 1×106 cells into each mouse mammary gland in situ (n=6 mice/group). On days 10, 12, and 14 after cell injection, mice were injected intraperitoneally with 2 mg/kg 0.9% saline (control), 2 mg/kg PTX, 2 mg/kg MG132, or 1 mg/kg PTX and 1 mg/kg MG132. Tumor growth was observed for a total of 15 days, at which time the mice were euthanized, photographed, and the tumors used for further analyses. Tumor volume was calculated using the following formula: (W(2)×L)/2, where W is tumor width and L is tumor length.
Immunohistochemistry (IHC)
Paraffin-embedded sections (4-μm thick) were prepared. The sections were deparaffinized using xylene and rehydrated in graded alcohol (100–80–50–30%). For antigen retrieval, sections were heated in citrate buffer (pH=6.0) (TRS; Dako, Kyoto, Japan) using the high-pressure method. Rabbit anti-pNF-κB or rabbit anti-NF-κB (both 1: 100) were incubated with the sections for 2 h. Then, an anti-rabbit antibody conjugated to horseradish peroxidase (Dako) was added for 1 h. The binding sites were visualized with diaminobenzidine staining. Sections were photographed using an LSM 700 confocal imaging system (Zeiss, Oberkochen, Germany).
Statistical analysis
Each experiment was carried out at least 3 times. Data are presented as means ± standard deviation and were analyzed using SAS-JMP 11 software (SAS, Cary, NC, USA). To measure the significance of differences in multiple comparisons, we used an analysis of variance and post hoc test. p<0.05 was regarded as statistically significant.
Results
Effect of PTX combined with MG132 on aggressive breast cancer cell phenotypes
To clarify the combined effect of PTX plus MG132 versus PTX or MG132 alone on breast cancer cells, optimized concentrations were first determined on the basis of minimal impact on the viability of HUVECs. Based on the MTT assay, treatment with PTX or MG132 alone reduced the viability of both EO771 and MCF-7 cells in a time- and dose-dependent manner (Figure 1A, 1C; p<0.05). A synergistic effect on EO771 and MCF-7 cell viability was observed following a combination of PTX and MG132 (Figure 1B, 1D; p<0.05). In contrast, 0.25 μM MG132 did not affect viability nor increase the effect of PTX on HUVECs (Figure 1E, 1F).
Figure 1.

Effect of paclitaxel (PTX) and/or MG132 on the viability of breast cancer cells and human umbilical vein endothelial cells (HUVECs). (A, C, E) Treatment with PTX or MG132 for 24, 48, and 72 h suppressed the viability of MCF-7 and EO771 cells, and HUVECs, in a concentration- and time-dependent manner. (B, D, F) Cell viability after treatment with PTX and/or MG132 for 24 or 48 h. MG132 (0.25 μM) combined with PTX (0.1 μM) had no effect on the viability of HUVECs compared with the PTX alone group (F). Data are expressed as means ± standard deviation of 3 independent experiments. P.1, PTX 0.1 μM; P.5, PTX 0.5 μM; M.25, MG132 0.25 μM; M.5, MG132 0.5 μM; P.1 + M.25, PTX 0.1 μM and MG132 0.25 μM; P.5 + M.5, PTX 0.5 μM and MG132 0.5 μM. * p<0.05, vs. single-treatment groups and control (ctrl). # p<0.05 vs. the MG132 alone and ctrl groups.
The CDI was calculated to assess the presence of a drug interaction. Results indicated that the CDI was less than 0.7, indicating a significant synergistic inhibitory effect of MG132 and PTX on EO771 and MCF-7 cells.
The effects of treatment with PTX and/or MG132 on cell cycle, apoptosis, migration, and invasion were analyzed. PTX and/or MG132 induced apoptosis and G2 arrest in a dose-dependent manner in both EO771 and MCF-7 cells (Figure 2A, 2B, 2D, 2E). In HUVECs, 0.25 μM MG132 did not induce G2 arrest or apoptosis, nor did it enhance the effect of PTX (Figure 2C, 2F). The Transwell invasion and wound healing assays revealed that PTX and/or MG132 weakened the invasion and migration abilities of these cells (Figure 3A–3D). In addition, PTX and MG132 acted synergistically to cause cell cycle arrest, induce apoptosis, and inhibit cell migration and invasion in EO771 and MCF-7 cells.
