

Abstract
Epilepsy is a common disease of the central nervous system. Tetanic spasms and convulsions are the key symptoms exhibited during epileptic seizures. However, the majority of patients have a significant post-seizure silence following a serious seizure; the underlying molecular neural mechanisms in this burst interval are unclear. The aim of the present study was to reveal the effect and role of calcium-activated potassium channels during this seizure interval silence period. Cyclothiazide (CTZ) was used to establish the seizure model in rat hippocampal cultured neurons, then the after-burst hyperpolarization (ABH) activities were recorded using the patch clamp technique. By comparing the amplitude and duration of hyperpolarizations, the present study analyzed the association between epileptiform bursts and ABHs when treated with different concentrations of CTZ. In addition, apamin and iberiotoxin were used for pharmacological tests. An intracranial electroencephalogram (EEG) recording was also performed when the CTZ experiments were repeated on animals. The experimental results revealed that treatment with high levels of CTZ induced larger ABHs and was associated with stronger burst activities, which suggested a positive correlation between ABH and epileptiform burst. Apamin, an antagonist of small conductance calcium-activated potassium (SK) channels, decreased the amplitude of ABH; however, reduced ABH was associated with enhanced burst activity, in burst probability and burst strength. These results revealed an important role of SK channels in the formation of ABH and in the inhibition of burst activity. Iberiotoxin, an antagonist of big conductance calcium-activated potassium (BK) channels, had no significant effect on ABH and burst activity. In addition, a positive correlation was identified between burst duration and ABH parameters. An intracellular calcium chelator impaired the amplitude of ABH; however, it did not affect the burst parameters. The rat cortical EEG recordings also exhibited a similar positive correlation between the duration of epileptic burst and after burst depression. Collectively, the results indicate that ABH may serve in the physiological feedback system to reduce the strength of epileptic hyperexcitation, a process in which SK channels are important.
Keywords: after burst hyperpolarization, epileptiform burst, cyclothiazide convulsant model, small conductance calcium-activated potassium channel, hippocampal culture
Introduction
Epilepsy is one of the most prevalent and serious central nervous system disorders that affects ~1% of the global population (1). In previous epilepsy studies, burst activities were often followed by a depression of spike discharges, termed after burst depression (ABD) or postictal depression (2–6). This low-amplitude neuronal activity is believed to result from the factors that lead to seizure auto-termination. This depression period is very important, as it can extend the interval duration of burst activity and also reduce the intensity of seizure onset. A number of antiepileptic drugs can enhance this depression period in patients with epilepsy (7).
Although a number of previous studies concentrated on the dynamics of ion channels and other proteins in epileptic seizure, little is known regarding the development of postictal depression (8,9). One possibility is that there may be a large influx of calcium ions during this burst activity, which may activate calcium-activated potassium channels and thus mediate the hyperpolarized current and lead to postictal depression (10,11). Calcium-activated potassium channels are a group of potassium channels that are different to voltage-gated potassium channels as they are activated by elevated intracellular calcium concentration. They can be divided into 2 types: Big conductance calcium-activated potassium channels known as BK channels, and the small conductance type known as SK channels (12). BK channels contribute to the fast and medium after hyperpolarization (AHP) activities that follow a single action potential (AP). In contrast, SK channels have a small conductance with a longer open duration, and they contribute to the slow AHP activities of neurons following a series of APs (13–15).
In addition to this hypothesis involving calcium-activated potassium channels, there are still a number of other potential mechanisms. One such mechanism may involve membrane shunting (16), which is associated with a decrease in neuronal input resistance, a consequence of seizure onset. Membrane shunting decreases the synaptic current effect on the postsynaptic membrane and also decreases the coupling effect of gap junctions. The energy failure hypothesis has also been suggested as another potential mechanism. Depletion of energy substances, such as ATP and glucose, may be an important factor in ABD. However, it has been previously established that hypoglycemia and hypoxia may lead to a seizure onset rather than seizure control (17).
An increasing number of previous studies have demonstrated that SK channels have an important role in epileptogenesis (18,19). In the central nervous system, SK channels couple directly with N-methyl-D-aspartate (NMDA) receptors in dendritic spines. Calcium influx through NMDA receptors during membrane depolarization can activate SK channels (20). In addition, activation of SK channels may reduce epileptiform activity in an acute model of epilepsy (21). By contrast, suppression of SK channels may increase burst activity in the hippocampal CA3 area of a brain slice (22,23). Thus, SK channels may be critical in the formation of postictal depression and spontaneous seizure control. In the present study, cyclothiazide (CTZ) was used to establish the epilepsy model. CTZ is a novel potent convulsant with the advantage of lower excitotoxicity (24) and it is capable of inducing a stable burst activity in vitro and in vivo (24,25). CTZ induces convulsion via a number of different physiological processes. The most important effect is the allosteric modulation of AMPA and GABA receptors (26). In addition, there are also a number of other mechanisms that contribute to the convulsant effect of CTZ, including glutamate depletion (27), dynamic changes in ion concentrations (28), persistent activation of hyperpolarization activated cation conductance (2,21,23) and activation of acid-sensitive ion channels (3). On the basis of these previous studies, it was hypothesized that SK channels may enhance the amplitude and duration of ABD and control the strength of seizures.
Materials and methods
Animals
The electroencephalographic experiments were performed on 250–300 g male Sprague Dawley rats, 8 weeks old. These rats were supplied by Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). In this study, 20 rats were used for the experiments. All animals were maintained in air-conditioned rooms with a controlled temperature at 23±2°C, 40–60% humidity, under a 12 h light/dark cycle with lights on from 7:00 a.m. to 7:00 p.m. Animals were housed separately in plastic cages and were supplied with water and food ad libitum. All experiments conformed to the guidelines provided by the Institutional Committee of Laboratory Animals, Fudan University and to Chinese government regulations. The present study was approved by the Ethics Committee of Fudan University (Shanghai, China).
Surgery and intracranial cortical electroencephalogram (EEG) quantification
The protocols applied for surgery and electroencephalography were performed as previously described (25,29,30). Briefly, as CTZ cannot pass through the blood-brain barrier, all studies were performed using an intraventricular injection of CTZ. Animals were mounted on stereotaxic apparatus, with a guide cannula (22GA; Plastics One, Roanoke, VA, USA) planted into the lateral ventricle (AP 0.3 mm, ML 1.3 mm, DP 4.0 mm). The intraventricular CTZ injection was performed through a soft plastic catheter and this guide cannula. To record the cortical EEG, two little screw electrodes were embedded into the skull to reach the surface of the brain. One screw was located in the left cortex above the hippocampus (AP-3.8 mm, ML 2.0 mm), which served as the recording electrode, while the other was located above the forehead, serving as the reference electrode. Finally, two screws were attached to a connector to create a link to the Neurolog system (Digitimer Ltd., Hertfordshire, UK). The signals were visualized by Spike2 software version 6.04 (Cambridge Electronic Design Ltd., Cambridge, UK) and recorded on a PC computer through an A-D converter, CED 1401 micro (Cambridge Electronic Design Ltd.).
