

Abstract
The aim of the present study was to investigate the effect of glucocorticoids in osteoblasts and to examine the role of β-ecdysterone in the pathogenesis of glucocorticoid-induced osteoporosis. Osteoblasts were induced from bone marrow mesenchymal stem cells, which were isolated from C57BL/6 mice. Cell viability and apoptosis of osteoblasts were measured by Cell Counting Kit-8 and flow cytometry analysis, respectively. The expression of related genes and proteins was measured by reverse transcription quantitative polymerase chain reaction and western blot analysis respectively. Dose-dependent decreases in the cell proliferation and differentiation were observed in dexamethasone (Dex)-treated osteoblasts, evidenced by downregulation in the activity of alkaline phosphatasedecreased expression levels of Runt-related transcription factor 2 and osteocalcin, and upregulated expression of RANK ligand. Dex also induced apoptosis and inhibited autophagy of osteoblasts, evidenced by upregulated B-cell lymphoma 2 (Bcl-2)-associated X protein/Bcl-2 ratio and the activation of mammalian target of rapamycin (mTOR), and decreased expression levels of Beclin-1, autophagy protein 5 and microtubule-associated protein 1 light chain 3 II. The effects on cell proliferation, apoptosis and autophagy induced by Dex were reversed by β-ecdysterone in a dose-dependent manner. Therefore, β-ecdysterone may be a promising candidate drug for the treatment of osteoporosis through inducing osteoblast autophagic activity by inactivating mTOR.
Keywords: steoporosis, β-ecdysterone, apoptosis, autophagy, mammalian target of rapamycin
Introduction
Glucocorticoids (GCs) are frequently used clinically to treat patients with organ transplantation and malignancies, in addition to diseases, including inflammation, cancer and autoimmune disorders (1,2). Patients exposed to GC therapy exhibit osteonecrosis and osteoporosis via the direct responses of osteoblasts, ultimately reducing bone formation (3,4) and resulting in changes to cell number and function. It has been reported that GCs can inhibit osteoblast proliferation and differentiation, and induce apoptosis (5,6); however the molecular mechanism underlying the involvement of GCs in the differentiation and apoptosis of osteoblasts remains to be fully elucidated. The effect of GCs on osteoblasts involves the suppression of Wnt/β-catenin signaling (7) and activation of caspase-3, a common downstream effector of several apoptotic signaling pathways (8).
Based on similarities in structure and function, β-ecdysterone, a major component of Chinese herbal medicines, is likely to have similar protective effects to estrogen, which is the primary therapeutic strategy for the treatment of osteoporosis (9). Although β-ecdysterone has various activities, including anabolic and hepatoprotective effects, promoting the synthesis of collagen protein, protecting cells from apoptosis and inducing their proliferation (10–13), the signaling pathway involved in theβ-ecdysterone-mediated protective effects remains to be elucidated.
Autophagy is a process of programmed cell death, which is essential for cell growth, survival, differentiation and homeostasis (14). Autophagy can protect cells from apoptosis by removing oxidatively damaged organelles, and excess autophagy can destroy cellular components. To date, it has been recognized that estradiol inhibits osteoblast apoptosis via the promotion of autophagy through the estrogen receptor (ER)-extracellular signal-regulated protein kinase-mammalian target of rapamycin (mTOR) pathway (15). GCs have been shown to induce autophagy in an animal model of osteoporosis through decreasing the ratio ofmicrotubule-associated protein 1 light chain 3 (LC3)-I to LC3-II (16). However, whether autophagy is present in GC-induced osteoblasts, and whether autophagy is involved in the protective effect of β-ecdysterone against apoptosis in osteoblasts remain to be elucidated.
The aim of the present study was to investigate whether β-ecdysterone can improve the dexamethasone (Dex)-induced effects on osteoblast differentiation and apoptosis, and whether this is dependent on mTOR signaling pathway-dependent autophagy. Preliminary investigations were also performed to determine the potential role of β-ecdysterone in the prevention of osteoporosis.