Figure 2.

Effect of paclitaxel (PTX) and/or MG132 on cell cycle and apoptosis of breast cancer cells and human umbilical vein endothelial cells (HUVECs). (A, B, D, E) PTX or MG132 treatment for 24 h induced G2 arrest and apoptosis in a concentration-dependent manner in MCF-7 and EO771 cells. (C, F) Effect of PTX and/or MG132 treatment for 24 h on cell cycle and apoptosis in HUVECs. MG132 (0.25 μM) had no effect on G2 arrest and apoptosis of HUVECs. When combined with PTX, MG132 had no significant effect compared with the PTX alone group. Data are expressed as means ± standard deviation of 3 independent experiments. P0.1, PTX 0.1 μM; P0.5, PTX 0.5 μM; M0.25, MG132 0.25 μM; M0.5, MG132 0.5 μM; P + M, PTX 0.1 μM and MG132 0.25 μM. * p<0.05 vs. single treatment and control (ctrl) groups. # p<0.05 vs. MG132 and ctrl groups.
Figure 3.

Effect of paclitaxel (PTX) and/or MG132 on the migration and invasion of breast cancer cells. Wound healing assays show that PTX and/or MG132 treatment decreased the ability of MCF-7 and EO771 cells to migrate in a concentration-dependent manner (A, B), and reduced the invasive potential of MCF-7 and EO771 cells (C, D). Data are expressed as means ± standard deviation of 3 independent experiments. P0.1, PTX 0.1 μM; P0.5, PTX 0.5 μM; M0.25, MG132 0.25 μM; M0.5, MG132 0.5 μM; P + M, PTX 0.1 μM and MG132 0.25 μM; * p<0.05 vs. single-treatment and control (ctrl) groups.
Protein expression of phenotype-related and NF-κB pathway-related molecules in EO771 and MCF-7 cells after exposure to PTX and/or MG132
Western blotting showed that Bax levels increased more in the combination groups than in PTX monotherapy groups, whereas the opposite was observed for cyclin B1, Cdc2, Bcl-2, and MMP2 in both EO771 and MCF-7 cells (Figure 4A).
Figure 4.

Expression of proteins in breast cancer cells treated with paclitaxel (PTX) and/or MG132. (A) Western blotting results showing expression of phenotype-related proteins in MCF-7 and EO771 cells following treatment with PTX and/or MG132. (B) Western blotting results showing pNF-κB levels within the cytoplasm and nucleus in E0771 and MCF-7 cells. (C) Immunofluorescence staining showing the expression of pNF-κB within the cytoplasm and nucleus in EO771 and MCF-7 cells. P0.1, PTX 0.1 μM; P0.5, PTX 0.5 μM; M0.25, MG132 0.25 μM; M0.5, MG132 0.5 μM; P + M, PTX 0.1 μM and MG132 0.25 μM.
To determine the participation of NF-κB signaling in breast cancer cells after exposure to PTX, the levels of NF-κB, pNF-κB, and IκBα were assessed by Western blotting. Compared with the PTX monotherapy groups, the levels of NF-κB and pNF-κB decreased significantly after treatment with PTX and MG132, whereas the expression of IκBα was remarkably higher in the combination group (Figure 4B). Cytoplasmic and nuclear translocation of pNF-κB was evaluated separately. Our findings demonstrate that expression of pNF-κB was markedly decreased in the combination group (Figure 4B, 4C). Together, these results support our hypothesis that MG132 influences PTX by regulating NF-κB signaling.