Primary hippocampal neuronal culture
Primary hippocampal neurons were collected from embryonic day 18 Sprague-Dawley rat fetuses, as described in our previous studies (31–33). Briefly, the fetuses were first dissected to collect the hippocampi. The tissues were rinsed in cold Hanks' balanced salt solution, then digested with 0.05% trypsin-EDTA solution for 15–20 min at 37°C. Following digestion, single cells were subsequently isolated by trituration with 1 ml plastic pipette tips in the plating medium (Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 10% F12 and 25 m/ml penicillin/streptomycin, termed DF12; purchased from Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA), and then a centrifugation step of 1,000 × g at 4°C, for 8 min. Then, following rinsing twice by DF12, cells were seeded onto poly-D-lysine (0.1 mg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) pre-coated coverslips at a density of 40,000–60,000 cells/cm2. Following cell culture for 1 day, half of the media were changed to neurobasal medium, containing 2% B27, 2 mM GlutaMAX™ and 25 ml/ml penicillin/streptomycin. AraC (1 µM; Sigma-Aldrich; Merck KGaA) was added at 6 days following initial plating. Cell cultures were fed once every 3 days by replacing half of the medium. All cultures were maintained at 37°C and in 5% CO2 incubators. Following 14–21 days after plating, cultured neurons were used for electrophysiology. All cell culture reagents were obtained from Invitrogen; Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Drug treatment
To induce robust burst activity in cultured neurons, stock CTZ (Tocris Bioscience, Bristol, UK) solution (20 mM in DMSO) was added into the culture medium to produce a final concentration of either 5 or 20 µM CTZ. A similar volume of DMSO (1:1,000) was added into the cultured medium as a control to form the DMSO group. The two CTZ groups (5 or 20 µM CTZ) and the DMSO group, with 12–32 neurons in each group, were incubated for 2 h at 37°C prior to patch clamp recordings. The SK channel antagonist, apamin (APM; Tocris Bioscience, Bristol, UK), and the BK channel antagonist, iberiotoxin (IBTX; Tocris Bioscience), were dissolved in water to produce two stock solutions (500 µM), which were then added into the bath solution to produce final concentrations of 500 nM, forming +APM and +IBTX groups of neurons cultured with CTZ and DMSO, respectively.
Electrophysiology
Whole-cell recordings were performed in current clamps and held at −70 mV as described previously (24,32,34,35). Patch pipettes were pulled from borosilicate glass with a pipette puller (P-97; Sutter Instrument, Novato, CA, USA), and then fire polished to a resistance of 3–6 MΩ. The pipette was filled with following internal solution: 125 mM K-gluconate, 10 mM KCl, 10 mM Hepes, 10 mM Tris-phosphocreatine, 4 mM MgATP, 0.5 mM Na2GTP (pH 7.3 adjusted with KOH) and ~305 mM Osm. An additional 30 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA) was added into the internal solution for experiments involving high EGTA recordings, with the same pH and osmolarity as normal; resulting in the EGTA and Normal groups in CTZ and DMSO cultured neurons, respectively. The recording chamber was continuously perfused with a bath solution, containing the following components: 128 mM NaCl, 30 mM Glucose, 25 mM Hepes, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2 (pH 7.3 adjusted with NaOH) and ~315 mM Osm. Electrical signals were digitized and sampled at a frequency of 5 kHz with Digidata 1440A and Multiclamp 700B amplifier (Axon Instruments; Molecular Devices, LLC, Sunnyvale, CA, USA), using pCLAMP version 10.2 (Axon Instruments; Molecular Devices, LLC). Data were filtered at 2 kHz and analyzed with Clampfix version 10 (Axon Instruments; Molecular Devices, LLC). A large depolarization shift was defined as a membrane potential ≥10 mV depolarizing shift, with a ≥300 msec time period. An epileptiform burst was defined by ≥5 consecutive APs overlaying on top of a large depolarization shift (24). A neuron with burst activities was defined by ≥2 repeated bursts occurring within 6 min of recording (25,32). Data within a group have ≥3 different groups of neurons. A bursting neuron was not calculated in the analysis if the recording time was <6 min, even if it had bursts ≥2. Only bursting neurons were used for further analyses, including burst frequency, burst duration, amplitude and duration of after burst hyperpolarization (ABH) and the AP frequency in burst.
Statistical analysis
All of the ABH and burst results recorded in cultured neurons were analyzed using Clampfit version 10 (Axon Instruments; Molecular Devices, LLC). A 5-min baseline was recorded for every neuron, and the baseline was calculated to confirm the membrane potential value of the mean and noise using the software. The value of the mean ± noise represents the baseline region. ABH amplitude is calculated by baseline mean minus the negative peak potential of ABH. ABH duration represents the lasting time of the hyperpolarizing activity. There was potential of high level of noise in the potential recording; therefore, the present study used the baseline region rather than the single baseline value to reduce the influence of noise as much as possible. Similarly, the baseline definition in burst duration is also the same as that in ABH duration. Finally, the AP frequency in burst was calculated by dividing the AP number by the burst duration.
Pooled data are all presented as the mean ± standard error mean and were analyzed using GraphPad Prism version 4 (GraphPad Software, Inc., La Jolla, CA, USA). An unpaired Student's t-test was used for comparisons between 2 groups. In Figs. 1C and D, and 2C-E, an unpaired Student's t-test was also performed for the comparisons group-by-group. A χ2 test was used for statistical analysis of percentage changes. Linear regression was also tested using GraphPad software in linear correlation analysis. P<0.05 was considered to indicate a statistically significant difference.
Figure 1.

ABH amplitude increased; however, the duration decreased following the induction of seizures with CTZ. Representative recording traces in the different groups, with (A) a single burst and (B) enlarged images of the ABH period indicated by the boxed area. (C) There was a significant increase in ABH amplitude in the 5 µM CTZ (n=35 bursts in 10 neurons) and 20 µM CTZ (n=30 bursts in 10 neurons) treatment groups when compared with the DMSO group (n=47 bursts in 11 neurons). There was also a significant increase when comparing the two CTZ groups. (D) Two CTZ groups had a significantly lower ABH duration when compared with the DMSO group. Data are presented as the mean ± standard error mean. *P<0.05, **P<0.01 and ***P<0.001 vs. DMSO; ##P<0.01 vs. 5 µM CTZ. ABH, after burst hyperpolarization; CTZ, cyclothiazide.