Materials and methods
Culture of bone marrow mesenchymal stem cells (BMSCs) and osteoblasts
The BMSCs were obtained from 4-5-week-old male C57BL/6 mice purchased from Slacom Experimental Animal Company (Shanghai, China), as previously described (9). When the cells reached 80% confluence, they were subcultured until the third passage. The BMSCs were cultured in DMEM/F12 (Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% FBS, 100 U/ml penicillin and 100 mg/l streptomycin (Thermo Fisher Scientific, Inc.) in an incubator at 37°C, 100% humidity and 5% CO2. The induction and culture of osteoblasts were performed as follows: Briefly, the BMSCs were seeded in 24-well plates at a density of 5×104 and cultured in modified DMEM/F12 containing 10% FBS, vitamin C (50 mg/l; Aladdin Shanghai Biochemical Technology Co., Ltd., Shanghai, China), β-sodium glycerophosphate (10−3 mol/l; Sigma-Aldrich; Merck Millipore, Darmstadt, Germany), 100 U/ml penicillin, 100 mg/l streptomycin and Dex (10−8 mol/l; Sigma-Aldrich; Merck Millipore) at 37°C in a humidified atmosphere of 5% CO2. After 14 days, the BMSCs were stained using a BCIP/NBT Alkaline Phosphatase Color Development kit (cat. no. C3206; Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer's instructions. The osteoblasts were cultured in DMEM/F12 in the same manner as the BMSCs. The present study was approved by the ethics committee of Xiaoshan TCM Hospital (Hangzhou, China).
CCK-8 assay
The osteoblasts were plated in 96-well plates (5×104 cells/well) and treated with Dex in the absence or presence of10−5 mol/l alendronate (Ale) or β-ecdysterone at various concentrations (10−7−10−5 mol/l) at 37°C. At the end of treatment (24, 48 and 72 h), 10 µl of CCK-8 solution was added to each well of the plate. Following incubation at 37°C for 1 h, the culture medium containing CCK-8 solution was removed. The absorbance at 450 nm was measured using a spectrophotometric microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Measurement of alkaline phosphatase (ALP) activity
The osteoblasts were plated in 24-well plates and treated with Dex in the absence or presence of Ale (10−5 mol/l) or β-ecdysterone at various concentrations (10−7−10−5 mol/l). At the end of treatment, ALP activity was measured using an ALP assay kit according to the manufacturer's instructions.
Cell apoptosis assay
To detect apoptosis, an APC Annexin V Apoptosis Detection kit (cat. no. 88-8007; eBioscience, San Diego, CA, USA) was used according to the manufacturer's instructions. Briefly, the osteoblasts were treated with Dex in the presence or absence of Ale (10−5 mol/l) or β-ecdysterone at various concentrations (10−7−10−5 mol/l), washed with PBS and incubated with 195 µl Annexin V-APC and 5 µl propidium iodide for 15 min at 4°C, prior to analysis by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
The osteoblasts were cultured in DMEM/F12 and treated with Dex in the presence or absence of Ale (10−5 mol/l) or β-ecdysterone at various concentrations (10−7−10−5 mol/l). Total RNA was isolated from cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and detected by agarose gel electrophoresis. As the template for PCR, cDNA was synthesized from 5 µg of RNA using AMV reverse transcriptase (Fermentas; Thermo Fisher Scientific, Inc.). The RT-qPCR procedure were performed using SYBR® Green 10X Supermix (Takara Bio, Inc., Otsu, Japan) in a 25 µl total volume (12.5 µl SYBR® Green 10X SuperMix, 0.5 µl forward primer, 0.5 µl reverse primer, 9.5 µl ddH2O and 2 µl cDNA) on a Roche Light Cycler® 480 II system (Roche Diagnostics, Basel, Switzerland). The primer sequences are shown in Table I. The gene expression levels were normalized to the levels of GADPH. The gene expression levels were calculated using the 2−ΔΔCq method (17).
Table I.
Primes sequences used in the present study.
| Gene | Sequence |
|---|---|
| Runx2 | F:5′-TTTGCCCTCATCCTTCAC-3′ |
| Runx2 | R:5′-GCTTCTGCTACCACTCTAAC-3′ |
| OCN | F:5′-GCCCTAAAGCCAAACTCTG-3′ |
| OCN | R:5′-GCTGCTGTGACATCCATAC-3′ |
| RANKL | F:5′-GGGCTTTCAAAGTTCAGG-3′ |
| RANKL | R:5′-GGGCTGTGAGTTTCATAC-3′ |
| Beclin-1 | F:5′-GCATCCTTTCCCTCTTTC-3′ |
| Beclin-1 | R:5′-CCACAAGCATCTCATCTC-3′ |
| ATG5 | F:5′-AACCACCTTGAGTCAGGACAAC-3′ |
| ATG5 | R:5′-TCGGCTGCATTGCATTTCAC-3′ |
| GAPDH | F:5′-ATCACTGCCACCCAGAAG-3′ |
| GAPDH | R:5′-TCCACGACGGACACATTG-3′ |
Runx2, Runt-related transcription factor 2; OCN, osteocalcin; RANKL, RANK ligand; ATG5, autophagy protein 5.