Effect of PTX and/or MG132 on tumor growth in vivo
To examine the effect of combined PTX and MG132 therapy on breast cancer in vivo, mice were inoculated with EO771 cells and treated with PTX and/or MG132. The tumor volumes of EO771 allografts were smaller following combined treatment than in the other groups (Figure 5A, 5B). After treatment with MG132 and PTX, breast cancer tissue showed a lower expression of NF-κB and pNF-κB, and higher expression of IκBα, than the single-treatment groups (Figure 5C). IHC staining of tumor tissue also showed lower expression of NF-κB and pNF-κB in the combination group than in the single-treatment groups (Figure 5D).
Figure 5.

Paclitaxel (PTX) and/or MG132 suppressed the growth of breast cancer cells in vivo. Growth of EO771 cells in mice treated with PTX and/or MG132 assessed by measurement of (A) tumor size and (B) growth curve. (C) Western blots showing the expression of NF-κB, pNF-κB, and IκBα. (D) Immunohistochemistry showing the expression of NF-κB and pNF-κB. PTX, 2 mg/kg; MG132, 2 mg/kg; P + M, PTX 1 mg/kg and MG132 1 mg/kg. * p<0.05 vs. PTX and control (ctrl) groups.
Discussion
Currently, PTX-based combination chemotherapy is considered a standard treatment for patients with intractable breast cancer [30]. Nevertheless, limited efficacy and high toxicity are still major concerns with this approach. Therefore, there is an increasing demand to discover novel agents with greater efficacy and reduced toxicity for the treatment of breast cancer.
PTX stimulates the phosphorylation and inactivation of IκB, leading to activation of the NF-κB pathway. This restores the aggressive phenotype of cancer cells associated with reduced treatment efficiency and overall survival [3]. Our results are consistent with previous studies demonstrating that the proteasome inhibitor MG132 exerted an anti-tumor effect by inhibiting activation of NF-κB [31]. MG132 inhibits IκB degradation and inactivates the NF-κB pathway [32]. In vitro, MG132 showed potent cytotoxicity against 2 breast cancer cell lines but, at least at low concentrations, it did not affect the viability of normal HUVECs. Data from the in vivo experiment confirmed the efficacy and safety of MG132, which was evidenced by a reduction in tumor volume in C57BL/6 mice. These findings are in agreement with previous reports, indicating that MG132 may have potential selectivity toward tumor cells, thus reducing undesirable cytotoxicity to normal cells [33].
Proteasome inhibitors not only have a significant anti-tumor effect, but they can also be combined with other chemotherapy drugs for better anti-tumor efficacy [34]. Dong et al. reported that proteasome inhibitors combined with cisplatin were more effective than cisplatin alone in killing ovarian cancer cells [35]. Nakano et al. reported that MG132 increased the sensitivity of tumor cells to radiotherapy [36]. Warren et al. found that MG132 increased the sensitivity of cancer cells to radiation therapy and improved the therapeutic outcome [37].
Héctor et al. reported that PTX-induced cytotoxicity was improved after combining PTX and the proteasome inhibitor MG132. Consistent with our study, MG132 appeared to enhance PTX cytotoxicity, particularly if administered after PTX [38]. A possible explanation for these results involves activation of NF-κB by PTX, which is otherwise suppressed by MG132. We report that PTX combined with MG132 significantly suppressed viability, migration, and invasion, and induced apoptosis and G2 arrest. The effect was much greater than in the PTX monotherapy group. In an in vivo tumor inhibition assay, PTX and MG132 together potently inhibited the growth of EO771 cells in C57BL/6 mice, surpassing the outcome of the PTX group. Mechanistic studies showed that the suppression of viability, migration, and invasion, as well as the induction of apoptosis and G2 arrest brought by a combination of PTX and MG132, might have involved inhibition of the NF-κB pathway. These findings not only indicate the therapeutic potential of combined PTX and MG132 therapy for breast cancer, but also provide valuable insight into their mechanism of action.