Figure 2.

CTZ induced burst activities and increased the bursting strength. (A) Representative current clamp recording traces in the control DMSO, 5 µM CTZ and 20 µM CTZ groups. (B) Percentage of neurons exhibiting burst activities in the three groups. Incubation with 20 µM CTZ significantly increased the percentage, whereas 5 µM CTZ produced no significant difference in burst activities. The numbers written in the bars represent the number of cells (number with burst activities/total number). (C) Frequency of bursting activities occurring. The average number of bursts every minute increased following incubation with 20 µM CTZ. A significant difference was observed between the 20 µM CTZ (n=10) and DMSO control (n=11) groups and also between the 5 µM CTZ (n=10) and 20 µM CTZ groups. (D) AP frequency in burst. The 5 µM CTZ (n=35) and 20 µM CTZ (n=30) groups were significantly increased when compared with the DMSO group (n=47). Additionally, there was a significant difference between the two CTZ groups. (E) Average burst duration in recording. The two CTZ groups had a shorter burst time when compared with the DMSO group [the same recording numbers as in (D)]. Data are presented as the mean ± standard error mean. *P<0.05, **P<0.01 and ***P<0.001 vs. DMSO; ##P<0.01 and ###P<0.001 vs. 5 µM CTZ. CTZ, cyclothiazide; AP, action potential.
Results
Epileptiform burst activities are followed by a slow after hyperpolarization in cultured hippocampal neurons
To investigate the underlying cellular mechanisms of postictal depression, cultured hippocampal neurons were used as the model. A relatively silent period was observed following burst activity. Each period of burst activity, whether it was spontaneous or CTZ-induced, was followed by a hyperpolarized potential (Fig. 1A and B). ABH activity was analyzed in the neurons that exhibited burst activity following every period burst activity. The results revealed that the amplitude of ABH significantly increased in the 5 µM CTZ treatment group (5.17±0.26 mV; n=35 ABH activities from 10 neurons; P<0.001; Fig. 1C) and in the 20 µM CTZ treatment group (7.03±0.65 mV; n=30 ABH activities from 10 neurons; P<0.001; Fig. 1C), when compared with DMSO group (3.24±0.26 mV; n=47 ABH activities from 11 neurons; Fig. 1C). The high concentration group (20 µM CTZ) also had a significantly greater amplitude when compared with the low concentration (5 µM) CTZ group (P<0.01; Fig. 1C). However, the ABH duration (length of the ABH period) significantly decreased in the CTZ treatment groups (5 µM CTZ: 5.00±0.39 sec; n=35 ABH activities; P<0.01; 20 µM CTZ: 5.47±0.27 sec; n=30 ABH activities; P<0.05) when compared with the DMSO group (8.69±1.04 sec; n=47 ABH activities; Fig. 1D).
CTZ also induced stronger epileptiform burst activities in cultured neurons. The percentage of neurons exhibiting burst activities was compared with the burst frequency (the average number of burst per minute), in neurons with and without CTZ (Fig. 2A). Treatment with a low concentration (5 µM) of CTZ did not significantly affect the percentage of bursting neurons (5 µM CTZ: 52.6%, n=19 neurons; DMSO: 34.4%, n=32 neurons; P>0.05; Fig. 2B), nor did it have an effect on the bursting frequency (5 µM CTZ: 2.07±0.35/min, n=10 bursting neurons; DMSO: 2.02±0.61/min, n=11 bursting neurons; Fig. 2C). However, a high concentration (20 µM) of CTZ significantly increased the percentage of bursting neurons to 83.3% (n=12 neurons; P<0.01; Fig. 2B) as well as the burst frequency (4.20±0.54/min; n=10 bursting neurons; P<0.05; Fig. 2C). The burst frequency between the two different CTZ concentrations was also significantly different (P<0.01). These results indicated that CTZ induced burst activity and increased the bursting frequency.
Subsequently, the changes in single burst activity were observed. The average burst duration was compared with the average AP frequency within each burst in all groups, to indicate the dynamics of burst strength. When compared with the control (DMSO: 9.45±0.63 Hz, n=47 bursting activities), the low and high CTZ concentration treatments significantly increased the AP frequency (5 µM CTZ: 14.04±0.68 Hz, n=35 bursting activities; 20 µM CTZ: 29.49±0.96 Hz, n=30 bursting activities; both P<0.001; Fig. 2D). The two CTZ groups were also significantly different from one another (P<0.001; Fig. 2D). These results indicate that CTZ may increase burst strength in a dose-dependent manner; however, as only two different concentrations were evaluated in the present study further investigation is required. In addition, the average burst duration of the two CTZ groups were significantly reduced when compared with the control (DMSO: 2.11±0.14 sec, n=47 bursting activities; 5 µM CTZ: 1.19±0.07 sec, n=35 bursting activities; 20 µM CTZ: 1.40±0.10 sec, n=30 bursting activities; both P<0.001; Fig. 2E).
APM-sensitive SK channels are involved in the formation of ABH
The intracellular mechanism underlying the ABH was also investigated. A large influx of calcium is known to occur in neurons during burst activity (11); therefore, it was hypothesized that calcium-activated potassium channels may be involved in the regulation of ABH. Previous studies have demonstrated that SK channels have a greater association with slow and long-lasting hyperpolarization activities than BK channels (13,36). Therefore, SK channels may have an important role in ABH. BK and SK channels were pharmacologically investigated using their respective antagonists.
To determine whether SK channels regulate ABH and burst activities, the SK channel-sensitive antagonist APM was used to treat the cells cultured with and without 5 µM CTZ pretreatment. SK channel inhibition with 500 nM APM induced a significant reduction in the amplitude of ABH, in the CTZ pretreatment (1.43±0.11 mV; n=61; P<0.001 vs. 5 µM CTZ; Fig. 3A-D) and the DMSO control (1.43±0.08 mV; n=64; P<0.001 vs. DMSO; Fig. 3A-D) groups. However, APM did not affect the duration of ABH in the presence of CTZ (4.93±0.36 sec; n=61; P>0.05; Fig. 3E), although it did reduce the duration in the DMSO control group (3.44±0.22 sec; n=64; P<0.001; Fig. 3E). These findings indicate that SK channels may mediate the majority of ABH activities. The inhibition of SK channels reduced the amplitude of ABH by ~50–70%. The APM insensitive potentials suggest there may be additional receptors and ion channels involved.
Figure 3.

