

Abstract
The chondrogenic differentiation of synovial mesenchymal stem cells (SMSCs) is regulated by essential transcription factors and signaling cascades. However, the precise mechanisms involved in this process remain unclear. MicroRNAs (miRs/miRNAs) are undersized non-coding RNAs responsible for the post-transcriptional regulation of gene expression, by binding to the 3′-untranslated regions (3′-UTRs) of their target mRNAs. miRNAs may constitute a promising tool to regulate SMSC differentiation and to advance the controlled differentiation of SMSCs in therapeutic applications. The aim of the present study was to examine the role of miR-218 in SMSC differentiation towards chondrocytes. The present study comparatively analyzed the expression profile of known miRNAs and specific target genes in SMSCs between early and late differentiation stages. Western blotting and reverse transcription-quantitative polymerase chain reaction analysis of gene expression demonstrated the upregulation of 15-hydroxyprostaglandin dehydrogenase [NAD(+)] (15-HPGD), prostaglandin E2 (PGE2) and rate limiting enzymes responsible for the synthesis of PGE2 precursors throughout chondrogenesis. Through correlation analysis, it was observed that there was a significant association between miR-128, 15-HPGD gene expression, 15-HPGD protein expression and microsomal prostaglandin E synthase 1. Further experiments demonstrated that miR-218 decreased PGE2 concentration by binding to the 3′-UTR of 15-HPGD. Using an immunofluorescence reporting system, it was observed that miR-218 regulated the expression of 15-HPGD during the differentiation of SMSCs into cartilage, and subsequently inhibited osteogenesis during chondrogenesis by acting on the 3′UTR of 15-HPGD. Therefore, miR-218 may be an important regulator targeting osteogenic factors and modulating cartilage formation and differentiation. The results of the present study provided a novel insight beneficial to cellular manipulation methods during cartilage regeneration, and in cartilage tissue engineering research.
Keywords: microRNA-218, 15-hydroxyprostaglandin dehydrogenase [NAD(+)], synovial mesenchymal stem cells, chondrogenesis, osteogenesis
Introduction
Mesenchymal stem cells (MSCs) are multipotent non-hematopoietic progenitor cells that may differentiate into a variety of mesenchymal lineages, including osteoblasts, chondrocytes and adipocytes. Compared with other sources of MSCs, the synovium is easier to access and exhibits an increased chondrogenic capacity, rendering it a potential candidate cell source for fibrocartilage engineering (1–4). Therefore, synovial MSCs (SMSCs) may be appropriate for cell-based therapies for fibrocartilage, including meniscus reconstruction. It has been reported that the differentiation capacity of SMSCs may be affected by a number of factors, including the composition of the culture medium and the culture conditions (4–6). In the last decade, studies have made noteworthy advances and progress in investigating the functions of MSCs in cartilage repair (7–10). However, the application of previous findings in clinical practice is challenging, as experimental results are difficult to repeat and the safety and reliability require further confirmation. At present, the primary experimental limitations include: Low chondrogenic differentiation ability of bone marrow MSCs; early cartilage differentiation, including bone formation factors; successfully engineered cartilage tissues may undergo osteogenic differentiation; requirement of long-term follow-up; and the emergence of bone tissue in the cartilage may result in the loss of cartilage function (11). Therefore, the improvement of the chondrogenic capability of MSCs and the concurrent inhibition of osteogenic differentiation constitute a challenge for cartilage tissue engineering. The involvement of bone morphogenetic protein 2 (BMP2) in the osteogenic differentiation of MSCs was conveyed in previous studies (12,13). Other studies have similarly demonstrated that prostaglandin E2 (PGE2) modulates the BMP2 pathway by possible downregulation of BMP2-mediated phosphorylation of mothers against decapentaplegic homolog 1, 5 and 8 in chondrocytes (12–14). This regulation occurs during the cellular maturation stage in the processes of chondrogenesis and osteogenesis, and may be associated with the initiation of osteogenesis in the later stages of cartilage formation. During preliminary experiments (Cong R et al unpublished data), it was observed that treatment with low doses of PGE2 (1 µM) significantly promoted cartilage differentiation, delayed cell maturation and inhibited osteogenesis. In order to inhibit osteogenic differentiation, PGE2 was added in vitro, and it was observed that that the expression of alkaline phosphatase (ALP) decreased; however, in order to maintain a high concentration of PGE2, maintenance of the administration of exogenous PGE2 was required, suggesting that an unknown mechanism maintains PGE2 at a low level in the late stage of the cartilage formation. Notably, it was demonstrated that the concentration of miR-218 was markedly associated with PGE2 and it may be hypothesized that miR-218 may be associated with the regulation of PGE2 concentration.
Therefore, the present study was designed in order to elucidate the regulatory role of miR-218 and its correlation with PGE2 in the differentiation of SMSCs toward chondrocytes.
Materials and methods
Materials
β-glycerophosphate, ascorbic acid, Alizarin red S stain and an ALP staining kit were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). BMP2 was purchased from R&D Systems, Inc. (Minneapolis, MN, USA). The NE-PER Nuclear and Cytoplasmic Extraction Reagent was obtained from Pierce (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The easy-BLUE™ and StarTaq™ reagents were purchased from Intron Biotechnology, Inc. (Seongnam, Korea), and the AccuPower RT-PreMix was purchased from Bioneer Corporation (Daejeon, Korea). The miR isolation kit was purchased from Ambion (Thermo Fisher Scientific, Inc.). The Mir-X™ miRNA First-Strand Synthesis kit and SYBR Advantage miRNA qRT-PCR kit were obtained from Clontech Laboratories, Inc. (Mountainview, CA, USA). The polymerase chain reaction (PCR) primers were synthesized by Takara Korea Biomedical, Inc. (Seoul, Korea). SYBR Premix Ex Taq™ was purchased from Takara Bio, Inc. (Otsu, Japan). Dual-Glo luciferase assay kit was obtained from Promega Corporation (Madison, WI, USA). The concentration of PGE2 was detected using a Prostaglandin E2 ELISA kit (ab133021; Abcam, Cambridge, UK).
