

Abstract
Cysteinyl leukotrienes (cysLTs) facilitate mucosal type 2 immunopathology by incompletely understood mechanisms. Aspirin-exacerbated respiratory disease (AERD), a severe asthma subtype, is characterized by exaggerated eosinophilic respiratory inflammation and reactions to aspirin, each involving the marked overproduction of cysLTs. Here we demonstrate that the type 2 cysLT receptor (CysLT2R), which is not targeted by available drugs, is required in two different models to amplify eosinophlilic airway inflammation via induced expression of IL-33 by lung epithelial cells. Endogenously generated cysLTs induced eosinophilia and expanded group 2 innate lymphoid cells (ILC2s) in AERD-like Ptges−/− mice. These responses were mitigated by deletions of either Cysltr2 or leukotriene C4 synthase (Ltc4s). Administrations of either LTC4 (the parent cysLT) or the selective CysLT2R agonist N-methyl LTC4 to allergen sensitized WT mice markedly boosted ILC2 expansion and IL-5/IL-13 generation in a CysLT2R-dependent manner. Expansion of ILC2s and IL-5/IL-13 generation reflected CysLT2R-dependent production of IL-33 by alveolar type 2 cells, which engaged in a bilateral feed-forward loop with ILC2s. Deletion of Cysltr1 blunted LTC4-induced ILC2 expansion and eosinophilia but did not alter IL-33 induction. Pharmacological blockade of CysLT2R prior to inhalation challenge of Ptges−/− mice with aspirin blocked IL-33-dependent mast cell activation, mediator release, and changes in lung function. Thus, CysLT2R signaling IL-33-dependent ILC2 expansion and IL-33-driven mast cell activation that are necessary for induction of type 2 immunopathology and aspirin sensitivity. CysLT2R-targeted drugs may interrupt these processes.
Keywords: Leukotrienes, type 2 immunity, asthma
Introduction
Mucosal type 2 immune responses elicit tissue eosinophilia, goblet cell metaplasia, and IgE synthesis. Such responses defend against intestinal helminths (1) and facilitate epithelial repair following respiratory viral infections (2, 3). Danger signals from pathogens or allergens (e.g., fungal or helminthic proteases, glycans, endotoxin) initiate type 2 responses and associated immunopathology by activating or damaging barrier cells, which generate and release of innate type 2 cytokines IL-33, IL-25, thymic stromal lymphopoietin (TSLP))(4). These barrier-derived cytokines facilitate durable type 2 adaptive immune responses through their effects on dendritic cells (DCs) (5), and directly activate innate hematopoietic tissue resident effector cells (e.g., mast cells (MCs), macrophages, group 2 innate lymphoid cells (ILC2s)) and conventional T cells (6) to generate IL-5, IL-13, and other cytokines that induce eosinophilic inflammation (4). Prolonged or exaggerated activation of these pathways elicits organ dysfunction and contributes to disease. Epithelial barrier cell nuclei consitituvely contain IL-33 that is released during necrosis (7), but these same cells can also upregulate IL-33 expression and actively secrete IL-33 during inflammation (8, 9). Increases in steady-state tissue levels of IL-33 and its receptor, suppressor of tumorogenicity 2 (ST2), are associated with chronic, non-resolving eosinophilic inflammation and end-organ dysfunction, as observed in refractory chronic rhinosinusitis and severe asthma (10, 11). Animal models suggest that endogenously generated alarmins (12, 13) and/or autocrine feed-forward amplification systems (14, 15) can prolong/enhance IL-33 production by structural cells, and/or induce IL-33 expression de novo by hematopietic cells such as macrophages (16). Due to the potential of IL-33 to drive immunopathology, the mechanisms and pathways responsible for its upregulation with severe disease are fundamentally important. However, these pathways and mechanisms are incompletely understood.
Cysteinyl leukotrienes (cysLTs) are potent lipid inflammatory mediators that abound in allergic inflammation and play validated roles in asthma (17, 18). They form in myeloid cells by 5-lipoxygenase (5-LO)-mediated oxidation of arachidonic acid to leukotriene A4 (LTA4), which is converted to LTC4 by conjugation to reduced glutathione by leukotriene C4 synthase (LTC4S)(19). After its export from the cells of origin (20), extracellular enzymes convert LTC4 to LTD4 (21), a powerful but unstable smooth muscle spasmogen, and then LTD4 to LTE4, a stable metabolite (22). Three G protein coupled receptors bind the cysLTs. The type 1 cysLT receptor (CysLT1R) binds LTD4 with high affinity and in preference to LTC4 when expressed in heterologous cells (23), whereas the type 2 cysLT receptor (CysLT2R) binds LTC4 and LTD4 equally and with lower affinity (24). CysLT3R (also designated GPR99), binds LTE4 in preference to LTC4 or LTD4, although it can mediate responses to all three ligands in vivo (25). Mice lacking LTC4S (Ltc4s−/− mice) show reduced eosinophilia, IgE production, and goblet cell metaplasia compared with WT controls when sensitized and challenged with dust mite allergens or ovalbumin (OVA) (26, 27), indicating a role for cysLTs in type 2 immunity. Intransal administration of LTD4 to Alternaria-challenged mice induces ILC2 proliferation in vivo, and induces secretion of IL-4, IL-5 and IL-13 by ILCs sorted from the lungs of Alternaria challenged mice (28). Both LTC4 and LTD4 activate lung ILC2s by a direct CysLT1R-dependent mechanism in which CysLT2R is dispensable (29, 30). LTC4 and LTD4 also synergize with IL-33 to induce cytokine generation by lung ILC2s, again via CysLT1R (29, 30). Although respiratory mucosal epithelial cells also express cysLT receptors (particularly CysLT2R and CysLT3R)(31, 32), it is unknown whether cysLTs can also participate in upstream regulation of IL-33 expression by barrier cells. Hypothetically, such an effect could synergize with direct CysLT1R-driven ILC2 activation to promote type 2 immunopathology in circumstances where cysLTs are abundant.
Aspirin exacerbated respiratory disease (AERD) is the prototypical disorder in which markedly elevated levels of cysLTs accompany robust type 2 respiratory immunopathology. AERD affects ~7% of all asthmatics and a significantly higher proportion (15–30%) of those with severe disease (33, 34). The dysregulated basal production of LTC4 (35, 36) increases further and abruptly in response to the ingestion of nonselective cyclooxygenase (COX) inhibitors (36, 37). The increase in cysLTs results in an idiosyncratic respiratory reaction associated with cryptic, cysLT-dependent mast cell activation (38). We previously demonstrated that nasal polyps from subjects with AERD, which are especially rich in eosinophils (39), contain markedly more IL-33 protein than tissues from aspirin tolerant controls (40), indicating dysregulated innate type 2 inflammation. Moreover, lung IL-33 levels and eosinophilic inflammation are markedly increased in AERD-like prostaglandin E2-deficient (Ptges−/−) mice compared with WT controls when primed by repetitive low-dose inhalation of an extract (Df) from the house dust mite Dermatophagoides farinae. Deletion of Ltc4s prevents the increases in both lung IL-33 expression and eosinophilic inflammation in Ptges−/− mice, indicating that these features depend on endogenous cysLTs (40, 41). Moreover, blockade of LTC4 production, IL-33, or ST2 each prevent increases in airway resistance (RL) and mast cell activation in response to challenges of Ptges−/− mice with nonselective COX inhibitors, suggesting an essential, proximal role for cysLTs in driving IL-33 release (and consequent mast cell activation) during aspirin challenges (40, 41). The cysLT receptor(s) and cell type(s) responsible for the induced expression of IL-33 and enhanced type 2 immunopathology in AERD are not known.
In the current study, we demonstrate in two different in vivo models that cysLTs drive ILC2 expansion and activation not only directly through CysLT1R signaling, but also indirectly through CysLT2R-mediated IL-33 expression by type 2 alveolar (AT2) cells. These effects are synergistic, and each is essential for cysLT-facilitated type 2 immunopathology. Moreover, “unbraking” of LTC4 synthesis by lysine-aspirin (Lys-ASA) challenges of Ptges−/− mice reveals that CysLT2R signaling mediates IL-33-dependent mast cell activation and pathognomonic changes in airway resistance that are characteristic of AERD. CysLT2R-mediated feed-forward systems may amplify type 2 immunopathology in general, may explain the severe, non-resolving nature of respiratory inflammation in AERD, and may offer several potential targets for therapy.
Materials and Methods
Reagents
Df was obtained from Greer Laboratories (XPB81D3A25; Lenoir, NC). Ovalbumin and PBS were obtained from Sigma-Aldrich (St. Louis, MO). The mMCP-1 EIA kit was purchased from eBiosciences (San Diego, CA). LTC4, LTD4, LTE4, and N-Me-LTC4 were from Cayman Chemical (Ann Arbor, MI). Histamine, TXB2, PGD2, and cysLT EIA kits were from Cayman. IL-5, IL-13, ICAM-1, and VCAM-1 EIA kits were from R&D systems (Minneapolis, MN). CXCL7 EIA kit was purchased from Abcam (Cambridge, MA). The HMGB1 EIA kit was from LifeSpan (Providence, RI). The following antibody reagents were purchased from the indicated vendors: Polyclonal goat anti-mouse IL-33 (R&D systems), Polyclonal rabbit anti-human proSPC (Millipore, Billerica, MA), Donkey anti-Goat IgG (H+L) Secondary Antibody, Alexa Fluor® 488 (Invitrogen, Carlsbad, CA), Donkey anti-rabbit IgG(H+L) Secondary Antibody, Alexa Fluor®594 (Invitrogen), DAKO Serum-Free Protein Block (Agilent, Santa Clara, CA), DAKO Target Retrieval (Agilent). FITC anti-mouse CD11c, FITC anti-mouse/human CD11b, FITC anti-mouse IgE, FITC anti-mouse CD3ε, FITC anti-mouse CD19, FITC anti-mouse CD8a, FITC anti-mouse NK-1.1, FITC anti-mouse Ly-6G/Ly-6C (Gr-1), APC anti-mouse CD45, APC/Cy7 anti-mouse/human CD44, PerCP/Cy5.5 anti-mouse CD90.2, PerCP/Cy5.5 anti-mouse IL-33Rα (IL1RL1, ST2), PE anti-mouse CD278 (ICOS), APC-anti-mouse CD41, PE/Cy7-anti-mouse CD62P, PE-anti-HMGB1, anti-HMGB1, anti-mouse-CD90.2, anti-mouse-CD4, anti-mouse-NK1.1, anti-mouse CD16/32, and isotype controls were all from BioLegend (San Diego, CA). A549 cells were purchased from the American Type Culture Collection.
Mice
C57BL/6 mice lacking mPGES-1 (ptges−/− mice) were from Dr. Shizuo Akira (Osaka University, Japan) (42). The mice were intercrossed with Ltc4s−/− mice (43), Cysltr1−/− (44), Cysltr2−/− (45), and Cysltr3−/− (25) to generate respective DKO strains. All of the mice and wild type C57BL/6 controls were housed at Charles River (Wilmington, MA). Six- to 8-wk-old male were used. All animal studies were approved by the Animal Care and Use Committee of the Dana-Farber Cancer Institute (Protocol 03-042).
