

Abstract
Recent work indicates that salivary glands are able to constitutively recruit CD8+ T cells and retain them as tissue resident memory T cells (TRM), independently of local infection, inflammation or antigen. To understand the mechanisms supporting T cell recruitment to the salivary gland, we compared T cell migration to the salivary gland in mice infected or not with murine cytomegalovirus (MCMV), a herpesvirus that infects the salivary gland and promotes the accumulation of salivary gland TRM. We found that acute MCMV infection increased rapid T cell recruitment to the salivary gland, but that equal numbers of activated CD8+ T cells eventually accumulated in both infected and uninfected glands. T cell recruitment to uninfected salivary glands depended on chemokines and the integrin α4. Several chemokines were expressed in the salivary glands of both infected and uninfected mice and many of these could promote the migration of MCMV-specific T cells in vitro. MCMV infection increased expression of chemokines that interact with the receptors CXCR3 and CCR5, but neither receptor was needed for T cell recruitment to the salivary gland during MCMV infection. Unexpectedly however, the chemokine receptor CXCR3 was critical for T cell accumulation in uninfected salivary glands. Together, these data suggest that CXCR3 and the integrin α4 mediate T cell recruitment to uninfected salivary glands, but that redundant mechanisms mediate T cell recruitment after MCMV infection.
Introduction
The salivary gland is an exocrine organ, composed mostly of acinar cells producing a water-based fluid that is mucous, ion, enzyme-rich, and released in the oral cavity via a network of epithelial ducts. Because of this, several viruses, including all human β- and γ-herpesviruses, infect the salivary gland and use the saliva as a major route of transmission to new hosts(1–3). How the immune system responds to pathogens in the salivary gland, and prevents or limits viral shedding, remains poorly defined. In addition, how pathogens alter the salivary gland environment by infection is unknown.
Cytomegalovirus (CMV) is a β-herpesvirus that infects most organs in the body but undergoes prolonged replication in the salivary gland. CMV infects the acinar and ductal epithelial cells in the salivary gland and is thus shed into saliva during replication(4–6). Using the mouse CMV (MCMV) model, it was shown over 40 years ago that lymphocytes enter the salivary gland, resulting in the death of MCMV-infected epithelial cells(7, 8). MCMV also establishes latency in the salivary gland and readily reactivates in this tissue during periods of immune suppression, particularly when CD8+ T cells have been depleted (9). It might be assumed that lymphocytes are drawn to the salivary gland in response to the CMV infection. Interestingly however, recent data from several labs including our own, suggested that antigen-stimulated CD8+ T cells could be recruited to the salivary gland constitutively after MCMV infection or even without specific infection of the gland(10–12). MCMV-specific T cell populations were abundant in the salivary gland after MCMV infection where they adopted an intra-epithelial localization and expressed CD69 and CD103(10, 11), markers of tissue-resident memory T cells (TRM). TRM are memory T cell subsets that are retained in tissues independently of the circulating pool of T cells, thus providing a rapid defense against reactivation of a latent infection or local reinfection of a previously encountered pathogen(13, 14). Surprisingly, even in vitro activated T cells could migrate to the salivary gland where they developed into protective TRM cells in the complete absence of infection or specific inflammation (10–12), leading to the description of the salivary gland as a “sink” for CD8+ TRM (10).
It is currently unknown whether local inflammation or antigen can enhance the recruitment or retention of CD8+ TRM in the salivary gland. However, this ability of salivary glands to attract and retain protective numbers of CD8+ TRM, without a local infection or inflammation, is quite unexpected. In most other sites in the body, tissue-localized inflammation and/or antigen is critical for the efficient recruitment of T cells, or their retention as TRM (15–24). The best-studied example of the interplay between antigen, inflammation and TRM formation is the skin where inflammation alone is sufficient to enable T cell egress from the blood and formation of TRM phenotype cells (22). Interestingly, while infection at one skin site could lodge T cells at distant skin locations(23, 25), the efficiency is very poor without local inflammation and local antigen enhanced the maintenance of TRM populations and shaped the specificity of the cells that were retained (16, 23, 24).Thus, although antigen and inflammation within a particular skin site are not absolutely required for TRM formation, they markedly enhance the number of protective TRM that are established in the skin. Other tissues have been less well studied, but a similar theme is repeated. In the vaginal mucosa, CD8+ T cell entry during Herpes Simplex Virus infection was poor unless CD4+ T cells in the tissue to promoted local chemokines in an IFN-γ dependent manner (17). In the lungs, the formation and maintenance of protective numbers of TRM after multiple infections, including after MCMV, depended on both antigen and infection of the lungs (26, 27). The brain is even more restrictive, requiring infection or antigen for any detectable TRM formation (20). In fact, other than the salivary gland, only the small intestine has been described as permissive of TRM formation and maintenance in an antigen- and infection-independent manner(28). Thus, the salivary gland and the small intestine may be uniquely capable of both recruiting and retaining T cells without any specific infection.
While many studies have addressed the mechanisms of T cell recruitment to the intenstine (e.g.(28–31)), very little is known about the mechanisms of T cell recruitment to the salivary gland. A recent study demonstrated that systemic inflammation could induce expression of the cellular adhesion molecule VCAM-1 on vascular endothelial cells in the salivary gland, and that this boosted the recruitment of activated T cells via the integrin α4 (32), which pairs with the β1 integrin to form the ligand for VCAM-1. At first glance, this supports the notion that inflammation will enhance T cell recruitment to the salivary gland. However, TRM formation and maintenance was not studied. Moreover, the chemokines that recruit T cells to the salivary gland remain undefined.
We examined CD8+ T cell recruitment to the salivary gland in the presence or absence of active MCMV infection. Our data confirm and extend recent observations that uninfected salivary glands were permissive to the recruitment and retention of activated CD8+ T cells in a manner dependent on the integrin α4. Moreover, active MCMV infection of the salivary glands increased the rapid recruitment of activated T cells. Remarkably however, inflammation induced by MCMV infection did not enhance the number of TRM that were ultimately lodged in the salivary gland. Indeed, many chemokines abundantly expressed in the salivary gland of both infected and uninfected mice could attract MCMV-specific T cells in vitro, including CXCL9 (Monokine induced by IFN-γ - Mig) and CXCL10 (IFN-inducible 10 KDa protein - IP10), ligands for the receptor CXCR3. CXCR3 is a chemokine receptor that has been described to be important for CD8+ T cell migration to a variety of tissues, typically in the context of inflammation and infection (33). Unexpectedly, we found that CXCR3 expression by T cells was critical for efficient T cell accumulation in the salivary gland in uninfected mice, but dispensable for their accumulation in the salivary gland during MCMV infection. These data establish a mechanism for the surprisingly efficient recruitment of activated T cells to the salivary gland even after a completely irrelevant infection.
Materials and Methods
Mice and infections
All mice were purchased from the Jackson laboratory and bred in house. B6 (C57BL/6), CD45.1 (B6.SJL-PtprcaPepcb/BoyJ), Thy1.1 (B6.PL-Thy1a/CyJ) and IFNγ KO (B6.129S7-Ifngtm1Ts/J) mice were used as recipients in the adoptive transfer experiments and to assess chemokine expression in the salivary gland. OT-Is on a B6 background [C57BL/6-Tg(TcraTcrb)1100Mjb/J] were bred to CD45.1, CXCR3 knock-out (KO) mice (B6.129P2-Cxcr3tm1Dgen/J) and CCR5 KO mice (B6.129P2-Ccr5tm1Kuz/J) to generate congenic CXCR3 KO or CCR5 KO OT-Is.
MCMV-K181 virus (kindly provided by Ed Mocarski) was used in fig. 1–3, 4A–6, fig. 8F. MCMV-SL8-015 or MCMV-K181-trf-OVA (MCMV-OVA), have been previously described (34, 35), and were used in fig. 7, fig. 8A-D and to expand OT-I T cells after the first adoptive transfer on fig. 4. Infections were performed using 2×105 pfu and the i.p. route (100 μl per injection) in all experiments except fig.1 where some of the infections were performed via intranasal (i.n.) or footpad (f.p.) routes in a total volume of 20-25 μL per inoculation after anesthesia with Isoflurane. MCMV-K181, MCMV-OVA and MCMV-SL8-015 were produced as described (34, 36–38). All protocols were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee.
Lymphocyte isolation and FACS Staining
For all the experiments intravenous antibody injections were performed, as described in (39, 40) without perfusion, to distinguish between vasculature-localized (I.V.+) and parenchyma-localized (I.V.−) CD8+ T cells. In brief, mice were injected intravenously (i.v.) with 3 μg of an anti-CD8α antibody (clone: 53-6.7 conjugated to BV650 or BV421) 3 minutes before sacrifice. Blood was collected from the retro-orbital sinus or from the chest cavity, after cutting the pulmonary vein at sacrifice and the organs were collected in media containing an unlabeled CD8α antibody (clone: 53-6.7). Total CD8+ T cells were identified by CD8β staining (± CD8α of the intravascular portion) during the phenotypic analyses as described below.
Lymphocytes from the blood, spleen, inguinal lymph nodes, sub-mandibular salivary glands (referred to as “salivary glands” throughout), kidneys and lungs were isolated as described previously (40) with minimal modifications. Briefly the mucosal organs were minced using the gentleMACS Dissociator (Miltenyi Biotec) and incubated at 37°C for 1-1.5 hours in digestion media containing 1 mg/ml collagenase type IV, 5 mM CaCl2, 50 mg/ml DNase I, and 10% FBS in RPMI. Salivary glands were suspended in 40% Percoll and overlayed on top of a 75% Percoll layer, while the kidneys and lungs were suspended in 40% Percoll. Suspensions were centrifuged at 600 ×g for 25-30 min and the lymphocytes were collected from the 75/40 interface (salivary glands) or pellets (kidney and lung).
Phenotypic analyses of T cells were performed using the following antibodies: CD8α (clone: 53-6.7); CD8β (YTS156.7.7); CD69 (clone: H1.2F3); CD103 (clone: 2E7); CD44 (clone: IM7); KLRG1 (clone: 2F1); CXCR3 (clone: CXCR3-173); CXCR4 (clone: L276F12); CXCR6 (clone: SA051D1); CX3CR1 (clone: SA011F11). OT-Is were identified by their congenic markers with CD45.1 (clone: A20) and/or CD45.2 (clone: 104) and by the TCR chains Vα2 (clone: B20.1) and Vβ5 (clone: MR9-4). In fig. 9E (exp. 2) donor cells where identified also by the Thy1.2 marker (clone: 30-H12). All antibodies were purchased from Biolegend or BD Bioscience. All MHC-tetramers, loaded with peptides from M38, were provided by the National Institutes of Health Tetramer Core Facility (http://tetramer.yerkes.emory.edu/) and used as described(41). A tetramer loaded with the B8R peptide derived from Vaccinia virus was used as a negative control for the tetramer staining following a MCMV-infection. All samples were collected on a BD LSRFortessa or LSR II flow cytometer and analyzed using FlowJo software (TReeStar). The gating strategy is represented in the sup. fig. 1.