Isolation and culture of SMSCs
A total of 5 rabbits (age, 18–25 months; 3 males, 2 females; weight, ~2 kg) were obtained from the animal center of Shanghai Tenth Hospital (Shanghai, China). The present study was approved by the ethics committee of Shanghai Tenth Hospital (Shanghai, China). SMSCs were isolated from the knee joint. Following anesthesia with 35 mg/kg ketamine hydrochloride (#32386, Sigma-Aldrich; Merck KGaA), an incision in the knee joint capsule was performed. Random biopsies of synovial membrane (SM; 5×5 mm) were obtained aseptically. SM specimens were placed into transport medium containing sterile PBS (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 1% (v/v) antibiotic/antimycotic solution (10,000 U/ml penicillin, 10,000 µg/ml streptomycin and 25 µg/ml amphotericin B; Gibco; Thermo Fisher Scientific, Inc.). Fragments were digested with 0.1% (v/v) bacterial collagenase type II (Gibco; Thermo Fisher Scientific, Inc.) in high-glucose Dulbecco's modified Eagle's medium (HG DMEM; Gibco; Thermo Fisher Scientific, Inc.) containing 1% (v/v) antibiotic/antimycotic solution. Following overnight incubation at 37°C and removal of undigested tissue, cells were collected by centrifugation at 150 × g for 7 min at 4°C using a 40-µm nylon cell strainer (Falcon; BD Biosciences, Franklin Lakes, NJ, USA), rinsed twice with DMEM, and resuspended in complete culture medium containing α-minimum essential medium (Biochrom Ltd., Cambridge, UK) supplemented with 20% (v/v) FBS (Gibco; Thermo Fisher Scientific, Inc.) and 1% (v/v) antibiotic/antimycotic solution. Cells were incubated at 37°C in a humidified 5% CO2 atmosphere and allowed to adhere for 72 h. Non-adherent cells were removed. When cells reached 80–90% confluence, adherent cells were trypsinized using 0.05% trypsin/EDTA (Gibco; Thermo Fisher Scientific, Inc.) at 37°C for 5 min and replated at a seeding density of 1,000 cells/cm2. Culture medium was replaced every 3 days.
SMSC differentiation
For chondrogenic differentiation, cells were cultured at low density (~20–30% confluence) and treated with 10 ng/ml transforming growth factor (TGF)-β1 (DIAsource ImmunoAssays SA, Louvain-la-Neuve, Belgium) in serum-free medium at 37°C for 72 h. Following induction, adjacent single cells proliferated and combined into high-density cell pellets. Pellets were cultured at a density of 2×105 cells/well in 6-well plates containing chondrogenic medium, consisting of HG DMEM supplemented with 50 µg/ml ascorbic acid, 0.9 mM sodium pyruvate, 1% ITS+ (6.25 µg/ml insulin, 6.25 µg/ml transferrin, 6.25 ng/ml selenious acid), 1.25 mg/ml bovine serum albumin, 5.35 mg/ml linolenic acid and 100 nM dexamethasone (Sigma-Aldrich; Merck KGaA). Cells cultured in chondrogenic medium without TGF-β1 or PGE2 served as the control.
In silico target prediction and analysis of miR-218 expression
TargetScan v5.1 (www.targetscan.org) target prediction software was used to pinpoint miRNAs that may potentially target the 3′UTR of 15-hydroxyprostaglandin dehydrogenase [NAD(+)] (15-HPGD).
Western blot analysis
Cell cultures were treated with lysis buffer (Gibco; Thermo Fisher Scientific, Inc.) containing protease inhibitors (Protease Inhibitor Cocktail Set III; Calbiochem; EMD Millipore, Billerica, MD, USA) and 1 mM sodium orthovanadate. Protein concentrations were determined using a Coomassie Plus protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). Protein extracts (20 µg) were separated by 10% SDS-PAGE, transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and blocked with 5% milk in PBS for 1 h at room temperature (or 4°C, overnight). The expression of 15-HPGD (ab115945), cyclooxygenase-1 (COX-1, ab695), microsomal prostaglandin E synthase 1 (mPGES-1, ab168621), phospholipase A2 (PLA2, ab188028) and β-actin (mAbcam8226) (all from Abcam) was detected via incubation with primary antibodies, diluted 1:3,000, for 1 h at room temperature. Membranes were subsequently incubated with the secondary antibody (1:4,000; goat anti-mouse horseradish peroxidase conjugated; Beyotime Institute of Biotechnology, Shanghai, China) for 1 h at room temperature. Immunodetection was performed using ECL Plus Western Blotting Substrate reagent (32134, Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. Semi-quantification was performed using Quantity One version 4.62 (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative PCR (RT-qPCR)
RT-qPCR was used for detecting the expression levels of candidate genes at the gene level. The sequences of all primers used were shown in Table I. A TRIzol extraction kit (Invitrogen; Thermo Fisher Scientific, Inc.) was employed for extracting total RNA from cells. Subsequently, 1.5 µg extracted total RNA was converted into cDNA using a First-Strand Synthesis System for RT-PCR (Invitrogen; Thermo Fisher Scientific, Inc.). The qPCR experiment was performed using a QuantiFast SYBR Green PCR kit (Qiagen GmbH, Hilden, Germany), according to the manufacturer's protocol, using a reaction volume of 20 µl. The fluorescence intensity was determined using the Bio-Rad CFX96™ Real-Time System (Bio-Rad Laboratories, Inc.). The relative level of mRNA for a specific gene was normalized to β-actin which was used as an endogenous gene (15).