Although the molecular mechanisms underlying these findings are still poorly researched, our results provide some insight into the signaling pathway leading to the synergistic effect of PTX and MG132 in breast cancer cells. Phosphorylation and degradation of IκB were shown to play a pivotal role in activating NF-κB, being closely related to decreased sensitivity to PTX-based chemotherapy [39]. Consistent with those findings, our results showed up-regulation of NF-κB-related proteins. Previous studies indicated that inactivation of NF-κB significantly inhibited tumor growth and development [40]. Our results also indicate that MG132 up-regulated IκB expression and down-regulated the protein levels of NF-κB and pNF-κB. This suggests that MG132 enhances the efficacy of PTX via inactivation of the NF-κB pathway. Furthermore, we report that expression of Bcl-2, cyclin B1, Cdc2, and MMP2 is down-regulated, but Bax is up-regulated, following treatment with PTX plus MG132 (Figure 4A). A schematic representation of the hypothesized effect of PTX and MG132 on NF-κB signaling is displayed in Figure 6.
Figure 6.
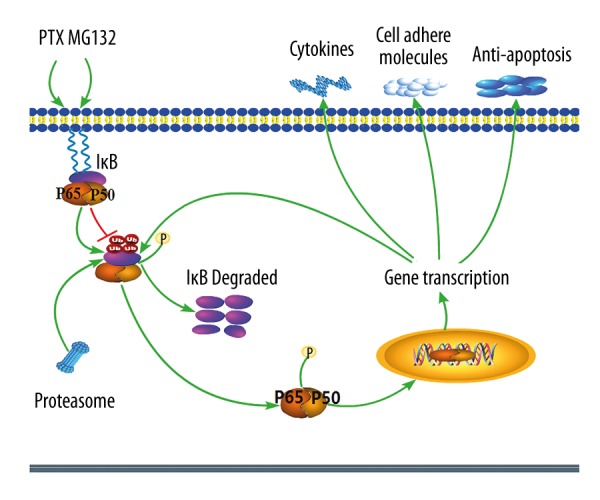
Schematic representation of the effect of paclitaxel (PTX) and MG132 on the NF-κB signaling pathway in breast cancer cells. PTX phosphorylates and activates the IκK complex, leading to the phosphorylation, ubiquitination, and degradation of IκBα. This frees NF-κB, which is then activated and translocated to the nucleus, thereby activating transcription of multiple downstream genes. This leads to an increased expression of proteins related to cell cycle, apoptosis, migration, and invasion, and decreases apoptosis. MG132 inhibits the degradation of IκBα, thereby inhibiting activation and output of NF-κB signaling, possibly by targeting IκBα and decreasing the nuclear and cytoplasmic levels of NF-κB. This leads to a decreased expression of proteins related to the cell cycle, apoptosis, migration, and invasion, and increases apoptosis. Stimulatory events are indicated with green arrows. Inhibitory events are indicated with red lines ending in bars.
Conclusions
The results of this study demonstrate the synergistic anti-tumor activity of PTX and MG132 against breast cancer both in vitro and in vivo. Mechanistic studies indicate that induction of apoptosis and G2 arrest, and suppression of viability, migration, and invasion play important roles in the synergistic anti-tumor activity of PTX and MG132, which is likely to be mediated by the NF-κB pathway. These findings suggest the potential usefulness of MG132 as an adjuvant in breast cancer treatment and support further preclinical and future clinical testing.
Footnotes
Conflicts of interest
None.
