

Abstract
Whereas 400 million distinct compounds are now purchasable within the span of a few weeks, the biological activities of most are unknown. To facilitate access to new chemistry for biology, we have combined the Similarity Ensemble Approach (SEA) with the maximum Tanimoto similarity to the nearest bioactive to predict activity for every commercially available molecule in ZINC. This method, which we label SEA+TC, outperforms both SEA and a naïve-Bayesian classifier via predictive performance on a 5-fold cross-validation of ChEMBL’s bioactivity data set (version 21). Using this method, predictions for over 40% of compounds (>160 million) have either high significance (pSEA ≥ 40), high similarity (ECFP4MaxTc ≥ 0.4), or both, for one or more of 1382 targets well described by ligands in the literature. Using a further 1347 less-well-described targets, we predict activities for an additional 11 million compounds. To gauge whether these predictions are sensible, we investigate 75 predictions for 50 drugs lacking a binding affinity annotation in ChEMBL. The 535 million predictions for over 171 million compounds at 2629 targets are linked to purchasing information and evidence to support each prediction and are freely available via https://zinc15.docking.org and https://files.docking.org.
Introduction
The purchasable chemical space has roughly doubled every two and a half years since 1990, owing to steady progress in efficient parallel synthesis1−8 and the synthesis of new building blocks. There are now over 400 million compounds one can easily purchase using ZINC,9 which covers 204 commercial catalogs from 145 companies. Each catalog is categorized by ease of purchase, and each compound in turn inherits a purchasability level from its catalog membership. The growth in catalog size is impressive, particularly among the make-on-demand catalogs. Purchasable compounds in the favored lead-like10 and fragment-like11 areas have grown from 3 million and a half million in 2007 to 124 million and 9.2 million today, respectively. Many vendors have incorporated the lessons of lead- and fragment-likeness in library design,47 often filtering for PAINS.48 About 340 million (85%) of these compounds are affordable enough for the average academic lab to conduct a ligand discovery project, retaining a price point around $100 per sample or less. A further 60 million compounds are available at higher building-block prices, often $400 USD or more and are included here for completeness. We find that synthesis plus delivery of make-on-demand screening compounds often takes little more than a month or so, just twice the time to source many in-stock compounds.
The molecular targets (proteins) that these purchasable compounds bind and modulate—if any—are rarely known. Fewer than 1 million compounds—less than 0.25%—have been reported active in a target-specific assay according to public databases such as ChEMBL12 or other annotated collections indexed by ZINC.13 Investigators searching for testable ligands might not consider the remaining readily available compounds, as they are not annotated for targets and the sheer number of options can be daunting. In the absence of target activity information, the process of selecting compounds for general purpose screening will often be target-naïve, relying on chemical or physical-property diversity to sample chemical and property space, respectively.14 If information on target bias—the likelihood that a compound is more disposed to bind to a particular target or class of targets—were readily available, libraries more likely to cover biological targets of interest could be designed.
Systematically assaying every commercially available compound against every target is experimentally impractical, so prioritizing compounds through computational predictions is a pragmatic alternative. There are many methods for predicting biological activities by chemical similarity;15−36 here, we use two. The Similarity Ensemble Approach (SEA)37,38 predicts biological targets of a compound based on its resemblance to ligands annotated in a reference database, such as ChEMBL.12 SEA relates proteins by their pharmacology by aggregating chemical similarity among entire sets of ligands. By leveraging extreme value statistics, SEA filters out unreliable signals and normalizes the aggregate results against a random chemical background to predict the significance of pharmacological similarity. SEA has successfully predicted targets of marketed drugs,37−39 toxicity targets,40 and mechanism of action targets for hits in zebrafish41 and C. elegans(42) phenotypic screens. We also use the maximum Tanimoto coefficient43 at 0.4044 or better based on ECFP4 fingerprints45 to inform predictions. Neither method generates models incorporating discrete chemotypes as do Naïve Bayes classifiers, for instance, but instead consider the molecule holistically. This is advantageous because the method can suggest molecules that do not conform to what has been highly weighted by precedent. Other methods such as Naïve Bayes46 can explicitly weight for chemical substructures that are potentially important to bioactivity (“warheads”), and thus a future version might use such an approach to complement this work.