Table I.
Primer sequences.
| Gene | Primer sequence |
|---|---|
| SOX-9 | Forward 5′-CATGAACGCCTTCATGGTGT-3′ |
| Reverse 5′-CCACACCTCCTCTTCTTTCT-3′ | |
| AGN | Forward 5′-ATCCCAGAAAACTTCTTTGG-3′ |
| Reverse 5′-TTCTACTCACTCCCCCTT-3′ | |
| Col II | Forward 5′-AACAGCCAAAGGACCCAAGT-3′ |
| Reverse 5′-AAATAACACAACCACCCTCT-3′ | |
| Col X | Forward 5′-TTCCATTTGATAAGATTTTG-3′ |
| Reverse 5′-ACCTACACTCTCTTTTACTC-3′ | |
| HPGD | Forward 5′-CGGGCATGAGTCCTGCTAAA-3′ |
| Reverse 5′-TCACCCCTTCCCTTTCTACC-3′ | |
| Col I | Forward 5′-TCTTTTTCCTCTTCTTCTTT-3′ |
| Reverse 5′-TTTTTATTTTATTTTTCCTA-3′ | |
| β actin | Forward 5′-CCAACCGCGAGAAGATGA-3′ |
| Reverse 5′-CCAGAGGCGTACAGGGATAG-3′ |
SOX-9, transcription factor Sox9; AGN, aggrecan; Col, collagen; HPGD, 15-hydroxyprostaglandin dehydrogenase [NAD(+)].
Overexpression of a miR-218 mimics
The gene coding for 15-HPGD short of its 3′UTR was cloned from rabbit genomic DNA with Takara LA Taq (Takara Biotechnology Co., Ltd., Dalian, China) and subcloned into the pcDNA3.1-vector (Invitrogen; Thermo Fisher Scientific, Inc.) to generate an expression vector for 15-HPGD. PrimeSTAR HS DNA Polymerase (Takara Biotechnology Co., Ltd.) was used to amplify the 3′UTR of the 15-HPGD mRNA, which was subcloned into the pMIR-REPORT vector (Applied Biosystems; Thermo Fisher Scientific, Inc.) with the luciferase gene immediately downstream. Constructs of pMiR-REPORT-mutant (mut)-15-HPGD 3′UTR were synthesized, containing the 15-HPGD 3′UTR with three point alterations in the seed sequence, using a QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies, Inc., Santa Clara, CA, USA) according to the manufacturer's instructions. Transfections of RNAs and plasmids were performed, using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) and a Fugene HD reagent (Promega Corporation), respectively, according to the manufacturers' protocol. Following transient transfection, the culture medium was replaced with fresh osteogenic medium, and the cells were incubated in the presence or absence of BMP2 for the indicated periods of time (3 days). A total of 50 nM miR-218 mimics, and their corresponding negative control RNAs (negative control (NC) sequence: 5′-UUCUCCGAACGUGUCACGUTT-3′) (Shanghai GenePharma Co., Ltd., Shanghai, China) were introduced into cells every 4 days.
Luciferase reporter assay
Each well of 96-well plates were seeded with 5×103 293 cells (CRL-1573; American Type Culture Collection, Mannassas, VA, USA) and incubated overnight. Subsequently, cells were co-transfected using Lipofectamine 2000 with 80 ng pMIR-REPORT-mut-5-HPGD 3′UTR plasmid or pMIR-REPORT-5-HPGD 3′UTR plasmid, 8 ng endogenous control pRL-TK-Renilla-luciferase plasmid (a gift from Shanghai Usen Biological Technology Co., Ltd., Shanghai, China) and 50 nM miR-218 mimics. A total of 200 ng pMIR-REPORT-5-HPGD 3′UTR plasmid or the pMIR-REPORT-mut-HPGD 3′UTR plasmid and 20 ng pRL-TK plasmid were transfected into the SMSCs in a 24-well plate. The luciferase activity was measured using the Dual-Luciferase Reporter Assay system (Promega Corporation), according to the manufacturer's protocol. The data were normalized by dividing the firefly luciferase activity by that of the Renilla luciferase.
Statistical analysis
SPSS version 11.0 (SPSS, Inc., Chicago, IL, USA) was used to perform the statistical analyses. All quantitative assays were calculated from at least 3 replicate samples. Error bars indicate 95% confidence intervals of the mean values (mean ± standard deviation). Replicate samples of each assays group were taken from cells of a single animal. One-way analysis of variance was used to perform multiple comparisons and t-tests were performed for pairwise comparisons. A Least Significant Difference test was used for post hoc analyses. P<0.05 was considered to indicate a statistically significant difference. Multiple linear regression was used to identify the association between PGE2 and mPGES-1, mPGES-2, cytosolic (c)PGES, PGH2, 15-HPGD and miR-218. The test standard was α=0.05, and stepwise linear regression was used to screen variables. The analysis was verified using residual analysis.