Immunization and challenge
To study potentiation of airway inflammation by exogenous cysLTs, mice were sensitized by intraperitoneal (i.p.) injections on days 0 and 5 with Alum-precipitated chicken egg OVA (10 µg). On days 16-18, the mice received intranasal challenge of 2.2 nmol LTC4 or vehicle. On days 17-19, mice were challenged by inhalation of 0.1% OVA (46). Twenty-four hours after the final OVA aerosol challenge, the mice were euthanized and exsanguinated. The lungs were lavaged three times with 0.7 ml PBS/5 mM EDTA. Bronchoalveolar lavage (BAL) fluid cells were cytocentrifuged onto slides, stained with Diff-Quick (Fisher Diagnostics, Middletown, VA), and differentially counted.
To study the effect of endogenous cysLTs, airway inflammation was induced by intranasal administration of Df (Greer, XPB81D3A25; containing <0.005 EU/mL of endotoxin, 3 µg dissolved in 30 µl PBS) to mice after anesthesia with isoflurane in a bell jar system on days 0, 4, 7, 10, 14, and 17 as described elsewhere (41). Control mice were treated with an equal volume of PBS alone. Mice were euthanized for studies 24 h after the last Df treatment. The dose of Df was titrated previously to elicit a sizable cysLT-dependent increment in inflammation in Ptges−/− mice over the minimal response of WT C57BL/6 controls (47).
Flow cytometry
Mice lungs (right lobes) were transferred into 6 well dish and teased tissue apart with forceps. Then the tissues were digested at RT for 45 min in 2 ml of dispase (2 U/ml), followed by adding 0.5 mg DNAse/mouse to the mixture and incubated for 10 min at RT with gently rocking on a shaker to 200 rpm. Cells were filtered through 70 µm nylon meshes, and pelleted by centrifugation for 10 min at 350 g at 4 oC. RBC lysis was performed by resuspending the pellet in 2 ml 1x RBC lysis buffer (Biolegend) and incubating on ice for 4 min, terminated by addition of 13 ml DMEM. Cells were centrifuged for 10 min 350 g at 4 oC, and then washed twice with FACS buffer (0.5% BSA in PBS). 1 × 106 cells were stained with antibodies in 100 µl FACS buffer for 20 min on ice in dark. The cells were washed and resuspended in 300 µl of 1% paraformaldehyde in PBS prior to analysis on a FACSCanto flow cytometer (BD Biosciences). ILC2s were quantitated as Lin-,CD45+,CD44+,CD278+,CD90.2+ cells in the lymphocyte gate . In the depletion experiments involving anti-CD90.2, anti-ST2 was substituted for anti-CD90.2 to enumerate ILC2s within the Lin-,CD45+,CD44+,CD278+ cells. The numbers of ILC2s measured by both sets of criteria were similar (not shown).
IL-33 staining
Lungs were fixed in 4% PFA (wt./vol) for 24 h, and embedded in paraffin. Four µm tissue sections were deparaffinized by heating to 60o C for 30 minutes and washing serially with xylene; ETOH at 100%, 95%, 70%, and 50%; and ddH2O. Slides were quenched with 1 mg/ml sodium borohydride solution on ice for 10 min. For target retrieval, slides were incubated in DAKO target retrieval citrate buffer at 95o C for 30 min, cooled for 20 min at RT, and washed with ddH20 and PBS. Slides were blocked with DAKO Protein Block + 5% donkey serum for 60 minutes, incubated with 100 µL of goat anti-mouse IL-33 at 1 µg/ml and Polyclonal rabbit anti-human proSPC at 1:750 overnight at 4° C, and with donkey secondary Abs at 1:500 with Hoechst nuclear dye at 1:10,000 in PBS-T at RT for 1 h. Images were acquired with a Nikon Eclipse E800 microscope at 20X and analyzed with Image J (NIH). To count IL-33+ cells, the threshold gray value of WT PBS-treated mice was set at 45. The mean number of IL-33+ cells per image and the mean intensity of IL-33+ staining was averaged from three images per mouse. The specificity of staining was validated using WT and Il33−/− mice (not shown).
Measurement of airway resistance
Airway resistance (RL) in response to Lys-ASA was assessed with an Invasive Pulmonary Function Device (Buxco, Sharon, CT) as described elsewhere (41). Briefly, mice were anesthetized 24 h after the last Df challenge, and a tracheotomy was performed. After allowing for RL to reach a stable baseline, Lys-ASA (12 µl of 100 mg/ml) was delivered to the lung via nebulizer, and RL was recorded for 45 min. This dose was based on the peak effect of Lys-ASA on RL in Ptges−/− mice, which generally is reached between 30-45 min after administration (41). The results were expressed as the peak RL as a percentage change from baseline.
Cell depletions, antibody blockade and CysLT2R antagonism
Mice were given i.p. injections with polyclonal goat anti-mouse IL-33 (3.6 µg/mouse), anti-CD90.2 (50 µg/mouse), anti-NK1.1 (50 µg/mouse), anti-CD4 (50 µg/mouse), or equivalent doses of isotype controls were administered on days 16 and 18 of the OVA protocol. In the Lys-ASA challenge experiments, mice were given i.p. injections of anti-HMGB1 antibody (25 µg/mouse) 1 day before the challenge. HAMI-3379 (0.2mg/kg) or buffer control (ethanol) were given by i.p. on 1 and 2 days before the challenge. In some experiments, HAMI-3379 was administered daily for 17 days during Df induction of lung inflammation.
Statistical analysis
Data are expressed as mean ±SEM from at least 10 mice from at least two experiments, except where otherwise indicated. Analyses were performed with Prism software (Graphpad). Differences between two treatment groups were assessed using Student t test, and differences among multiple groups were assessed using one-way ANOVA and Bonferroni post hoc test. P<0.05 was considered statistically significant.
Results
CysLT2R is essential for type 2 immunopathology induced by endogenous cysLTs in PGE2-deficient mice
To identify the receptors through which endogenously generated cysLTs drive the features of type 2 immunopathology in Ptges−/− mice, we intercrossed the parent Ptges−/− strain (42) with Cysltr1−/− (44), Cysltr2−/− (45), and Cysltr3−/− (25) strains to generate respective strains of receptor double knockout (DKO) mice. All three receptor DKO strains, along with WT and Ptges/Ltc4s−/− DKO mice, were treated with six doses of intranasal Df to induce type 2 pulmonary inflammation. Twenty-four hours after the final dose of Df, BAL fluid was collected and single lung cell suspensions were analyzed by flow cytometry. Df weakly induced BAL fluid eosinophilia (Fig. 1A) and lung ILC2 expansion (Fig. 1B) in WT mice in this low-dose protocol, but strongly induced these parameters in Ptges−/− mice (Fig. 1A, 1B). Df-treated Ptges−/− mice displayed significantly stronger expression of IL-13 mRNA in the lung than did WT controls (Supplemental Fig. 1A). Levels of IL-5 and IL-13 proteins were at or below limits of detection by ELISA of whole lung lysates.
Figure 1. Contributions of individual cysLT receptors and ligands to features of type 2 immunopathology.
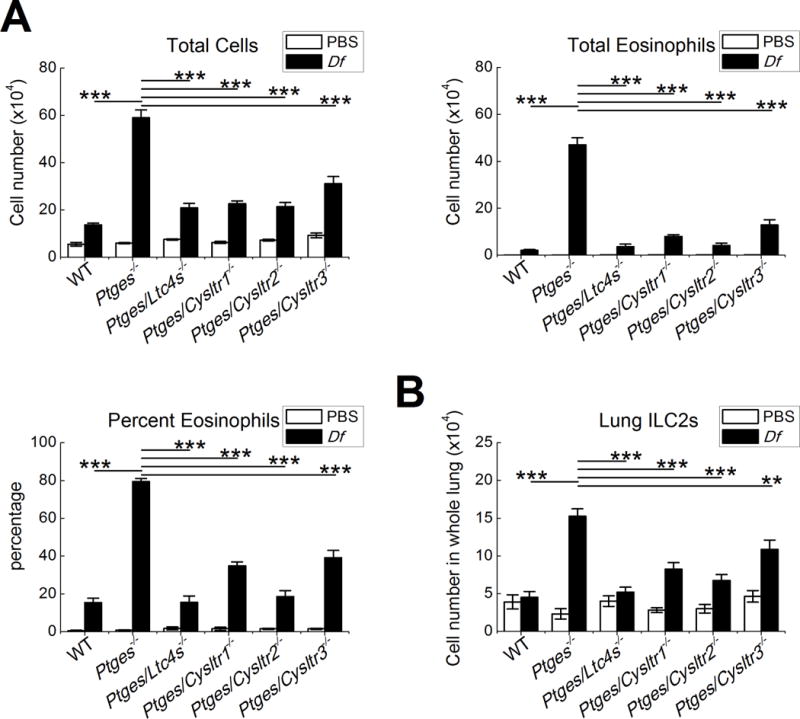
Mice of the indicated genotypes were treated on six occasions with Df (3 µg) or PBS over a 17-day period. Twenty-four hours after the last dose, mice were euthanized for studies. A. BAL fluid total cell counts (left), eosinophil counts (right), and percentages of eosinophils (bottom). B. Numbers of ILC2s (Lin-CD45+,CD44+,CD278+,CD90.2+ cells) in the lungs of the indicated strains. Results from 10 mice/group. *** P < 0.001, ** P < 0.01.
When compared with Df-treated Ptges−/− mice, Ptges/Ltc4s−/− DKO mice and each of the receptor DKO strains showed significantly diminished BAL fluid eosinophilia 24 hours after treatment with Df (Fig. 1A). The Ptges/Ltc4s−/− and Ptges/Cysltr2−/− DKO strains showed the greatest decreases, with significant reductions in eosinophil numbers compared to both the parent Ptges−/− strain and to the Ptges/Cysltr1−/− and Ptges/Cysltr3−/− DKO strains. Deletion of Ltc4s eliminated the Df-induced increase in ILC2s observed in Ptges−/− single KO mice, whereas the Cysltr1, Cysltr2, and Cyslt3r in the DKO strains also all showed significantly reduced the numbers of lung ILC2s relative to the Ptges−/− strain (Fig. 1B). Deletions of Ltc4s, Cysltr2, and Cysltr3 all sharply reduced the Df-induced expression of IL-13 mRNA in the lungs of Df-treated Ptges−/− mice, whereas deletion of Cysltr1 did not (Supplemental Fig. 1A). None of the receptor deletions affected expression of the other receptors (Supplemental Fig. 1B), though Cysltr3 transcripts were below detection limits in all strains. Deletion of Ltc4s, but not of any of the receptors, significantly reduced the levels of cysLTs detected in BAL fluid at 24 hours after the last dose of Df (Supplemental Fig. 1C). The numbers of BAL fluid eosinophils (Supplemental Fig. 2A) and lung ILC2s (Supplemental Fig. 2B) in PGE2-sufficient Cysltr1−/−, Cyslt2r−/− and Cyslt3r−/− single KO mice were low and not different from the WT controls.