Protein extraction and western blot analysis
The osteoblasts were lysed in ice-cold radioimmunoprecipitation assay buffer (Beyotime Institute of Biotechnology) containing 0.01% protease and phosphatase inhibitor (Sigma-Aldrich; Merck Millipore), and incubated on ice for 30 min. The protein concentration was assessed using a bicinchoninic acid protein assay kit (cat. no. PICPI23223; Thermo Fisher Scientific, Inc.). The cell lysate was centrifuged at 12,000 × g at 4°C for 10 min, and the proteins in the supernatant were obtained (20–30 µg) and separated on a 10% SDS-PAGE gel, followed by electrophoretic transfer onto a polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). Following blocking with 5% skim milk, the membrane was incubated at 4°C overnight with the following rabbit monoclonal antibodies: Anti-runt-related transcription factor 2 (Runx2; cat. no. ab23981, 1:1,000), anti-osteocalcin (OCN; cat. no. ab93876, 1:500), anti-RANK ligand (RANKL; cat. no. ab45039, 1:500), anti-Beclin 1 (cat. no. ab55878, 1:500) and anti-autophagy protein 5 (ATG5; cat. no. ab108327, 1:1,000) (all from Abcam, Cambridge, MA, USA), anti-B-cell lymphoma 2 (Bcl-2; cat. no. sc-492, 1:100; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-Bcl-2-associated X protein (Bax; cat. no. sc-493, 1:100; Santa Cruz Biotechnology, Inc.), anti-LC3-II (cat. no. 2775, 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-phosphorylated (p)-mTOR (cat. no. 2971, 1:1,000), anti-mTOR (cat. no. 2983, 1:1,000) and anti-GAPDH (cat. no. 5174, 1:1,500) (all from Cell Signaling Technology, Inc.). The blots were then incubated for 1 h at 37°C with goat anti-mouse or anti-rabbit secondary antibody (cat. nos. A0216 and A0208; Beyotime Institute of Biotechnology) and intensities were measured using enhanced chemiluminescence (Thermo Fisher Scientific, Inc.). The signal intensity was determined using ImageJ software version 1.46 (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation. One-way analysis of variance, followed by an unpaired two-tailed t-test was used for statistical analysis (GraphPad Prism 5 software; GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of β-ecdysterone on Dex-induced osteoblast differentiation
At 14 days post-induction, osteoblasts development was observed, evidenced by purple staining present in the extracellular matrix, which was measured using an inverted phase contrast microscope (magnification, ×100; Fig. 1A). This suggested that the osteoblast had been successfully induced from the BMSCs, which were then used in the following experiments.
Figure 1.
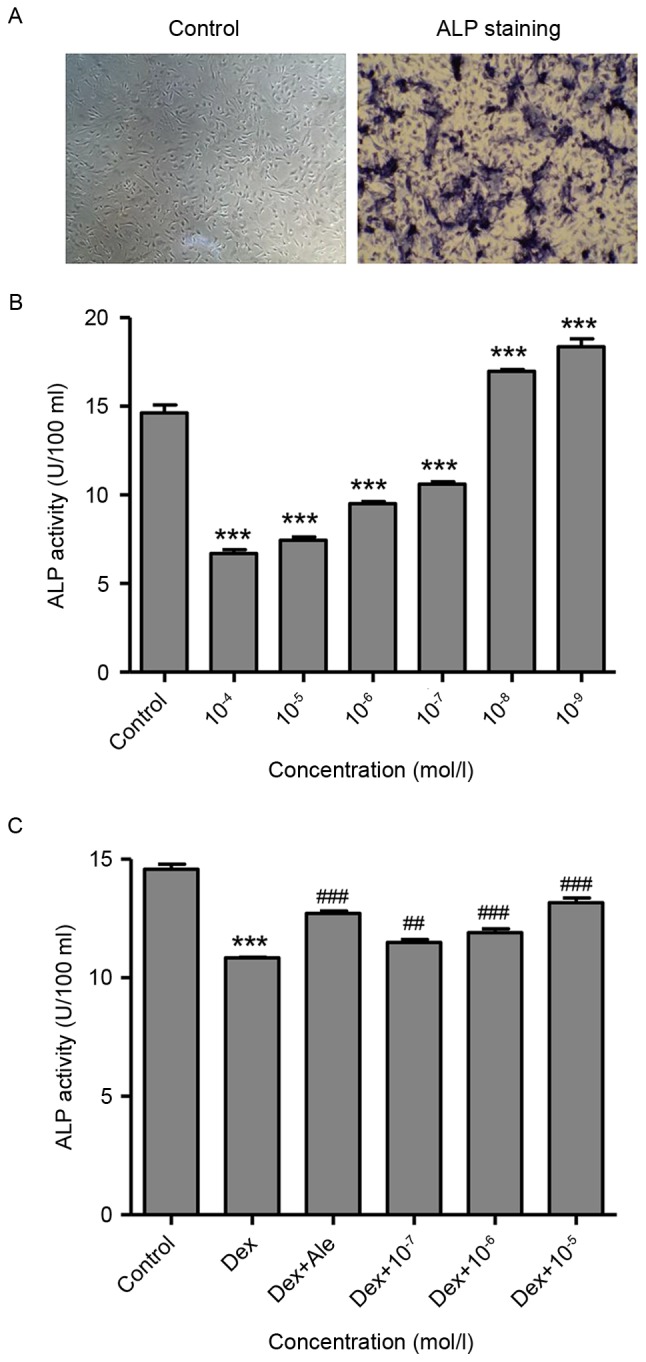
