

Abstract
A series of copper complexes bearing new 6-substituted tris(2-pyridylmethyl)amine ligands (LR) appended with NH(p-R-C6H4) groups (R=H, CF3, OMe) were prepared. These ligands are electronically tunable (ΔE1/2 = 160 mV) and CuI(LR)+ complexes react with oxygen to form hydrogen bonded (trans-1,2-peroxo)dicopper species.
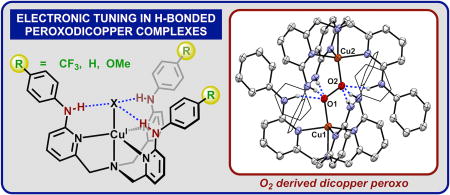
Graphical abstract
Copper-containing oxygenase and oxidase enzymes are an important class of metalloenzymes whose diverse O2 activation pathways facilitate a wide variety of biological functions.1 Although challenging to study in the native enzymes, the study of O2 binding and activation by copper within easily modifiable synthetic systems has greatly expanded our understanding of these metalloenzymes,2 and the first crystallized Cu-O2 adduct was the (trans-1,2-peroxo)dicopper complex of the tris(2-pyridylmethyl)amine (tpa) ligand.3 In the nearly 30 years since that structure, modifications to tpa and similar ligand frameworks have been targeted to elucidate the key factors responsible for O2 binding and activation. Although critical to the function of many metalloenzymes,4 the impact of secondary coordination sphere hydrogen bonding (H-bonding) interactions on copper-oxygen species is not well understood.5 For example, H-bonding interactions have been demonstrated to either stabilize6 or destabilize7 Cu-O2(H) adducts. For this reason, synthetic systems bearing tunable second sphere H-bonding groups provide a facile means to study their influence on Cu-O2 binding and activation.
Our lab recently introduced the tris(6-hydroxyl-2-pyridylmethyl)amine (H3thpa) ligand that incorporates three –OH groups within the secondary sphere of tpa (Fig. 1A).8 The pendent –OH groups are potent H-bond donors to metal-coordinated substrates and facilitate proton and electron transfer reactions.9 However, these complexes underwent facile formation of dinuclear copper species in the presence of weak bases, which limited investigations of subsequent reactivity. To overcome this limitation, we targeted pendent phenylamino groups as less acidic, sterically protected H-bond donors (Fig 1A).10 In contrast to previously reported –NHCOtBu substituted tpa variants, the phenylamino group provides both steric and electronic flexibility that allows them to act as highly tunable H-bond donors. In this communication we present a series of aniline-appended tripodal ligands that feature highly directed H-bonding interactions of varied acidity and highlight the design concept by demonstrating H-bond dictated O2 reactivity.
Figure 1.

CuICl complexes derived from H3thpa and LR (A). Cyclic voltammetry of 1H, 1CF3 and 1OMe ((B) 0.1 M NBu4PF6 CH2Cl2). Crystal structure of 1H ((C) 30% ellipsoids, H atoms not involved in H-bonding omitted for clarity).
The ligand tris(6-phenylamino-2-pyridylmethyl)amine (LH) was prepared via a Buchwald-Hartwig coupling of tris(6-bromo-2-pyridylmethyl)amine (Br3tpa) with aniline, Pd(OAc)2, and rac-BINAP. The addition of CuCl to LH in THF afforded the yellow complex Cu(LH)Cl (1H) in 59% yield after 48 h. A crystal of 1H suitable for X-ray diffraction was grown by layering pentane over a concentrated toluene solution at −30°C (Fig 1C). The solid-state structure revealed C3-symmetry (R-3 space group) with directed H-bonds from the pendent –NHPh units to the Cl ligand (N–Cl: 3.311(4) Å). These H-bonds are significantly longer than the previous Cu(H3thpa)Cl complex bearing pendent –OH groups (Navg–Cl: 3.048 Å) consistent with weaker H-bond interactions.8 In addition, the Cu–Naxial and Cu–Cl bonds in 1H, at 2.252(4) Å and 2.3398(14) Å respectively, are shortened compared to Cu(H3thpa)Cl (2.283(2) Å and 2.5661(6) Å respectively). Overall, the phenylamino groups in 1H provide a sterically protected pocket for the Cl ligand while allowing for further electronic tuning by modifying the identity of the aniline.