Source of support: This work was supported by the First Affiliated Hospital of Jinzhou Medical University and the National Natural Science Foundation of China (grant no. 81472460)
References
- 1.Fitzmaurice C, Allen C, Barber RM, Barregard L. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 2017;3:524–48. doi: 10.1001/jamaoncol.2016.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hortobagyi GN, Holmes FA, Theriault RL, Buzdar AU. Use of Taxol (paclitaxel) in breast cancer. Oncology. 1994;51(Suppl 1):29–32. doi: 10.1159/000227413. [DOI] [PubMed] [Google Scholar]
- 3.Spencer W, Kwon H, Crepieux P, et al. Taxol selectively blocks microtubule dependent NF-kappaB activation by phorbol ester via inhibition of IkappaBalpha phosphorylation and degradation. Oncogene. 1999;18:495–505. doi: 10.1038/sj.onc.1202335. [DOI] [PubMed] [Google Scholar]
- 4.Alkalay I, Yaron A, Hatzubai A, et al. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1995;92:10599–603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldi L, Brown K, Franzoso G, Siebenlist U. Critical role for lysines 21 and 22 in signal-induced, ubiquitin-mediated proteolysis of I kappa B-alpha. J Biol Chem. 1996;271:376–79. doi: 10.1074/jbc.271.1.376. [DOI] [PubMed] [Google Scholar]
- 6.Roff M, Thompson J, Rodriguez MS, et al. Role of IkappaBalpha ubiquitination in signal-induced activation of NFkappaB in vivo. J Biol Chem. 1996;271:7844–50. doi: 10.1074/jbc.271.13.7844. [DOI] [PubMed] [Google Scholar]
- 7.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol Cell Biol. 2000;20:2687–95. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang CY, Mayo MW, Korneluk RG, et al. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–83. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 9.Wang CY, Guttridge DC, Mayo MW, Baldwin AS. NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol Cell Biol. 1999;19:5923–29. doi: 10.1128/mcb.19.9.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zong WX, Edelstein LC, Chen C, et al. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 1999;13:382–87. doi: 10.1101/gad.13.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chitra S, Nalini G, Rajasekhar G. The ubiquitin proteasome system and efficacy of proteasome inhibitors in diseases. Int J Rheum Dis. 2012;15:249–60. doi: 10.1111/j.1756-185X.2012.01737.x. [DOI] [PubMed] [Google Scholar]
- 12.Almagro MC, Vucic D. The inhibitor of apoptosis (IAP) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp Oncol. 2012;34:200–11. [PubMed] [Google Scholar]
- 13.Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Ann Rev Biophys. 2013;42:443–68. doi: 10.1146/annurev-biophys-083012-130338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–36. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 15.McNulty SE, del Rosario R, Cen D, et al. Comparative expression of NFkappaB proteins in melanocytes of normal skin vs. benign intradermal naevus and human metastatic melanoma biopsies. Pigment Cell Res. 2004;17:173–80. doi: 10.1111/j.1600-0749.2004.00128.x. [DOI] [PubMed] [Google Scholar]
- 16.Kordes U, Krappmann D, Heissmeyer V, et al. Transcription factor NF-kappaB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia. 2000;14:399–402. doi: 10.1038/sj.leu.2401705. [DOI] [PubMed] [Google Scholar]
- 17.Tricot G. New insights into role of microenvironment in multiple myeloma. Lancet. 2000;355:248–50. doi: 10.1016/S0140-6736(00)00019-2. [DOI] [PubMed] [Google Scholar]
- 18.Jeremias I, Kupatt C, Baumann B, et al. Inhibition of nuclear factor kappaB activation attenuates apoptosis resistance in lymphoid cells. Blood. 1998;91:4624–31. [PubMed] [Google Scholar]
- 19.Patel NM, Nozaki S, Shortle NH, et al. Paclitaxel sensitivity of breast cancer cells with constitutively active NF-kappaB is enhanced by IkappaBalpha super-repressor and parthenolide. Oncogene. 2000;19:4159–69. doi: 10.1038/sj.onc.1203768. [DOI] [PubMed] [Google Scholar]