To be useful for research, predictions should be accessible, searchable, and downloadable. An interface should allow access to predictions for each compound, as well as for each target, vendor, and gene. A mechanism to select more novel or more conservative predictions would cater to a wide range of requirements. And libraries should be downloadable in 2D formats for chemoinformatics as well as in popular 3D formats for docking screens.
The prospective user of such a resource expects some way to evaluate the predictions. As one proxy to assess this data set, we performed a retrospective 5-fold cross-validation on the ChEMBL bioactivity data set for our method as compared to SEA and a naïve-Bayesian classifier, at a variety of threshold parameters (Figure 1; Supporting Information Figures S1 and S2). Second, in assessing performance, we reencountered the observation that whereas the canonical targets of all but a few drugs are known,47 hundreds of established drugs and investigational compounds nonetheless lack their respective target annotations in ChEMBL. We turned this deficit to our advantage, by testing the method’s prediction of targets for several such drugs, corroborating our predictions with the literature when available. Finally, as these predictions are based on protein−ligand annotations derived from ChEMBL, we expect that this method will be silent about chemotypes and targets not contained in this approximation of the public pharmacopeia.
Figure 1.
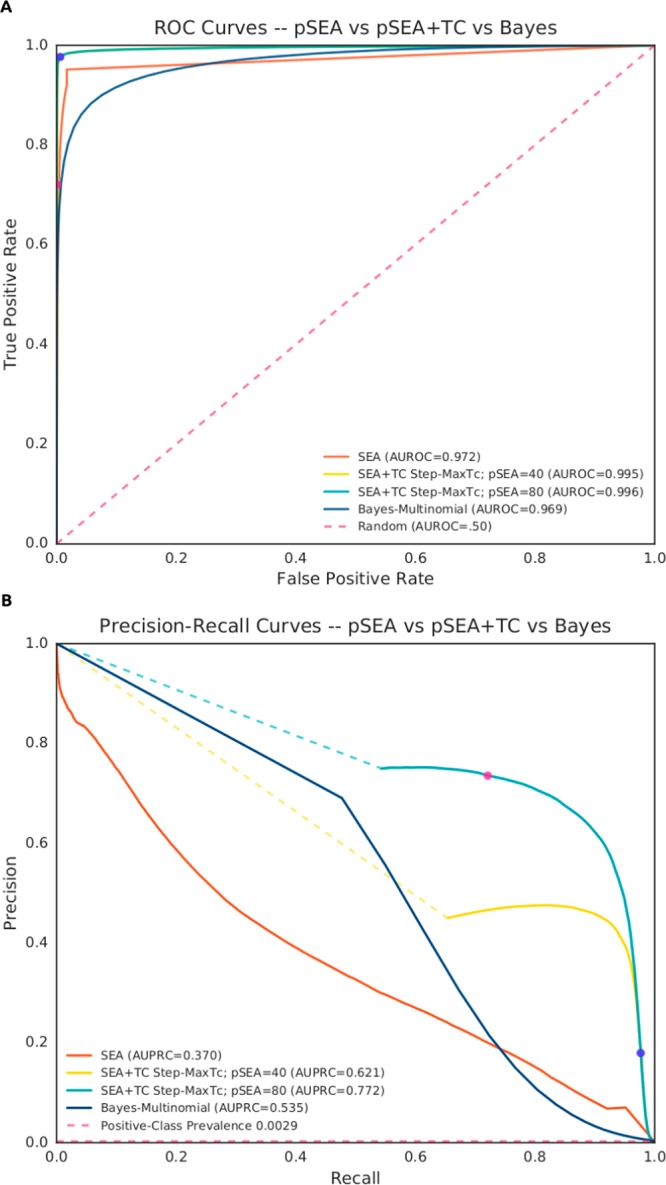
Comparative performance of SEA, SEA+TC, and a multinomial naive-Bayesian classifier (NBC) on ChEMBL cross-validation sets. (A) Receiver operating characteristic (ROC) curves from independent 5-fold cross-validation runs for each method. Methods are evaluated on independent cross-validation sets filtered for >5 ligands per ChEMBL protein target (equivalent analyses at >50 ligands per target reported in Supporting Information Figure S2). Overall performance is gauged by the area under the ROC curve (AUROC). Note, for SEA+TC cross-validation sets, ROC curves are the result of stepping a decision threshold across MaxTc values, while holding a separate pSEA decision threshold at 40 (yellow curve) or 80 (cyan curve) (see Methods). Complementary curves stepping across SEA p-values are available in Supporting Information Figures S1 and S2. Dotted lines span the distance between a fully stratified classifier (TPR = 0; FPR = 0) and the minimum point at which both SEA+TC decision thresholds begin to affect performance. Pink and blue circles indicate the recommended upper and lower bounds for MaxTc thresholding on their respective pSEA-threshold curves, respectively (upper = 0.80; lower = 0.40). (B) Corresponding precision-recall curves (PRCs) for cross-validation runs described in part A. Positive-class prevalence (dashed red line) indicates the chance of selecting a positive association from the data set at random (0.0014). Performance is measured by the area under the PRC (AUPRC).