Results
PGE related protein seeking
According to the statistical analysis of existing models (15), HPGD expression appears to be significantly associated with the regulation of PGE2 (Table II). In addition, Pearson correlation analysis revealed further potential proteins associated with PGE2 expression (Table III), including COX1, PLA and PTGES. Analysis of variables associated with PGE linearity after filtering, correlation coefficient and equation fitting suggested miR-218 and HPGD were most closely associated with PGE expression (Table IV).
Table II.
Correlation of PGE2 with the abundance of its upstream and downstream control factors.
| Model | S-coefficient (β) | t | P-value |
|---|---|---|---|
| (Constant) | 0.035 | 0.974 | |
| miR | 1.114 | 6.842 | 0.000 |
| HPGD gene | −2.006 | −2.723 | 0.035a |
| HPGD protein | 2.189 | 3.131 | 0.02a |
| mPGES | −0.059 | −0.993 | 0.359 |
miR, microRNA; HPGD, 15-hydroxyprostaglandin dehydrogenase [NAD(+)]; mPGES, microsomal prostaglandin E synthase 1.
P<0.05.
Table III.
Pearson correlation analysis of PGE2 with other variables.
| Factor | PGE2 | miR-218 | HPGD gene | PTGES | PLA2G6 | COX1 | HPGD protein | mPGES | PLA | COX |
|---|---|---|---|---|---|---|---|---|---|---|
| PGE2 | 1.000 | 0.989 | −0.949 | 0.099 | −0.547 | 0.245 | −0.940 | −0.774 | −0.254 | 0.245 |
| miR-218 | 0.989 | 1.000 | −0.974 | 0.023 | −0.547 | 0.209 | −0.970 | −0.784 | −0.243 | 0.209 |
| HPGD gene | −0.949 | −0.974 | 1.000 | 0.125 | 0.448 | −0.216 | 0.999 | 0.764 | 0.253 | −0.216 |
| PTGES | 0.099 | 0.023 | 0.125 | 1.000 | −0.246 | −0.154 | 0.141 | −0.341 | 0.366 | −0.154 |
| PLA2G6 | −0.547 | −0.547 | 0.448 | −0.246 | 1.000 | 0.248 | 0.448 | 0.467 | −0.005 | 0.248 |
| COX1 | 0.245 | 0.209 | −0.216 | −0.154 | 0.248 | 1.000 | −0.217 | −0.240 | 0.041 | 1.000 |
| HPGD protein | −0.940 | −0.970 | 0.999 | 0.141 | 0.448 | −0.217 | 1.000 | 0.772 | 0.227 | −0.217 |
| mPGES | −0.774 | −0.784 | 0.764 | −0.341 | 0.467 | −0.240 | 0.772 | 1.000 | −0.109 | −0.240 |
| PLA | −0.254 | −0.243 | 0.253 | 0.366 | −0.005 | 0.041 | 0.227 | −0.109 | 1.000 | 0.041 |
| COX | 0.245 | 0.209 | −0.216 | −0.154 | 0.248 | 1.000 | −0.217 | −0.240 | 0.041 | 1.000 |
PGE2, prostaglandin E2; miR, microRNA; HPGD, 15-hydroxyprostaglandin dehydrogenase [NAD(+)]; PTGES, prostaglandin E synthase; PLA2G6, 85/88 kDa calcium-independent phospholipase A2; COX1, cyclooxygenase 1; mPGES, microsomal prostaglandin E synthase 1; PLA, phospholipase A2.
Table IV.
Variables associated with PGE linearity after filtering, correlation coefficient and equation fitting.
| Correlations | ||||||||
|---|---|---|---|---|---|---|---|---|
| PGE2 | miR-218 | HPGD gene | HPGD protein | Model | S-coefficients (β) | t | P-value | |
| PGE | 1.000 | 0.989 | −0.949 | −0.940 | Constant | −1.071 | 0.320 | |
| miR-218 | 0.989 | 1.000 | −0.974 | −0.970 | miR-218 | 1.167 | 7.583 | 0.000a |
| HPGD gene | −0.949 | −0.974 | 1.000 | 0.999 | HPGD gene | −1.777 | −2.542 | 0.039a |
| HPGD protein | −0.940 | −0.970 | 0.999 | 1.000 | HPGD protein | 1.966 | 2.973 | 0.021a |
PGE, prostaglandin E; miR, microRNA; HPGD, 15-hydroxyprostaglandin dehydrogenase [NAD(+)].
P<0.05.
PGE2 regulates SMSC chondrogenic differentiation
Previous studies (16,17) demonstrated that SMSCs exhibit the potential to differentiate into a chondrogenic lineage. In the present study, to investigate the effects of PGE2 on SMSC chondrogenesis, SMSCs were incubated in chondrogenic medium for 22 days. The concentration of PGE2, in addition to the expression of its biosynthesis enzymes mPGES, PLA2 and COX-1 were examined over differentiation time. The expression of 15-HPGD and proteins associated with PGE2 catabolism were additionally measured. The results demonstrated that chondrogenic induction led to a rapid increase in PGE2 concentration (with the maximum level recorded at 2 days), followed by a progressive decrease until the end of the experiment (Fig. 1A). The protein expression levels of mPGES, PLA2 and COX-1 equally increased rapidly during the first 2 days following chondrogenic induction, and remained largely constant thereafter (Fig. 1B and C). By contrast, the levels of 15-HPGD increased throughout the osteogenic differentiation process. In order to further understand the effect of PGE2 on SMSC chondrogenesis, the isolated SMSCs were cultured in the chondrogenic medium with or without the addition of PGE2. The results demonstrated that the exogenous addition of PGE2 inhibited the expression of osteogenic markers in a dose-dependent manner and retarded cartilage maturation (Fig. 1D). The results of the present study suggested the implication of an unknown 15-HPGD/PGE2 regulatory mechanism during chondrogenesis in SMSCs.