β-ecdysterone reverse Dex-induced suppression of osteoblast differentiation. (A) After 14 days, osteoblast were fixed and stained, and images were captured (magnification, ×100). (B) Osteoblasts were treated with Dex at various concentrations (10−9−10−4 mol/l), and ALP activity was measured. (C) Osteoblasts were treated with Dex (10−6 mol/l) in the presence or absence of β-ecdysterone at various concentrations (10−7−10−5 mol/l), and ALP activity was measured. ***P<0.001, vs. control group; ##P<0.01 and ###P<0.001 vs. Dex group. Dex, dexamethasone; Ale, alendronate; ALP, alkaline phosphatase.
To investigate the effect of Dex on the activity of ALP, which is an early marker of osteogenic differentiation, the osteoblasts were treated with various concentrations of Dex (10−9−10−4 mol/l) for 72 h, following which the activity of ALP was measured. As shown in Fig. 1B, Dex significantly stimulated the activity of ALP at lower concentrations (10−9−10−8 mol/l) and suppressed the activity of ALP at higher concentrations (10−7−10−4 mol/l), compared with the untreated control osteoblasts, and these effects were dose-dependent. However, the cells treated with β-ecdysterone at concentrations of 10−7−10−5 mol/l significantly stimulated the activity of ALP, compared with the cells treated with 10−6 mol/l of Dex alone, which was similar to activity in the cells treated with 10−5 mol/l of alendronate (Ale), which has long been used as a drug for the treatment of osteoporosis and was included as a positive control (Fig. 1C).
To confirm the effect of β-ecdysterone on the induction of osteogenic differentiation, RT-qPCR and western blot analyses were performed to examine gene expression. The results showed that 10−6 mol/l of Dex significantly decreased the expression levels of Runx2 and OCN, but increased the expression of RANKL at the mRNA and protein levels (Fig. 2A-D). However, treatment of the cells with β-ecdysterone at various concentrations (10−7−10−5 mol/l) dose-dependently reversed the changes in the expression of Runx2, OCN and RANKL induced by 10−6 mol/l of Dex. This was similar to the effects of 10−5 mol/l Ale on the osteoblasts (Fig. 2A-D). These results indicated that β-ecdysterone reversed the Dex-induced suppression of osteoblast differentiation.
Figure 2.

β-ecdysterone reverses Dex-induced expression levels of Runx2, OCN and RANKL. Osteoblasts were treated with Dex (10−6 mol/l) in the presence or absence of β-ecdysterone at various concentrations (10−7−10−5 mol/l), and the expression levels of (A) Runx2, (B) OCN and (C) RANKL were measured using reverse transcription-quantitative polymerase chain reaction and (D) western blot analyses. ***P<0.001 vs. control group; #P<0.05, ##P<0.01 and ###P<0.001 vs. Dex group. Runx2, Runt-related transcription factor 2; OCN, osteocalcin; RANKL, RANK ligand; Dex, dexamethasone; Ale, alendronate.