Exogenous LTC4 induces eosinophilia and expands lung ILC2s in PGE2-sufficient mice by a mechanism requiring CysLT2R
To verify that CysLT2R signaling also drives features of type 2 immunopathology in a context where PGE2 synthesis was unimpaired, we used a reductionist model in which intranasal cysLTs are administered to ovalbumin (OVA)-sensitized WT mice 12 h prior to low dose (0.1%) OVA challenges on three successive days (46). We administered LTC4, LTD4, LTE4, or N-methyl LTC4, a hydrolysis-resistant synthetic cysLT that stimulates CysLT2R in preference to CysLT1R (48) (2.2 nmol each) to respective cohorts of naïve and OVA sensitized mice. LTC4, N-methyl LTC4, and (to a lesser extent) LTE4 each increased OVA-induced BAL fluid total cell counts, eosinophil percentages, and total eosinophils in sensitized mice (Fig. 2A). In contrast, LTD4 did not induce eosinophilia (Fig. 2A). Despite differences in the capacity to induce eosinophilia, all three cysLTs increased the numbers of lung ILC2s to comparable extents, as did N-methyl LTC4 (Fig. 2B). LTC4 and N-methyl LTC4 significantly increased the quantities of IL-5 and IL-13 proteins in the lysates of lungs from OVA-challenged mice, whereas LTD4 and LTE4 were less active (Fig. 2C). LTC4 and N-methyl LTC4 -induced increases in total cells and total eosinophils were reduced by >80% in Cysltr2−/− mice compared to WT controls (Fig. 3A). ILC2 expansion (Fig 3B) and expressions of IL-5 and IL-13 (Fig. 3C) were reduced in Cysltr2−/− mice as well. In naive WT mice, intranasal LTC4 weakly induced airway inflammation (0 and 4.4 ± 1.2% BAL fluid eosinophils in PBS- and LTC4-challenged mice, P = 0.01) and ILC2 expansion (1.74 ± .09 and 2.28 ±.11 × 104 cells/lung in PBS- and LTC4-challenged mice, P = 0.006, n = 5, not shown).
Figure 2. Exogenous cysLTs induce type 2 immunopathology in PGE2-sufficient mice.
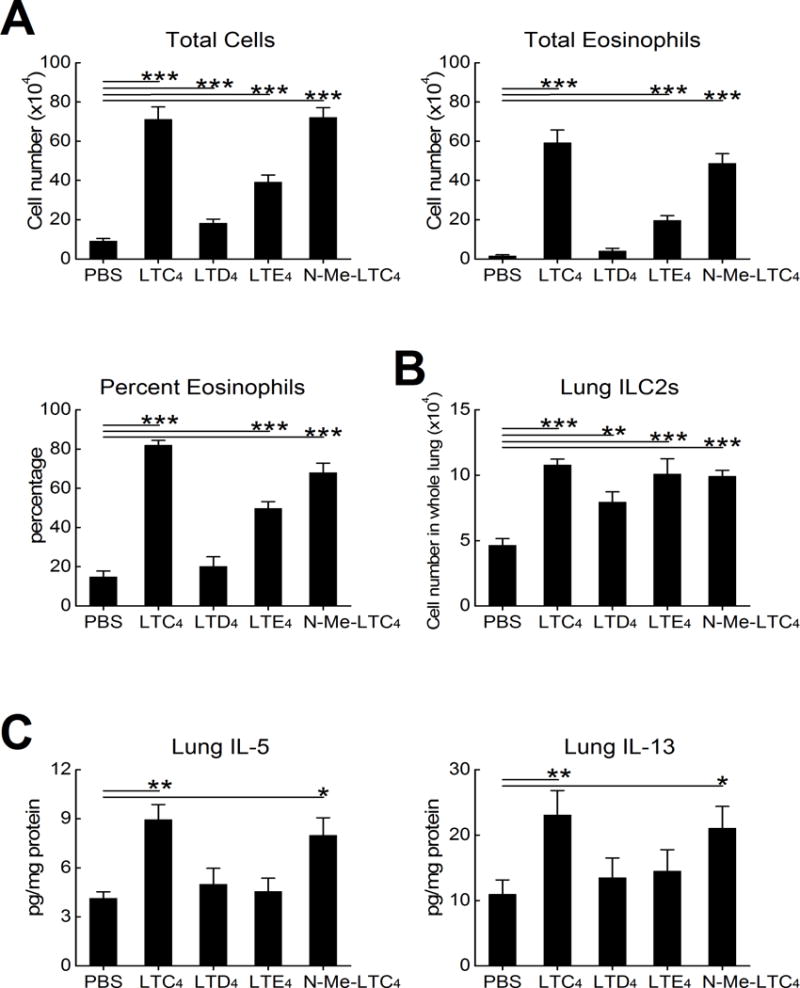
OVA sensitized WT C57BL/6 mice received challenges with 0.1% OVA on three successive days. The indicated groups of mice received 2.2 nmol of the indicated cysLTs, N-methyl LTC4, or a vehicle control intranasally 12 h prior to each OVA challenge. A. BAL fluid total cell counts (left), percentage eosinophils (right) and total eosinophil counts (below) from mice euthanized 24 h after the last challenge. B. Numbers of ILC2s in the lungs of the indicated groups. C. Levels of IL-5 and IL-13 proteins in lysates of the lungs from the same mice as in B. Results are from 10 mice/group. *** P < 0.001, ** P < 0.01, * P < 0.05.
Figure 3. CysLT2R mediates responses to exogenous LTC4.
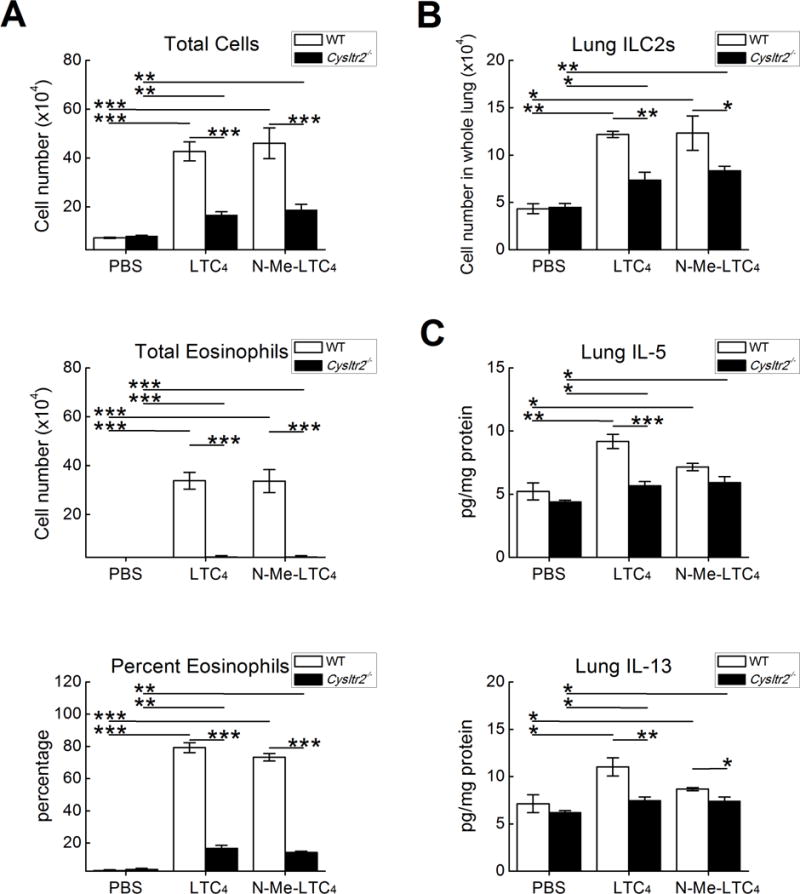
OVA sensitized WT C57BL/6 mice received challenges with 0.1% OVA on three successive days. The indicated groups of mice received 2.2 nmol of LTC4, N-methyl LTC4, or a vehicle control intranasally 12 h prior to each OVA challenge. A. BAL fluid total cell counts (top), percentage eosinophils (middle) and total eosinophil counts (bottom) from mice euthanized 24 h after the last challenge. B. Numbers of ILC2s in the lungs of the indicated groups. C. Levels of IL-5 (top) and IL-13 (bottom) proteins in lysates of the lungs from the same mice as in B. Results are from 10 mice/group. *** P < 0.001, ** P < 0.01, * P < 0.05.
CysLT2R signaling drives IL-33 expression
Compared with WT controls, Df-treated Ptges−/− mice display sharply increased levels of IL-33 in the lung. These levels are reduced in Ptges/Ltc4s−/− mice, implying a dependence on endogenous cysLTs (40). Because IL-33 can expand lung ILC2s and synergize with CysLT1R signaling to drive IL-5/IL13 production (30), we examined lysates of whole lung for IL-33 protein in each DKO strain to determine which cysLT receptors might account for the induction of IL-33 expression in Ptges−/− mice. Df treatment induced significantly higher levels of lung IL-33 protein in Ptges−/− mice than WT controls, and also strongly induced IL-33 expression in the Ptges/Cysltr1−/− and Ptges/Cysltr3−/− DKO mice. In sharp contrast, Df failed to significantly induce IL-33 expression in either the Ptges/Cyslt2r−/− or Ptges/Ltc4s−/− DKO strains (Fig. 4A). None of the strains displayed significant induction of TSLP protein, and IL-25 was undetectable (not shown). To exclude potential epistatic influences of Cysltr2 deletion on IL-33 induction, we treated Ptges−/− mice with a CysLT2R selective antagonist, HAMI-3379, to pharmacologically block CysLT2R during Df priming. Compared with vehicle treatment, the daily administration of HAMI-3379 during the Df exposure period prevented BAL fluid eosinophilia (Fig. 4B) and the upregulation of IL-33 protein (Fig. 4C). Immunofluorescent staining (Fig. 4D) revealed that the nuclei of surfactant protein C (SPC)+ cells were the dominant site of IL-33 immunoreactivity. Compared with saline-treated controls, Df-treated Ptges−/− mice displayed a 3-fold increases in the numbers of IL-33+ cells. These increases were sharply reduced in the lungs of Df-treated Ptges/Ltc4s−/− and Ptges/Cysltr2−/− DKO mice (Fig. 4D,4E). IL-33 expression on a per cell basis was also increased in Df-treated Ptges−/− mice, as indicated by the intensity of nuclear staining, and decreased in both Df-treated Ptges/Ltc4s−/− and Ptges/Cysltr2−/− mice (Fig. 4E). Similar results were observed in HAMI-3379-treated Ptges−/− mice (not shown). There were rare CD45+IL-33+ cells with cytoplasmic IL-33 expression. These cells were not different in number or intensity across Df-treated Ptges−/−, Ptges/Ltc4s−/− and Ptges/Cysltr2−/− DKO mouse strains, and were not altered by HAMI-3379. Deletions of Ltc4s, Cysltr1, Cysltr2, or Cysltr3 failed to alter the weak IL-33 expression, ILC2 expansion, or lung eosinophilia in PGE2-sufficient mice (Supplemental Fig. 2C).
Figure 4. CysLT2R is necessary for IL-33 expression induced by endogenous or exogenous cysLTs.