Figure 1.

15-HPGD is the rate limited degradation enzyme of PGE2, and 15-HPGD expression is negatively associated with PGE2. (A) PGE2 concentration during SMSC differentiation. In the early differentiation period (10 days ago), PGE2 expression was significantly different compared with the undifferentiated group. (B) Representative western blot images. (C) Expression of proteins at different time points during SMSC differentiation, determined by western blot analysis. (D) Gene expression at different time points during SMSC differentiation determined by reverse transcription-quantitative polymerase chain reaction. *P<0.05 vs. undifferentiated cells. 15-HPGD, 15-hydroxyprostaglandin dehydrogenase [NAD(+)]; PGE2, prostaglandin E2; COX-1, cyclooxygenase-1; mPGES-1, microsomal prostaglandin E synthase 1; PLA2, phospholipase A2; SMSC, synovial mesenchymal stem cells; miR, microRNA; undiff, undifferentiated.
PGE2 expression is correlated with miR-218 expression levels
In order to further elucidate the mechanism underlying PGE2 catabolism and maintaining the concentration at low level in the late differentiation stages, the bioinformatics tool TargetScan was used to identify candidate miRNAs targeting 15-HPGD. Systematic bioinformatics analysis demonstrated that miR-4465, miR-26a, miR-26b, miR-1297 and miR-218 were potential posttranscriptional regulators of 15-HPGD (Fig. 2) with miR-218 presenting highly-conserved regions with the 3′-UTR of 15-HPGD at positions UACUUGAA and AAGCACAA. In order to investigate the expression profiles of these miRNAs, RT-qPCR experiments were performed. The results confirmed that the mRNA expression of miR-218 decreased gradually, consistent with the PGE2 concentration (Fig. 3). Unlike miR-218, no similar expression trend was observed for other predicted miRNAs, which exhibited decreased expression over the differentiation course compared with miR-218. Correlation analyses (Table II) demonstrated that the expression of miR-218, and the protein and mRNA expression levels of 15-HPGD and mPGES were significantly correlated, without a linear correlation between mPGES and PGE2 concentration (data not shown).
Figure 2.

Identification of miRs targeting 15-HPGD using TargetScan. miR, microRNA; UTR, untranslated region; 15-HPGD, 15-hydroxyprostaglandin dehydrogenase [NAD(+)]; UTR, untranslated region.
Figure 3.
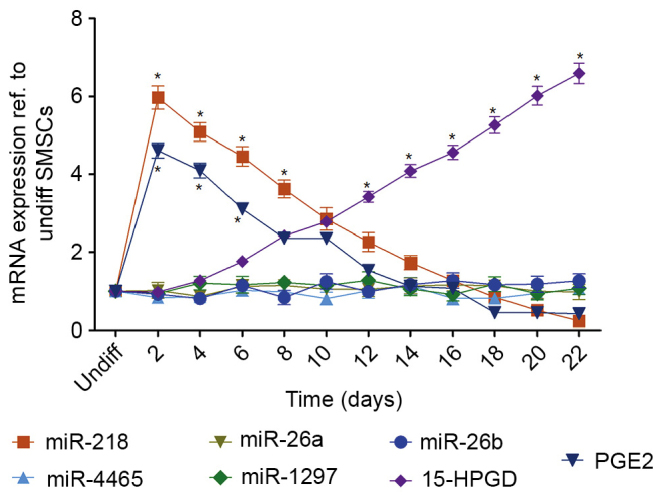
mRNA expression of 15-HPGD, PGE2 and miRs targeting 15-HPGD. Targeting miRs were identified using TargetScan and analyzed via reverse transcription-quantitative polymerase chain reaction. miRNA218 and PGE2 expression were significantly higher compared with undifferentiated cells at baseline. Following 10 days of differentiation, 15-HPGD was significantly increased compared with undifferentiated cells. *P<0.05 vs. Undifferentiated cells. miR, microRNA; 15-HPGD, 15-hydroxyprostaglandin dehydrogenase [NAD(+)]; PGE2, prostaglandin E2; undiff, undifferentiated.
The expression of 15-HPGD promotes induction of SMSC osteogenesis
The correlation between the expression of miR-218 and the mRNA and protein expression levels of 15-HPGD suggested that miR-218 may be a catabolic regulator of PGE2, by targeting the 3′-UTR region of 15-HPGD. In order to confirm the relationship between 15-HPGD with PGE2 or other osteogenesis markers, a 15-HPGD cDNA coding sequence was inserted into the pcDNA3.1 vector (pcDNA3.1-15-HPGD). The pcDNA3.1-15-HPGD and the empty control vectors were transfected into undifferentiated SMSCs separately. Notably, the concentration of PGE2 in the group transfected with 15-HPGD was significantly decreased compared with the control group at the initial stage of chondrogenic induction (Fig. 4A). Further analysis demonstrated that the expression of differentiation markers, including transcription factor SOX-9 and aggrecan (AGN) were markedly delayed while the expression of 15-HPGD at the gene and protein levels were markedly increased compared with those in the control group. No significant statistical difference was observed between groups in the later differentiation period (Fig. 4B).