In vitro T cell activation and expansion
OT-Is were activated in vitro based on the protocol described (12) with modifications. Briefly, splenocytes from OT-I mice were harvested and 4×106 cells/mL were cultured with 1 μg/mL of the SIINFEKL peptide for 2 days. On the second day the cells were resuspended to 5×105 cells/mL and incubated with 0.03 U/mL of IL-2 that was renewed every 2 days for a total of 4-5 days until the adoptive transfer.
Adoptive transfers
All the adoptive transfers were performed via retro-orbital injections in a volume of 100 μL between congenic donor and recipient mice (differing in CD45.1/.2 or Thy1.1/1.2 expression).
In vitro activated T cells
For the adoptive transfers in fig. 2–4 (C), fig. 8E-F, fig. 9 and sup. fig. 2, in vitro activated, CD8+ CD44+ OT-I T cells were transferred to congenic naïve recipients or recipients infected for 9 weeks (fig.2) or 11 days (fig. 3) with MCMV-K181 (lacking Ova). For treatment with Pertussis Toxin, OT-Is were suspended at a concentration of 1.5×107cells/mL and treated or not with 50 ng/ml Pertussis Toxin (Sigma-Aldrich) for 1h at 37°C prior to transfer. In fig. 8F, WT and CCR5 KO OT-I T cells were mixed and co-transferred to B6 mice that have been previously infected with MCMV-K181 for 11 days. The mixture of donor cells was treated with anti-CXCR3 blocking antibody (clone: CXCR3-173) or isotype control antibody (Polyclonal Armenian Hamster IgG) at a concentration of 30 μg per 4×107 cells for 15 min. Recipient mice were also treated with the anti-CXCR3 antibody or isotype control (250 μg/mouse) via i.p. injections on days -2; day 0 (the day of transfer) and day 2. Blocking antibodies and isotype controls were purchased from Bio X cell. In fig. 9 the same approach was used but only WT OT-Is were transferred to naïve recipients.
In vivo activated T cells
For in vivo activation (fig. 4A-B, fig. 7, fig. 8 A-D), naïve OT-I T cells were transferred into naive congenic recipients, followed by infection with MCMV expressing the cognate SIINFEKL peptide (either MCMV-SL8-015 or K181-MCMV-OVA) 1-3 days after transfer. The number of cells transferred is indicated in each figure legend. In fig. 4A-B, 5 × 104 OT-Is were transferred into congenic recipients to produce large numbers of OT-Is for a secondary transfer by day 5 post infection. In this experiment, OT-Is were recovered from the spleen 5 days after infection, treated for 30 min with 60 μg/mL of either anti-α4 (clone PS/2), anti-α4β7 (clone DATK32) or the respective isotype controls and transferred to a new group of infection-matched or naïve recipients. The recipients were treated on the day of the transfer with 300 μg of each antibody or isotype control via i.p. injection. Organs were collected 2 days after the secondary transfer and the tissues were processed as described above.
Cell proliferation assays
To assess proliferation of OT-I T cells in naïve or MCMV-infected mice (sup. fig. 2), OT-Is were labeled with a cell tracer dye (CellTrace Violet or CFSE) following manufacture’s instructions before the adoptive transfer. Briefly, cells were suspended at a concentration of 106 cells/mL and incubated with 5 μM of the Cell Trace Violet dye in for 20 min or suspended at a concentration of 1×107 cells/mL and incubated with 1 μM CFSE for 10min.
Transwell migration assays
For Transwell migration assays, CD8+ T splenocytes were isolated from MCMV-K181 infected mice (7 days) using the EasyStep Biotin selection kit (StemCell Technologies) and biotinylated antibodies against erythrocytes (Ter119), CD19 (6D5), NK1.1(PK136), I-A/I-E (M5/114.15.2) and CD4 (GK1.4) following the manufacture’s protocol. Typically, CD8+ T cells were 80-90% pure following this protocol. Purified cells were resuspended in RPMI media containing 2% BSA and 25 nM of Hepes buffer (migration media) at a concentration of 5×106 cells/mL and incubated for 1h at 37°C. Subsequently, 5×105 cells, in a total volume of 100 μL were added to the upper chamber of a 6.5 mm Polycarbonate Transwell system (from Corning Inc.) with a pore diameter of 5.0 μm. Chemokines (all from Biolegend) were diluted in migration media and added to the lower chamber at titrating concentrations in a total volume of 600 μL (3-4 replicate wells per concentration). Control (media alone) samples without chemokines in the lower chamber of the Transwell plates, were included on every plate to account for plate-to-plate variations in T cell migration. All tests were run in duplicate, or triplicate on every plate. Chemokine concentrations shown in fig. 6 represent the optimal migration over control (media alone) wells based on replicate titrations of each chemokine. Cells in the Transwell plates were incubated for 1.5h at 37°C. At the end of the incubation, 200 μL of cells from the bottom chamber were mixed with counting beads (CountBright Absolute Counting Beads, Invitrogen), while the remaining volume was used for FACS analyses of tetramer-binding cells. Cells were collected by flow cytometry as above.
qRT-PCR
For assessment of chemokine receptors expressed by T cells in fig. 7 C-D, OT-I T cells were transferred to B6 mice that were then infected with MCMV-K181-Ova (n=3 mice). Seven days after infection, spleen and salivary glands were collected and CD8+ T cells were enriched using the EasySep Biotin selection kit (Stemcell Technologies), as described above for the Transwell assays. OT-Is were sorted based on the congenic markers CD45.1 and CD45.2. RNA was extracted from the sorted OT-Is (fig. 7 C-D) and whole salivary glands of naïve B6 or IFN-γ KO mice (fig. 9F) using the RNeasy Mini Kit (QIAGEN) cDNA was recovered using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). In both cases the β-actin and chemokine receptor transcripts expressed were detected on a StepOnePlus system (Applied Biosystems) using predesigned qPCR assays from Integrated DNA Technologies and FAM for detection. The relative concentration of chemokine receptors on these cells was determined by comparing the chemokine receptor signal to the β-actin signal for the same sample on the same plate and the data is expressed as the 2−ΔCT value (e.g. 2 −(CT value for chemokine A – CT value internal reference control A)).
RNA-Seq
For the RNA-Seq analysis salivary glands from mice that were infected, with 2×105 pfu of MCMV-K181 i.p., for 14 days or naïve mice were collected and RNA was obtained with the miRCURY RNA Isolation kit Tissues (EXIQON).
RNA Clean-up and ribosomal RNA (rRNA) depletion for the RNA-Seq analysis
Prior to cDNA library preparation, RNA samples were purified using RNA Clean & Concentrator (Zymo research, R1015) and treated with DNase I to remove contaminating genomic DNA. Validation of RNA quality and concentration was determined using the RNA 6000 Pico Kit (Agilent Technologies, catalog number 5067-1513) for the BioAnalyzer 2100 instrument (Agilent Technologies). Four micrograms of total RNA per sample were used as input for the depletion of ribosomal RNA (RiboMinus kit; Ambion, A15020), yielding 6% recovery of input RNA on average.
RNA-Seq Library Preparation and Sequencing
Library preparation was performed using the Ion Total RNA Seq Kit v2 (Thermo Fisher Scientific) according to the manufacturer’s protocol. The yield and size scatter of the cDNA libraries were evaluated using High Sensitivity DNA Chips (Agilent Technologies, catalog number 5067-4626) on the BioAnalyzer 2100 instrument. The barcoded cDNA libraries were diluted to a final concentration of 100 pM, and used for template preparation on the Ion Chef instrument using the Ion PI Hi-Q Chef Kit (Thermo Fisher Scientific, catalog number A27198). Sequencing was performed on the Ion Proton sequencer system from Thermo Fisher Scientific using Ion Torrent PI v3 chips (Thermo Fisher Scientific, catalog number A26771). Sequenced data were preprocessed on the Ion Torrent server with Torrent Suite version 4.4.3.
RNA-seq data Analysis
IonTorrent single-end sequence reads were mapped to the mm10 genome using the IonTorrent Torrent Server tmap aligner, version 4.4.11. Gene abundances were estimated by counting strand-specific reads using the Subread package featureCounts tool, where reads with mapq of 0 or multiple mappings were filtered out(42). Reads overlapping more than one gene feature were not counted because the transcript of origin could not be confidently determined, as recommended by the Subread/featureCounts manual. Resulting read counts were then used to analyze differential expression between infected and uninfected samples using the DESeq2 package for R/bioconductor, with default settings(43). For additional data exploration and visualization, RPKM estimates were calculated from featureCounts results.
Gene Set Enrichment Analysis (GSEA) was also performed to identify up- and down-regulated gene groups among available pathway and gene ontology annotations(44). Mouse gene symbols were converted to their human orthologs, obtained from MGI on March 3rd 2017, for compatibility with GSEA. GSEA preranked analysis was performed using the list of available human orthologs and the DESeq2 test statistic as the ranking metric, and GSEA enrichment score set to “classic” as described in the GSEA FAQ.
The data has been submitted to the NCBI-GEO database (https://www.ncbi.nlm.nih.gov/geo/). The accession number is: GSE107338.
ELISAs for chemokine expression
Whole salivary glands of naïve B6 and IFN-γ KO mice were collected in PBS with 10 μL/mL of the Halt™ Protease Inhibitor Cocktail (Thermo Scientific) and homogenized for 2 cycles of 30sec in a mini bead beater. After centrifugation (14000 ×g for 15 min) the supernatant was stored at -80C. The total protein concentration was determined using the Quick Start™ Bradford Protein Assay from Bio-Rad. Samples were normalized to 8 mg/mL of total protein. The concentration of CXCL9 in each sample was quantified in duplicate with the CXCL9 (MIG) ELISA kit (Thermo scientific) according to the manufacture’s specifications.
Statistical analysis
Differences in absolute numbers were determined after Log10 transformation and the fold change was determined based on the ratios of the geometric mean. The specific statistical test used for each experiment is indicated in the figure legend. Prism 6 for Mac OS X was used to determine the Log10 transformed values, geometric mean and to perform the statistical analysis.
Results
The route of MCMV infection impacts the CD8+ T cell migration to the salivary gland but not the rate of TRM differentiation
We have previously shown that MCMV infection by the intraperitoneal (i.p.) route lead to robust formation of TRM in the salivary gland after infection (11). However, factors outside of the tissue such as the site of T cell priming may influence CD8+ T cell trafficking and TRM formation in some tissues (45–48). To determine whether the route of MCMV infection influenced the migration of CD8+ T cells and formation of MCMV-specific TRM, we compared TRM cell numbers 14 days after infection via the i.p., intranasal (i.n.) and footpad (f.p.) routes. Here, and throughout, cells that were still within the circulation were distinguished from those that had reached the parenchyma by intravascular staining with anti-CD8 antibodies as described in the materials and methods. As shown in fig. 1, MCMV-specific T cells reached the parenchyma of the salivary gland after all three routes of infection and developed the TRM phenotype. Interestingly, infections by the i.n. and f.p. routes were associated with lower overall numbers of MCMV-specific T cells in the salivary gland, which reached statistical significance after f.p. infection. Notably, this effect was not due to an overall difference between circulating populations, as seen by the similar number of MCMV-specific T cells in the spleen at the same time point after all 3 infections. Importantly however, the frequency of T cells that developed the TRM phenotype in the salivary gland was identical after all 3 routes of infection (fig. 1C), implying that once T cells arrived in the gland, they were equally capable of differentiating into TRM. In contrast, MCMV-specific TRM cells only formed in the lung after i.n. infection (fig. 1B and 1C), which confirms the results of a recent study(19). These data suggest that the route of infection impacts the ability of T cells to reach the salivary gland but not their differentiation into TRM cells.