- 20.Spencer E, Jiang J, Chen ZJ. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev. 1999;13:284–94. doi: 10.1101/gad.13.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scherer DC, Brockman JA, Chen Z, et al. Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc Natl Acad Sci USA. 1995;92:11259–63. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winston JT, Strack P, Beer-Romero P, et al. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999;13:270–83. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009;30:191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Tseng LM, Liu CY, Chang KC, et al. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012;14:R68. doi: 10.1186/bcr3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan BZ, Chapman JA, Reynolds SH. Proteasome inhibitor MG132 induces apoptosis and inhibits invasion of human malignant pleural mesothelioma cells. Transl Oncol. 2008;1:129–40. doi: 10.1593/tlo.08133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zanotto-Filho A, Braganhol E, Battastini AM, Moreira JC. Proteasome inhibitor MG132 induces selective apoptosis in glioblastoma cells through inhibition of PI3K/Akt and NFkappaB pathways, mitochondrial dysfunction, and activation of p38-JNK1/2 signaling. Invest New Drugs. 2012;30:2252–62. doi: 10.1007/s10637-012-9804-z. [DOI] [PubMed] [Google Scholar]
- 27.Xu SP, Sun GP, Shen YX, et al. Antiproliferation and apoptosis induction of paeonol in HepG2 cells. World J Gastroenterol. 2007;13:250–56. doi: 10.3748/wjg.v13.i2.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin JL, Gong J, Yin TJ, et al. PTD4-apoptin protein and dacarbazine show a synergistic antitumor effect on B16-F1 melanoma in vitro and in vivo. Eur J Pharmacol. 2011;654:17–25. doi: 10.1016/j.ejphar.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 29.Chen S, Zhao Y, Gou WF, et al. The anti-tumor effects and molecular mechanisms of suberoylanilide hydroxamic acid (SAHA) on the aggressive phenotypes of ovarian carcinoma cells. PLoS One. 2013;8:e79781. doi: 10.1371/journal.pone.0079781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bishop JF, Dewar J, Toner GC, et al. Paclitaxel as first-line treatment for metastatic breast cancer. The Taxol Investigational Trials Group, Australia and New Zealand. Oncology. 1997;11:19–23. [PubMed] [Google Scholar]
- 31.Ding WX, Ni HM, Chen X, et al. A coordinated action of Bax, PUMA, and p53 promotes MG132-induced mitochondria activation and apoptosis in colon cancer cells. Mol Cancer Ther. 2007;6:1062–69. doi: 10.1158/1535-7163.MCT-06-0541. [DOI] [PubMed] [Google Scholar]
- 32.Letoha T, Somlai C, Takacs T, et al. The proteasome inhibitor MG132 protects against acute pancreatitis. Free Radic Biol Med. 2005;39:1142–51. doi: 10.1016/j.freeradbiomed.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 33.He Q, Huang Y, Sheikh MS. Proteasome inhibitor MG132 upregulates death receptor 5 and cooperates with Apo2L/TRAIL to induce apoptosis in Bax-proficient and -deficient cells. Oncogene. 2004;23:2554–58. doi: 10.1038/sj.onc.1207351. [DOI] [PubMed] [Google Scholar]
- 34.Lauricella M, Emanuele S, D’Anneo A, et al. JNK and AP-1 mediate apoptosis induced by bortezomib in HepG2 cells via FasL/caspase-8 and mitochondria-dependent pathways. Apoptosis. 2006;11:607–25. doi: 10.1007/s10495-006-4689-y. [DOI] [PubMed] [Google Scholar]
- 35.Dong X, Liu J, Zheng H, et al. In situ dynamically monitoring the proteolytic function of the ubiquitin-proteasome system in cultured cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;287(3):H1417–25. doi: 10.1152/ajpheart.01233.2003. [DOI] [PubMed] [Google Scholar]
- 36.Nakano H, Nakajima A, Sakon-Komazawa S, et al. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006;13:730–37. doi: 10.1038/sj.cdd.4401830. [DOI] [PubMed] [Google Scholar]
- 37.Warren G, Grimes K, Xu Y, et al. Selectively enhanced radiation sensitivity in prostate cancer cells associated with proteasome inhibition. Oncol Rep. 2006;15:1287–91. [PubMed] [Google Scholar]
- 38.Héctor HV, Cayetano K, Carolina SE, et al. Inhibition of paclitaxel-induced proteasome activation influences paclitaxel cytotoxicity in breast cancer cells in a sequence-dependent manner. Cell Cycle. 2007;6:2662–68. doi: 10.4161/cc.6.21.4821. [DOI] [PubMed] [Google Scholar]
- 39.Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest. 2001;107:241–46. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baud V, Jacque E. [The alternative NF-κB activation pathway and cancer: Friend or foe]. Med Sci (Paris) 2008;24:1083–88. doi: 10.1051/medsci/200824121083. [in French] [DOI] [PubMed] [Google Scholar]