APM impaired ABH; however, it also enhanced burst activities. (A-C) Comparisons between the original recording traces with and without 500 nM APM in the control and 5 µM CTZ group for 2 h. The boxes indicate the area that has been enlarged in the subsequent images. (D) APM significantly suppressed the amplitude of ABH in the control group (n=47 without APM; n=64 with APM) and the CTZ group (n=35 without APM; n=61 with APM). (E) APM reduced the duration of ABH in the control group; however, this was not observed in the CTZ group. (F) APM increased the percentage of neurons exhibiting burst activities in the control group; however, not in the CTZ group. The numbers written in the bars represent the number of cells (number with burst activities/total number). (G) APM significantly increased the burst number per minute, in the control group (n=11 without APM; n=10 with APM) and the CTZ group (n=10 without APM; n=9 with APM). (H) AP frequency in burst increased with the application of APM, in the control and CTZ groups. (I) Burst duration significantly decreased in the control and CTZ groups when the bath solution contained APM. Data in D, E, H and I represent the number of burst activities, whereas that in (G) represent the number of neurons. Data are presented as the mean ± standard error mean. *P<0.05, ***P<0.001 DMSO vs. DMSO + APM; ##P<0.01, ###P<0.001 CTZ vs. CTZ + APM. APM, apamin; ABH, after burst hyperpolarization; CTZ, cyclothiazide; AP, action potential.
Given the inhibitory effect of APM on ABH, the present study investigated the influence of this effect on burst activity itself. Although it is thought that SK channels are able to mediate the amplitude of ABH, the effect of SK channels on burst activities remains to be elucidated. In this series of experiments, a 5 µM dose of CTZ was selected to treat cultured neurons. The effects of APM/IBTX were assessed through the comparison of DMSO/DMSO + APM/DMSO + IBTX and the similar comparison of CTZ/CTZ + APM/CTZ + IBTX. Following CTZ (5 µM) treatment, whole cell recordings were performed in bath solution containing 500 nM APM to block SK channels. Once the SK channels were blocked, the percentage of neurons exhibiting burst activities increased in the control and CTZ groups (from 34.4 to 71.4% in control and 52.6 to 75.0% in CTZ), however, only the control group presented significant differences (P<0.05 in χ2 test; Fig. 3F). Burst frequency and burst strength also significantly increased following blocking of the SK channels with APM. Burst frequency increased from 2.02±0.61 to 6.08±0.81/min in the control groups (P<0.001) and from 2.07±0.35 to 5.31±1.10/min in the CTZ groups (P<0.01; Fig. 3G). The frequency of APs in burst also increased from 9.45±0.63 to 11.14±0.40 Hz in control (P<0.05) and from 14.04±0.68 to 20.12±0.79 Hz in CTZ group (P<0.001; Fig. 3H). However, the burst duration significantly decreased in the control and CTZ groups (both P<0.001; Fig. 3I), which was similar to the results observed following the initial application of CTZ (Fig. 2E). The results suggest that APM may enhance intrinsic excitability by increasing the burst number per minute and also burst strength.
In contrast to APM, the BK channel antagonist IBTX did not alter the majority of the parameters in ABH and burst activities. In the DMSO control and 5 µM CTZ treated groups, the ABH amplitude, ABH duration, burst probability and percentage, burst frequency and burst inner AP frequency all had no statistically significant differences, when comparing those with and without IBTX, respectively (Fig. 4A-H). The burst duration was the only parameter that was significantly different. In the presence of IBTX, the duration increased from 2.11±0.14 sec (n=47) to 2.55±0.12 sec (n=22) in the control group (P<0.05) and from 1.19±0.07 sec (n=35) to 2.02±0.20 sec (n=23) in the CTZ group (P<0.001; Fig. 4I). A previous study has reported similar results (37). However, the results of the present study revealed that blockage of the SK channels is the principal induction of this excitatory effect.
Figure 4.

IBTX did not alter the majority of burst and ABH parameters, with the exception of burst duration. (A-C) Representative original traces with and without 500 nM IBTX in the DMSO control and 5 µM CTZ groups for 2 h. The boxes indicate the area that has been enlarged in the subsequent images. No statistical difference was identified between groups treated with and without IBTX in terms of (D) ABH amplitude, (E) ABH duration, (F) bursting neuron percentage [the numbers written in the bars represent the number of cells (number with burst activities/total number)], (G) bursting number per minute and (H) AP frequency in burst, in the control (n=47 bursts in 11 neurons without IBTX; n=22 bursts in 5 neurons with IBTX) and CTZ groups (n=35 bursts in 10 neurons without IBTX; n=23 bursts in 5 neurons with IBTX). (I) IBTX significantly increased the burst duration in the two groups. Data are presented as the mean ± standard error mean. *P<0.05 DMSO vs. DMSO + IBTX; ###P<0.001 CTZ vs. CTZ + IBTX. IBTX, iberiotoxin; ABH, after burst hyperpolarization; CTZ, cyclothiazide; AP, action potential.
Correlations between epileptiform bursts and ABH
Further analysis highlighted the associations between burst and ABH. The present study compared the associations between burst duration (the time a burst continued) and the remaining three parameters: ABH amplitude, ABH duration and the product of ABH amplitude and duration (Fig. 5). The last parameter was used to represent the total hyperpolarizing charges in ABH activity, although the unit is mV*s, not the unit of electric charge, coulomb. The patch clamp recording mode used was ‘current clamp’, which injects current to hold the membrane potential at −70 mV. Therefore, the original data collected are the membrane potential values. However, it is difficult to accurately calculate the electrical charge value as the accurate calculation of neuronal electrical charge requires a measurement of cellular membrane conductance of each recorded neuron. As this membrane conductance is highly dispersed and fluctuated in different neurons, so actually the assessment parameter mV*s is only a carefully selected approximation to the electrical charge value of neurons.
Figure 5.

A positive correlation was identified for burst duration, ABH amplitude, ABH duration, and the product of ABH amplitude and duration, in the DMSO and CTZ groups. (A-C) Correlation analyses in the DMSO group, of burst duration with either ABH amplitude, ABH duration or the product. A significant positive linear correlation was observed in all parameters when using linear regression. (D-F) Correlation analyses in the 5 µM CTZ group. Only ABH amplitude and the product exhibited a significant positive linear correlation with burst duration. (G-I) Correlation analyses in 20 µM CTZ group. All analyses produced significant positive correlations. Data points in each figure represent all bursting activities from the neurons of each group. ABH, after burst hyperpolarization; CTZ, cyclothiazide.