Figure 4.
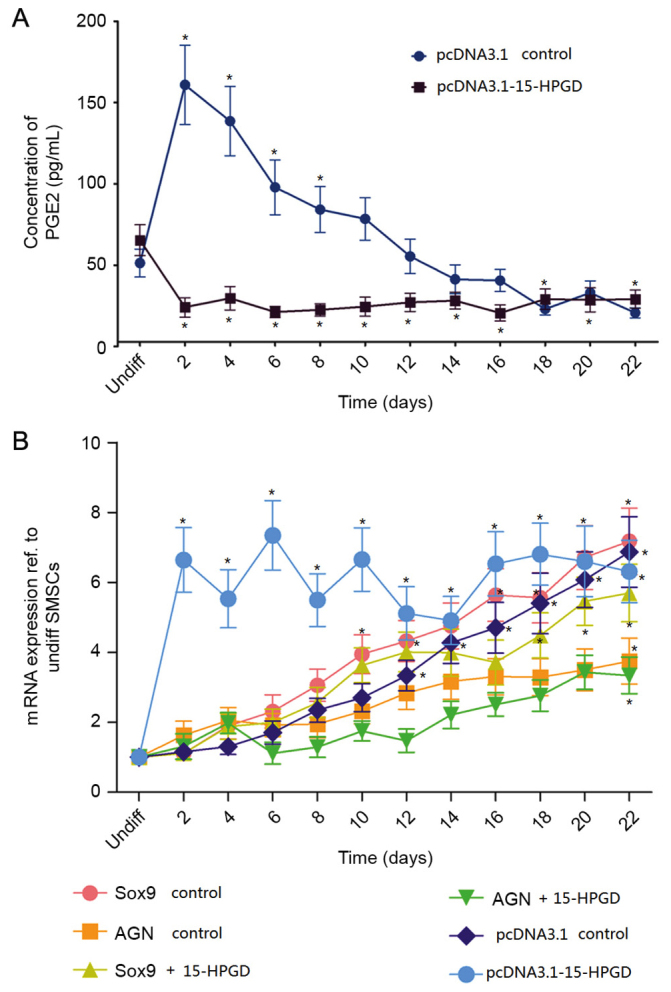
15-HPGD decreases PGE2 concentration. (A) Comparison of the concentration of PGE2 in SMSCs transfected with the wide type 15-HPGD coding sequence (pcDNA3.1-15-HPGD) or normal SMSCs (empty control vector). (B) mRNA expression of differentiation-associated genes and 15-HPGD in SMSCs transfected with the 15-HPGD coding sequence (pcDNA3.1-15-HPGD) or empty control vector SMSCs. *P<0.05 vs. Undifferentiated cells. PGE2, prostaglandin E2; 15-HPGD, 15-hydroxyprostaglandin dehydrogenase [NAD(+)]; SMSCs, synovial mesenchymal stem cells; UTR, untranslated region; nom, normal; Sox9, transcription factor Sox9; AGN, aggrecan.
Notably, the gene and protein expression of 15-HPGD was significantly decreased following the addition of miR-218 post-induction, while the expression of PGE2 was increased compared with the group without external adjunction of miR-218. These results suggested that miR-218 may impact upon PGE2 concentration by regulating 15-HPGD. As the expression of PGE2 in SMSCs was decreased in the late stage of differentiation compared with the early stage, and the expression of 15-HPGD exhibited the opposite trend, it was important to identify the transcriptional regulation mechanism of this process.
In order to further ascertain that the 3′-UTR of 15-HPGD contained binding sites for mir-218, luciferase reporting systems containing either wild-type 3′-UTR or a mutant 3′-UTR of 15-HPGD were designed (Fig. 5A). The luciferase reporting systems and mir-218 mimics were co-transfected into SMSCs. As presented in Fig. 5B, significantly decreased luciferase activity was recorded in the reporter vector containing the wild-type 3′-UTR of 15-HPGD, while high luciferase activity was recorded for the mutant 3′-UTR. These data indicated that 3′-UTR of 15-HPGD may be a target of mir-218 and contains binding sites for miR-218. It was anticipated that miR-218 may regulate the expression of 15-HPGD during chondrogenic differentiation by acting on the 3′-UTR region of 15-HPGD.
Figure 5.