Fig. 1. MCMV-specific CD8+ T cells accumulate in the salivary gland after several routes of MCMV infection.
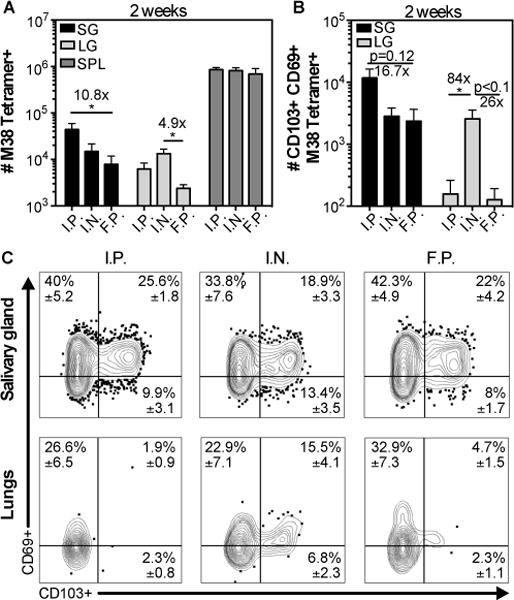
Mice were infected by intraperithoneal (i.p.), intranasal (i.n.) and footpad (f.p.) inoculation. Cells in the parenchyma or vasculature of each tissue were distinguished by I.V. staining as described in the materials and methods. A) Shown is the absolute number of M38 tetramer+ CD8+ T cells from the parenchyma of the salivary gland (SG) and lungs (LG), and from the overall CD8β+ population of the spleen two weeks after infection. B) Absolute number of CD103+ CD69+ M38 Tetramer+ cells in the SG and LG from the data shown in (A). Data are from 2 independent experiments (n=6 for f.p. and i.p.; n=5 for i.n.). Error bars represent the SEM and statistical significance was measured by one-way ANOVA after log10 transformation of the absolute numbers (*p<0.05). (C) Concatenated FACS plots from one representative experiment (n=2 i.n.; n=3 for i.p. and f.p.) of the CD69 and CD103 expression of M38 tetramer+ CD8+ T cells from the salivary gland (top panel), and the lungs (bottom panel). The mean frequency ± SEM in the indicated quadrant were calculated considering both experiments.
Activated CD8+ T cells can enter the salivary gland and develop the TRM phenotype even when a pre-established TRM population is already present
Previous studies suggested that activated T cells could enter and reside in naïve, uninfected salivary glands. Moreover, we previously suggested that persistent MCMV infection resulted in continuous recruitment and retention of new MCMV-specific TRM, long after the primary infection had been resolved(11). To directly compare these two conditions, we activated OT-I T cells in vitro and transferred them to congenic mice that were either naïve, or latently infected with wild-type MCMV lacking Ova (9 weeks after infection, fig. 2A). Wild-type MCMV was used in these experiments to assess the impact of viral infection and the presence of unrelated TRM, and avoid the complication of antigen-driven T cell expansion of the donor OT-Is. Two weeks after transfer, the OT-Is had reached the salivary gland in similar numbers in the naïve and latently infected recipients (fig. 2B), and similar frequencies of OT-Is expressing CD69 and CD103, the markers of bonafide TRM cells in the salivary gland, were observed in naïve and infected salivary glands (fig. 2C and 2D). Likewise, when naïve or MCMV-infected mice were infected with vaccinia virus (VACV), VACV-specific TRM cells (B8R tetramer+) were present in the salivary glands in similar numbers and the frequency of TRM-phenotype VACV-specific T cells was equivalent between groups (data not shown). Thus, our data suggest that naïve, acutely-infected and latently-infected salivary glands are all capable of recruiting activated CD8+ T cells and supporting their differentiation into TRM.
Fig. 2. CD8+ T cells with a TRM phenotype can form in the salivary glands of naïve mice and mice latently infected with MCMV.
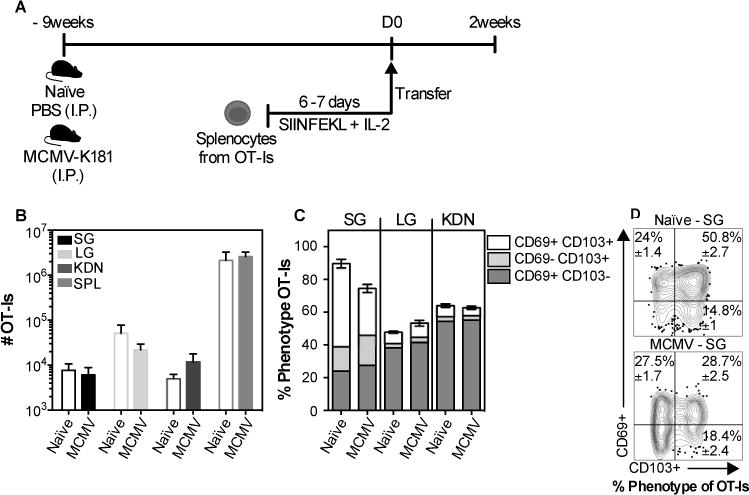
(A) Schematic of the experimental design. Naïve mice or mice that had been infected (i.p.) for 9 weeks with wild-type MCMV (lacking Ova) were seeded with 3×106 in vitro activated OT-Is and sacrificed 2 weeks after transfer. B) Absolute number of OT-Is from the parenchyma of the salivary gland (SG), lungs (LG), kidneys (KDN) and from the overall CD8β+ population of the spleen (SPL). (C) Frequency of CD103 and CD69 expression on OT-Is from the parenchyma of the SG, LG and KDN. Data combined from 2 independent experiments (n=7). (D) Concatenated FACS plots from a representative experiment with frequency mean ± SEM values considering all the experiments. An unpaired t-test was used to test for statistical significance.
Acute MCMV infection promotes rapid CD8+ T cell recruitment to the salivary gland but does not affect the overall numbers of TRM phenotype T cells
Although the above experiments demonstrate that infection and inflammation are not needed for T cell recruitment to the salivary gland and development of the TRM phenotype, MCMV infects the salivary gland directly and our data do not exclude a role for acute inflammation in promoting the process. To directly compare T cell migration to naïve salivary glands and salivary glands with active MCMV replication, in vitro activated OT-Is were transferred to naïve mice or mice infected 11 days previously with MCMV (fig. 3), a time when replicating MCMV can be detected in the salivary glands after i.p. infection (11). For these experiments, we infected mice with wild-type MCMV (lacking Ova) to test the role of inflammation in the gland, again without the confounding issue of the transferred cells undergoing antigen-driven proliferation in infected mice. An increased recruitment of OT-Is to the salivary gland, but not other organs, was observed in the infected recipients early after transfer (fig.3B), which was not due to higher T cell proliferation rates in the infected glands (sup. fig. 2). At later time points, 31 days after transfer, there was a reduction in the number of OT-Is in the lung and the kidneys as expected due to the lack of antigen, but not in the salivary gland. Remarkably, the early advantage in T cell numbers recruited to the infected salivary gland was no longer evident (fig. 3B). Moreover, the frequency of OT-I TRM cells present in the salivary gland was similar in both infected and naïve recipients 31 days after transfer (fig. 3C-D). Thus, surprisingly, our data suggest that although inflammation of the salivary gland, caused by MCMV infection, resulted in an accelerated accumulation of activated OT-Is in the gland shortly after transfer, infection and inflammation provided no long-term advantage for the formation or maintenance of CD8+ TRM cells.
Fig. 3. CD8+ TRM phenotype cells can form and persist at similar numbers in salivary glands from MCMV infected and naïve mice.
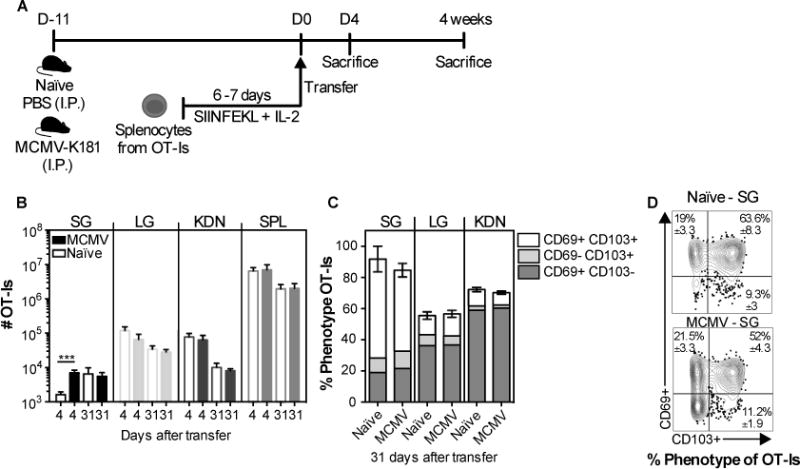
(A) Schematic of the experimental design. Naïve mice, or mice infected 11 days earlier with MCMV (via the i.p. route) were seeded with 3×106 in vitro activated OT-Is. The recipients were sacrificed at 4 and 31 days after transfer. B) Absolute number of OT-Is in the parenchyma of the SG, LG, KDN and from the CD8β+ cells of the SPL. C and D) Frequency of CD103- and CD69-expressing OT-Is in the parenchyma of the SG, LG and KDN at 31 days after transfer. Data are combined from 2 experiments (n= 7 naïve and 6 infected recipients at day; n=6 naïve and 6 infected at day 31). D) Concatenated FACS plots, of CD103 and CD69 expression of the OT-I T cells in the SG from one representative experiment (n=3 naïve and n=3 infected recipients) with mean ± SEM values considering all the experiments. Error bars represent the SEM and the statistical significance in (B) was measured by unpaired t-test after log10 transformation of absolute numbers (***p<0.001).
CD8+ T cell homing to the salivary gland is mediated by α4β1 and chemokines at steady state
Recent data from Woyciechowski et al have suggested that the integrin α4β1 plays a critical role in T cell recruitment to the salivary gland during systemic inflammation induced by intravenous poly (I:C) treatment (32). We wondered whether this mechanism would also contribute to T cell recruitment to naïve salivary glands. To test this, naïve, WT OT-Is were transferred into congenic mice and driven to expand with MCMV-OVA infection. Five days after infection, splenic T cells containing activated OT-Is were transferred to MCMV-K181 infection-matched or naïve recipients in the presence or absence of antibodies to block the α4-integrin or the α4β7 integrin. As expected, the α4 blockade greatly reduced the number of OT-Is in the salivary gland of infected recipients 2 days after transfer (fig. 4A)(32). Likewise, in naïve recipients, the α4 blockade reduced the numbers of OT-Is in the salivary gland and in the lamina propria of the small intestine compared to the group that received the isotype control antibody, differences that were not observed in the spleens of the same animals (fig. 4B). In contrast, the blockade of α4β7 had no significant impact on T cells in the salivary gland, despite inhibiting OT-I migration to the lamina propria of the small intestine (fig. 4B), as expected (30, 32, 49). In addition, pre-incubation of the OT-I T cells with retinoic acid, which strongly induces the α4β7 integrin(50), had no effect on salivary gland migration (data not shown). The α4 integrin is known to pair with either β1 or β7 chain. Therefore, these data suggest that α4β1 is important for the migration of activated CD8+ T cells to the salivary gland regardless of infection. These data also imply that the salivary gland expresses sufficient VCAM-1, the ligand for α4β1, independently of local MCMV infection.