Effect of β-ecdysterone on Dex-induced osteoblast proliferation
The present study also examined the effects of β-ecdysterone on Dex-induced osteoblast proliferation using a CCK-8 assay. The results showed that Dex treatment for 24 h markedly increased cell proliferation at lower concentrations (10−9−10−8 mol/l; Fig. 3A) and suppressed cell proliferation at higher concentrations (10−6−10−4 mol/l) after 48 and 72 h treatment, compared with the control (Fig. 3B and C). With the exception of the results over 24 h (Fig. 3D), significant increases in cell proliferation were observed in the cells treated with β-ecdysterone at various concentrations (10−7−10−5 mol/l) for 48 and 72 h (Fig. 3E and F), compared with the cells treated with 10−6 mol/l of Dexalone. This was similar to the effect of treatment with 10−5 mol/l Ale.
Figure 3.

β-ecdysterone reverses the Dex-induced suppression of osteoblast proliferation. Osteoblasts were treated with Dex for (A) 24, (B) 48 and (C) 72 hat various concentrations (10−9−10−4 mol/l), and cell proliferation was measured using a CCK-8 assay. Osteoblasts were treated with Dex (10−6 mol/l) in the presence or absence of β-ecdysterone for (D) 24, (E) 48 and (F) 72 h at various concentrations (10−7−10−5 mol/l), and cell proliferation was measured using a CCK-8 assay. *P<0.05, **P<0.01 and ***P<0.001 vs. control group; #P<0.05 and ##P<0.01 vs. Dex group. Dex, dexamethasone; OD, optical density.
Effect of β-ecdysterone on Dex-induced osteoblast apoptosis
To investigate the effect of β-ecdysterone on Dex-induced osteoblast apoptosis, the osteoblasts were treated with 10−6 mol/l of Dex for 72 h, and cell apoptosis was measured. As shown in Fig. 4A and B, Dex increased cell apoptosis 7-fold compared with that in the control. However, the cells treated with β-ecdysterone at various concentrations (10−7−10−5 mol/l) exhibited significantly reduced cell apoptosis, compared with the cells treated with 10−6 mol/l of Dex alone, which was similar to the effect of treatment with 10−5 mol/l of Ale.
Figure 4.

β-ecdysterone reverses Dex-induced osteoblast apoptosis. Osteoblasts were treated with Dex (10−6 mol/l) in the presence or absence of β-ecdysterone at various concentrations (10−7−10−5 mol/l), and (A) cell apoptosis was measured using Annexin V-FITC/propidium iodide and flow cytometry. (B) Quantification of percentages of apoptotic cells. (C) Expression levels of Bax and Bcl-2 were measured using western blot analysis. ***P<0.001 vs. control group; ###P<0.001 vs. Dex group. Dex, dexamethasone; Ale, alendronate; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein.
To confirm the inducive effect of β-ecdysterone on osteogenic apoptosis, western blot analysis was performed to examine protein expression levels. The results showed that 10−6 mol/l of Dex significantly increased the ratio of Bax/Bcl-2, compared with that in the control (Fig. 4C). However, in the cells treated with β-ecdysterone at various concentrations (10−7−10−5 mol/l), the alteration of the Bax/Bcl-2 ratio induced by 10−6 mol/l of Dex was reversed in a dose-dependent manner, which was similar to the effect of treatment with 10−5 mol/l of Ale (Fig. 4C). These results suggested that β-ecdysterone reversed the effect of Dex-induced osteoblast apoptosis.
Effect of β-ecdysterone on Dex-induced osteoblast autophagy
To investigate the effect of β-ecdysterone on Dex-induced osteoblast autophagy, the osteoblasts were treated with 10−6 mol/l of Dex for 48 h, following which the expression of autophagy-related genes were measured using RT-qPCR and western blot analyses. As shown in Fig. 5A-C, 10−6 mol/l of Dex significantly activated p-mTOR, and decreased the expression levels of Beclin-1, ATG5 and LC3-II, compared with those in the control. However, in the cells treated with β-ecdysterone at various concentrations (10−7−10−5 mol/l), the changes in the expression levels of p-mTOR, Beclin-1, ATG5 and LC3-II induced by 10−6 mol/l of Dex were reversed in a dose-dependent manner, which was similar to the results in the osteoblasts treated with 10−5 mol/l of Ale. These results suggested that β-ecdysterone reversed Dex-induced osteoblast autophagy, possibly through inactivation of the TOR signaling pathway.