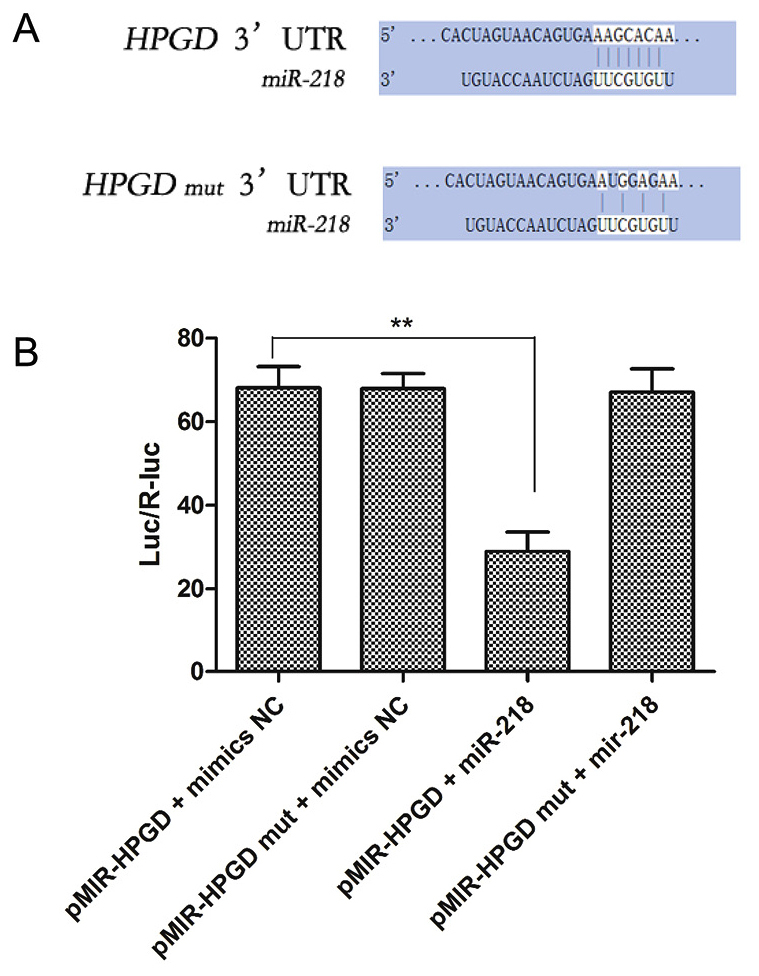
miR-218 targets the 15-HPGD 3′-UTR. (A) The three point mutations induced in the 3′UTR of 15-HPGD. (B) Overexpression of exogenous miR-218 suppressed the 15-HPGD expression, with the opposite effect observed with mutant 15-HPGD. **P<0.01. miR, microRNA; 15-HPGD, 15-hydroxyprostaglandin dehydrogenase [NAD(+)]; UTR, untranslated region; SMSCs, synovial mesenchymal stem cells; mut, mutant.
Discussion
The purpose of the present work was to determine the mechanism underlying the regulation of PGE2, which has been proven to modulate the expression of a major osteogenic factor, BMP-2 (17,18), in osteosarcoma cell lines and human MSCs (14). In preliminary studies, it was observed that treatment with small doses of PGE2 (1 µM) was able to markedly promote cartilage differentiation, delay cell maturation and inhibit osteogenesis during differentiation of SMSCs (Cong R et al unpublished data). These data indicated that PGE2 regulation may be of importance in cartilage repair, since the in vitro external adjunction of PGE2 led to decreased activity of ALP, an osteogenic indicator. However, in order to maintain a high endogenous concentration of PGE2, it was necessary to maintain the adjunction of exogenous PGE2, suggesting that an unknown mechanism stimulates PGE2 catabolism in the late stage of the SMSC chondrogenesis. To clarify this aspect, the present study investigated the expression of 15-HPGD, an enzyme involved in PGE2 catabolism, and that of rate limiting enzymes responsible for the synthesis of PGE2 precursors. miRNAs targeting 15-HPGD were identified, and their expression was additionally evaluated. Notably, it was observed that the expression of miR-218 was correlated with that of PGE2, and it was hypothesized that miR-218 may be associated with the regulation of PGE2 concentration. However, our results demonstrated that it was impossible to find suitable binding sites between miR-218 and the 3′UTR region of the rate limiting enzymes involved in the synthesis of PGE2. In addition, the high complementarity of the expression of miR-218 and that of PGE2 suggested an indirect association between miR-218 and PGE2. The confirmation of the potential of miR-218 to bind to the 3′UTR region of 15-HPGD was demonstrated by the following findings: i) Luciferase assays using wild type or mutant vectors for the seed matched region of miR-218 in the 3′UTR region of 15-HPGD, in addition to miR-218 mimics, revealed that only co-transfection with the wild type reporter and the miR-218 mimic was able to noticeably reduce luciferase activity; ii) overexpression of miR-218 decreased 15-HPGD expression in SMSCs; and iii) inhibition of miR-218 in SMSCs increased the expression of PGE2, as verified by western blot analysis. The opposing expression levels of miR-218 and PGE2 suggest a possible regulatory mechanism of SMSCs differentiation through miR-218-mediated post-transcriptional regulation of 15-PGDH. The results of the present study are consistent with previous findings demonstrating that the expression of miR-218 is down-regulated during osteoclast differentiation, and that the overexpression of miR-218 negatively regulates osteoclastogenesis by suppressing the tumor necrosis factor ligand superfamily member 11-induced activation of p38 mitogen-activated protein kinase/proto-oncogene c-Fos/nuclear factor of activated T-cells cytoplasmic 1 signaling (18). The present study, to the best of our knowledge, is the first to demonstrate the association between miR-218, 15-HPGD and PGE2 in the differentiation of SMSCs.
In conclusion, the results of the present study are the first to illustrate the important role of miR-218 in SMSC differentiation into chondrocytes and osteocytes, and may provide a novel insight beneficial to cell manipulation methods during cartilage regeneration and in cartilage tissue engineering research.
Acknowledgements
The present study was supported by the Fund of Action Plan for Young Talents Training of Tongji University (grant no. 2014KJ091) and the Fund of Key Disciplines of Shanghai Tenth People's Hospital (grant no. 2015021).