Fig. 4. CD8+ T cell accumulation in infected and uninfected salivary glands is dependent on α4 integrin and chemokines.
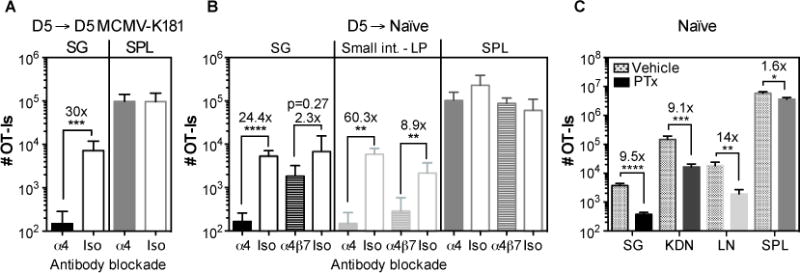
A and B) Naive OT-Is (5×104) were transferred into congenic mice that were infected one day later, with MCMV expressing OVA via the i.p. route. Splenocytes containing expanded OT-Is were recovered from the spleen 5 days later and transferred to (A) mice infected with wild-type MCMV (lacking Ova) or (B) naïve mice. Recipients were either treated or not with anti-α4 blocking antibody on the day of the transfer and mice were sacrificed 2 days after the transfer. Data show the absolute number of OT-I T cells recovered from the parenchya of salivary gland (SG), small intestine lamina propria (LP) and from the CD8β+ fraction of the spleen (SPL). Results were combined from 2 independent experiments (n= 5-6 mice per group). C) Naïve B6 mice were seeded with 8×106 in vitro activated OT-Is that had been treated with PTx or vehicle as a control before the adoptive transfer. Shown are the absolute numbers of OT-Is recovered from the parenchyma of salivary gland (SG), kidneys (KDN) lymph nodes (LN) and from the overall CD8β+ T cell population of the spleen 4 days after the adoptive transfer. Results from 2 independent experiments were combined (n=7). Error bars represent the SEM and statistical significances in (A-C) were measured by unpaired t-test after log10 conversion of the absolute numbers (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001).
Chemokines are important for activating integrins and promoting lymphocyte transendothelial migration. However, several recent reports have implicated chemokine-independent mechanisms of T cell recruitment (e.g. CD44, IL-33, or antigen (51–53)). To test whether chemokines are mediators of CD8+ T cell recruitment to the naïve salivary gland, OT-I T cells were activated in vitro and treated or not with Pertussis toxin (PTx), which irreversibly inhibits signalling through G-coupled protein receptors like chemokine receptors (54–56). Untreated, activated OT-I T cells migrated into all organs, including the salivary gland, within 4 days of transfer as expected (fig. 4C). In contrast, PTx treatment substantially reduced the numbers of OT-I T cells that migrated into the lymph nodes (fig. 4C), as previously shown (55, 56) as well as the salivary gland and kidney (fig. 4C). These data suggest that activated CD8+ T cells migrate to naive salivary glands in response to a chemokine signal.
Chemokines expressed in the salivary gland with or without MCMV infection
To explore the chemokines expressed by the salivary gland in the presence or absence of MCMV infection, we used RNA-Seq. Salivary glands were extracted from naïve mice or mice that had been infected for 2 weeks with MCMV strain K181. Perhaps not surprisingly, the dominant changes in overall gene expression could be traced back to immune responses, Type-I IFN, and IFN-γ, including increases in genes encoding MHC molecules and antigen processing machinery, Stat1 and Stat2, Irf1, Irf7 and Ifitm3 (Table S1A, which shows all genes that increased or decreased significantly). Interestingly, a number of IFN-γ-induced guanylate-binding proteins (Gbp2, 3, 6, and 7 and Ifi47) and IFN-γ-induced GTPases (Iigp1, Irgm1, Irgm2, Igtp) were among the most increased genes after infection, suggesting a dominant IFN-induced change in gene expression. Gene Set Enrichment Analyses (GSEA) using the Gene Ontology Biological Process (GOBP) database revealed that the top 20 enriched gene sets were all related to innate and adaptive immune responses, cytokine signaling, Type-I IFN, IFN-γ and leukocyte activation (Table S1C). In contrast, very few genes were significantly downregulated after MCMV infection and many of these were small nucleolar RNAs (SNORDs, Table S1A). There were no significant changes in expression of integrins or cell adhesion molecules that survived correction for multiple testing errors (FDR < 0.05), although expression of the α4, αL (CD11a), αX (CD11c) and β2 integrins was increased in the infected salivary glands when the gene set was filtered by a raw p value of lower than 0.05 (Table S1B).
Of the chemokines, expression of CCL5 and CXCL9 were significantly increased by infection (FDR < 0.05) and CCL7, CCL8, CCL12, and CXCL10 were also detected when we used a raw p-value cutoff of 0.05 (fig. 5 and table S1A and S1B). In contrast, several chemokines were abundantly expressed in the salivary gland, regardless of infection, including CCL28, CXCL12, CXCL14, CXCL16, CXCL17 and CX3CL1 (fig.5 and Table S1B). All of these chemokines, with the exception of CXCL17, have been described to recruit T cells in various settings(29, 57–69). These data show that MCMV infection of the salivary gland induces a dominant inflammatory response centered on IFN-stimulated gene expression. However, several chemokines were abundantly expressed in the salivary gland regardless of infection.
Fig. 5. Chemokine profile of the salivary gland after MCMV infection.
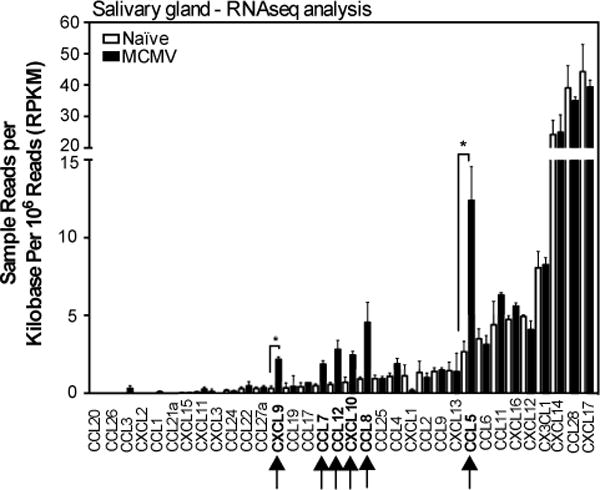
The array of chemokines expressed in the salivary glands of uninfected mice, or 14 days after MCMV infection. Shown are the sample reads per kilobase per million reads (RPKM) of chemokines to illustrate the relative abundance of different transcripts. The complete gene list of genes that were significantly differentially expressed is shown in Table S1A. The complete gene list sorted by fold change, regardless of significance, is shown in Table S1B. Data are from one experiment (n=3 mice per group). Error bars represent the SEM (↑=p< 0.05; *=FDR<0.05).
MCMV-specific T cells express multiple chemokine receptors and are able to migrate towards multiple chemokines
To test whether MCMV-specific T cells could migrate to some of the chemokines that were present in the salivary gland independently of infection, we conducted Transwell migration assays in vitro with the top 6 most abundantly expressed chemokines, along with CCL5, CXCL9, CXCL10 and CCL19. T cells were harvested from the spleens of mice seven days after MCMV infection, which represents the peak of T cell clonal expansion (34, 70) and a time at which splenic T cells migrate readily to the salivary gland in adoptive transfer experiments (not shown). In multiple experiments, CCL28 and CXCL16 failed to induce any migration of splenic CD8+ T cells (data not shown). In contrast, robust CD8+ T cell migration was induced by CCL19 (~9-fold increased over background, not shown), but most of these cells were CD44low as expected (not shown) and there was no increase in tetramer-binding MCMV-specific T cells among the migrated cells (fig. 6A). All of the other tested chemokines induced significant migration of MCMV-specific T cells (fig. 6A, 6B). These included chemokines that were expressed at high levels constitutively (CXCL12, CXCL14, CXCL17 and CX3CL1), as well as chemokines induced in the salivary gland by infection (CXCL9, CXCL10 and CCL5) (fig.6). These data show that MCMV-specific T cells can migrate in vitro, toward several chemokines that are either induced by MCMV infection or constitutively expressed in the salivary gland.
Fig. 6. MCMV-specific CD8+ T cells migrate towards multiple chemokines.
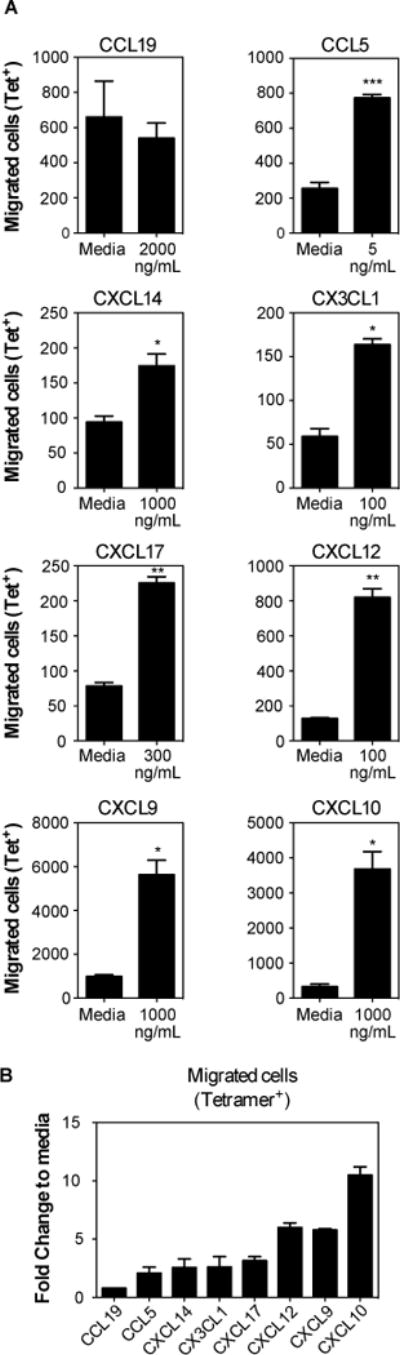
Mice were sacrificed 7 days after MCMV infection (i.p.). CD8+ T cells were enriched and used to performed transwell migration assays with multiple chemokines. Assays were performed with replicates and media control was included in each assay to account for plate-to-plate variation. (A) Absolute numbers of MCMV-specific T cells (M45-Tetramer+ plus M38-Tetramer+) that migrated towards the indicated chemokine in comparison to control wells with media alone. Data are from a single experiment performed in triplicate or quadruplicate, and representative of 2 to 5 experiments per chemokine. Statistical significance was determined using paired t-test (*p<0.05; **p<0.01; ***p<0.001). (B) Fold change of migration in comparison to media. Pooled data from 2 to 4 independent experiments indicating the fold change of migration of MCMV-specific cells (M45-Tetramer+ plus M38-Tetramer+) in response to the indicated chemokines at the concentrations shown in (A).