Results
The ZINC database contains 400 million commercially available organic molecules with molecular weight between 50 and 1000 Da, sourced from 204 commercial catalogs published by 145 companies. We have created a database of predicted biological activities for the 171 million compounds that had predictions and have made it freely accessible via ZINC (https://zinc15.docking.org) and our file server (https://files.docking.org). All predictions were computed using a combination of the Similarity Ensemble Approach (SEA)37 and Tanimoto similarity calculations based on compound annotations derived from ChEMBL Version 2112 (see Methods). We refer to this combinatorial approach as SEA+TC throughout the text.
To enhance this resource’s applicability to a broad audience, we sought to increase the specificity of predictions by using more stringent criteria for what constitutes an annotated ligand. In prior work we had used a 10 μM affinity cutoff, but at this scale, we encountered flawed predictions that appeared to arise from similarity to weak binders, possible PAINS, or promiscuous aggregator compounds. Based on our experience with these encounters, we changed the baseline affinity threshold to 1 μM and further required activities of at least 100 nM for compounds containing PAINS patterns or being Tc 0.70 to any compound observed to aggregate.48−50
We adopted a statistical significance threshold of negative log SEA p-value54 (pSEA) ≥ 40 and a MaxTc cutoff ≥0.40 guided by the work on belief theory from the Abbvie group.34 MaxTc is complementary to pSEA as it provides a single-nearest-neighbor-molecule view of similarity, compared to SEA’s global view arising from the ensemble of annotated ligands. To quantify how this bivariate threshold improves predictive capability, we evaluated the performance of SEA, SEA+TC, and a Naïve-Bayesian classifier (NBC) via 5-fold cross-validation of ChEMBL’s bioactivity data set (version 21; Figure 1). SEA+TC’s ability to correctly predict compound−target interactions as either positive (does bind) or negative (does not bind) outperformed both SEA and the NBC, as measured by the area under the receiver operating characteristic (AUROC) curve, (AUROC = 0.995, Figure 1A). Further, when predicting a compound−target interaction as positive, SEA+TC was correct in its prediction more often than SEA or the NBC, as indicated by its area under the precision-recall (AUPRC) curve (AUPRC = 0.684, Figure 1B). In performing this analysis, we additionally identified a more stringent bivariate threshold, which some users may wish to adopt. At a threshold of MaxTc ≥ 0.80 with pSEA ≥ 80, the retrospective analyses achieve higher precision than the baseline threshold (Figure 1A and B, blue circle) at acceptable recall (pink circle). Users of the ZINC interface may choose thresholds to suit their needs.
In addition to controlling the sensitivity and specificity of predictions, the significance threshold (i.e., pSEA and MaxTc values)17 also influences the novelty of the predictions. Novel compounds can be desirable because they likely have unrelated off-target effects, which can help establish the signaling and toxicity role of a receptor, as well as selectively activate downstream signaling, which is important for many receptors such as GPCRs.38 Accordingly, we designed the ZINC interface to help users rapidly identify predictions with their desired precision. The user can control the MaxTc and pSEA limits, and each prediction can be compared with the most similar annotated actives (Figure 2) allowing side-by-side comparison. Each SEA prediction is accompanied by a pSEA to the set of actives and MaxTc to the nearest active. Clicking on the MaxTc value in the interface performs a real-time search for the most similar ligands annotated at 10 μM or better for that target.
Figure 2.