Figure 5.

β-ecdysterone reverses Dex-induced osteoblast autophagy. Osteoblasts were treated with Dex (10−6 mol/l) in the presence or absence of β-ecdysterone at various concentrations (10−7−10−5 mol/l), and the expression levels of (A) Beclin-1, (B) ATG, and (C) LC3-II and p-mTOR/mTOR were measured using reverse transcription-quantitative polymerase chain reaction and/or western blot analyses. ***P<0.001 vs. control group; ##P<0.01 and ###P<0.001 vs. Dex group. Dex, dexamethasone; Ale, alendronate; ATG5, autophagy protein 5; mTOR, mammalian target of rapamycin; p-mTOR, phosphorylated mTOR; LC3-II, microtubule-associated protein 1 light chain 3 II.
Discussion
All diseases involving bone loss have a common pattern, in that osteoclasts are specifically responsible for bone resorption and, when osteoporosis occurs, osteoblast activity is overcome by osteoclast activity. In the present study, it was demonstrated that Dex inhibited the osteogenic differentiation, proliferation and autophagy of osteoblasts in vitro, and induced apoptosis. The protective effect of β-ecdysterone on Dex-induced osteoporosis was also shown. These findings provided the first evidence, to the best of our knowledge, that β-ecdysterone isolated from traditional Chinese medicine can induce osteogenic differentiation and autophagy, and this effect is dependent on the inactivation of the mTOR signaling pathway.
During differentiation in vitro, osteoblast phenotypic markers appear in the following order: Formation of type I collagen matrix, expression of ALP and then Runx2, secretion of OCN, and mineralization of bone nodules. Runx2 is an important transcription factor, which controls several subsequent osteogenic differentiation-related gene expression events, and mice with Runx2-knockout have severe bone defects (18). OCN is another marker expressed later during differentiation (19). RANKL is a membrane-bound tumor necrosis factor receptor, which is expressed on osteoblast precursor cells and recognize RANK on the osteoclast surface through direct cell-cell interaction (20). The results of the present study showed that Dex at higher concentrations inhibited osteoblast differentiation through decreasing the activity of ALP, reducing the expression of Runx2 and OCN, and increasing the expression of RANKL, which was in accordance with the results of other studies (21). However, higher concentrations of Dex promoted osteoblast differentiation with increased ALP activity and expression of Runx2 (22). These findings suggest that the effects of Dex on impairing or favoring osteoblastic differentiation appear to be dependent on its concentration and experimental conditions. β-ecdysterone reversed the effect of Dex on osteoblast differentiation and proliferation in a dose-dependent manner, which was similar to previous findings that β-ecdysterone stimulates the activity of ALP, and induce the expression of Runx2 and OCN (9).
Apoptosis and autophagy are two important programmed mechanisms to control cell death and survival. Autophagy can either delay the occurrence of apoptosis (23) or promote apoptosis (24). The balance between apoptosis and autophagy is critical to the cellular homeostasis. In the present study, it was found that Dex induced osteoblast apoptosis and inhibited autophagy, with an increased Bax/Bcl-2 ratio and decreased expression of autophagy-related markers, including Beclin-1, ATG5 and LC3-II. In partial agreement with these findings, there are other studies that a number of apoptotic cells and increased autophagic activity are generated in response to Dex, suggesting that autophagy is a self-protective response to GCs (16,25). Previous studies have shown that the overexpression of Beclin-1 in HeLa cells results in an increased number of autophagosome (26), and that Beclin-1 deficiency in mice reduces autolysosome production (27). This indicated that the expression of Beclin-1 accompanies autophagy. The deletion of ATG5 has been found to reduce the protein expression of LC3-II in cortical bone of mice, indicating decreased autophagic activity (28).
β-ecdysterone has previously been shown to mediate the neuroprotective effect in SH-SY5Y cells through preventing mitochondria-dependent apoptosis via inhibiting the activation of p53, which was limited within concentrations of 1–10 µM (13). In agreement, the results of the present study showed that β-ecdysterone at different concentrations (10−7−10−5 mol/l) reduced osteoblastic apoptosis in a dose-dependent manner. In addition, a sustained state of cellular stress, including exposure to a high dose or chronic GC treatment, can result in failure of the induction of autophagy. Based on the results of the present study, it was hypothesized that osteoblasts initially responded to the Dex-induced stress by increasing the number of cells undergoing autophagy, which was in accordance with a previous report that β-ecdysterone prevented the GC-induced increase in autophagy of BMSCs and whole bone (29).