Next we used flow cytometry to explore chemokine receptor expression by OT-I T cells in the spleen and salivary gland, 7-9 days after MCMV-Ova infection. Cells were further differentiated by KLRG1 expression since our previous data showed that most KLRG1 expressing T cells failed to accumulate in the parenchyma of the salivary gland (11). Seven to nine days after MCMV-Ova infection, the KLRG1 expressing splenic OT-Is were mostly CX3CR1+ CXCR3−, which is consistent with recent work (71, 72). Additionally, we found that these KLRG1+ cells expressed CXCR6, but mostly lacked CXCR4 (fig. 7A). In contrast, the KLRG1− portion of the splenic OT-Is contained subsets expressing or lacking CX3CR1 and a higher frequency of KLRG1− cells expressed CXCR3, CXCR4 and CXCR6 (fig 7A). Of note, these phenotypes were consistent: at 7 months after MCMV infection, KLRG1 expression on MCMV-specific T cells still correlated with the expression of CX3CR1 and a lack of CXCR3 (data not shown).
Fig. 7. Most CD8+ T cells in the parenchyma of the salivary gland lack KLRG1 and CX3CR1, but express multiple other chemokine receptors.
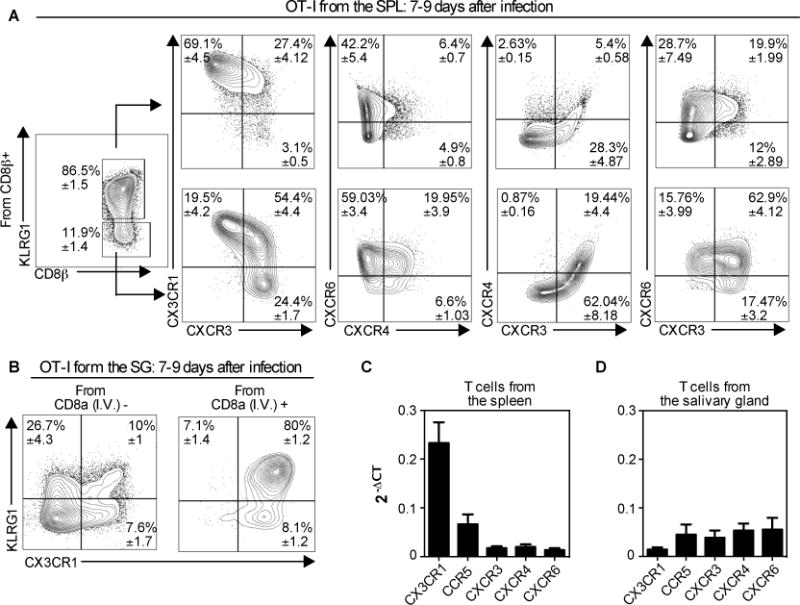
Naïve OT-Is (2×103) were transferred to naïve B6 mice that were infected via the i.p. route one day later with MCMV expressing OVA. Mice were sacrificed 7 or 9 days after infection. (A) Chemokine receptor expression within the KLRG1+ and KLRG1− subsets of the OT-I T cells in the spleen. (B) KLRG1 and CX3CR1 expression of OT-Is from the vasculature (I.V.+) and parenchyma (I.V.−) portions of the salivary gland. In A and B, the data show concatenated FACS plots from one representative experiment at day 9 (n= 3) with mean ± SEM values in each quadrant derived from all experiments (total n=8). (C and D) Naive OT-Is were transferred as in A and sorted from the spleen (B) and the salivary gland (C) 7 days after infection with MCMV expressing OVA. Chemokine receptor expression was assessed on sorted T cells by RT-qPCR. Data were combined from 2-5 independent RT-qPCR assays per sample, with cDNA from OT-I T cells sorted from 2-3 independent mice. Error bars represent SEM.
Within the parenchyma of the salivary gland, the majority of cells lacked CX3CR1 and KLRG1 (fig 7B, left). In sharp contrast, T cells found in the vasculature of the salivary gland (i.e. stained by the intravascular antibody) were almost entirely CX3CR1+ and most also expressed KLRG1 (fig. 7B, right). We have had difficulty getting consistent staining of any of other chemokine receptors on T cells extracted from the salivary glands. In our hands, CXCR3 has been sensitive to the collagenase used to extract T cells from the salivary gland and attempts at extracting T cells without collagenase have failed to result in sufficient T cell recovery. Therefore, we measured the expression of chemokine receptors on T cells sorted from the spleen and salivary gland using qRT-PCR. Naïve, wild-type OT-Is were transferred into congenic mice and driven to expand with a MCMV-OVA infection. Seven days later, OT-I T cells sorted from the spleen and salivary gland expressed CX3CR1, CCR5, CXCR3, CXCR4 and CXCR6 (fig. 7C-D). In contrast, CCR10 (receptor for CCL28) and CXCR7 (one receptor for CXCL14) were undetectable on either spleen or salivary gland localized OT-I T cells (data not shown). Interestingly, while salivary gland and spleen-localized T cells expressed comparable amounts of CXCR3 and CCR5, salivary gland-localized T cells expressed more CXCR4 (p=0.016) and CXCR6 (p=0.023) than spleen-localized T cells. In contrast, spleen-localized T cells expressed much more CX3CR1 than salivary gland localized T cells (p<0.0001, fig. 7C-D), consistent with our FACS data.
CXCR3 is critical for T cell migration to uninfected salivary glands, but is dispensable after MCMV infection
Since MCMV infection induced chemokines that bind CCR5 and CXCR3, we directly tested whether these receptors were critical for the accumulation of MCMV-specific T cells in the salivary gland. To this end, OT-I T cells that expressed or lacked either receptor were mixed with their wild-type counterparts and co-transferred to congenic B6 recipients. Recipient mice were infected on the following day with MCMV virus expressing OVA. Two weeks after the infection, there were only small reductions in the numbers of CXCR3 KO or CCR5 KO OT-Is in the salivary gland (fig. 8A and 8C) and these small differences were mirrored in the spleens of the same animals, implying no impairment in the recruitment of T cells lacking either CXCR3 (fig. 8A) or CCR5 (fig. 8C) receptor. Moreover, there were no differences in the absolute numbers of OT-Is that expressed the tissue-resident markers CD69 and CD103 in the gland (fig. 8B and 8D).
Fig. 8. Lack of CXCR3 or CCR5 does not impact the accumulation of CD8+ T cells in the salivary gland after MCMV infection.
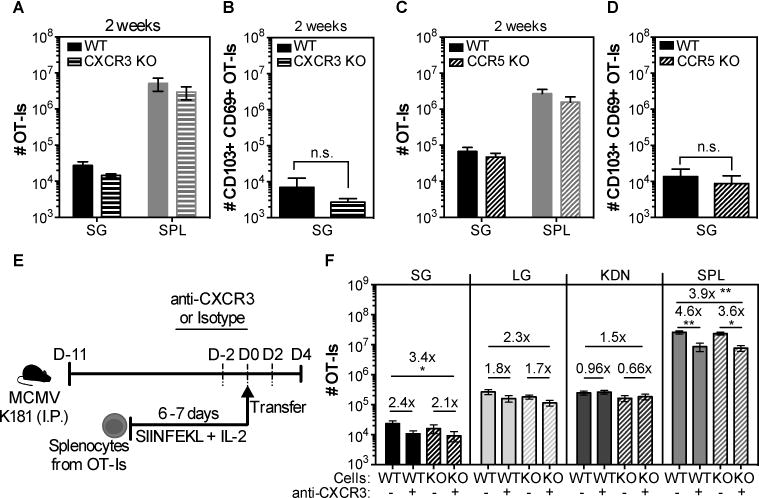
(A and C) Wild-type (WT) OT-Is and OT-Is that lacked either the chemokine receptor CXCR3 or CCR5 were mixed (1×103 of each) and co-transferred to naïve B6 mice that were then infected with MCMV-Ova. Shown are the overall numbers of CXCR3 KO (A) or CCR5 KO (C) versus wild-type (WT) OT-Is from the spleen (SPL) and the parenchyma of the salivary gland (SG) 14 days after infection. (B and D) Shown are the absolute numbers of CD103+ CD69+ OT-I T cells from the SG. Data are combined data from 2 independent experiments for each KO (dashed bars)/WT (filled bars) pair (n=7 for the WT/CXCR3 and n=5 for the WT/CCR5 experiments). E) Experimental design of the OT-I adoptive transfer for F. WT and CCR5 KO OT-Is were activated in vitro, and 4×106 of each were mixed. Mixed cells were treated with either anti-CXCR3 antibody or an isotype control and co-transferred to MCMV infected mice that were treated with anti-CXCR3 (+) or an isotype control antibody (-) via i.p. injections every other day starting 2 days before the adoptive transfer. F) Shown are the absolute numbers of WT (filled bars) or CCR5 KO (dashed bars) OT-I T cells in the parenchyma of the salivary gland (SG), lungs (LG), kidneys (KDN) and from the overall CD8β+ T cells from the spleen (SPL) of CXCR3 blocked (+) or isotype control-treated (-) mice. Data from 2 independent experiments (n=7 for the isotype treated group and n=8 for the CXCR3 treated group). Error bars represent that SEM. The statistical significance was measured by unpaired t-test after log10 conversion of the absolute numbers (A-D) and One-way ANOVA (F).
It was possible that CCR5 and CXCR3 played redundant roles in migration of T cells to the salivary gland during MCMV infection. To address this possibility, wild-type and CCR5 KO OT-I T cells were activated in vitro and transferred into infected mice, with or without an antibody specific for CXCR3 that has been reported to block CXCR3-dependent cell migration(73, 74). For infection, we used the K181 strain of MCMV lacking OVA, again to avoid the influence of antigen and additional T cell expansion after adoptive transfer. However, donor OT-I T cells still migrated to the salivary gland in all cases and the absolute number of OT-Is in the gland was similar in both groups independently of the CXCR3 blockade (fig. 8F). There was a subtle difference on the overall number between the WT OT-Is in the unblocked group and the CCR5KO OT-Is in the CXCR3 blocked group, possibly suggesting a combined impact of CXCR3 and CCR5 on T cell migration. However, the effect was subtle and the CXCR3 blockade had an impact in the spleen in all mice. Therefore, it is difficult to distinguish if this effect was partially due to differences on T cells in circulation (fig. 8F). There was no impact of CCR5 deficiency with or without CXCR3 blockade on T cell migration to lungs or kidneys. Although it is possible that the CXCR3 blockade was poorly effective in vivo in infected mice, these data suggest that CCR5 and CXCR3 are not required for T cell accumulation in infected salivary glands.