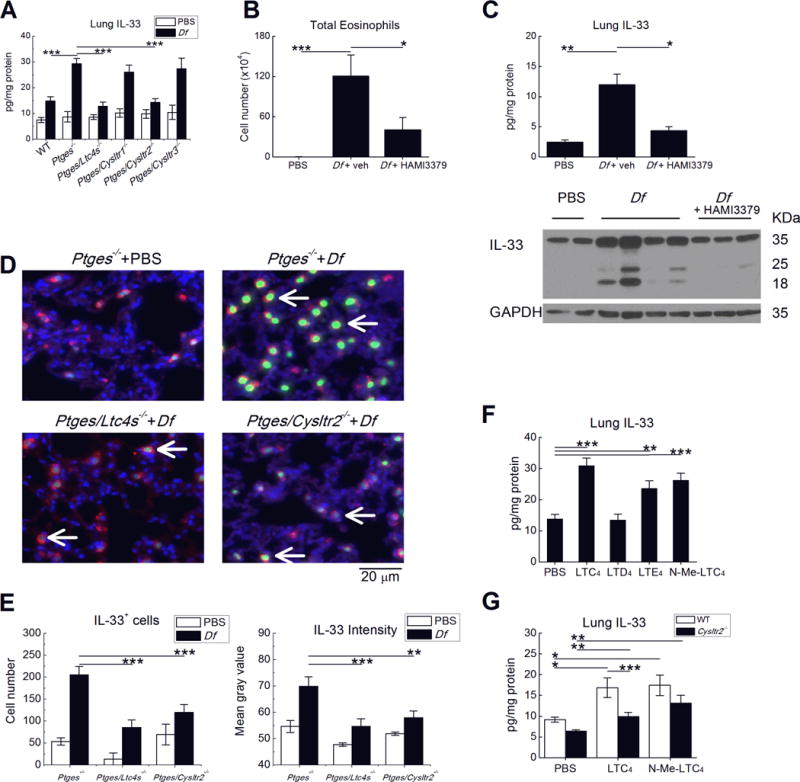
A. Levels of IL-33 protein detected in the lysates of lungs from the indicated strains of mice treated on six occasions with Df (3 µg) intranasally, or with PBS. B. Total BAL fluid eosinophils and C. Lung IL-33 levels (top) in Ptges−/− mice treated daily with the CysLT2R-selective antagonist HAMI-3379 during priming with Df. Western blot (bottom) showing full length (~35 kDa) and cleaved forms of IL-33 in whole lung lysates from Ptges−/− mice of the indicated treatment groups. D. Immunofluorescent stains for IL-33 (green) localizing to the nuclei (blue) of SPC (red)-expressing lung AT2 cells from representative mice of the indicated strains and treatment groups. E. Quantitative analysis of numbers (left) and per cell intensity (right) of IL-33 staining in AT2 cells. F. Effects of exogenous cysLTs on lung IL-33 protein in OVA-sensitized and challenged PGE2 sufficient mice. G. Effect of CysLT2R deletion on lung IL-33 expression induced by the indicated exogenous cysLTs. Results in A-C and E-G are from 10 mice/group. *** P < 0.001, ** P < 0.01, * P <0.05.
To verify that LTC4-induced CysLT2R signaling could also promote IL-33 expression in PGE2-sufficient mice, we examined the lungs of WT mice treated with exogenous cysLTs. The administration of LTC4, N-Methyl LTC4, and (to a lesser degree) LTE4 to WT OVA-sensitized mice increased the levels of lung IL-33 protein (Fig. 4F), again localizing to SPC+ cells (not shown). As was the case for eosinophilia, LTD4 failed to elicit IL-33 expression. Compared to WT mice, Cysltr2−/− mice showed reduced induction of IL-33 expression in response to both LTC4 and N-methyl LTC4 (Fig. 4G). The administration of LTC4 to Df-primed WT mice also significantly increased both lung eosinophilia and lung IL-33 levels (Supplemental Fig. 3).
To determine whether CysLT2R signaling could directly induce IL-33 expression in AT2 cells, we stimulated a human AT2 cell line, A549, with LTC4 or with N-methyl LTC4. Some samples were treated simultaneously with IL-13 (2 ng/ml), which upregulates the expression of IL-33 mRNA in A549 cells (14). Neither LTC4 nor N-methyl LTC4 significantly increased expression levels of IL-33 mRNA when provided alone or in combination with IL-13 (not shown).
CysLT1R and CysLT2R signaling synergize to promote LTC4-induced ILC2 expansion and immunopathology
To distinguish between the contributions of CysLT1R, CysLT2R, and CysLT3R to LTC4-induced immunopathology, we compared the responses of OVA-sensitized WT, Cysltr1−/−, Cysltr2−/− and Cysltr3−/− single KO mice to exogenous LTC4. Deletions of either CysLT1R or CysLT2R both reduced the numbers of total cells and eosinophils in the BAL fluids of LTC4-challenged mice (Fig. 5A), and sharply reduced the numbers of ILC2s (Fig. 5B). The deletion of CysLT2R was significantly more efficacious than the deletion of CysLT1R for both responses. In contrast to its effects on eosinophilia and ILC2 numbers, deletion of CysLT1R did not alter the induction of IL-33 expression, whereas deletion of CysLT2R did (Fig. 5C). CysLT1R deletion modestly reduced the levels of lung IL-5 protein, but not IL-13 protein, whereas CysLT2R deletion substantially reduced both (Fig. 5D). Notably, deletion of CysLT3R had no effect on the induction of eosinophilia, ILC2 expansion, IL-33 induction, or IL-5/IL-13 expression in response to exogenous LTC4 (Fig. 5A–D). In contrast, deletion of CysLT3R abrogated the LTE4-induced increments in inflammation, ILC2 expansion, and IL-5 expression in OVA sensitized mice without completely eliminating the modest IL-33 increase (not shown).
Figure 5. CysLT1R and CysLT2R contribute separately to LTC4-induced ILC2 expansion.
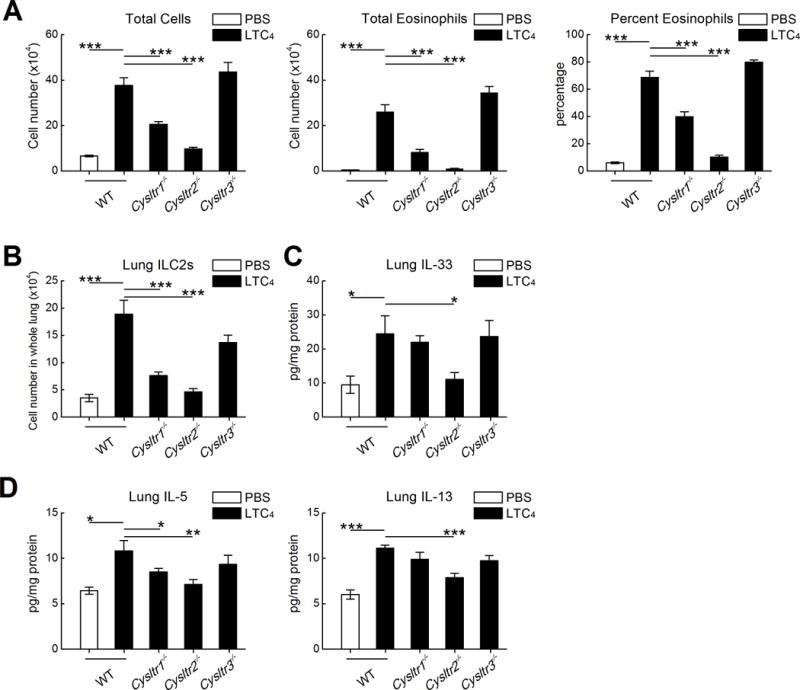
OVA sensitized mice of the indicated genotypes received challenges with 0.1% OVA on three successive days. The indicated groups of mice received 2.2 nmol of LTC4 or control. A. Total BAL fluid cell counts (left), eosinophil counts (middle) and percentages of eosinophils (right). B. Total numbers of lung ILC2s from the same mice as in A. C. Levels of IL-33 detected in whole lung lysates from the indicated mice. D. Levels of IL-5 (left) and IL-13 (right) proteins in the same samples as in C. Results are from 10 mice/group. *** P < 0.001, ** P < 0.01, * P <0.05.
CysLT2R-driven immunopathology requires both ILC2s and CD4+ T cells, but only ILC2s engage in a feed-forward loop with IL-33
We used Ab-based neutralization and cell depletion strategies to determine whether LTC4-elicited IL-33 was necessary for the accompanying increase in airway eosinophilia, and to identify the lymphocyte subsets most essential to covey the responses. Treatment of the mice with anti-IL-33 eliminated the LTC4-induced increment in lung IL-33 (Supplemental Fig 4), blocked the LTC4-induced increase in eosinophilia by >80% (Fig. 6A), and reduced the numbers of ILC2s by ~40% (Fig. 6B). Anti-IL-33 also prevented the LTC4-induced increases in both IL-5 and IL-13 proteins in the lung (Fig. 6C). The administration of an anti-CD90.2 Ab, which depletes ILC2s (3), NK cells, and T cells (49), completely prevented the LTC4-induced potentiation of BAL fluid total cells and eosinophils (Fig. 6D) and expansion of ILC2s by >80% (Fig. 6E), while also depleting NK1.1+ NK cells (not shown) compared with an isotype control Ab. In contrast, selective NK cell depletion with anti-NK1.1 (95% depletion, not shown) had no effect on cellularity, eosinophilia (Fig. 6D) or ILC2 expansion (Fig. 6E). The increases in IL-5 and IL-13 elicited by LTC4 were eliminated by anti-CD90.2, but not by anti-NK1.1 or anti-CD4 (Fig. 6F), indicating ILC2s as the most likely relevant target. Because ILC2s can sustain lung IL-33 expression in a feed-forward loop (14), we examined the lungs of the Ab-treated mice for the levels of IL-33 protein. Notably, anti-CD90.2 prevented the LTC4-induced induced expression of IL-33 completely, whereas anti-NK.1.1 had no effect (Fig. 6G). Although anti-CD4 also prevented the LTC4-mediated increases in total BAL fluid cells and eosinophils (Fig. 6D), it did not alter LTC4-induced expansion of ILC2s (Fig. 6E) or IL-33 expression (Fig. 6G).
Figure 6. LTC4 elicits a bilateral feed-forward system between IL-33 and CD90.2+ cells.