The results suggest that there may be a correlation between ABH duration and burst duration. Despite the different concentrations of CTZ treatment (Figs. 1D and 2E), or APM blockage (Fig. 3E and I), ABH duration exhibited similar changes to those of burst duration. Therefore, it was hypothesized that there may be a positive correlation between single cell activities. The experimental results revealed a significant positive correlation between burst duration and ABH amplitude (R2=0.39; P<0.001; Fig. 5A), ABH duration (R2=0.18; P<0.01; Fig. 5B), and the product of ABH amplitude and duration (R2=0.34; P<0.001; Fig. 5C) in the DMSO group. The 5 µM CTZ-treated group also exhibited a significant positive correlation in the majority of the parameters; however, the differences were not as significant as those exhibited by the DMSO group. The correlation between burst duration and ABH amplitude (R2=0.15; P<0.05; Fig. 5D), and burst duration and the product (R2=0.15; P<0.05 Fig. 5F) were significant; however, there was no significant association between burst duration and ABH duration (R2=0.09; P=0.08; Fig. 5E). In the 20 µM CTZ group, all three parameters were significant (burst duration vs. ABH amplitude: R2=0.62; P<0.001; Fig. 5G; burst duration vs. ABH duration: R2=0.28; P<0.01; Fig. 5H; burst duration vs. the product: R2=0.56; P<0.001; Fig. 5I). These results indicate that there is normal physiological feedback between ABH activities and burst activities, as after a long burst a large period of ABH follows in order to terminate the burst and prevent any new burst activity.
EGTA-induced reduction of intracellular calcium concentration reduces ABH; however, it does not affect burst activities
To determine the mechanisms underlying neuronal regulation in ABH and burst, the present study investigated intracellular calcium. Intracellular calcium is involved in the ABH process and burst activities. The [Ca2+]i concentration is associated with the strength of AP and burst and with the activation of BK and SK channels (38–41). EGTA, a calcium selective chelator, was added to the pipette solution to reduce the intracellular calcium concentration. The ABH and bursts with and without 30 mM EGTA, in the DMSO control and 5 µM CTZ pretreatment groups were subsequently compared. The results demonstrated that there was a significant reduction in ABH amplitude in the control (from 3.24±0.26 to 1.10±0.08 mV; P<0.001) and CTZ treatment groups (from 5.17±0.26 to 2.39±0.22 mV; P<0.001; Fig. 6A-D). However, burst duration was affected differently. The application of 30 mM EGTA decreased the burst duration in the DMSO control (Normal: 8.69±1.04 sec, n=47; 30 mM EGTA: 2.17±0.15 sec, n=35; P<0.001); however, no decrease was observed in the CTZ group (Normal: 5.00±0.39 sec, n=35; 30 mM EGTA: 4.19±0.60 sec, n=22; Fig. 6E). In parallel to the reduction of ABH, 30 mM EGTA did not alter the majority of the burst activity parameters, including burst neuron percentage (Fig. 6F), burst frequency (Fig. 6G) and the burst inner AP frequency (Fig. 6H). The only significant difference identified was in the burst duration of the control group (from 2.11±0.14 to 1.04±0.07 sec; P<0.001; Fig. 6I).
Figure 6.

High EGTA pipette solution reduced the ABH; however, it did not alter the burst probability and strength. (A-C) Representative original recording traces of neurons treated with normal and 30 mM EGTA pipette solution in the DMSO control and 5 µM CTZ groups. The boxes indicate the area that has been enlarged in the subsequent images. (D) High EGTA significantly reduced the amplitude of ABH in the control (normal: n=47 bursts in 11 neurons; 30 mM EGTA: n=35 bursts in 6 neurons) and CTZ groups (normal: n=35 bursts in 10 neurons; 30 mM EGTA: n=22 bursts in 5 neurons). (E) High EGTA only reduced ABH duration in the control group, not in the CTZ group. There were no significant differences identified between two types of pipette solution, in control and CTZ groups for (F) bursting neuron percentage [the numbers written in the bars represent the number of cells (number with burst activities/total number)], (G) bursting number per minute and (H) AP frequency in burst. (I) High EGTA significantly decreased the burst duration in the control group; however, not in the CTZ group. Data are presented as the mean ± standard error mean. ***P<0.001 DMSO/normal vs. DMSO/EGTA; ###P<0.001 CTZ/normal vs. CTZ/EGTA. EGTA, ethylene glycol-bis(b-aminoethyl ether)-N,N,N',N'-tetraacetic acid; ABH, after burst hyperpolarization; CTZ, cyclothiazide; AP, action potential.
Epileptiform burst activities are followed by a long-lasting ABD in the hippocampal neurons of anaesthetized rats
CTZ has been previously reported to generate epileptiform burst activities in hippocampal neurons in vitro and in vivo (24,30) and it also induced seizure behavior in rats (29). The present study investigated whether the results from the cultured neurons also occurred in the rat model. CTZ (5 µmol; 5 µl; intracerebroventricular injection) induced epileptiform burst activities in all 6 rats tested, which is in agreement with previous reports (24,30). During the 3 h intracranial EEG recording, CTZ induced a total of 20 bursts in all 6 rats, with a mean burst duration of 15.26±1.04 sec (range, 10.37 to 26.04 sec; Fig. 7A). Immediately following burst activities, there was a long period of depression, or ABD, which lasted for 8.80±1.08 sec on average (range, 2.59 to 22.06 sec; n=20 bursts in 6 rats; Fig. 7B and C). A decrease in electroencephalographic power occurred following burst activity. Prior to burst activity, the power was recorded between 5–20 Hz in 20 sec, which was larger than that observed following burst activities (Fig. 7D). Further analysis revealed a significant positive correlation between burst duration and ABD duration in all bursts tested (Fig. 7E), which is similar to the correlation identified in cultured neurons. The goodness of fit in the two groups has a R2 value of 0.26 and P=0.022. These results indicate that the strength of the ABD may be directly associated with the strength of the epileptiform burst activities in seizure.
Figure 7.

CTZ induced epileptiform burst activities in the hippocampal area followed by a period of activity depression in anaesthetized rats. (A-C) Representative electroencephalogram recordings revealing the period of ABD following burst activities (indicated by arrows). The box in recording (B) indicates the area that has been enlarged in image (C). (D) Power analysis of baseline activities in 20 sec demonstrated that there was a decrease in power after burst activities when compared with before burst activities. (E) A positive correlation was identified between burst duration and ABD duration. Bursts with a longer burst duration also had a longer ABD duration. Data points in (E) represent the values of single burst activities. CTZ, cyclothiazide; ABD, after burst depression.
Discussion
The results of the present study are schematically represented in Fig. 8. Burst activities elevate the concentration of intracellular calcium, which can conversely enhance the strength of burst. In addition, the elevated calcium concentration activates the SK channels to enhance the amplitude of ABH. This enhanced hyperpolarization activity can inhibit or reduce burst activity. In the present study, when CTZ was applied a number of excitatory processes were activated, inducing burst activities. Bursts increased the calcium influx, enhanced the intracellular calcium concentration and produced a greater ABH. When APM was applied, the SK channels were blocked and thus ABH was impaired. As a result, the neurons had more excitatory activities and generated stronger bursts. However, when IBTX was applied and the BK channels were blocked, no significant differences were identified in the different parameters. Therefore, the blockage of the SK channels was identified as the principal induction of this excitatory effect. EGTA (30 mM) reduced intracellular calcium concentration, and impaired ABH and burst activities. However, the reduction in ABH may have elevated burst activities. Thus, the effects of ABH and intracellular calcium may offset each other. The majority of the burst activity parameters were not altered (Fig. 8). In our experimental results, the relationship between BK channel and bursting activities were not identified. Thus, IBTX and BK channels were excluded from Fig. 8.