As a control for these experiments, we had transferred in vitro activated WT OT-Is to naïve recipients, with or without CXCR3 blockade. Surprisingly, in naïve mice, the CXCR3 blockade had a striking impact on T cell accumulation to the salivary gland (fig. 9), resulting in approximately a 9-fold reduction in the numbers of OT-Is that reached the parenchyma (fig. 9B). Although the blockade had an impact on the numbers of OT-I T cells in the spleen, as in infected mice (fig 8F), it was lower than the impact on the salivary gland in naïve mice and there was no effect on the T cells recovered from the lungs or kidneys (fig. 9B). These data imply that the CXCR3 blockade was effective in naïve mice and suggest that the CXCR3 blockade was having a specific impact on T cell accumulation in the salivary glands of naïve mice, despite the absence of an effect in infected mice.
Fig. 9. CXCR3 blockade reduces the recruitment of CD8+ T cells to salivary glands in uninfected mice independently of IFNγ.
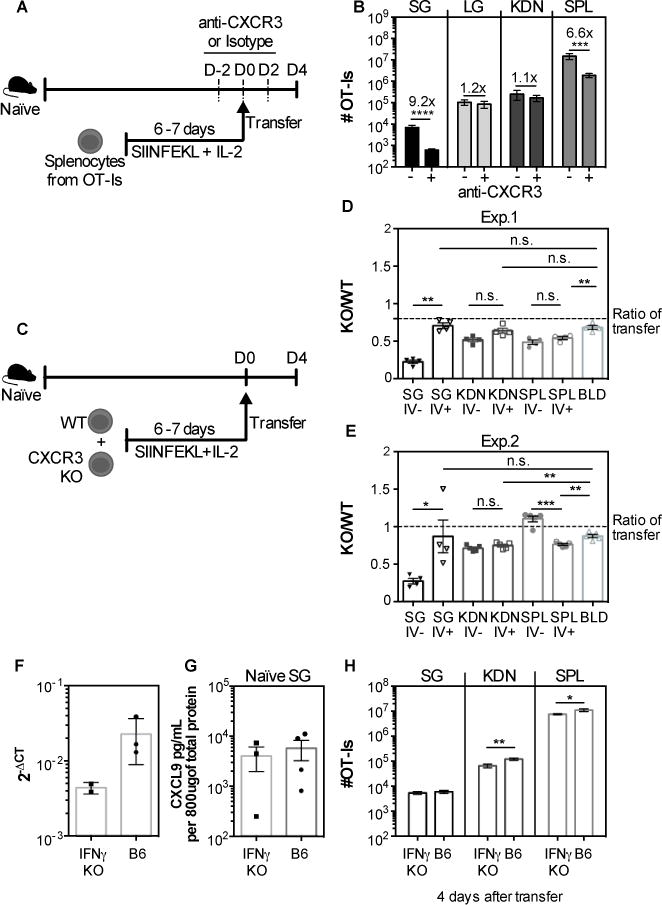
A) WT OT-I T cells were activated in vitro, treated with either anti CXCR3 antibody or isotype control and 4×106 cells were transferred to naïve mice. The recipients were also treated with anti CXCR3 antibody or isotype control every other day starting 2 days before transfer until sacrifice. (B) The absolute numbers of OT-I T cells that reached the parenchyma of the salivary gland (SG), lungs (LG) and kidneys (KDN) and from the CD8β+ population of the spleen (SPL) are shown. Data are from 2 independent experiments (n=7 for the isotype treated group and n=8 for the group treated with anti-CXCR3). (C) Experimental design for figures D and E. WT and CXCR3 KO OT-Is were activated in vitro, and 4×106 of each were mixed and co-transferred to naïve mice. (D-E) Shown is the ratio of KO/WT OT-Is in the vasculature (I.V.−) and parenchyma (I.V.+) of the SG, KDN, SPL, and from the overall CD8β+ cells in the blood. Two independent experiments are shown in D and E (n=4 in D and n=5 in E). The dotted lines represent the ratio of KO to WT cells in the transferred pool (as assessed by FACS on the day of transfer). Error bars represent SEM. Statistical significance was measured by unpaired t-test after log10 conversion of the absolute numbers (B) and ratio paired t-test in (D-E). The levels of CXCL9 in the SG of naïve B6 and IFN-γ KO mice were determined by qPCR (F) and ELISA (G). Data from one experiment (n=2-3 in F; n=3-4 in G). H) WT OT-I were activated in vitro and 4×106 T cells were transferred to naïve B6 or IFN-γ KO mice (similarly to A and B). Four days after transfer the organs were collected and the overall number of OT-I in the SG, KDN and SPL are shown. Data are pooled from 2 independent experiments (n=6 B6 mice and 7 IFN-γ KO mice). Error bars represent the SEM and the statistical significance was measured by unpaired t-test after log10 conversion of the absolute numbers (*p<0.05; **p<0.01).
To confirm these surprising results, and determine whether CXCR3 was needed on T cells specifically, CXCR3 KO and WT OT-Is were activated in vitro, mixed together, and co-transferred to naïve mice. Four days after transfer, the absolute numbers of each OT-I population was assessed and the ratio of KO to WT cells was calculated within the parenchyma and the circulation. In line with our blocking antibody data, accumulation of CXCR3 KO OT-Is in the salivary gland, but not the kidneys or spleen, was markedly impaired, in two separate experiments (fig. 9D-E). Together, these data strongly imply that T cell migration to and accumulation in uninfected salivary glands depends critically on CXCR3.
CXCL9 is expressed in the salivary gland at steady state even in the absence of IFN-γ
Because the ligands for CXCR3 (CXCL9 and CXCL10 in B6 mice) are strongly induced by IFN-γ and upregulated by MCMV infection (fig. 5), we wished to confirm that these ligands were present in naïve mice and test whether IFN-γ was required for T cell migration to the salivary gland. The chemokine CXCL9 was readily detectable by ELISA and qRT-PCR in the salivary glands of naïve B6 and was also evident in IFN-γ KO mice (fig. 9F-G). In addition, when in vitro activated OT-Is were transferred to naïve IFN-γ KO or B6 mice they reached the salivary gland at similar numbers (fig 9H). Thus, CXCL9 is available for T cell recruitment to uninfected salivary glands with or without infection or IFN-γ. Collectively, these data suggest that the integrin α4β1 and the chemokine receptor CXCR3 play critical roles in the recruitment of T cells to uninfected or non-inflamed salivary glands but that CXCR3 is redundant during inflammation induced by MCMV infection.
Discussion
The salivary gland is an important site of viral infection for transmission to new hosts. Indeed, all β- and γ-herpesviruses infect the salivary gland and are shed into saliva. Moreover, herpesviruses like CMV establish latency in the salivary gland and reactivate readily in this site during periods of immune suppression, particularly when CD8+ T cells have been depleted(9). Therefore, it is important to understand the mechanisms that promote and establish CD8+ T cell residency in the salivary gland.
Recent studies have shown that salivary gland localized T cells can become TRM that reside near or within the epithelial layer (10, 11). Interestingly, the mechanisms governing TRM formation and maintenance differ in different tissues(75). While TGF-β is described as critical for promoting CD103-expressing TRM in most tissues tested to date, including the salivary gland, a role for antigen and inflammation is more variable(10, 11, 15, 21, 25, 76). Antigen seems to be required for the formation of CD103+ TRM cells in the central nervous system and lungs(19, 20, 77). In the skin, the efficiency of T cell recruitment is poor without local inflammation and antigen improves the TRM maintenance as well as the selection of TRM specificities (16, 24). In contrast, TRM can form in the small intestine independently of antigen, infection, or commensal microbiota. In fact, antigen in the small intestine may reduce expression of CD103+ on T cells that reach this tissue(28). Remarkably, like the small intestine, the salivary gland seems capable of recruiting activated T cells in the absence of a specific infection or inflammation (10–12). In fact, our data show that naïve, uninfected salivary glands and those with an active MCMV infection were equally capable of recruiting activated T cells over a month of time (fig. 3). These data suggest that the salivary gland and small intestine may represent a set of tissues that are continuously surveyed by activated T cells and capable of inducing their retention through constitutive TGF-β expression. T cell recruitment to both naïve and infected salivary glands depended on the α4 integrin (fig. 4) while recruitment to naïve salivary glands depended on the chemokine receptor CXCR3 (fig. 9). These results imply that sufficient levels of VCAM-1 and CXCR3 ligands are expressed at steady state in the salivary gland for T cell recruitment. Indeed, CXCL9 was detectable in the salivary glands of naïve B6 mice and its expression was not dependent on IFN-γ (fig 9). These data establish a mechanism for the recruitment of activated T cells to the salivary gland, regardless of infection.
Given that CXCL9 is classically thought of as IFN-γ-induced chemokine, it is interesting that it was constitutively expressed in the salivary gland. It is possible that animal colony conditions or husbandry practices within our animal colony contribute to the constitutive expression of CXCL9 that we observed (fig 9) and the constitutive ability of naïve salivary glands to recruit T cells (fig 3). However, the recruitment of T cells to the salivary gland in naïve mice has been demonstrated in 2 other laboratories besides our own (10, 12), suggesting that our results cannot be explained by housing conditions in the Thomas Jefferson University animal facility. It must be noted that the salivary gland ducts are open to the oral cavity and therefore likely to be colonized by the oral microbiome. Thus, it is possible that CXCL9 expression is a direct response to oral microbiota. Interestingly, several chemokines including CXCL9, CXCL10 and CXCL11, can have antimicrobial functions (78–80). Therefore, it is possible that T cell recruitment is secondary to the anti-microbial role of chemokines in the salivary gland, and perhaps other digestive tissues. It will be fascinating in future work to determine whether CXCL9 levels, and the steady-state recruitment of activated T cells to the salivary gland, are controlled by the oral microbiome.
Although CXCR3 appears to play a crucial role in the recruitment of T cells to naïve salivary glands, it is important to note that our experiments did not test whether CXCL9 and CXCL10 were required to recruit activated T cells to the naïve or infected salivary glands. Indeed, it is possible that CXCR3 was being activated by alternative ligands in naïve mice or that CXCL9 and CXCL10 could use alternative chemokine receptors in infected mice. Moreover, heterodimerization of chemokines as well as heterodimerization of receptors have been described and can increase the breadth of potential ligand/receptor interactions (81, 82). Therefore, extensive future work will be needed to tease apart the specific ligand/receptor interactions involved in T cell recruitment in different settings.
Although infected salivary glands recruited activated OT-Is more rapidly, we observed a plateau in recruitment (fig 3). It is possible that the ultimate numbers of TRM that accumulated in the salivary gland may have depended on the potential of the T cells induced by in vitro activation. In our hands, a subset of in vitro activated OT-Is expressed CXCR3, CXCR4 and/or CXCR6, and the population, as a whole, almost completely lacked CX3CR1 (not shown), much like our MCMV-specific T cells in the salivary gland (fig. 7). However, we cannot exclude the possibility that only a subset of activated OT-I T cells was capable of migrating to the salivary gland and becoming TRM, which could simulate a plateau in OT-I TRM cell numbers in the salivary gland over time. If such limit in cell numbers existed however, it was unlikely to be caused by competition for space or environmental cues in the salivary gland. New OT-I or VacV-specific TRM were able to form in the salivary glands of mice previously infected with MCMV (fig 2 and data not shown), despite the fact that infected salivary glands contained 20 to 30-fold more T cells than naïve salivary glands (data not shown). Together, these experiments suggest that the salivary gland can accommodate more TRM than were induced in our experimental systems. Nevertheless, it is possible that pre-existing TRM in the salivary gland would be reduced over time in animals exposed to repeated infections. Thus, it will be exciting to learn how salivary gland-localized TRM populations would evolve over time in response to multiple different infections.