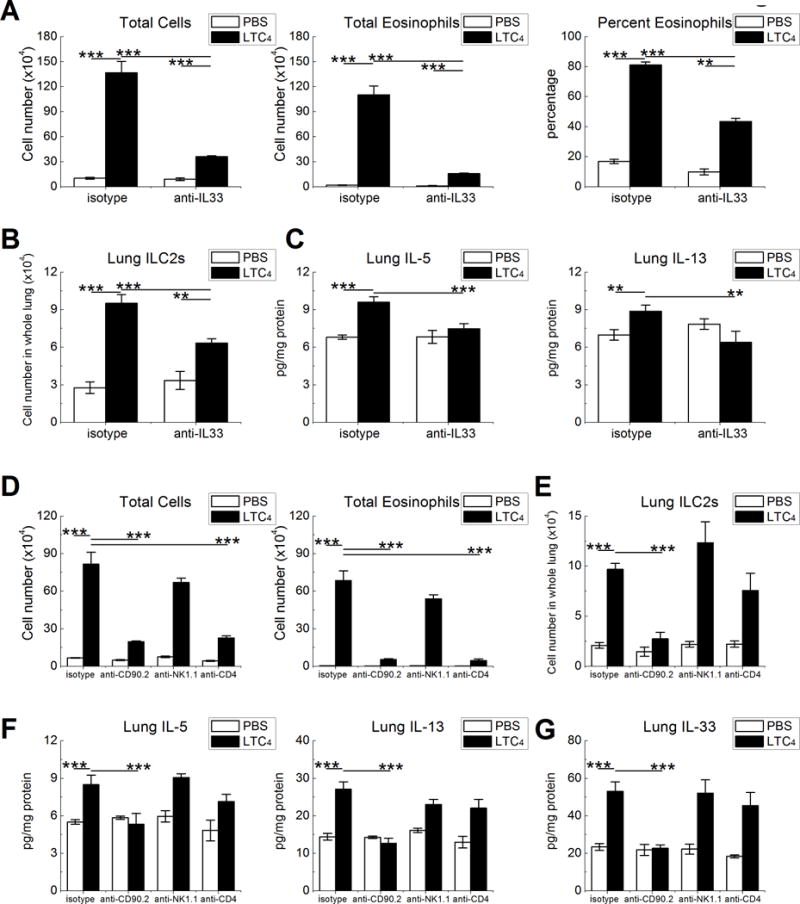
OVA sensitized CD57/BL6 mice were challenged three times with LTC4 prior to OVA inhalation. Abs against the indicated proteins or isotype controls were administered to cohorts of mice before the 2nd and 3rd challenges. A. Effect of treatment with a neutralizing anti-IL-33 Ab on BAL fluid total cell counts (left) and eosinophil counts (middle) and percentages (right). B. Effect IL-33 neutralization on lung ILC2 numbers. C. Effect of IL-33 neutralization on IL-5 and IL-13 levels in lung lysates. D. BAL fluid total cell (left) and total eosinophil (right) counts in mice treated with the Abs against the indicated cell surface markers. E. Lung ILC2 counts from the same mice as in D. F. Levels of IL-5 and IL-13 detected by ELISA of lung lysates. G. IL-33 protein levels from the same samples as in E. Results are from at least 10 mice per group. *** P < 0.001, ** P < 0.01.
CysLT2R-mediated pathways are necessary for aspirin sensitivity
We next sought to determine whether CysLT2R was necessary for aspirin sensitivity in Df-primed Ptges−/− mice. Reactions to Lys-ASA in this model depend on unbraked cysLT production, cysLT-driven IL-33 release, and consequent mast cell activation. Df-primed Ptges−/− mice were treated intraperitoneally with HAMI-3379 at 24 h before and again immediately before challenge by inhalation of Lys-ASA. Lys-ASA challenged mice treated with vehicle or isotype control Ab exhibited sharply increased RL (Fig. 7A), along with increases in cysLTs (Fig. 7B), and MC activation products (mouse mast cell protease -1 (mMCP-1), histamine, and PGD2) (Fig. 7C–E). Blockade of CysLT2R attenuated the Lys-ASA-induced increase in RL (Fig. 7A), along with increases in BAL fluid cysLTs (Fig. 7B) and the MC activation signatures mMCP-1, histamine, and PGD2 (Fig. 7C–E). Administration of anti-CD90.2 24 h prior to the Lys-ASA challenge attenuated both the physiologic response and the release of mediators (Fig. 7F–J).
Figure 7. CysLT2R blockade prevents physiologic responses to aspirin challenge in AERD-like mice.
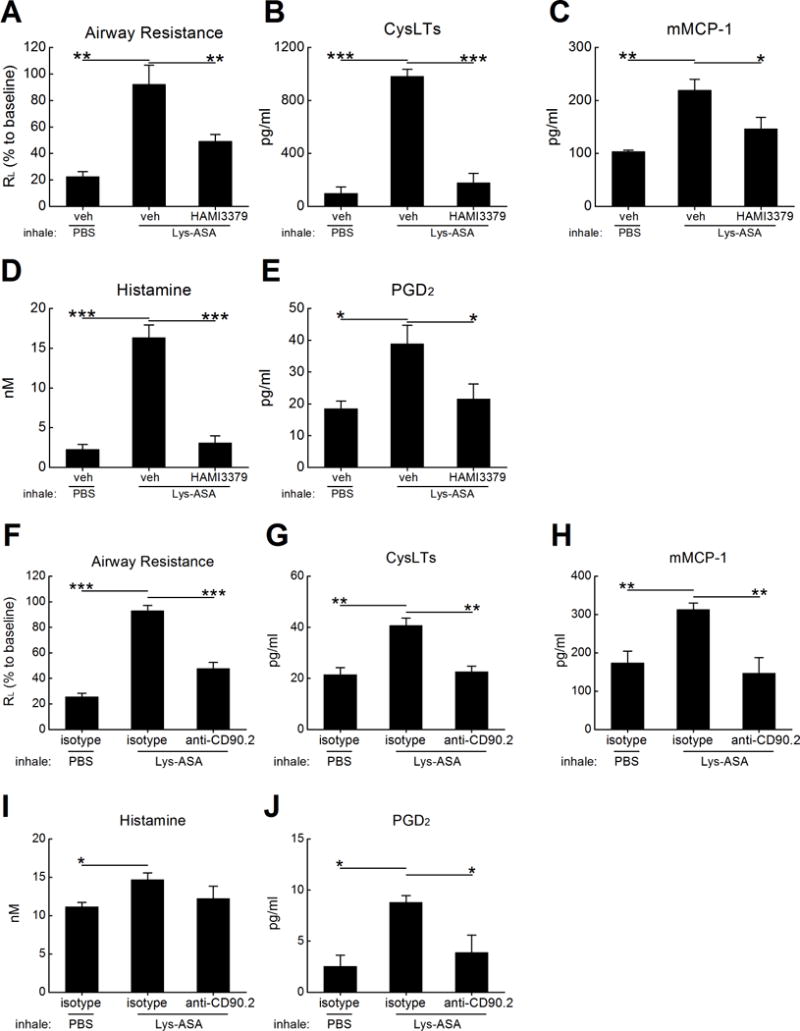
Df-primed Ptges−/− mice were treated overnight with IP doses of HAMI-3379 or vehicle control. Sedated and mechanically ventilated mice were challenged by inhalation with Lys-ASA or PBS. A. Peak changes in RL monitored over a 45-min period during the challenge. B. BAL fluid levels of cysLTs in the same mice. C. BAL fluid levels of mMCP-1, D. histamine and E. PGD2 from the same mice. F. Effects of depleting CD90.2+ cells on RL. G. BAL fluid levels of cysLTs, H. mMCP-1, I. histamine, and J. PGD2 from the same mice as in F. Results in A-E are from 10 mice/group, and those in F-J are from five per group. *** P < 0.001, ** P < 0.01, * P < .05.
Discussion
Long recognized as constrictors of human airway smooth muscle in asthma (50, 51), cysLTs are now known to also act directly on cellular effectors of type 2 immunity (27, 28, 30). Ltc4s−/− mice display markedly impaired eosinophilic pulmonary inflammation and type 2 recall responses to OVA (26) and dust mite (27), supporting the biological importance of these immunologic effects. LTC4 and LTD4 can both elicit CysLT1R-dependent ILC2 proliferation and nuclear factor of activated T cells (NFAT) activation that strongly synergizes with IL-33 to drive IL-5 and IL-13 expression (30). Although LTE4 is inactive on ILC2s in vitro (30), it does activate lung ILC2s in vivo by a mechanism that resists CysLT1R blockade (28). Thus, cysLTs may affect ILC2 homeostasis by eliciting synergy between direct CysLT1R-dependent and heretofore uncharacterized indirect mechanisms. Although ILC2s express both Cysltr1 and Cysltr2 transcripts, only Cysltr1−/− ILC2s show impaired proliferation or activation with direct stimulation by cysLTs (30). No prior studies had demonstrated a role for CysLT2R in ILC2 homeostasis. Although CysLT2R binds LTC4 and LTD4 equally avidly when expressed in heterologous cell systems (24), it exhibits a clear preference for activation by LTC4 over LTD4 in platelets (46) and dermal fibroblasts (52), suggesting a functional modification of the receptor when expressed in its native context, or in settings where the other receptors are also present. The relatively low affinity of CysLT2R for LTC4 (EC50 10-70 nM (24))(53) suggests that it can mediate autocrine signals (54), or can function in paracrine signaling when local concentrations of LTC4 are elevated due to increased synthesis. Such increased synthesis is a hallmark of AERD, where severe respiratory dysfunction is accompanied by marked tissue eosinophilia (55, 56) and strong expression of IL-33 in the respiratory tract (40). Our previous studies demonstrated that endogenous cysLTs were necessary to drive the robust Df-induced lung eosinophilia and high level steady-state IL-33 expression in the lungs of AERD-like Ptges−/− mice (40). We therefore undertook this study to determine the ligand, receptor, and cellular requirements responsible for cysLT-driven IL-33 expression, and to determine its contribution to cysLT-mediated pathobiology, including ILC2 homeostasis.
To study the role of each receptor in a context where endogenous cysLTs play a major pathogenic role, we introduced the Cysltr1, Cysltr2, and Cysltr3 null alleles into Ptges−/− mice. CysLTs play a dominant role in driving Df-induced immunopathology in this strain (40, 41). All three receptor deletions decreased the induction of pulmonary eosinophilia in Ptges−/− mice (Fig. 1A), but had minimal effects in the PGE2-sufficient controls (Supplemental Fig. 2). The Ptges/Cysltr2−/− DKO strain showed the greatest reduction, equaling the efficacy of Ltc4s deletion. All three receptor null strains (and the Ptges/Ltc4s−/− strain) showed similarly impaired ILC2 expansion relative to the Ptges−/− strain (Fig. 1B). The blunted ILC2 expansion observed in the Ptges/Cysltr1−/− mice (Fig. 1B), the ILC2 expansion induced by the preferred CysLT1R ligand LTD4 in sensitized WT mice (Fig. 2B), and the reduced LTC4-induced ILC2 expansion observed in Cysltr1−/− mice (Fig. 5B) are each consistent with mitogenic effects mediated by CysLT1R on ILC2s reported elsewhere (28, 30). Nonetheless, the lack of eosinophilia induced by exogenous LTD4 (Fig. 2A), its lack of effect on whole lung IL-5/IL-13 levels (Fig. 2C)), and the inability of Cysltr1 deletion to impair Df-induced IL-13 expression in Ptges−/− mice (Supplemental Fig. 1) suggest that direct CysLT1R-mediated ILC2 expansion is insufficient alone to drive cysLT-induced type 2 immunopathology. In contrast, both exogenous LTC4 and N-methyl LTC4 elicited lung eosinophilia (Fig. 3A), ILC2 expansion (Fig. 3B) and IL-5/IL-13 production (Fig. 3C), all of which were markedly diminished in Cysltr2−/− mice. The unexpectedly marked effects of Cysltr2 deletion on both eosinophilic inflammation and ILC2 expansion in both models (Fig. 1, Fig. 3) prompted a focus on potential mechanisms by which CysLT2R signaling might promote ILC2 effector function indirectly.