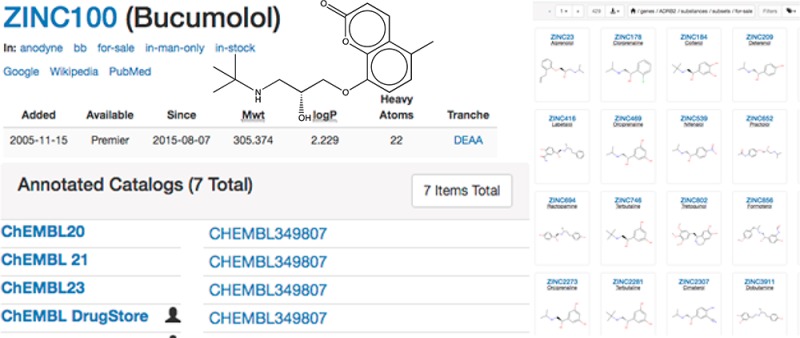
Predictions supported by evidence. (A) Here, Bucumolol (ZINC100) is shown with a SEA prediction for ADRB2 at a pSEA = 33 and MaxTc to the nearest annotated compound of 0.44. The user may click on the “44” to go to the URL shown, which lists bucumolol’s closest-match known ADRB2 ligands in decreasing order of similarity (the first four are shown). The user may also click on “Run SEA” to rerun a SEA calculation on the molecule, providing comprehensive statistics.
To find predictions for a given target using ZINC15 (zinc15.docking.org), the user may select Genes from the Biological dropdown menu to browse a listing of all genes and predictions (Figure 3A). In this work, we use genes and their identifiers as convenient shorthand for their protein products—or molecular targets. To find a specific gene, the user may type part of the gene name in the top right search bar, here SLC6, and click the blue search button on the top right. To display predictions for this gene, the user clicks on the link in the predictions column, here for SLC6A1 (Figure 3B). The user may for example use the subset selector to specify strong predictions (which we chose to mean pSEA = 80) and purchasability (Figure 3C). Some advanced features are currently only accessible by hand-editing the URL. Here, the user adds table.html?sort=-maxtc and &maxtc-between=40+45 to display the information in a tabular format, to sort by decreasing MaxTc, and to select only predictions between MaxTc of 40 and 45, respectively (Figure 3D). We plan to make these API-level features available via a point and click interface soon. Documentation is available via the help pages https://zinc15.docking.org/genes/help and https://zinc15.docking.org/predictions/help.
Figure 3.

Tools to display predictions for a gene and filter and sort them by MaxTc and pSEA. (A) Gene page showing predictions, with search bar to locate genes by name, top right. https://zinc15.docking.org/genes. (B) Gene listings for genes matching “SLC6” https://zinc15.docking.org/genes/search?q=SLC6. (C) Strongly predicted ligands for SLC6A11, showing the popup for subset selections https://zinc15.docking.org/genes/SLC6A11/predictions/subsets/strong. (D) Individual predictions, showing MaxTc and pSEA for each prediction, sorted by pSEA, with a MaxTc (novelty/similarity) limit specified https://zinc15.docking.org/genes/SLC6A1/predictions/subsets/strong/table.html?sort=-pvalue&maxtc-between=40+45.
Predictions are available for 2629 genes51 (Figure 4). The number of predictions per gene varies substantially, reflecting both the diversity of annotated ligands for the target as well as how well these chemotypes are represented in current vendor catalogs. For example, natural products and their analogs are often difficult to access synthetically and are therefore generally sparsely represented. At the high end of predictions per gene, the eukaryotic GPCRs D4 dopamine receptor (DRD4), C−C chemokine receptor type 3 (CCR3), and the voltage gated ion channels KCNK3 and KCNK9 each have over 4.8 million purchasable predicted ligands. The number of strong predictions (pSEA ≥ 80) varies from over 500 000 for KCNK3 to as few as 9181 for DRD4. Filtering at MaxTc ≥ 0.60 instead, corresponding to a precision exceeding 0.334 using ECFP4 fingerprints,44 the predictions for these four genes varied from as many as 25 728 for DRD4 to as few as 8912 for KCNK9. At the other extreme of predictions per gene, fungal laccase-2 precursor (LCC2), human C−C chemokine receptor type 6 (CCR6), voltage-gated sodium channel Nav1.9 (SCN11A), and fruit fly DNA topoisomerase 2 (TOP2) each had fewer than 50 predicted commercially available ligands. The small number of predicted ligands can often be explained by a paucity of reference ligands; here, SCN11A and CCR6 have only 1 ligand each at 10 μM or better. Another reason for the lack of ligands is that the knowns are in an area of chemical space that is difficult to access synthetically, such as natural products for both SCN11A and CCR6.