Two ligand derivatives featuring electronically distinct, yet sterically similar H-bond donors were prepared. 4-Trifluoromethylaniline and 4-methoxyaniline afforded ligands LCF3 and LOMe respectively, which were metalated with CuCl to provide Cu(LCF3)Cl (1CF3) and Cu(LOMe)Cl (1OMe). The electronic influence of each ligand variant was interrogated by analysis of the CuI/II redox couple (Fig 1B). Complex 1H exhibits a reversible CuI/II redox couple at −470 mV vs Fc (CH2Cl2; 0.1 M NBu4PF6). This value is shifted to more negative potentials relative to the –OH complex Cu(H3thpa)Cl (−365 mV vs Fc), consistent with increased electron releasing properties of LH than H3thpa. The CuI/II redox couple in 1OMe features the most reducing Cu center at −510 mV vs Fc. The electronically deficient 1CF3, however, exhibits a CuI/II redox couple at −350 mV vs Fc, which highlights the electronic tunability of the LR ligands with simple substitution on the parent aniline. To contextualize these electrochemically obtained values with a well-defined metric, the potential difference of 1CF3 (120 mV) and 1OMe (−40 mV) from the parent aniline (1H) were plotted against Hammett values (p-substitution) of 0.54 and −0.27 respectively (see SI). The electrochemical shifts show a good correlation (R2 = 0.98) with these Hammett values, indicating that they may be used to provide a predictive measure ligand field effects within the tpa scaffold.
The electronic variability in the LR ligand series was also evident by 1H NMR and IR spectroscopy. All three complexes are C3-symmetric by 1H NMR spectroscopy (CD2Cl2) and show a significantly downfield shifted –NH resonance consistent with H-bonding interactions between the –NH and the Cl ligand. The –NH peak appears at δ=9.88 in 1H whereas it shifts downfield (δ=10.17) in 1CF3 and upfield (δ=9.72) in 1OMe. The magnitude of the two shifts, +0.29 and −0.16 respectively, are again consistent with the expected proportion based on Hammett parameters (R2 = 0.99, see SI). The weakening of the –NH bond upon H-bonding is also evident by infrared spectroscopy (CH2Cl2) where the –NH stretching frequency shifts from 3431 cm−1 for LH to 3223 cm−1 in 1H. However, the value of the –NH stretch for 1CF3 and 1OMe does correlate directly with the Hammett value. The –NH stretch in 1OMe shifts to lower energy (3220 cm−1) consistent with a stronger H-bond interaction, while the –NH stretch in 1CF3 falls between 1H and 1OMe at 3221 cm−1. The IR data demonstrate the difficulty of assigning trends in a complex system where electronic character of the metal and M-X unit is highly coupled to both H-bond donor strength and H-bond acceptor strength.
The –NHPh appended tpa derivatives provide a tunable framework for studying O2 binding to Cu and the resulting H-bonded (trans-1,2-peroxo)dicopper complexes. The complex [CuI(LH)]BAr’ (BAr’= [B(C6F5)4]−) was prepared by mixing LH and Cu(MeCN)4BAr’ in CH2Cl2 under an inert atmosphere. 1H NMR spectroscopy was used to verify formation of a C3-symmetric Cu(I) complex. Cooling a CH2Cl2 solution of [CuI(LH)]BAr’ to −70°C and introducing dry O2 afforded the (trans-1,2-peroxo)dicopper complex [(Cu(LH))2(O2)][BAr’]2 (2H). The reaction was confirmed by a color change in solution (colorless to brown) and the appearance of a band at 457 nm (ε= 3000 M−1cm−1) in the electronic absorption spectrum, assigned as the oxygen to copper ligand-to-metal charge transfer (LMCT) band. This contrasts with the unsubstituted complex [(Cu(tpa))2(O2)][PF6]2 which exhibits a primary LMCT band at 525 nm (ε= 11500 M−1cm−1) and a shoulder at 590 nm (ε= 7600 M−1cm−1).3 In 2H we propose that the directed H-bonding interactions to the peroxide unit reduces peroxide electron donation into Cu. This effect weakens the Cu–O bond and results in a hypsochromic shift of the LMCT. Furthermore, the hypochromic shift and loss of the shoulder band in 2H was also observed by Masuda and co-workers in a similar H-bonded system and was postulated to be due to decreased rotational freedom of the peroxo unit.5f Complex 2H was subjected to additional spectroscopic characterization.11 The EPR (X-band) spectrum for 2H is silent, consistent with other S=1 integer spin (trans-1,2-peroxo)dicopper complexes. Upon warming to room temperature, solutions of 2H convert to a green, EPR active species, suggesting decomposition to monomeric Cu(II) byproducts. The UV-Vis and EPR data were corroborated by solid-state characterization of 2H.