We focused our attention to IL-33 for several reasons. First, it potently activates resident ST2+ ILC2s and mast cells (57, 58) to generate IL-5, IL-13, and other type 2 cytokines. Second, it synergizes with cysLTs (acting at CysLT1R) to promote type 2 cytokine generation by ILC2s ex vivo (30). Third, IL-33 expression in the lungs of Df-primed Ptges−/− mice is upregulated by a pathway requiring endogenous cysLTs (40). Studies in the receptor DKO strains revealed that endogenous cysLT-driven expression of IL-33 in Df-primed Ptges−/− mice depended entirely on CysLT2R (Fig. 4A). Pharmacologic blockade of CysLT2R yielded similar results (Fig. 4B, 4C), indicating that the effects of CysLT2R deletion on IL-33 expression were not due to epistasis. Moreover, full induction of IL-33 expression by exogenous LTC4 and N-methyl LTC4 required CysLT2R in PGE2-sufficient mice as well (Fig. 4G, Fig. 5C). Conversely, studies in both Ptges/Cysltr1 DKO mice (Fig. 4A), in WT mice treated with LTD4 (Fig. 4F), and in LTC4-challenged Cysltr1−/− mice (Fig. 5C) verify that CysLT1R plays no direct role in inducing IL-33 expression, contrasting with its role in expansion of ILC2s (Fig. 1B, Fig. 2B, Fig. 5B).
Although deletion of the epithelially-dominant CysLT3R also reduced eosinophilia and ILC2 accumulation (Fig. 1), it did not suppress IL-33 induction by Df (Fig. 4A), suggesting the involvement of an additional to-be characterized type 2 immune pathway. This is further supported by the fact that eosinophilia, ILC2 expansion, and IL-5 generation induced by exogenous LTE4 was abrogated in Cysltr3−/− mice, even though the modest increment in IL-33 remained largely intact. Thus, cysLTs control ILC2 homeostasis by both direct (CysLT1R-dependent) and indirect (CysLT2R and possibly CysLT3R-mediated) effects. These effects at anatomically distinct targets may synergistically promote type 2 immunopathology in contexts where cysLT production is increased. Moreover, CysLT2R and CysLT3R in the lung show sharp respective preferences for LTC4 and LTE4, respectively.
CysLT2R-dependent IL-33 expression in our study was restricted primarily to SPC+ AT2 cells (Fig. 4D), which are alveolar stem cells that constitutively express IL-33 and participate in epithelial regeneration (49, 59). Both the studies using gene deleted mice and pharmacologic blockade indicate that CysLT2R controls both the total number of IL-33+AT2 cells and the amount of IL-33 protein per cell. Although AT2 cells from rat lung were previously shown to express CysLT2R in preference to CysLT1R (60), cysLTs failed to induce IL-33 expression directly by A549 cells (an AT2 cell line). While this may reflect a limitation of the cell line used, AT2 cells could be the target of additional inductive factors that either synergize with or result from CysLT2R signaling to upregulate IL-33 expression in vivo. Because CysLT2R is expressed broadly by hematopoietic (platelets, macrophages, eosinophils) and non-hematopoietic (fibroblasts, endothelium), such cell-cell interactions seem plausible.
While IL-33 elicits cytokine generation by both ILC2s and CD4+ effector Th2 cells, only ILC2s can drive an IL-13-mediated epithelial feed-forward system that sustains innate type 2 inflammation through IL-33 (14). Blocking experiments supported the importance of IL-33 for full LTC4-induced eosinophilic inflammation (Fig. 6A), ILC2 expansion (Fig. 6B), and IL-5 and IL-13 production (Fig. 6C). Depletion of either ILC2s (with anti-CD90.2) or conventional T cells (with anti-CD4) both dramatically suppressed the increase in total BAL fluid cell counts and the induction of eosinophilia by LTC4 (Fig. 6D), whereas anti-NK1.1 was inactive. Although anti-CD90.2 was highly effective for depletion of ILC2s (Fig. 6E), its effects on eosinophilia could well reflect depletion of one or more additional relevant populations (including T cells) that also express this marker. However, the marked reductions in lung levels of IL-5, IL-13 and IL-33 protein levels induced by this depletion was not replicated by T cell or NK cell-depleting antibodies (Fig. 6F,6G). It is likely that LTC4, by both directly targeting ILC2s (via CysLT1R) and inducing IL-33 expression by AT2 cells (via CysLT2R), facilitates a bilateral feed-forward pathway between IL-33 and ILC2s noted in other models (14). The suppression of eosinophilia by anti-CD4 likely reflects a requirement for allergen-specific sensitization to OVA in this protocol to induce maximal eosinophilia, and could also reflect synergy between conventional T cells and ILC2s recently reported in other models (61, 62).
CysLT-driven reactions to the ingestion of nonselective COX inhibitors are pathognomonic of AERD. Such reactions putatively result from depletion of PGE2, which “unbrakes” 5-LO in granulocytes and other cells in the respiratory tract. Whereas selective CysLT1R antagonists can attenuate the decline in lung function occurring in these reactions (63), the 5-LO inhibitor zileuton prevents additional sinonasal, gastrointestinal, and cutaneous manifestations (37), suggesting that receptors other than CysLT1R may contribute prominently to the pathophysiology of reactions. Curiously, 5-LO inhibition also blocks the cryptic, pharmacologically-mediated activation of MCs that occurs during such reactions in both humans (38) and Ptges−/− mice (41), indicating that 5-LO products (likely cysLTs) are essential to this activation mechanism. We previously verified that changes in RL induced by Lys-ASA challenges of Df-primed Ptges−/− mice recapitulated the cysLT-dependent pharmacology of reactions in humans, including blockade of changes in lung function with CysLT1R antagonists (41). We also demonstrated that 5-LO inhibition prevented the activation of MCs (41), and that the latter depended on cysLT-driven IL-33 release(40). Our current study demonstrates that CysLT2R signaling is essential for the downstream pathophysiologic consequences of incremental cysLT generation, activation of MCs (and potentially basophils), and increases in RL (Fig. 7A–E). Moreover, endogenous cysLTs act through CysLT2R to amplify their own synthesis (Fig. 7B) (likely via downstream IL-33-mediated mast cell activation) when the PGE2-dependent brake is removed. Given that other cells (e.g., endothelium, granulocytes) also express CysLT2R, cysLTs could well have additional relevant targets in these reactions that further amplify their severity and/or contribute to their resolution. Notably, the effects of CD90.2 depletion (Fig. 7F–J) support the likely interposition of cysLT-activated ILC2s (via IL-13 or other cytokines) in maintaining IL-33 necessary for Lys-ASA-induced MC activation. The cysLTs that initiate the reactions may derive from platelets, as suggested by our previous studies (41).
Our findings illustrate distinct and previously unrecognized functions for CysLT2R in type 2 immunopathology. These functions promote a feed-forward system between ILC2s and IL-33. CysLTs released in response to allergen challenges in atopic subjects elicit bronchoconstriction primarily through CysLT1R (64). Although no studies have directly contrasted the effects of CysLT1R antagonists with those of 5-LO inhibition, a recent study demonstrated that a dual CysLT1R/CysLT2R antagonist, but not a CysLT1R-selective antagonist, prevented the increase in sputum eosinophils occurring in atopic asthmatic subjects following allergen inhalation, even though both drugs blocked the attendant changes in lung function (65). The markedly dysregulated cysLT synthesis, strong IL-33 expression, and high levels of tissue eosinophilia observed in AERD may reflect, in part, immunopathologic contributions from CysLT2R. The findings support the potential therapeutic utility of drugs with the capacity to block CysLT2R, alone or in combination with the other cysLT receptors, in AERD and other conditions characterized by robust type 2 immunopathology and cysLT production.
Supplementary Material
Acknowledgments
We thank H. Cirka and H. Raff for technical contributions, and members of the Boyce lab for helpful discussions.
Abbreviations
- AERD
aspirin exacerbated respiratory disease
- AT2
type 2 alveolar cell
- COX
cyclooxygenase
- CysLTs
cysteinyl LTs
- CysLT1R
type 1 cysLT receptor
- CysLT2R
type 2 cysLT receptor
- CysLT3R
type 3 cysLT receptor
- Df
extract from Dermatophagoides farinae
- DKO
double knockout
- ILC2s
group 2 innate lymphoid cells
- LT
leukotriene
- Lys-ASA
lysine aspirin
- LTC4S
leukotriene C4 synthase
- MC
mast cell
- MMCP
mouse MC protease
- PGE2
prostaglandin E2
- SPC
surfactant protein C
- ST2
suppressor of tumorogenicity 2
- TXA2
thromboxane A2
- 5-LO
5-lipoxygenase
Footnotes
This work was supported by generous contributions from the Vinik Family, the Kaye Family, and National Institutes of Health Grants AI078908, AI095219, AT002782, AI082369, HL111113, HL117945, and HL36110.
The authors have declared that no conflict of interest exists
Author contributions
T. Liu designed the experiments and performed them with C. Feng and D. Garofalo, and analyzed the results with J. Boyce. N. Barrett conducted the analyses of IL-33 immunolocalization and identified the effect of CysLT2R blockade on type 2 immunity with E. Yoshimoto. Y. Kanaoka developed the compound KO mouse strains. J. Boyce conceived of the study and composed the manuscript with N. Barrett and T. Liu.