Figure 4.

Predictions available for 2629 genes. (A) The web interface allows genes and their predictions to be found by name or gene symbol: https://zinc15.docking.org/genes. Enter the gene name in the search field (1). Click on the predictions link (2) to display the predicted ligands. (B) Predictions and purchasable compounds for 2629 genes. The horizontal axis is genes, sorted by number of predictions. The vertical axis is number of compounds, log scale, labeled by exponent. Dark gray circles indicate the number of predicted purchasable compounds for a gene. Green triangles represent the number of purchasable annotated compounds for the same gene.
Access by Gene Groupings
In addition to individual genes, predictions may also be accessed by groups of genes. This could be helpful if the investigator is looking for new aminergic GPCR ligands or ligands for voltage gated ion channels or simply wishes to ensure balanced coverage of major target classes in a library. The interface offers convenient ways to access gene groupings based on a protein classification scheme inherited from ChEMBL. There are 15 major target classes (Figure 5A) further organized into 42 target subclasses (Figure 5B). Thus, there are 67 million predictions for membrane proteins, of which 1 million are strong (pSEA ≥ 80). Considered separately, there are 873,000 less chemically novel predictions having a Tanimoto coefficient ≥0.60 to an annotated active. At a higher level of granularity, there are 4.7 million predictions for epigenetic reader proteins, of which 2.4 million are strong predictions (pSEA ≥ 80) and 38 000 are highly similar (Tc ≥ 0.60). At the organism level (Figure 5C), 18 million ligands are predicted for specific bacterial targets, 1.0 million of which are stronger (pSEA ≥ 80) and 92 000 of which are highly similar (Tc ≥ 0.60). The user may select purchasable compounds based on this classification. These compounds will resemble precedented bacterial protein inhibitors far more strongly than compounds selected at random. Ligands predicted for specific bacterial targets are available to browse interactively at https://zinc15.docking.org/organisms/bacteria/genes/ or to download by gene at https://files.docking.org/predictions/current/. A plot of predictions per gene vs annotated ligands per gene shows a general trend toward more predicted ligands when more known ligands are available (see Supporting Information Figure S3).
Figure 5.


Prediction counts and purchasable compounds. The gray line indicates the number of predictions, and the green line represents the number of annotated compounds. (A) By major target class. Data from https://zinc15.docking.org/majorclasses. (B) By target subclass. Most target predictions have a maximum tanimoto coefficient between 0.30 and 0.39 and 0.40−0.49. Percent of predictions for each target subclass relative to MaxTc are plotted in the inset to show the full spread of prediction across bins. (C) By Kingdom, called organism class in ChEMBL and ZINC. Data from https://zinc15.docking.org/organisms.
Benchmarks
We predicted the targets of established drugs that nonetheless lack a protein binding affinity annotation in ChEMBL to benchmark our approach. We found hundreds of drugs, withdrawn drugs, and investigational compounds with target predictions that agreed with the literature. Fifty of these were selected and tabulated as illustration of our predictions (Table 1). Thus, the beta blocker bufetolol52 (ZINC101) is predicted to be a β2 adrenergic receptor ligand with pSEA = 47 and MaxTc = 0.46 and to be a β1 adrenergic receptor ligand with pSEA = 51and MaxTc = 0.44. Aranidipine53 (ZINC600803) is predicted for the calcium voltage-gated ion channel CACNA1C with pSEA = 121 and MaxTc = 0.75. Ancarolol (ZINC39) illustrates the discriminatory value of the SEA prediction, with pSEA = 59 and MaxTc = 0.43 for ADRB1: 255 656 purchasable ligands have higher MaxTc than ancarolol to this target while only 46 753 have a higher pSEA score.