It has been reported that mTOR is a negative regulator of autophagy and that the inhibition of p-mTOR has been used widely as an indicator of activation of the autophagy pathway (30,31). AMPK controls the osteogenic differentiation of human mesenchymal stem cells through early mTOR inhibition-mediated autophagy and late activation of the Akt/mTOR signaling axis (32). The results of the present study showed that Dex dose-dependently increased the expression of p-mTOR, which was reversed by β-ecdysterone treatment. This suggested that β-ecdysteron may act upon the autophagic pathway via an mTOR-dependent mechanism.
In conclusion, the present study demonstrated that Dex inhibited proliferation, differentiation and autophagy, and induced apoptosis of osteoblasts. β-ecdysterone significantly reversed Dex-inducedapoptosis through inducing autophagy via mTOR signaling. These results provide pre-clinical support for further assessment of the ability of β-ecdysterone treatment to improve skeletal fragility resulting from excess GC.
Acknowledgements
This study was supported by the Science and Technology Project from Traditional Chinese Medicine of Zhejiang Province (grant no. 2015ZA174) and the Social Development Major Scientific and Technological Projects in Xiaoshan District of Hangzhou City (grant no. 2014207).
References
- 1.Whittier X, Saag KG. Glucocorticoid-induced Osteoporosis. Rheum Dis Clin North Am. 2016;42:177–189. doi: 10.1016/j.rdc.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Buehring B, Viswanathan R, Binkley N, Busse W. Glucocorticoid-induced osteoporosis: An update on effects and management. J Allergy Clin Immunol. 2013;132:1019–1030. doi: 10.1016/j.jaci.2013.08.040. [DOI] [PubMed] [Google Scholar]
- 3.Fukushima W, Fujioka M, Kubo T, Tamakoshi A, Nagai M, Hirota Y. Nationwide epidemiologic survey of idiopathic osteonecrosis of the femoral head. Clin Orthop Relat Res. 2010;468:2715–2724. doi: 10.1007/s11999-010-1292-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol. 2010;6:82–88. doi: 10.1038/nrrheum.2009.259. [DOI] [PubMed] [Google Scholar]
- 5.Yun SI, Yoon HY, Jeong SY, Chung YS. Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3beta. J Bone Miner Metab. 2009;27:140–148. doi: 10.1007/s00774-008-0019-5. [DOI] [PubMed] [Google Scholar]
- 6.Li H, Qian W, Weng X, Wu Z, Li H, Zhuang Q, Feng B, Bian Y. Glucocorticoid receptor and sequential P53 activation by dexamethasone mediates apoptosis and cell cycle arrest of osteoblastic MC3T3-E1 cells. PLoS One. 2012;7:e37030. doi: 10.1371/journal.pone.0037030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun. 2005;329:177–181. doi: 10.1016/j.bbrc.2005.01.117. [DOI] [PubMed] [Google Scholar]
- 8.O'Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–1841. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 9.Gao L, Cai G, Shi X. Beta-ecdysterone induces osteogenic differentiation in mouse mesenchymal stem cells and relieves osteoporosis. Biol Pharm Bull. 2008;31:2245–2249. doi: 10.1248/bpb.31.2245. [DOI] [PubMed] [Google Scholar]
- 10.Cahlíková L, Macáková K, Chlebek J, Host'álková A, Kulhánková A, Opletal L. Ecdysterone and its activity on some degenerative diseases. Nat Prod Commun. 2011;6:707–718. [PubMed] [Google Scholar]
- 11.Syrov VN, Khushbaktova ZA, Nabiev AN. An experimental study of the hepatoprotective properties of phytoecdysteroids and nerobol in carbon tetrachloride-induced liver lesion. Eksp Klin Farmakol. 1992;55:61–65. (In Russian) [PubMed] [Google Scholar]
- 12.Zou Y, Wang R, Guo H, Dong M. Phytoestrogen β-ecdysterone protects PC12 cells against MPP+−induced neurotoxicity in vitro: Involvement of PI3K-Nrf2-regulated pathway. Toxicol Sci. 2015;147:28–38. doi: 10.1093/toxsci/kfv111. [DOI] [PubMed] [Google Scholar]