References
- 1.Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, McIlgorm A, Jolin HE, McKenzie AN. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203(4):1105–16. doi: 10.1084/jem.20051615. doi: jem.2005-15 [pii];10.1084/jem.20051615 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104(6):777–85. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12(11):1045–54. doi: 10.1031/ni.2131. doi: ni.2131 [pii];10.1031/ni.2131 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16(4):343–53. doi: 10.1038/ni.3123. doi: ni.3123 [pii];10.1038/ni.3123 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, Qin FX, Yao Z, Cao W, Liu YJ. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202(9):1213–23. doi: 10.1084/jem.20051135. doi: jem.20051135 [pii];10.1084/jem.20051135 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo L, Huang Y, Chen X, Hu-Li J, Urban JF, Jr, Paul WE. Innate immunological function of TH2 cells in vivo. Nat Immunol. 2015;16(10):1051–9. doi: 10.1038/ni.3244. doi: ni.3244 [pii];10.1038/ni.3244 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lefrancais E, Cayrol C. Mechanisms of IL-33 processing and secretion: differences and similarities between IL-1 family members. Eur Cytokine Netw. 2012;23(4):120–7. doi: 10.1684/ecn.2012.0320. doi: ecn.2012.0320 [pii];10.1684/ecn.2012.0320 [doi] [DOI] [PubMed] [Google Scholar]
- 8.Hong J, Bae S, Jhun H, Lee S, Choi J, Kang T, Kwak A, Hong K, Kim E, Jo S, Kim S. Identification of constitutively active interleukin 33 (IL-33) splice variant. J Biol Chem. 2011;286(22):20078–86. doi: 10.1074/jbc.M111.219089. Epub 2011/04/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gordon ED, Simpson LJ, Rios CL, Ringel L, Lachowicz-Scroggins ME, Peters MC, Wesolowska-Andersen A, Gonzalez JR, MacLeod HJ, Christian LS, Yuan S, Barry L, Woodruff PG, Ansel KM, Nocka K, Seibold MA, Fahy JV. Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proc Natl Acad Sci U S A. 2016;113(31):8765–70. doi: 10.1073/pnas.1601914113. Epub 2016/07/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo Z, Wu J, Zhao J, Liu F, Chen Y, Bi L, Liu S, Dong L. IL-33 promotes airway remodeling and is a marker of asthma disease severity. J Asthma. 2014;51(8):863–9. doi: 10.3109/02770903.2014.921196. [DOI] [PubMed] [Google Scholar]
- 11.Reh DD, Wang Y, Ramanathan M, Jr, Lane AP. Treatment-recalcitrant chronic rhinosinusitis with polyps is associated with altered epithelial cell expression of interleukin-33. Am J Rhinol Allergy. 2010;24(2):105–9. doi: 10.2500/ajra.2010.24.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186(7):4375–87. doi: 10.4049/jimmunol.1003020. doi: jimmunol.1003020 [pii];10.4049/jimmunol.1003020 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ullah MA, Loh Z, Gan WJ, Zhang V, Yang H, Li JH, Yamamoto Y, Schmidt AM, Armour CL, Hughes JM, Phipps S, Sukkar MB. Receptor for advanced glycation end products and its ligand high-mobility group box-1 mediate allergic airway sensitization and airway inflammation. J Allergy Clin Immunol. 2014;134(2):440–50. doi: 10.1016/j.jaci.2013.12.1035. doi: S0091-6749(13)02939-4 [pii];10.1016/j.jaci.2013.12.1035 [doi] [DOI] [PubMed] [Google Scholar]
- 14.Christianson CA, Goplen NP, Zafar I, Irvin C, Good JT, Jr, Rollins DR, Gorentla B, Liu W, Gorska MM, Chu H, Martin RJ, Alam R. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. 2015;136(1):59–68. doi: 10.1016/j.jaci.2014.11.037. doi: S0091-6749(14)01740-0 [pii];10.1016/j.jaci.2014.11.037 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oczypok EA, Milutinovic PS, Alcorn JF, Khare A, Crum LT, Manni ML, Epperly MW, Pawluk AM, Ray A, Oury TD. Pulmonary receptor for advanced glycation end-products promotes asthma pathogenesis through IL-33 and accumulation of group 2 innate lymphoid cells. J Allergy Clin Immunol. 2015;136(3):747–56. doi: 10.1016/j.jaci.2015.03.011. doi: S0091-6749(15)00409-1 [pii];10.1016/j.jaci.2015.03.011 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamazumi Y, Sasaki O, Imamura M, Oda T, Ohno Y, Shiozaki-Sato Y, Nagai S, Suyama S, Kamoshida Y, Funato K, Yasui T, Kikutani H, Yamamoto K, Dohi M, Koyasu S, Akiyama T. The RNA Binding Protein Mex-3B Is Required for IL-33 Induction in the Development of Allergic Airway Inflammation. Cell Rep. 2016 doi: 10.1016/j.celrep.2016.07.062. doi: S2211-1247(16)30993-7 [pii];10.1016/j.celrep.2016.07.062 [doi] [DOI] [PubMed] [Google Scholar]
- 17.Israel E, Chervinsky PS, Friedman B, Van BJ, Skalky CS, Ghannam AF, Bird SR, Edelman JM. Effects of montelukast and beclomethasone on airway function and asthma control. J Allergy Clin Immunol. 2002;110(6):847–54. doi: 10.1067/mai.2002.129413. doi: S0091674902014653 [pii] [DOI] [PubMed] [Google Scholar]
- 18.Israel E, Cohn J, Dube L, Drazen JM. Effect of treatment with zileuton, a 5-lipoxygenase inhibitor, in patients with asthma. A randomized controlled trial. Zileuton Clinical Trial Group. JAMA. 1996;275(12):931–6. [PubMed] [Google Scholar]
- 19.Lam BK, Penrose JF, Freeman GJ, Austen KF. Expression cloning of a cDNA for human leukotriene C4 synthase, an integral membrane protein conjugating reduced glutathione to leukotriene A4. Proc Natl Acad Sci U S A. 1994;91(16):7663–7. doi: 10.1073/pnas.91.16.7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mueller CF, Wassmann K, Widder JD, Wassmann S, Chen CH, Keuler B, Kudin A, Kunz WS, Nickenig G. Multidrug resistance protein-1 affects oxidative stress, endothelial dysfunction, and atherogenesis via leukotriene C4 export. Circulation. 2008;117(22):2912–8. doi: 10.1161/CIRCULATIONAHA.107.747667. doi: CIRCULATIONAHA.107.747667 [pii];10.1161/CIRCULATIONAHA.107.747667 [doi] [DOI] [PubMed] [Google Scholar]
- 21.Carter BZ, Shi ZZ, Barrios R, Lieberman MW. gamma-glutamyl leukotrienase, a gamma-glutamyl transpeptidase gene family member, is expressed primarily in spleen. J Biol Chem. 1998;273(43):28277–85. doi: 10.1074/jbc.273.43.28277. [DOI] [PubMed] [Google Scholar]
- 22.Lee CW, Lewis RA, Corey EJ, Austen KF. Conversion of leukotriene D4 to leukotriene E4 by a dipeptidase released from the specific granule of human polymorphonuclear leucocytes. Immunology. 1983;48(1):27–35. [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch KR, O’Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, Coulombe N, Abramovitz M, Figueroa DJ, Zeng Z, Connolly BM, Bai C, Austin CP, Chateauneuf A, Stocco R, Greig GM, Kargman S, Hooks SB, Hosfield E, Williams DL, Jr, Ford-Hutchinson AW, Caskey CT, Evans JF. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399(6738):789–93. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- 24.Heise CE, O’Dowd BF, Figueroa DJ, Sawyer N, Nguyen T, Im DS, Stocco R, Bellefeuille JN, Abramovitz M, Cheng R, Williams DL, Jr, Zeng Z, Liu Q, Ma L, Clements MK, Coulombe N, Liu Y, Austin CP, George SR, O’Neill GP, Metters KM, Lynch KR, Evans JF. Characterization of the human cysteinyl leukotriene 2 receptor. J Biol Chem. 2000;275(39):30531–6. doi: 10.1074/jbc.M003490200. doi: 10.1074/jbc.M003490200 [doi];M003490200 [pii] [DOI] [PubMed] [Google Scholar]
- 25.Kanaoka Y, Maekawa A, Austen KF. Identification of GPR 99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J Biol Chem. 2013;288(16):10967–72. doi: 10.1074/jbc.C113.453704. doi: C113.453704 [pii];10.1074/jbc.C113.453704 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim DC, Hsu FI, Barrett NA, Friend DS, Grenningloh R, Ho IC, Al-Garawi A, Lora JM, Lam BK, Austen KF, Kanaoka Y. Cysteinyl leukotrienes regulate Th2 cell-dependent pulmonary inflammation. J Immunol. 2006;176(7):4440–8. doi: 10.4049/jimmunol.176.7.4440. doi: 176/7/4440 [pii] [DOI] [PubMed] [Google Scholar]
- 27.Barrett NA, Rahman OM, Fernandez JM, Parsons MW, Xing W, Austen KF, Kanaoka Y. Dectin-2 mediates Th2 immunity through the generation of cysteinyl leukotrienes. J Exp Med. 2011;208(3):593–604. doi: 10.1084/jem.20100793. doi: jem.20100793 [pii];10.1084/jem.20100793 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol. 2013;132(1):205–13. doi: 10.1016/j.jaci.2013.03.048. doi: S0091-6749(13)00588-5 [pii];10.1016/j.jaci.2013.03.048 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lund SJ, Portillo A, Cavagnero K, Baum RE, Naji LH, Badrani JH, Mehta A, Croft M, Broide DH, Doherty TA. Leukotriene C4 Potentiates IL-33-Induced Group 2 Innate Lymphoid Cell Activation and Lung Inflammation. Journal of immunology (Baltimore, Md : 1950) 2017;199(3):1096–104. doi: 10.4049/jimmunol.1601569. Epub 2017/07/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von MJ, O’Leary CE, Barrett NA, Kanaoka Y, Austen KF, Locksley RM. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J Exp Med. 2016 doi: 10.1084/jem.20161274. doi: jem.20161274 [pii];10.1084/jem.20161274 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corrigan C, Mallett K, Ying S, Roberts D, Parikh A, Scadding G, Lee T. Expression of the cysteinyl leukotriene receptors cysLT(1) and cysLT(2) in aspirin-sensitive and aspirin-tolerant chronic rhinosinusitis. J Allergy Clin Immunol. 2005;115(2):316–22. doi: 10.1016/j.jaci.2004.10.051. doi: S0091674904031227 [pii];10.1016/j.jaci.2004.10.051 [doi] [DOI] [PubMed] [Google Scholar]
- 32.Bankova LG, Lai J, Yoshimoto E, Boyce JA, Austen KF, Kanaoka Y, Barrett NA. Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein-coupled receptor, GPR99. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1605957113. doi: 1605957113 [pii];10.1073/pnas.1605957113 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JH, Haselkorn T, Borish L, Rasouliyan L, Chipps BE, Wenzel SE. Risk factors associated with persistent airflow limitation in severe or difficult-to-treat asthma: insights from the TENOR study. Chest. 2007;132(6):1882–9. doi: 10.1378/chest.07-0713. doi: 132/6/1882 [pii];10.1378/chest.07-0713 [doi] [DOI] [PubMed] [Google Scholar]
- 34.Rajan JP, Wineinger NE, Stevenson DD, White AA. Prevalence of aspirin-exacerbated respiratory disease among asthmatic patients: A meta-analysis of the literature. J Allergy Clin Immunol. 2015;135(3):676–81. doi: 10.1016/j.jaci.2014.08.020. doi: S0091-6749(14)01192-0 [pii];10.1016/j.jaci.2014.08.020 [doi] [DOI] [PubMed] [Google Scholar]