Figure 8.
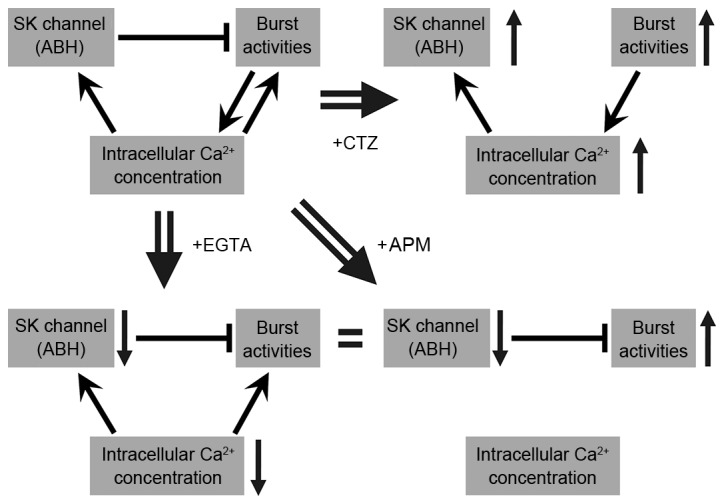
A schematic representation of the associations identified among burst activities, ABH and intracellular calcium concentration in the present study. The schematic summarizes and analyzes the results from all of the experiments performed in the present study. The three different conditions, CTZ, APM and EGTA, are listed separately within the schematic. ABH, after burst hyperpolarization; CTZ, cyclothiazide; APM, apamin; EGTA, ethylene glycol-bis (β-aminoethyl ether)-N,N,N',N'-tetraacetic acid; SK, small conductance calcium-activated potassium channels.
In the present study, cultured hippocampal neurons treated with DMSO or without any treatment also exhibited spontaneous burst activities (DMSO: 34.4%, n=32; Fig. 2B; No treatment: 38.6%, n=44; data not shown). This is likely due to the environment changing from culture media to the recording bath solution, which may have stimulated the cultured neurons. However, the experimental protocol included measures to counter this limitation to a certain degree, such as heating the bath solution to ~37°C prior to recording.
The results of the present study appear to be paradoxical as when the ABH duration and burst duration are reduced, ABH amplitude and burst strength are increased; a phenomenon that is difficult to account for. Cellular regulation may exist in order to reduce the ion influx when the activity strength is enhanced in ABH and burst. One possible explanation is that enhanced ABH and burst may alter the membrane potential faster than in the control, thereby shortening the duration. The currents increase in burst and ABH, producing faster depolarization and hyperpolarization, shortening the duration.
SK channels are Ca2+−activated K+ channels with small conductance (10–20 pS) and are widely expressed in vertebrate neurons and other tissues (42). SK channel-mediated hyperpolarized K+ currents have an important role in regulating neuronal excitability in physiological (43) and pathological conditions (18,23). In the present study, the cultured neurons exhibited a depression in ABH amplitude when treated with APM. This suggested that AHP may be mediated by SK channels as APM can block the majority of SK channel activation. This finding has also been observed in different epilepsy models (22,23). For example, Fernández de Sevilla et al (23) found that in 4-aminopyridine (4-AP) epilepsy model and in Mg(2+)-free induced model, slow AHP (sAHP) was also inhibited, particularly in hippocampal CA3 pyramidal neurons. 1-Ethyl-2-benzimidazolinone (1-EBIO), a modulator of SK channels, can slow the deactivation of SK channels and prolong their opening time (44). It has been previously demonstrated that 1-EBIO may potentially inhibit burst activity in vitro (21) and in vivo (45). The results of the present study demonstrated this same effect in the opposite manner. The previous study activated SK channels by agonist, and then assessed the excitability, positively regulated the function of SK channels, whereas the present study inhibited SK channels by antagonist apamin, and then assessed the bursting activities through a negative regulation.
Small conductance calcium-activated potassium channels are the most important factor in the generation of ABH. The elevation in ABH amplitude is dependent on the increase in intracellular calcium concentration; stronger burst activity requires more calcium ions, thereby inducing a larger ABH.
Although the association between burst strength and ABH is relatively well-established, there are different hypotheses to explain the underlying mechanisms. One such hypothesis is that burst activities may activate some intracellular protein kinases (25) such as protein kinase A (PKA), which can regulate the kinetics of SK channels. A previous study has demonstrated that SK channel activity is regulated by PKA (46). Therefore, the future studies will focus on intracellular protein kinases, in order to elucidate which protein kinase regulates the dynamics of SK channel function during the process of epileptic seizure.
In the present study, the correlation between the strength of burst and ABH is, to the best of our knowledge, a relatively new discovery in the field, but not a totally novel finding. These positive correlations reveal that ABH may aid neurons to reduce the damage of hyperexcitation during bursts. This process is potentially mediated by SK channels. However, in the control and CTZ groups, there was still a part of hyperpolarization (~1.4 mV) that was insensitive to APM. These APM insensitive potentials indicate that other receptors and ion channels may participate, such as cyclic nucleotide-gated nonspecific cation channels. Previous studies (15,36) have suggested that SK channels may only contribute to those ABHs with the duration of ~100 msec and not to those slow ABHs with the duration of several sec. Based on these new experimental results, it is possible that SK channels contribute, at least in part, to second-level slow ABHs. Thus, mice with complete knockout of all SK channels may still have second-level slow ABHs. In addition, SK channels as intrinsic auto-balancers, may have an important role in suppressing burst activity. Collectively, these findings suggest that SK channels may be potential targets for designing novel antiepileptic drugs.
The results in this animal model provide novel in vivo evidence. However, the in vivo postictal ABD in EEG and the in vitro ABH in a single neuron are clearly distinct phenomena. One is neuronal activity and the other is the activity of the brain region. Although they hold similar characteristics, a straightforward association between them cannot be identified. The EEG represents the population activities of neurons in a brain region, whereas the neuronal population activities are based on the activity of a single neuron. Thus, the recordings from the EEG and patch clamp were compared to determine the similarities and differences between them. This comparison may reveal the characteristics of ABD and ABH; however, this may be insufficient or incomplete. In addition, the present study, and those performed previously, lack sufficient in vivo evidence to suggest that blocking SK channels enhances burst activity. The in vivo evidence represents the results from model animals and also those from human patients (47,48). For example, Beck et al (47) investigated 34 dentate gyrus granule cells from 11 patients with temporal lobe epilepsy and found that APM (50 nM) was only able to inhibit ~13% Ca(2+)-dependent K+ currents. However, it remains to be elucidated, with further patient-based studies that investigate the dynamics of SK channels in epileptic seizures.