Our data showed that most KLRG1 expressing MCMV-specific T cells upregulated CX3CR1, but lost CXCR3 expression and failed to migrate into the salivary gland, even during MCMV infection (fig. 7). However, CXCR3 deficiency did not preclude MCMV-specific T cells from the salivary gland during infection (fig. 8). Thus, presumably the loss of CXCR3 alone was not responsible for the inability of these T cells to migrate into the salivary gland. Moreover, a lack of the α4-integrin cannot explain the failure of these cells to enter the salivary gland as we have seen comparable expression of α4 integrin on KLRG1+ and KLRG− MCMV-specific T cells (data not shown). These results were surprising to us, but also broadly consistent with work from the Pircher lab, which suggested that CXCR3 was dispensable for CD8+ T cell migration to the salivary gland after LCMV-WE infection(32). LCMV-WE is reported to not infect the salivary gland directly (12) and it is unclear whether CXCR3 ligands in the salivary gland are increased by LCMV-WE infection. Regardless, it is certain that other changes induced by infection will contribute to the efficiency and speed of T cell recruitment. Indeed, LCMV-WE infection was associated with an increase in VCAM-1 on the salivary gland vascular endothelium that likely facilitated the recruitment of T cells. Thus, it is possible that MCMV infection reduced the burden on CXCR3 by increasing additional molecules involved in recruitment of T cells. Another possible explanation is that MCMV infection induced an array of chemokines that could redundantly recruit T cells. We tested whether CCR5 and CXCR3 were acting together to recruit T cells by blocking CXCR3 on CCR5 KO T cells. However, these results should be interpreted cautiously. Although the CXCR3 blockade reduced T cell recruitment to the salivary gland in naïve mice, it is unclear whether the antibody was fully able to block the CXCR3 receptor in infected mice, which likely have many more CXCR3-expressing cells. Future work will need to explore the specific mechanisms used by T cells to enter the salivary gland during acute MCMV infection.
In conclusion, our data show that the salivary gland is able to constitutively recruit CD8+ T cells in a α4-integrin and CXCR3-dependent manner, and subsequently induce and sustain TRM populations in the absence of infection, antigen or inflammation. Thus, it is plausible to think that the TRM populations in the salivary gland, and perhaps other tissues in the digestive tract, will retain a record of previous infections and their specificities, regardless of whether those infections were related to the salivary gland. Moreover, the fact that CXCR3 was critical for recruitment of T cells to naïve salivary glands should be useful for guiding the future development of vaccines that aim to establish or boost the mucosal immune responses in the salivary gland.
Supplementary Material
Footnotes
This work was supported by a grant from the NIH (AI106810) awarded to C.M.S. and by a grant from the Portuguese Foundation for Science and Technology (SFRH-BD-52319-2013), awarded to S.C.D.
References
- 1.Cannon MJ, Stowell JD, Clark R, Dollard PR, Johnson D, Mask K, Stover C, Wu K, Amin M, Hendley W, Guo J, Schmid DS, Dollard SC. Repeated measures study of weekly and daily cytomegalovirus shedding patterns in saliva and urine of healthy cytomegalovirus-seropositive children. BMC Infect Dis. 2014;14:1–10. doi: 10.1186/s12879-014-0569-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harnett GB, Farr TJ, Pietroboni GR, Bucens MR. Frequent shedding of human herpesvirus 6 in saliva. J Med Virol. 1990;30:128–130. doi: 10.1002/jmv.1890300209. [DOI] [PubMed] [Google Scholar]
- 3.Miller CS, Berger JR, Mootoor Y, Avdiushko SA, Zhu H, Kryscio RJ. High prevalence of multiple human herpesviruses in saliva from human immunodeficiency virus-infected persons in the era of highly active antiretroviral therapy. J Clin Microbiol. 2006;44:2409–2415. doi: 10.1128/JCM.00256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddehase MJ, Balthesen M, Rapp M, Jonjic S, Pavic I, Koszinowski UH. The Conditions of Primary Infection Define the Load of Latent Viral Genome in Organs and the Risk of Recurrent Cytomegalovirus Disease. J Exp Med. 1994;179:185–193. doi: 10.1084/jem.179.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonjić S, Mutter W, Weiland F, Reddehase MJ, Koszinowski UH. Site-restricted persistent cytomegalovirus infection after selective long-term depletion of CD4+ T lymphocytes. J Exp Med. 1989;169:1199–212. doi: 10.1084/jem.169.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crough T, Khanna R. Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev. 2009;22:76–98. doi: 10.1128/CMR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henson D, Strano AJ. Necrosis of infected and morphologically normal submaxillary gland acinar cells during termination of chronic infection. Am J Pathol. 1972;68:183–202. [PMC free article] [PubMed] [Google Scholar]
- 8.Krmpotic A, Bubic I, Polic B, Lucin P, Jonjic S. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 2003;5:1263–1277. doi: 10.1016/j.micinf.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Polic B, Hengel H, Krmpotic A, Trgovcich J, Pavic I, Lucin P, Jonjic S, Koszinowski UH. Hierarchical and Redundant Lymphocyte Subset Control Precludes Cytomegalovirus Replication during Latent Infection. J Exp Med. 1998;188 doi: 10.1084/jem.188.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thom JT, Weber TC, Walton SM, Torti N, Oxenius A. The Salivary Gland Acts as a Sink for Tissue-Resident Memory CD8+ T Cells, Facilitating Protection from Local Cytomegalovirus Infection. Cell Rep. 2015;13:1125–1136. doi: 10.1016/j.celrep.2015.09.082. [DOI] [PubMed] [Google Scholar]
- 11.Smith CJ, Caldeira-Dantas S, Turula H, Snyder CM. Murine CMV Infection Induces the Continuous Production of Mucosal Resident T Cells. Cell Rep. 2015;13:1137–1148. doi: 10.1016/j.celrep.2015.09.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofmann M, Pircher H. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. PNAS. 2011;108:16741–16746. doi: 10.1073/pnas.1107200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. 2014;41:886–897. doi: 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol. 2015;16:1–11. doi: 10.1038/nri.2015.3. [DOI] [PubMed] [Google Scholar]
- 15.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T. The developmental pathway for CD103+ CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14:1294–301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 16.Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med. 2016;213:951–66. doi: 10.1084/jem.20151855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature. 2009;462:2–6. doi: 10.1038/nature08511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hogan RJ, Usherwood EJ, Zhong W, Roberts AD, Dutton RW, Harmsen AG, Woodland DL. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J Immunol. 2001;166:1813–1822. doi: 10.4049/jimmunol.166.3.1813. [DOI] [PubMed] [Google Scholar]
- 19.Morabito KM, Ruckwardt TR, Redwood AJ, Moin SM, Price DA, Graham BS. Intranasal administration of RSV antigen-expressing MCMV elicits robust tissue-resident effector and effector memory CD8+ T cells in the lung. Mucosal Immunol. 2016;10:1–10. doi: 10.1038/mi.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. PNAS. 2010;107:17872–9. doi: 10.1073/pnas.1010201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee YT, Suarez-Ramirez JE, Wu T, Redman JM, Bouchard K, Hadley GA, Cauley LS. Environmental and antigen receptor-derived signals support sustained surveillance of the lungs by pathogen-specific cytotoxic T lymphocytes. J Virol. 2011;85:4085–94. doi: 10.1128/JVI.02493-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, Gebhardt T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. PNAS. 2012;109:7037–42. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davies B, Prier JE, Jones CM, Gebhardt T, Carbone FR, Mackay LK. Cutting Edge: Tissue-Resident Memory T Cells Generated by Multiple Immunizations or Localized Deposition Provide Enhanced Immunity. J Immunol. 2017;198:2233–2237. doi: 10.4049/jimmunol.1601367. [DOI] [PubMed] [Google Scholar]
- 24.Muschaweckh A, V, Buchholz R, Fellenzer A, Hessel C, König PA, Tao S, Tao R, Heikenwälder M, Busch DH, Korn T, Kastenmüller W, Drexler I, Gasteiger G. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8 + T cells. J Exp Med. 2016 doi: 10.1084/jem.20160888. jem.20160888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature. 2012;483:227–231. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, Cauley LS. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. 2014;95:215–24. doi: 10.1189/jlb.0313180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venturi V, Nzingha K, Amos TG, Wisler CC, Dekhtiarenko I, Cicin-sain L, Davenport MP, Rudd BD. The Neonatal CD8+ T Cell Repertoire Rapidly Diversifies during Persistent Viral Infection. J Immunol. 2016:199. doi: 10.4049/jimmunol.1501867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, Vezys V, Masopust D. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. 2012;188:4866–75. doi: 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato T, Thorlacius H, Johnston B, Staton TL, Xiang W, Littman DR, Butcher EC. Role for CXCR6 in recruitment of activated CD8+ lymphocytes to inflamed liver. J Immunol. 2005;174:277–283. doi: 10.4049/jimmunol.174.1.277. [DOI] [PubMed] [Google Scholar]
- 30.Hamann A, Andrew DP, Jablonski-Westrich D, Holzmann B, Butcher EC. Role of alpha 4-integrins in lymphocyte homing to mucosal tissues in vivo. J Immunol. 1994;152:3282–3293. [PubMed] [Google Scholar]
- 31.Campbell DJ, Butcher EC. Intestinal attraction : CCL25 functions in effector lymphocyte recruitment to the small intestine. J Clin Invest. 2002;110:1079–1081. doi: 10.1172/JCI16946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woyciechowski S, Hofmann M, Pircher H. α4β1 integrin promotes accumulation of tissue-resident memory CD8 + T cells in salivary glands. Eur J Immunol. 2016;1:244–250. doi: 10.1002/eji.201646722. [DOI] [PubMed] [Google Scholar]