A crystal of 2H suitable for X-ray diffraction was grown in CH2Cl2 in a −80° freezer under an atmosphere of oxygen over 3 days (Fig 2B), constituting the first crystallographically characterized H-bonded (trans-1,2-peroxo)dicopper complex. For each ‘Cu(LH)’ in 2H only one –NHPh group engages in H-bonding with the proximal oxygen of the peroxo unit (N5–O1: 2.723(7) Å) while the other two –NHPh groups engage the distal oxygen (N6–O2: 2.859(7) Å and N7–O2: 2.851(7) Å). Previous examples of H-bonded Cu-O2 species have shown that H-bonding to the proximal oxygen generally increases stability of the species while H-bonding to the distal oxygen decreases stability.5g 2H contains both proximal and distal H-bonds and stable in solution at −70°C for at least 8 hours, consistent with a net stabilizing effect to the O–O unit. The weakening of the Cu–O bond covalency in 2H, observed by UV-Vis, was also corroborated by the solid-state data. The Cu–O bond at 1.902(3) Å is elongated compared to the unsubstituted complex [(Cu(tpa))2(O2)][PF6]2 (1.852(4) Å). The O–O bond in 2H (1.477(5) Å) is also elongated compared to the parent tpa complex (1.433(9) Å). Although the O–O bond might be expected to shorten as a result of H-bonding interactions with the peroxide,5e, 5f additional structural factors may be responsible for the bond elongation. The steric profile provided by the –NHPh groups on LH limit the possible interatomic distance between Cu centers. The Cu…Cu distance in 2H (4.691(1) Å) is 0.3 Å longer than that observed for [(Cu(tpa))2(O2)][PF6]2 (4.358(3) Å). Despite these steric considerations, stability may also be augmented by π-π interactions between the pendent –NHPh groups and the opposing pyridine rings (π-π distance: 3.4–3.6 Å).
Figure 2.

Reactivity of Cu(I) complexes toward O2 (A). Crystal structure of 2H ((B) 30% ellipsoids, some atoms represented in wireframe, H atoms not involved in H-bonding and BAr’ counteranions omitted for clarity).
The electronic tuning provided by LCF3 and LOMe regulates the energy of the O to Cu LMCT. [Cu(LCF3)]BAr’ and [Cu(LOMe)]BAr’ readily bind O2 at −70°C to form the analogous (trans-1,2-peroxo)dicopper complexes [(Cu(LCF3))2(O2)][BAr’]2 (2CF3) and [(Cu(LOMe))2(O2)][BAr’]2 (2OMe). While the series of complexes (2R) are all brown in solution, the LMCT band shifts as a function of the electronic character at the metal.12 2CF3 features a 7 nm hypsochromic shift of the LMCT, while 2OMe features a 3 nm bathochromic shift from the parent 2H. The observed shifting of the LMCT contrasts with previously reported substituted tpa ligands, where the addition of 4-OMe groups to the pyridine backbone had no effect on the LMCT.13 In 2R, the shift of the LMCT bands is in accordance with the ligands’ respective electronic (Hammett) parameter (R2 = 0.99, see SI) and may be a product of both the Cu effective charge and H-bond donor strength. The oxidation potential of the halide-free [CuI(LR)]BAr’ complexes, obtained by square-wave voltammetry, provided an additional description of the electronic character at Cu. [Cu(LH)]BAr’ in MeCN (0.1M NBu4PF6) exhibits an irreversible oxidation event at +110 mV vs Fc. The associated LCF3 and LOMe Cu(I) complexes feature redox potentials shifted by +90 mV and −20 mV from LH, respectively. Importantly, these values are significantly more positive than [Cu(tpa)]PF6 (Eox = −386 mV vs Fc, see SI). O2 binding to [Cu(LCF3)]BAr’ at potentials as high as +200 mV vs Fc contrasts with most known Cu(I)-tpa variants that exhibit diminished O2 reactivity at more positive potentials.14 These data indicate that the Cu(I) centers in [Cu(LR)]BAr’ are only modestly reducing and might be anticipated to engage in very weak binding to O2. To account for the facile O2 reactivity, we propose that the H-bonding groups on LR provide additional stabilizing interactions for O2 binding.