In conclusion, although there were a number of limitations in the present study, one being that APM could only be administered in in vitro experiments, it is evident that APM may be able to suppress ABH to enhance the excitability of the neuronal network in cultured neurons. However, the effect of APM on ABD in anesthetic animal models remains to be elucidated. Future studies will determine the effect of APM on the duration of ABD and elucidate any changes in the CTZ epilepsy animal model.
Acknowledgements
The present study was supported by grants from the Nature Science Foundation of China (grant nos. 81171224, 31471027 and 31771188) and the Science and Technology Commission of Shanghai Municipality (grant nos. 13DJ1400302) to Professor Yun Wang. The authors would like to acknowledge the technical support provided by Dr Zheng Wu (Fudan University) for whole cell patch clamp recording.
References
- 1.Browne TR, Holmes GL. Epilepsy. N Engl J Med. 2001;344:1145–1151. doi: 10.1056/NEJM200104123441507. [DOI] [PubMed] [Google Scholar]
- 2.Lado FA, Moshé SL. How do seizures stop? Epilepsia. 2008;49:1651–1664. doi: 10.1111/j.1528-1167.2008.01669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziemann AE, Schnizler MK, Albert GW, Severson MA, Howard MA, III, Welsh MJ, Wemmie JA. Seizure termination by acidosis depends on ASIC1a. Nat Neurosci. 2008;11:816–822. doi: 10.1038/nn.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Gompel JJ, Bower MR, Worrell GA, Stead M, Chang SY, Goerss SJ, Kim I, Bennet KE, Meyer FB, Marsh WR, et al. Increased cortical extracellular adenosine correlates with seizure termination. Epilepsia. 2014;55:233–244. doi: 10.1111/epi.12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Afra P, Jouny CC, Bergey GK. Termination patterns of complex partial seizures: An intracranial EEG study. Seizure. 2015;32:9–15. doi: 10.1016/j.seizure.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferastraoaru V, Schulze-Bonhage A, Lipton RB, Dümpelmann M, Legatt AD, Blumberg J, Haut SR. Termination of seizure clusters is related to the duration of focal seizures. Epilepsia. 2016;57:889–895. doi: 10.1111/epi.13375. [DOI] [PubMed] [Google Scholar]
- 7.Mula M, Sander JW. Negative effects of antiepileptic drugs on mood in patients with epilepsy. Drug Saf. 2007;30:555–567. doi: 10.2165/00002018-200730070-00001. [DOI] [PubMed] [Google Scholar]
- 8.Spencer SS, Spencer DD. Implications of seizure termination location in temporal lobe epilepsy. Epilepsia. 1996;37:455–458. doi: 10.1111/j.1528-1157.1996.tb00591.x. [DOI] [PubMed] [Google Scholar]
- 9.Bragin A, Penttonen M, Buzsáki G. Termination of epileptic afterdischarge in the hippocampus. J Neurosci. 1997;17:2567–2579. doi: 10.1523/JNEUROSCI.17-07-02567.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alger BE, Nicoll RA. Epileptiform burst afterhyperpolarization: Calcium-dependent potassium potential in hippocampal CA1 pyramidal cells. Science. 1980;210:1122–1124. doi: 10.1126/science.7444438. [DOI] [PubMed] [Google Scholar]
- 11.Albowitz B, König P, Kuhnt U. Spatiotemporal distribution of intracellular calcium transients during epileptiform activity in guinea pig hippocampal slices. J Neurophysiol. 1997;77:491–501. doi: 10.1152/jn.1997.77.1.491. [DOI] [PubMed] [Google Scholar]
- 12.Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/S0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, McBain CJ. Potassium conductances underlying repolarization and after-hyperpolarization in rat CA1 hippocampal interneurones. J Physiol. 1995;488:661–672. doi: 10.1113/jphysiol.1995.sp020998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- 15.Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Llinas R, Baker R, Sotelo C. Electrotonic coupling between neurons in cat inferior olive. J Neurophysiol. 1974;37:560–571. doi: 10.1152/jn.1974.37.3.560. [DOI] [PubMed] [Google Scholar]
- 17.Auer RN. Progress review: Hypoglycemic brain damage. Stroke. 1986;17:699–708. doi: 10.1161/01.STR.17.4.699. [DOI] [PubMed] [Google Scholar]
- 18.Oliveira MS, Skinner F, Arshadmansab MF, Garcia I, Mello CF, Knaus HG, Ermolinsky BS, Otalora LF, Garrido-Sanabria ER. Altered expression and function of small-conductance (SK) Ca(2+)-activated K+ channels in pilocarpine-treated epileptic rats. Brain Res. 2010;1348:187–199. doi: 10.1016/j.brainres.2010.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz R, Kirschstein T, Brehme H, Porath K, Mikkat U, Köhling R. Network excitability in a model of chronic temporal lobe epilepsy critically depends on SK channel-mediated AHP currents. Neurobiol Dis. 2012;45:337–347. doi: 10.1016/j.nbd.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 20.Luján R, Maylie J, Adelman JP. New sites of action for GIRK and SK channels. Nat Rev Neurosci. 2009;10:475–480. doi: 10.1038/nrn2668. [DOI] [PubMed] [Google Scholar]
- 21.Lappin SC, Dale TJ, Brown JT, Trezise DJ, Davies CH. Activation of SK channels inhibits epileptiform bursting in hippocampal CA3 neurons. Brain Res. 2005;1065:37–46. doi: 10.1016/j.brainres.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 22.Empson RM, Jefferys JG. Ca(2+) entry through L-type Ca(2+) channels helps terminate epileptiform activity by activation of a Ca(2+) dependent afterhyperpolarisation in hippocampal CA3. Neuroscience. 2001;102:297–306. doi: 10.1016/S0306-4522(00)00494-2. [DOI] [PubMed] [Google Scholar]
- 23.de Sevilla Fernández D, Garduño J, Galván E, Buño W. Calcium-activated afterhyperpolarizations regulate synchronization and timing of epileptiform bursts in hippocampal CA3 pyramidal neurons. J Neurophysiol. 2006;96:3028–3041. doi: 10.1152/jn.00434.2006. [DOI] [PubMed] [Google Scholar]