- 33.Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. 2011;317:620–631. doi: 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turula H, Smith CJ, Grey F, Zurbach KA, Snyder CM. Competition between T cells maintains clonal dominance during memory inflation induced by MCMV. Eur J Immunol. 2013;43:1252–1263. doi: 10.1002/eji.201242940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cunningham PT, Lloyd ML, Harvey NL, Williams E, Hardy CM, Redwood AJ, Lawson MA, Shellam GR. Promoter control over foreign antigen expression in a murine cytomegalovirus vaccine vector. Vaccine. 2011;29:141–151. doi: 10.1016/j.vaccine.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 36.Farrington LA, Smith TA, Grey F, Hill AB, Snyder CM. Competition for Antigen at the Level of the APC Is a Major Determinant of Immunodominance during Memory Inflation in Murine Cytomegalovirus Infection. J Immunol. 2013;190:3410–3416. doi: 10.4049/jimmunol.1203151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zurbach KA, Moghbeli T, Snyder CM. Resolving the titer of murine cytomegalovirus by plaque assay using the M2-10B4 cell line and a low viscosity overlay. Virol J. 2014;11:71. doi: 10.1186/1743-422X-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snyder CM, Cho KS, Bonnett EL, Allan JE, Hill AB. Sustained CD8+ T cell memory inflation after infection with a single-cycle Cytomegalovirus. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, Masopust D. Cutting Edge: Intravascular Staining Redefines Lung CD8 T Cell Responses. J Immunol. 2012;189:2702–2706. doi: 10.4049/jimmunol.1201682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith CJ, Turula H, Snyder CM. Systemic Hematogenous Maintenance of Memory Inflation by MCMV Infection. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Memory Inflation during Chronic Viral Infection Is Maintained by Continuous Production of Short-Lived, Functional T Cells. Immunity. 2008;29:650–659. doi: 10.1016/j.immuni.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao Y, Smyth GK, Shi W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 43.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallichan WS, Kenneth LR. Long-lived Cytotoxic T Lymphocyte Memory in Mucosal Tissues After Mucosal but Not Systemic Immunization. J Exp Med. 1996;184:1879–1890. doi: 10.1084/jem.184.5.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takamura S, Roberts AD, Jelley-Gibbs DM, Wittmer ST, Kohlmeier JE, Woodland DL. The route of priming influences the ability of respiratory virus-specific memory CD8+ T cells to be activated by residual antigen. J Exp Med. 2010;207:1153–1160. doi: 10.1084/jem.20090283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrançois L. Oral infection drives a distinct population of intestinal resident memory CD8+ T cells with enhanced protective function. Immunity. 2014;40:747–757. doi: 10.1016/j.immuni.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oduro JD, Redeker A, Lemmermann NaW, Ebermann L, Marandu TF, Dekhtiarenko I, Holzki JK, Busch DH, Arens R, Čičin-Šain L. Murine cytomegalovirus (CMV) infection via the intranasal route offers a robust model of immunity upon mucosal CMV infection. J Gen Virol. 2016;97:185–195. doi: 10.1099/jgv.0.000339. [DOI] [PubMed] [Google Scholar]
- 49.Salmi M, Jalkanen S. Lymphocyte homing to the gut: Attraction, adhesion, and commitment. Immunol Rev. 2005;206:100–113. doi: 10.1111/j.0105-2896.2005.00285.x. [DOI] [PubMed] [Google Scholar]
- 50.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 51.Walch JM, Zeng Q, Li Q, Oberbarnscheidt MH, Hoffman RA, Williams AL, Rothstein DM, Shlomchik WD, Kim JV, Camirand G, Lakkis FG. Cognate antigen directs CD8+ T cell migration to vascularized transplants. J Clin Invest. 2013;123:2663–2671. doi: 10.1172/JCI66722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Komai-Koma M, Gracie JA, Wei XQ, Xu D, Thomson N, McInnes IB, Liew FY. Chemoattraction of human T cells by IL-18. J Immunol. 2003;170:1084–1090. doi: 10.4049/jimmunol.170.2.1084. [DOI] [PubMed] [Google Scholar]
- 53.Katakai T, Hara T, Sugai M, Gonda H, Nambu Y, Matsuda E, Agata Y, Shimizu A. Chemokine-independent preference for T-helper-1 cells in transendothelial migration. J Biol Chem. 2002;277:50948–50958. doi: 10.1074/jbc.M204133200. [DOI] [PubMed] [Google Scholar]
- 54.Mangmool S, Kurose H. Gi/o protein-dependent and -independent actions of pertussis toxin (PTX) Toxins (Basel) 2011;3:884–899. doi: 10.3390/toxins3070884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spangrude GJ, Braaten BA, Daynes RA. Molecular mechanisms of lymphocyte extravasation. I. Studies of two selective inhibitors of lymphocyte recirculation. J Immunol. 1984;132:354–362. [PubMed] [Google Scholar]
- 56.Cyster JG, Goodnow CC. Pertussis Toxin Migration of B and T Lymphocytes into Splenic White Pulp Cords. J Exp Med. 1995;182:581–586. doi: 10.1084/jem.182.2.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kohlmeier JE, Miller SC, Smith J, Lu B, Gerard C, Cookenham T, Roberts AD, Woodland DL. The Chemokine Receptor CCR5 Plays a Key Role in the Early Memory CD8+ T Cell Response to Respiratory Virus Infections. Immunity. 2008;29:101–113. doi: 10.1016/j.immuni.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crispe IN. Migration of lymphocytes into hepatic sinusoids. J Hepatol. 2012;57:218–220. doi: 10.1016/j.jhep.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 59.Galkina E, Thatte J, Dabak V, Williams MB, Ley K, Braciale TJ. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. J Clin Invest. 2005;115:3473–3483. doi: 10.1172/JCI24482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bardina SV, Michlmayr D, Hoffman KW, Obara CJ, Sum J, Charo IF, Lu W, Pletnev AG, Lim JK. Differential Roles of Chemokines CCL2 and CCL7 in Monocytosis and Leukocyte Migration during West Nile Virus Infection. J Immunol. 2015;195:4306–18. doi: 10.4049/jimmunol.1500352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, Lira SA, Charo IF, Luster AD. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ TH2 cells. Nat Immunol. 2011;12:167–77. doi: 10.1038/ni.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Loetscher P, Seitz M, Clark-Lewis I, Marco B, Moser B. Monocyte chemotactic proteins MCP-1, MCP-2 and MCP-3 are major attractants for human CD4+ and CD8+ T lymphocytes. FASEB J. 1994;8:1055–1060. doi: 10.1096/fasebj.8.13.7926371. [DOI] [PubMed] [Google Scholar]
- 63.Olive AJ, Gondek DC, Starnbach MN. CXCR3 and CCR5 are both required for T cell-mediated protection against C. trachomatis infection in the murine genital mucosa. Mucosal Immunol. 2011;4:208–16. doi: 10.1038/mi.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR, Yewdell JW. CXCR3 chemokine receptor enables local CD8+ T cell migration for the destruction of virus-infected cells. Immunity. 2015;42:524–537. doi: 10.1016/j.immuni.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang W, Soto H, Oldham ER, Buchanan ME, Homey B, Catron D, Jenkins N, Copeland NG, Gilbert DJ, Nguyen N, Abrams J, Kershenovich D, Smith K, McClanahan T, Vicari AP, Zlotnik A. Identification of a novel chemokine (CCL28), which binds CCR10 (GPR2) J Biol Chem. 2000;275:22313–22323. doi: 10.1074/jbc.M001461200. [DOI] [PubMed] [Google Scholar]
- 66.Kitchen SG, LaForge S, Patel VP, Kitchen CM, Miceli MC, Zack JA. Activation of CD8 T cells induces expression of CD4, which functions as a chemotactic receptor. Blood. 2002;99:207–212. doi: 10.1182/blood.v99.1.207. [DOI] [PubMed] [Google Scholar]
- 67.Zhang T, Somasundaram R, Berencsi K, Caputo L, Gimotty P, Rani P, Guerry D, Swoboda R, Herlyn D. Migration of cytotoxic T lymphocytes toward melanoma cells in three-dimensional organotypic culture is dependent on CCL2 and CCR4. Eur J Immunol. 2006;36:457–467. doi: 10.1002/eji.200526208. [DOI] [PubMed] [Google Scholar]
- 68.Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, Babb JS, Schneider RJ, Formenti SC, Dustin ML, Demaria S. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181:3099–107. doi: 10.4049/jimmunol.181.5.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, Patel DD. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. 1998;188:1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. Four Distinct Patterns of Memory CD8 T Cell Responses to Chronic Murine Cytomegalovirus Infection. J Immunol. 2006;177:450–458. doi: 10.4049/jimmunol.177.1.450. [DOI] [PubMed] [Google Scholar]
- 71.Bottcher JP, Beyer M, Meissner F, Abdullah Z, Sander J, Hochst B, Eickhoff S, Rieckmann JC, Russo C, Bauer T, Flecken T, Giesen D, Engel D, Jung S, Busch DH, Protzer U, Thimme R, Mann M, Kurts C, Schultze JL, Kastenmuller W, Knolle PA. Functional classification of memory CD8+ T cells by Cx3CR1 expression. Nat Commun. 2015 doi: 10.1038/ncomms9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, von Andrian UH. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity. 2016;45:1270–1284. doi: 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Glennie ND, V, Yeramilli A, Beiting DP, Volk SW, Weaver CT, Scott P. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J Exp Med. 2015;212:1405–14. doi: 10.1084/jem.20142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jacquelot N, Enot DP, Flament C, Vimond N, Blattner C, Pitt JM, Yamazaki T, Roberti MP, Daillère R, Vétizou M, Poirier-Colame V, Semeraro M, Caignard A, Slingluff CL, Sallusto F, Rusakiewicz S, Weide B, Marabelle A, Kohrt H, Dalle S, Cavalcanti A, Kroemer G, DI Giacomo AM, Maio M, Wong P, Yuan J, Wolchok J, Umansky V, Eggermont A, Zitvogel L. Chemokine receptor patterns in lymphocytes mirror metastatic spreaDing in melanoma. J Clin Invest. 2016;126:921–937. doi: 10.1172/JCI80071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shin H, Iwasaki A. Tissue-resident memory T cells. Immunol Rev. 2013;255:165–181. doi: 10.1111/imr.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang D, Yuan R, Feng Y, El-Asady R, Farber DL, Gress RE, Lucas PJ, Hadley GA. Regulation of CD103 expression by CD8+ T cells responding to renal allografts. J Immunol. 2004;172:214–21. doi: 10.4049/jimmunol.172.1.214. [DOI] [PubMed] [Google Scholar]
- 77.Ely KH, Cookenham T, Roberts AD, Woodland DL. Memory T cell populations in the lung airways are maintained by continual recruitment. J Immunol. 2006;176:537–543. doi: 10.4049/jimmunol.176.1.537. [DOI] [PubMed] [Google Scholar]
- 78.Wolf M, Moser B. Antimicrobial activities of chemokines: not just a side effect? Front Immunol. 2012;3:1–12. doi: 10.3389/fimmu.2012.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cole AM, Ganz T, Liese AM, Burdick M, Liu L, Strieter RM. Cutting Edge : IFN-Inducible ELR-CXC Chemokines Display Defensin-Like Antimicrobial Activity. J Immunol. 2001 doi: 10.4049/jimmunol.167.2.623. [DOI] [PubMed] [Google Scholar]
- 80.Crawford MA, Zhu Y, Green CS, Burdick MD, Sanz P, Alem F, O’Brien AD, Mehrad B, Strieter RM, Hughes MA. Antimicrobial Effects of Interferon-Inducible CXC Chemokines against Bacillus anthracis Spores and Bacilli. Infect Immun. 2009;77:1664–1678. doi: 10.1128/IAI.01208-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stephens B, Handel TM. Chemokine receptor oligomerization and allostery. 1st. Elsevier Inc; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salanga CL, Handel TM. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: The role of structural dynamics in function. Exp Cell Res. 2011;317:590–601. doi: 10.1016/j.yexcr.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.