A sterically identical ligand to LH with no H-bond donors was prepared in order to determine the role of H-bonding on the formation of the (trans-1,2-peroxo)dicopper complexes. The ligand tris(6-phenoxy-2-pyridylmethyl)amine (tpaOPh) containing pendent phenylether groups was prepared via an Ullmann coupling of phenol and Br3tpa. When tpaOPh and Cu(MeCN)4BAr’ were dissolved in CH2Cl2 and cooled to −70°C the resulting complex [Cu(tpaOPh)]BAr’ did not react with O2. 1H NMR spectra of [Cu(LH)]BAr’ and [Cu(tpaOPh)]BAr’ in CD2Cl2 revealed an almost identical chemical shift of the methylene protons, at δ=4.07 and 4.05 respectively, consistent with a similar electronic environment at copper.15 1H NMR spectra of [Cu(LH)]BAr’ and [Cu(tpaOPh)]BAr’ exhibit C3-symmetry at both 25°C and −80°C, however, at −80°C the methylene proton peak on [Cu(tpaOPh)]BAr’ broadens by 16.8 Hz, consistent with a fluxional process. This observation of dynamic ligand binding may further contribute to the lack of O2 reactivity with tpaOPh.16 Although steric and electronic properties of [Cu(LR)]BAr’ would suggest that formation of 2R is unfavorable, these hindrances were overcome by introducing favorable hydrogen bonding interactions.
In conclusion, we have introduced a new variation on the tpa framework that incorporates highly tunable –NHPh groups in the secondary sphere. These groups act as H-bond donors while providing steric protection that can be used to isolate H-bonded CuICl complexes. Cu(I) complexes bearing the new ligands react with O2 to form H-bonded (trans-1,2-peroxo)dicopper complexes whose spectroscopic characteristics are dictated by the ligand electronics.
Supplementary Material
Figure 3.

UV-Vis overlay of 2H, 2CF3, 2OMe (1:1 CH2Cl2/acetone, −70°C), and the reaction of [Cu(tpaOPh)]BAr’ with O2 at −70°C. Inlay shows λmax of O to Cu LMCT in 2R.
Acknowledgments
This work was supported by an NIH grant (1R01GM111486-01A1) and NSF grant CHE- 0840456 for X-ray instrumentation. N.K.S. is a Camille Dreyfus Teacher−Scholar. We thank Dr. Jeff Kampf for crystallographic assistance, Prof. Kristin Koutmou for allowing use of a −80°C freezer, and Prof. Kyle Lancaster and James Lukens for performing Resonance Raman spectroscopy.
Footnotes
Electronic Supplementary Information (ESI) available: Synthetic and experimental procedures and spectral data. CCDC 1584330 and 1584331. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/x0xx000
Notes and references
- 1.(a) Klinman JP. Chem. Rev. 1996;96:2541–2562. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]; (b) Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Chem. Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Solomon EI, Sundaram UM, Machonkin TE. Chem. Rev. 1996;96:2563–2606. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 2.(a) Suzuki M. Acc. Chem. Res. 2007;40:609–617. doi: 10.1021/ar600048g. [DOI] [PubMed] [Google Scholar]; (b) Hatcher LQ, Karlin KD. In: Adv. Inorg. Chem. van Eldik R, Reedijk J, editors. Vol. 58. Academic Press; 2006. pp. 131–184. [Google Scholar]; (c) Elwell CE, Gagnon NL, Neisen BD, Dhar D, Spaeth AD, Yee GM, Tolman WB. Chem. Rev. 2017;117:2059–2107. doi: 10.1021/acs.chemrev.6b00636. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; (e) Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013–1046. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]; (f) Quist DA, Diaz DE, Liu JJ, Karlin KD. J. Biol. Inorg. Chem. 2017;22:253–288. doi: 10.1007/s00775-016-1415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kitajima N, Moro-oka Y. Chem. Rev. 1994;94:737–757. [Google Scholar]
- 3.Jacobson RR, Tyeklar Z, Farooq A, Karlin KD, Liu S, Zubieta J. J. Am. Chem. Soc. 1988;110:3690–3692. [Google Scholar]