- 24.Qi J, Wang Y, Jiang M, Warren P, Chen G. Cyclothiazide induces robust epileptiform activity in rat hippocampal neurons both in vitro and in vivo. J Physiol. 2006;571:605–618. doi: 10.1113/jphysiol.2005.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Qi JS, Kong S, Sun Y, Fan J, Jiang M, Chen G. BDNF-TrkB signaling pathway mediates the induction of epileptiform activity induced by a convulsant drug cyclothiazide. Neuropharmacology. 2009;57:49–59. doi: 10.1016/j.neuropharm.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng L, Chen G. Cyclothiazide potently inhibits gamma-aminobutyric acid type A receptors in addition to enhancing glutamate responses. Proc Natl Acad Sci USA. 2003;100:13025–13029. doi: 10.1073/pnas.2133370100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kullmann DM, Semyanov A. Glutamatergic modulation of GABAergic signaling among hippocampal interneurons: Novel mechanisms regulating hippocampal excitability. Epilepsia. 2002;43(Suppl 5):S174–S178. doi: 10.1046/j.1528-1157.43.s.5.12.x. [DOI] [PubMed] [Google Scholar]
- 28.Krishnan GP, Bazhenov M. Ionic dynamics mediate spontaneous termination of seizures and postictal depression state. J Neurosci. 2011;31:8870–8882. doi: 10.1523/JNEUROSCI.6200-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kong S, Qian B, Liu J, Fan M, Chen G, Wang Y. Cyclothiazide induces seizure behavior in freely moving rats. Brain Res. 2010;1355:207–213. doi: 10.1016/j.brainres.2010.07.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian B, Sun Y, Wu Z, Wan L, Chen L, Kong S, Zhang B, Zhang F, Wang ZY, Wang Y. Epileptiform response of CA1 neurones to convulsant stimulation by cyclothiazide, kainic acid and pentylenetetrazol in anaesthetized rats. Seizure. 2011;20:312–319. doi: 10.1016/j.seizure.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 31.Chen B, Jiang M, Zhou M, Chen L, Liu X, Wang X, Wang Y. Both NMDA and non-NMDA receptors mediate glutamate stimulation induced cofilin rod formation in cultured hippocampal neurons. Brain Res. 2012;1486:1–13. doi: 10.1016/j.brainres.2012.08.054. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Chen B, Chen L, Ren WT, Liu J, Wang G, Fan W, Wang X, Wang Y. U-shape suppressive effect of phenol red on the epileptiform burst activity via activation of estrogen receptors in primary hippocampal culture. PLoS One. 2013;8:e60189. doi: 10.1371/journal.pone.0060189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen L, Wan L, Wu Z, Ren W, Huang Y, Qian B, Wang Y. KCC2 downregulation facilitates epileptic seizures. Sci Rep. 2017;7:156. doi: 10.1038/s41598-017-00196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Huang Y, Liu X, Wang G, Wang X, Wang Y. Estrogen suppresses epileptiform activity by enhancing Kv4.2-mediated transient outward potassium currents in primary hippocampal neurons. Int J Mol Med. 2015;36:865–872. doi: 10.3892/ijmm.2015.2287. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Huang Y, Wang G, Wang X, Wang Y. Inhibition of 17-beta-estradiol on neuronal excitability via enhancing GIRK1-mediated inwardly rectifying potassium currents and GIRK1 expression. J Neurol Sci. 2017;375:335–341. doi: 10.1016/j.jns.2017.02.034. [DOI] [PubMed] [Google Scholar]
- 36.Faber ESL, Sah P. Functions of SK channels in central neurons. Clin Exp Pharmacol Physiol. 2007;34:1077–1083. doi: 10.1111/j.1440-1681.2007.04725.x. [DOI] [PubMed] [Google Scholar]
- 37.Kleiman-Weiner M, Beenhakker MP, Segal WA, Huguenard JR. Synergistic roles of GABAA receptors and SK channels in regulating thalamocortical oscillations. J Neurophysiol. 2009;102:203–213. doi: 10.1152/jn.91158.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartzkroin PA, Stafstrom CE. Effects of EGTA on the calcium-activated afterhyperpolarization in hippocampal CA3 pyramidal cells. Science. 1980;210:1125–1126. doi: 10.1126/science.6777871. [DOI] [PubMed] [Google Scholar]
- 39.Lancaster B, Adams PR. Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. J Neurophysiol. 1986;55:1268–1282. doi: 10.1152/jn.1986.55.6.1268. [DOI] [PubMed] [Google Scholar]
- 40.Abel HJ, Lee JC, Callaway JC, Foehring RC. Relationships between intracellular calcium and afterhyperpolarizations in neocortical pyramidal neurons. J Neurophysiol. 2004;91:324–335. doi: 10.1152/jn.00583.2003. [DOI] [PubMed] [Google Scholar]
- 41.Scutt G, Allen M, Kemenes G, Yeoman M. A switch in the mode of the sodium/calcium exchanger underlies an age-related increase in the slow afterhyperpolarization. Neurobiol Aging. 2015;36:2838–2849. doi: 10.1016/j.neurobiolaging.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 42.Köhler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- 43.Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci. 2002;22:10163–10171. doi: 10.1523/JNEUROSCI.22-23-10163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pedarzani P, Mosbacher J, Rivard A, Cingolani LA, Oliver D, Stocker M, Adelman JP, Fakler B. Control of electrical activity in central neurons by modulating the gating of small conductance Ca2+-activated K+ channels. J Biol Chem. 2001;276:9762–9769. doi: 10.1074/jbc.M010001200. [DOI] [PubMed] [Google Scholar]
- 45.Anderson NJ, Slough S, Watson WP. In vivo characterisation of the small-conductance KCa (SK) channel activator 1-ethyl-2-benzimidazolinone (1-EBIO) as a potential anticonvulsant. Eur J Pharmacol. 2006;546:48–53. doi: 10.1016/j.ejphar.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Kernig K, Kirschstein T, Würdemann T, Rohde M, Köhling R. The afterhyperpolarizing potential following a train of action potentials is suppressed in an acute epilepsy model in the rat Cornu Ammonis 1 area. Neuroscience. 2012;201:288–296. doi: 10.1016/j.neuroscience.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 47.Beck H, Clusmann H, Kral T, Schramm J, Heinemann U, Elger CE. Potassium currents in acutely isolated human hippocampal dentate granule cells. J Physiol. 1997;498:73–85. doi: 10.1113/jphysiol.1997.sp021842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garduño J, Galván E, de Sevilla Fernández D, Buño W. 1-Ethyl-2-benzimidazolinone (EBIO) suppresses epileptiform activity in in vitro hippocampus. Neuropharmacology. 2005;49:376–388. doi: 10.1016/j.neuropharm.2005.03.021. [DOI] [PubMed] [Google Scholar]