- 35.Christie PE, Tagari P, Ford-Hutchinson AW, Charlesson S, Chee P, Arm JP, Lee TH. Urinary leukotriene E4 concentrations increase after aspirin challenge in aspirin-sensitive asthmatic subjects. Am Rev Respir Dis. 1991;143(5 Pt 1):1025–9. doi: 10.1164/ajrccm/143.5_Pt_1.1025. [DOI] [PubMed] [Google Scholar]
- 36.Daffern PJ, Muilenburg D, Hugli TE, Stevenson DD. Association of urinary leukotriene E4 excretion during aspirin challenges with severity of respiratory responses. J Allergy Clin Immunol. 1999;104(3 pt 1):559–64. doi: 10.1016/s0091-6749(99)70324-6. doi: S0091674999005035 [pii] [DOI] [PubMed] [Google Scholar]
- 37.Israel E, Fischer AR, Rosenberg MA, Lilly CM, Callery JC, Shapiro J, Cohn J, Rubin P, Drazen JM. The pivotal role of 5-lipoxygenase products in the reaction of aspirin-sensitive asthmatics to aspirin. Am Rev Respir Dis. 1993;148(6 Pt 1):1447–51. doi: 10.1164/ajrccm/148.6_Pt_1.1447. [DOI] [PubMed] [Google Scholar]
- 38.Fischer AR, Rosenberg MA, Lilly CM, Callery JC, Rubin P, Cohn J, White MV, Igarashi Y, Kaliner MA, Drazen JM. Direct evidence for a role of the mast cell in the nasal response to aspirin in aspirin-sensitive asthma. J Allergy Clin Immunol. 1994;94(6 pt 1):1046–56. doi: 10.1016/0091-6749(94)90123-6. doi: S0091674994001570 [pii] [DOI] [PubMed] [Google Scholar]
- 39.Adamjee J, Suh YJ, Park HS, Choi JH, Penrose JF, Lam BK, Austen KF, Cazaly AM, Wilson SJ, Sampson AP. Expression of 5-lipoxygenase and cyclooxygenase pathway enzymes in nasal polyps of patients with aspirin-intolerant asthma. J Pathol. 2006;209(3):392–9. doi: 10.1002/path.1979. [DOI] [PubMed] [Google Scholar]
- 40.Liu T, Kanaoka Y, Barrett NA, Feng C, Garofalo D, Lai J, Buchheit K, Bhattacharya N, Laidlaw TM, Katz HR, Boyce JA. Aspirin-Exacerbated Respiratory Disease Involves a Cysteinyl Leukotriene-Driven IL-33-Mediated Mast Cell Activation Pathway. J Immunol. 2015;195(8):3537–45. doi: 10.4049/jimmunol.1500905. doi: jimmunol.1500905 [pii];10.4049/jimmunol.1500905 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu T, Laidlaw TM, Katz HR, Boyce JA. Prostaglandin E2 deficiency causes a phenotype of aspirin sensitivity that depends on platelets and cysteinyl leukotrienes. Proc Natl Acad Sci U S A. 2013;110(42):16987–92. doi: 10.1073/pnas.1313185110. doi: 1313185110 [pii];10.1073/pnas.1313185110 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uematsu S, Matsumoto M, Takeda K, Akira S. Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E(2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J Immunol. 2002;168(11):5811–6. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- 43.Kanaoka Y, Maekawa A, Penrose JF, Austen KF, Lam BK. Attenuated zymosan-induced peritoneal vascular permeability and IgE-dependent passive cutaneous anaphylaxis in mice lacking leukotriene C4 synthase. J Biol Chem. 2001;276(25):22608–13. doi: 10.1074/jbc.M103562200. doi: 10.1074/jbc.M103562200 [doi];M103562200 [pii] [DOI] [PubMed] [Google Scholar]
- 44.Maekawa A, Austen KF, Kanaoka Y. Targeted gene disruption reveals the role of cysteinyl leukotriene 1 receptor in the enhanced vascular permeability of mice undergoing acute inflammatory responses. J Biol Chem. 2002;277(23):20820–4. doi: 10.1074/jbc.M203163200. doi: 10.1074/jbc.M203163200 [doi];M203163200 [pii] [DOI] [PubMed] [Google Scholar]
- 45.Beller TC, Maekawa A, Friend DS, Austen KF, Kanaoka Y. Targeted gene disruption reveals the role of the cysteinyl leukotriene 2 receptor in increased vascular permeability and in bleomycin-induced pulmonary fibrosis in mice. J Biol Chem. 2004;279(44):46129–34. doi: 10.1074/jbc.M407057200. doi: 10.1074/jbc.M407057200 [doi];M407057200 [pii] [DOI] [PubMed] [Google Scholar]
- 46.Cummings HE, Liu T, Feng C, Laidlaw TM, Conley PB, Kanaoka Y, Boyce JA. Cutting edge: Leukotriene C4 activates mouse platelets in plasma exclusively through the type 2 cysteinyl leukotriene receptor. J Immunol. 2013;191(12):5807–10. doi: 10.4049/jimmunol.1302187. doi: jimmunol.1302187 [pii];10.4049/jimmunol.1302187 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu T, Laidlaw TM, Feng C, Xing W, Shen S, Milne GL, Boyce JA. Prostaglandin E2 deficiency uncovers a dominant role for thromboxane A2 in house dust mite-induced allergic pulmonary inflammation. Proc Natl Acad Sci U S A. 2012;109(31):12692–7. doi: 10.1073/pnas.1207816109. doi: 1207816109 [pii];10.1073/pnas.1207816109 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan D, Stocco R, Sawyer N, Nesheim ME, Abramovitz M, Funk CD. Differential signaling of cysteinyl leukotrienes and a novel cysteinyl leukotriene receptor 2 (CysLT(2)) agonist, N-methyl-leukotriene C(4), in calcium reporter and beta arrestin assays. Mol Pharmacol. 2011;79(2):270–8. doi: 10.1124/mol.110.069054. doi: mol.110.069054 [pii];10.1124/mol.110.069054 [doi] [DOI] [PubMed] [Google Scholar]
- 49.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–8. doi: 10.1038/ni.2045. doi: ni.2045 [pii];10.1038/ni.2045 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soter NA, Lewis RA, Corey EJ, Austen KF. Local effects of synthetic leukotrienes (LTC4, LTD4, LTE4, and LTB4) in human skin. J Invest Dermatol. 1983;80(2):115–9. doi: 10.1111/1523-1747.ep12531738. [DOI] [PubMed] [Google Scholar]
- 51.Weiss JW, Drazen JM, McFadden ER, Jr, Weller PF, Corey EJ, Lewis RA, Austen KF. Comparative bronchoconstrictor effects of histamine, leukotriene C, and leukotriene D in normal human volunteers. Trans Assoc Am Physicians. 1982;95:30–5. [PubMed] [Google Scholar]
- 52.Oyoshi MK, He R, Kanaoka Y, ElKhal A, Kawamoto S, Lewis CN, Austen KF, Geha RS. Eosinophil-derived leukotriene C4 signals via type 2 cysteinyl leukotriene receptor to promote skin fibrosis in a mouse model of atopic dermatitis. Proc Natl Acad Sci U S A. 2012;109(13):4992–7. doi: 10.1073/pnas.1203127109. Epub 2012/03/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hui Y, Yang G, Galczenski H, Figueroa DJ, Austin CP, Copeland NG, Gilbert DJ, Jenkins NA, Funk CD. The murine cysteinyl leukotriene 2 (CysLT2) receptor. cDNA and genomic cloning, alternative splicing, and in vitro characterization. J Biol Chem. 2001;276(50):47489–95. doi: 10.1074/jbc.M107556200. doi: 10.1074/jbc.M107556200 [doi];M107556200 [pii] [DOI] [PubMed] [Google Scholar]
- 54.Carnini C, Accomazzo MR, Borroni E, Vitellaro-Zuccarello L, Durand T, Folco G, Rovati GE, Capra V, Sala A. Synthesis of cysteinyl leukotrienes in human endothelial cells: subcellular localization and autocrine signaling through the CysLT2 receptor. FASEB J. 2011;25(10):3519–28. doi: 10.1096/fj.10-177030. doi: fj.10-177030 [pii];10.1096/fj.10-177030 [doi] [DOI] [PubMed] [Google Scholar]
- 55.Cowburn AS, Sladek K, Soja J, Adamek L, Nizankowska E, Szczeklik A, Lam BK, Penrose JF, Austen FK, Holgate ST, Sampson AP. Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma. J Clin Invest. 1998;101(4):834–46. doi: 10.1172/JCI620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nasser SM, Pfister R, Christie PE, Sousa AR, Barker J, Schmitz-Schumann M, Lee TH. Inflammatory cell populations in bronchial biopsies from aspirin-sensitive asthmatic subjects. Am J Respir Crit Care Med. 1996;153(1):90–6. doi: 10.1164/ajrccm.153.1.8542168. [DOI] [PubMed] [Google Scholar]
- 57.Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179(4):2051–4. doi: 10.4049/jimmunol.179.4.2051. doi: 179/4/2051 [pii] [DOI] [PubMed] [Google Scholar]
- 58.Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, Matsushita K, Taki Y, Futatsugi-Yumikura S, Tsutsui H, Ishii KJ, Yoshimoto T, Akira S, Nakanishi K. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci U S A. 2012;109(9):3451–6. doi: 10.1073/pnas.1201042109. doi: 1201042109 [pii];10.1073/pnas.1201042109 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509(7500):371–5. doi: 10.1038/nature13173. doi: nature13173 [pii];10.1038/nature13173 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sloniewsky DE, Ridge KM, Adir Y, Fries FP, Briva A, Sznajder JI, Sporn PH. Leukotriene D4 activates alveolar epithelial Na, K-ATPase and increases alveolar fluid clearance. Am J Respir Crit Care Med. 2004;169(3):407–12. doi: 10.1164/rccm.200304-472OC. doi: 10.1164/rccm.200304-472OC [doi];200304-472OC [pii] [DOI] [PubMed] [Google Scholar]
- 61.von Burg N, Turchinovich G, Finke D. Maintenance of Immune Homeostasis through ILC/T Cell Interactions. Front Immunol. 2015;6:416. doi: 10.3389/fimmu.2015.00416. Epub 2015/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li BW, de Bruijn MJ, Tindemans I, Lukkes M, KleinJan A, Hoogsteden HC, Hendriks RW. T cells are necessary for ILC2 activation in house dust mite-induced allergic airway inflammation in mice. European journal of immunology. 2016;46(6):1392–403. doi: 10.1002/eji.201546119. Epub 2016/04/12. [DOI] [PubMed] [Google Scholar]
- 63.Stevenson DD, Simon RA, Mathison DA, Christiansen SC. Montelukast is only partially effective in inhibiting aspirin responses in aspirin-sensitive asthmatics. Ann Allergy Asthma Immunol. 2000;85(6 Pt 1):477–82. doi: 10.1016/S1081-1206(10)62575-6. doi: S1081-1206(10)62575-6 [pii];10.1016/S1081-1206(10)62575-6 [doi] [DOI] [PubMed] [Google Scholar]
- 64.Hamilton A, Faiferman I, Stober P, Watson RM, O’Byrne PM. Pranlukast, a cysteinyl leukotriene receptor antagonist, attenuates allergen-induced early- and late-phase bronchoconstriction and airway hyperresponsiveness in asthmatic subjects. J Allergy Clin Immunol. 1998;102(2):177–83. doi: 10.1016/s0091-6749(98)70083-1. doi: S0091674998002887 [pii] [DOI] [PubMed] [Google Scholar]
- 65.Gauvreau GM, Boulet LP, Fitzgerald JM, Cockcroft DW, Davis BE, Leigh R, Tanaka M, Fourre JA, Tanaka M, Nabata T, O’Byrne PM. A Dual CysLT1/2 Antagonist Attenuates Allergen-induced Airway Responses in Subjects with Mild Allergic Asthma. Allergy. 2016 doi: 10.1111/all.12987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.