- 4.Borovik AS. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 5.(a) Fujii T, Yamaguchi S, Funahashi Y, Ozawa T, Tosha T, Kitagawa T, Masuda H. Chem. Commun. 2006:4428–4430. doi: 10.1039/b609673e. [DOI] [PubMed] [Google Scholar]; (b) Fujii T, Yamaguchi S, Hirota S, Masuda H. Dalton Trans. 2008:164–170. doi: 10.1039/b712572k. [DOI] [PubMed] [Google Scholar]; (c) Yamaguchi S, Wada A, Funahashi Y, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Eur. J. Inorg. Chem. 2003;2003:4378–4386. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]; (d) Kim S, Saracini C, Siegler MA, Drichko N, Karlin KD. Inorg. Chem. 2012;51:12603–12605. doi: 10.1021/ic302071e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Maiti D, Woertink JS, Narducci Sarjeant AA, Solomon EI, Karlin KD. Inorg. Chem. 2008;47:3787–3800. doi: 10.1021/ic702437c. [DOI] [PubMed] [Google Scholar]; (f) Wada A, Honda Y, Yamaguchi S, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Inorg. Chem. 2004;43:5725–5735. doi: 10.1021/ic0496572. [DOI] [PubMed] [Google Scholar]; (g) Peterson RL, Ginsbach JW, Cowley RE, Qayyum MF, Himes RA, Siegler MA, Moore CD, Hedman B, Hodgson KO, Fukuzumi S, Solomon EI, Karlin KD. J. Am. Chem. Soc. 2013;135:16454–16467. doi: 10.1021/ja4065377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wada A, Harata M, Hasegawa K, Jitsukawa K, Masuda H, Mukai M, Kitagawa T, Einaga H. Angew. Chem. Int. Ed. 1998;37:798–799. doi: 10.1002/(SICI)1521-3773(19980403)37:6<798::AID-ANIE798>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi S, Nagatomo S, Kitagawa T, Funahashi Y, Ozawa T, Jitsukawa K, Masuda H. Inorg. Chem. 2003;42:6968–6970. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]
- 8.Moore CM, Quist DA, Kampf JW, Szymczak NK. Inorg. Chem. 2014;53:3278–3280. doi: 10.1021/ic5003594. [DOI] [PubMed] [Google Scholar]
- 9.(a) Moore CM, Szymczak NK. Chem. Commun. 2015;51:5490–5492. doi: 10.1039/c4cc06832g. [DOI] [PubMed] [Google Scholar]; (b) Moore CM, Szymczak NK. Chem. Sci. 2015;6:3373–3377. doi: 10.1039/c5sc00720h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Dahl EW, Louis-Goff T, Szymczak NK. Chem. Commun. 2017;53:2287–2289. doi: 10.1039/c6cc10206a. [DOI] [PubMed] [Google Scholar]; (b) Dahl EW, Szymczak NK. Angew. Chem. Int. Ed. 2016;55:3101–3105. doi: 10.1002/anie.201511527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Resonance Raman spectroscopy was also used to interrogate the O–O and Cu–O stretching frequencies in 2H; however no 18O active modes were defined.
- 12.Complex 2CF3 precipitates from CH2Cl2 at −70°C over 30 sec. Therefore UV-Vis spectra of 2R were collected in a 1:1 CH2Cl2:acetone solution by first forming the complexes in pure CH2Cl2 followed by addition of cold acetone.
- 13.Zhang CX, Kaderli S, Costas M, Kim E-i, Neuhold Y-M, Karlin KD, Zuberbühler AD. Inorg. Chem. 2003;42:1807–1824. doi: 10.1021/ic0205684. [DOI] [PubMed] [Google Scholar]
- 14.(a) Chuang C-l, Lim K, Chen Q, Zubieta J, Canary JW. Inorg. Chem. 1995;34:2562–2568. [Google Scholar]; (b) Schatz M, Becker M, Thaler F, Hampel F, Schindler S, Jacobson RR, Tyeklár Z, Murthy NN, Ghosh P, Chen Q, Zubieta J, Karlin KD. Inorg. Chem. 2001;40:2312–2322. doi: 10.1021/ic000924n. [DOI] [PubMed] [Google Scholar]; (c) Karlin KD, Wei N, Jung B, Kaderli S, Niklaus P, Zuberbuehler AD. J. Am. Chem. Soc. 1993;115:9506–9514. [Google Scholar]
- 15.The chemical shift of the methylene protons is highly sensitive to the electronic environment. For instance, the methylene protons shift from +0.04 ppm in 1CF3 to −0.04 ppm in 1OMe compared to 1H. Voltammetry was uninformative (see ref 16).
- 16.No discernable redox events could be obtained for [Cu(tpaOPh)]BAr’. We propose that a fluxional ligand dissociation process (shown from VT NMR studies) is responsible for the observed poor voltammetry response on the timescale of the electrochemical measurements.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.