References
- 1.Sakaguchi Y, Sekiya I, Yagishita K, Muneta T. Comparison of human stem cells derived from various mesenchymal tissues: Superiority of synovium as a cell source. Arthritis Rheum. 2005;52:2521–2529. doi: 10.1002/art.21212. [DOI] [PubMed] [Google Scholar]
- 2.Shirasawa S, Sekiya I, Sakaguchi Y, Yagishita K, Ichinose S, Muneta T. In vitro chondrogenesis of human synovium-derived mesenchymal stem cells: Optimal condition and comparison with bone marrow-derived cells. J Cell Biochem. 2006;97:84–97. doi: 10.1002/jcb.20546. [DOI] [PubMed] [Google Scholar]
- 3.Mochizuki T, Muneta T, Sakaguchi Y, Nimura A, Yokoyama A, Koga H, Sekiya I. Higher chondrogenic potential of fibrous synovium- and adipose synovium-derived cells compared with subcutaneous fat-derived cells: Distinguishing properties of mesenchymal stem cells in humans. Arthritis Rheum. 2006;54:843–853. doi: 10.1002/art.21651. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimura H, Muneta T, Nimura A, Yokoyama A, Koga H, Sekiya I. Comparison of rat mesenchymal stem cells derived from bone marrow, synovium, periosteum, adipose tissue and muscle. Cell Tissue Res. 2007;327:449–462. doi: 10.1007/s00441-006-0308-z. [DOI] [PubMed] [Google Scholar]
- 5.Alm JJ, Heino TJ, Hentunen TA, Väänänen HK, Aro HT. Transient 100 nM dexamethasone treatment reduces inter- and intraindividual variations in osteoblastic differentiation of bone marrow-derived human mesenchymal stem cells. Tissue Eng Part C Methods. 2012;18:658–666. doi: 10.1089/ten.tec.2011.0675. [DOI] [PubMed] [Google Scholar]
- 6.Xiao Y, Peperzak V, van Rijn L, Borst J, de Bruijn JD. Dexamethasone treatment during the expansion phase maintains stemness of bone marrow mesenchymal stem cells. J Tissue Eng Regen Med. 2010;4:374–386. doi: 10.1002/term.250. [DOI] [PubMed] [Google Scholar]
- 7.Chang CH, Kuo TF, Lin FH, Wang JH, Hsu YM, Huang HT, Loo ST, Fang HW, Liu HC, Wang WC. Tissue engineering-based cartilage repair with mesenchymal stem cells in a porcine model. J Orthop Res. 2011;29:1874–1880. doi: 10.1002/jor.21461. [DOI] [PubMed] [Google Scholar]
- 8.Mobasheri A, Csaki C, Clutterbuck AL, Rahmanzadeh M, Shakibaei M. Mesenchymal stem cells in connective tissue engineering and regenerative medicine: Applications in cartilage repair and osteoarthritis therapy. Histol Histopathol. 2009;24:347–366. doi: 10.14670/HH-24.347. [DOI] [PubMed] [Google Scholar]
- 9.Ray K. Therapy. Tissue engineering: Harnessing stem cells in cartilage repair. Nat Rev Rheumatol. 2012;8:308. doi: 10.1038/nrrheum.2012.62. [DOI] [PubMed] [Google Scholar]
- 10.Zhang B, Yang S, Sun Z, Zhang Y, Xia T, Xu W, Ye S. Human mesenchymal stem cells induced by growth differentiation factor 5: An improved self-assembly tissue engineering method for cartilage repair. Tissue Eng Part C Methods. 2011;17:1189–1199. doi: 10.1089/ten.tec.2011.0011. [DOI] [PubMed] [Google Scholar]
- 11.Xian CJ, Foster BK. Repair of injured articular and growth plate cartilage using mesenchymal stem cells and chondrogenic gene therapy. Curr Stem Cell Res Ther. 2006;1:213–229. doi: 10.2174/157488806776956904. [DOI] [PubMed] [Google Scholar]
- 12.Rui YF, Lui PP, Ni M, Chan LS, Lee YW, Chan KM. Mechanical loading increased BMP-2 expression which promoted osteogenic differentiation of tendon-derived stem cells. J Orthop Res. 2011;29:390–396. doi: 10.1002/jor.21218. [DOI] [PubMed] [Google Scholar]
- 13.Clark CA, Li TF, Kim KO, Drissi H, Zuscik MJ, Zhang X, O'Keefe RJ. Prostaglandin E2 inhibits BMP signaling and delays chondrocyte maturation. J Orthop Res. 2009;27:785–792. doi: 10.1002/jor.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arikawa T, Omura K, Morita I. Regulation of bone morphogenetic protein-2 expression by endogenous prostaglandin E2 in human mesenchymal stem cells. J Cell Physiol. 2004;200:400–406. doi: 10.1002/jcp.20031. [DOI] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Moore AE, Young LE, Dixon DA. MicroRNA and AU-rich element regulation of prostagland in synthesis. Cancer Metastasis Rev. 2011;30:419–435. doi: 10.1007/s10555-011-9300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virdi AS, Cook LJ, Oreffo RO, Triffitt JT. Modulation of bone morphogenetic protein-2 and bone morphogenetic protein-4 gene expression in osteoblastic cell lines. Cell Mol Biol (Noisy-le-grand) 1998;44:1237–1246. [PubMed] [Google Scholar]
- 18.Qu B, Xia X, Yan M, Gong K, Deng S, Huang G, Ma Z, Pan X. miR-218 is involved in the negative regulation of osteoclastogenesis and bone resorption by partial suppression of p38MAPK-c-Fos-NFATc1 signaling: Potential role for osteopenic diseases. Exp Cell Res. 2015;338:89–96. doi: 10.1016/j.yexcr.2015.07.023. [DOI] [PubMed] [Google Scholar]