- 13.Pan Z, Niu Y, Liang Y, Zhang X, Dong M. β-Ecdysterone protects SH-SY5Y cells against 6-hydroxydopamine-induced apoptosis via mitochondria-dependent mechanism: Involvement of p38MAPK-p53 signaling pathway. Neurotox Res. 2016;30:453–466. doi: 10.1007/s12640-016-9631-7. [DOI] [PubMed] [Google Scholar]
- 14.Yang Z, Klionsky DJ. Eaten alive: A history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang YH, Chen K, Li B, Chen JW, Zheng XF, Wang YR, Jiang SD, Jiang LS. Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway. Apoptosis. 2013;18:1363–1375. doi: 10.1007/s10495-013-0867-x. [DOI] [PubMed] [Google Scholar]
- 16.Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, Biswas SK, Lo WK, Jiang JX. Glucocorticoid-induced autophagy in osteocytes. J Bone Miner Res. 2010;25:2479–2488. doi: 10.1002/jbmr.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Komori T. Regulation of osteoblast differentiation by Runx2. Adv Exp Med Biol. 2010;658:43–49. doi: 10.1007/978-1-4419-1050-9_5. [DOI] [PubMed] [Google Scholar]
- 19.Choo MK, Yeo H, Zayzafoon M. NFATc1 mediates HDAC-dependent transcriptional repression of osteocalcin expression during osteoblast differentiation. Bone. 2009;45:579–589. doi: 10.1016/j.bone.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473:139–146. doi: 10.1016/j.abb.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu FB, Wang JY, Zhang YL, Quan RF, Yue ZS, Zeng LR, Zheng WJ, Hou Q, Yan SG, Hu YG. Curculigoside regulates proliferation, differentiation, and pro-inflammatory cytokines levels in dexamethasone-induced rat calvarial osteoblasts. Int J Clin Exp Med. 2015;8:12337–12346. [PMC free article] [PubMed] [Google Scholar]
- 22.Hamidouche Z, Haÿ E, Vaudin P, Charbord P, Schüle R, Marie PJ, Fromigué O. FHL2 mediates dexamethasone-induced mesenchymal cell differentiation into osteoblasts by activating Wnt/beta-catenin signaling-dependent Runx2 expression. FASEB J. 2008;22:3813–3822. doi: 10.1096/fj.08-106302. [DOI] [PubMed] [Google Scholar]
- 23.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Canu N, Tufi R, Serafino AL, Amadoro G, Ciotti MT, Calissano P. Role of the autophagic-lysosomal system on low potassium-induced apoptosis in cultured cerebellar granule cells. J Neurochem. 2005;92:1228–1242. doi: 10.1111/j.1471-4159.2004.02956.x. [DOI] [PubMed] [Google Scholar]
- 25.Conradie MM, de Wet H, Kotze DD, Burrin JM, Hough FS, Hulley PA. Vanadate prevents glucocorticoid-induced apoptosis of osteoblasts in vitro and osteocytes in vivo. J Endocrinol. 2007;195:229–240. doi: 10.1677/JOE-07-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268–277. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qu X, Yu J, Bhagat G, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nollet M, Santucci-Darmanin S, Breuil V, Al-Sahlanee R, Cros C, Topi M, Momier D, Samson M, Pagnotta S, Cailleteau L, et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy. 2014;10:1965–1977. doi: 10.4161/auto.36182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai W, Jiang L, Lay YA, Chen H, Jin G, Zhang H, Kot A, Ritchie RO, Lane NE, Yao W. Prevention of glucocorticoid induced bone changes with beta-ecdysone. Bone. 2015;74:48–57. doi: 10.1016/j.bone.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: How pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fiorini C, Menegazzi M, Padroni C, Dando I, Dalla Pozza E, Gregorelli A, Costanzo C, Palmieri M, Donadelli M. Autophagy induced by p53-reactivating molecules protects pancreatic cancer cells from apoptosis. Apoptosis. 2013;18:337–346. doi: 10.1007/s10495-012-0790-6. [DOI] [PubMed] [Google Scholar]
- 32.Pantovic A, Krstic A, Janjetovic K, Kocic J, Harhaji-Trajkovic L, Bugarskib D, Trajkovica V. Coordinated time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cellsc. Bone. 2013;52:524–531. doi: 10.1016/j.bone.2012.10.024. [DOI] [PubMed] [Google Scholar]