Table 1. Drugs with No Binding Data in ChEMBL, Predicted by SEA or MaxTc, Corroborated by the Literature.
| drug(ref) | ZINC ID | target | pSEA | MaxTc |
|---|---|---|---|---|
| Acemetacin63 | 601272 | PTGS2 | 40 | 0.76 |
| Afeletecan64 | 150339966 | TOP1 | 69 | 0.41 |
| Alclometasone65 | 4172330 | NR3C1 | 15 | 0.58 |
| Alminoprofen66 | 22 | PTGS2 | 0.47 | |
| Amisulpride67 | 1846088 | DRD3 | 22 | 0.66 |
| Ancarolol68 | 39 | ADRB2 | 42 | 0.44 |
| ADRB1 | 59 | 0.43 | ||
| ADRB3 | 29 | 0.44 | ||
| Aranidipine53 | 600803 | CACNA1C | 121 | 0.75 |
| CACNA1D | 132 | 0.51 | ||
| Azasetron69 | 4132 | HTR3A | 25 | 0.61 |
| Azelnidipine70 | 38141706 | CACNA1C | 91 | 0.56 |
| CACNA1D | 124 | 0.57 | ||
| Azetirelin71 | 3804057 | TRHR | 95 | 0.59 |
| TRHR2 | 0.61 | |||
| Besifloxacin72 | 3787097 | PARC | 0.46 | |
| Bevantolol73 | 1542891 | ADRB1 | 89 | 0.51 |
| ADRB2 | 73 | 0.58 | ||
| ADRB3 | 73 | 0.53 | ||
| Bilastine74 | 3822702 | HRH1 | 48 | 0.51 |
| Binospirone75 | 1999423 | HTR1A | 0.48 | |
| Bufetolol52 | 101 | ADRB1 | 51 | 0.44 |
| ADRB2 | 47 | 0.46 | ||
| Bunazosin76 | 601249 | ADRA1B | 52 | 0.61 |
| Bupranolol77 | 106 | ADRB2 | 45 | 0.44 |
| ADRB1 | 19 | 0.45 | ||
| Butofilolol78 | 112 | ADRB1 | 50 | 0.40 |
| ADRB2 | 34 | 0.46 | ||
| Calcifediol79 | 12484926 | VDRA | 0.79 | |
| GC | 0.79 | |||
| Camazepam80 | 2008504 | GABARA5 | 25 | 0.53 |
| GABARA2 | 15 | 0.53 | ||
| Cellcept81 | 21297660 | IMPDH1 | 0.70 | |
| IMPDH2 | 0.70 | |||
| Ciprokiren82 | 8214528 | REN | 178 | 0.68 |
| Dasotraline83 | 2510873 | SLC6A3 | 25 | 0.63 |
| SLC6A2 | 29 | 0.63 | ||
| Demecarium84 | 3875376 | ACHE | 0.71 | |
| Dienesterol85 | 4742540 | ESR1 | 26 | 0.46 |
| ESR2 | 15 | 0.46 | ||
| Edaglitazone86 | 1483899 | PPARG | 83 | 0.66 |
| PPARA | 83 | 0.65 | ||
| Efonidipine87 | 38139973 | CACNA1C | 81 | 0.51 |
| CACNA1D | 118 | 0.51 | ||
| Eptazocine88 | 1846076 | OPRD1 | 30 | 0.42 |
| OPRK1 | 30 | 0.46 | ||
| OPRM1 | 32 | 0.46 | ||
| Etanterol89 | 263 | ADRB1 | 23 | 0.47 |
| ADRB2 | 47 | 0.40 | ||
| Ethylmorphine90 | 3629718 | OPRD1 | 28 | 0.62 |
| OPRK1 | 24 | 0.62 | ||
| OPRM1 | 32 | 0.75 | ||
| OPRL1 | 0.57 | |||
| Etomoxir91 | 1851171 | CPT1 | 0.47 | |
| Fiduxosin92 | 29747110 | ADRA1A | 30 | 0.53 |
| ADRA1B | 45 | 0.53 | ||
| ADRA1D | 38 | 0.46 | ||
| Floxacillin93 | 4102187 | BLAACC-4 | 0.80 | |
| Flurazepam94 | 537752 | GABARA5 | 28 | 0.50 |
| GABARA1 | 17 | 0.49 | ||
| Granisetron95 | 347 | HTR3A | 25 | 0.75 |
| Halobetasol96 | 4214603 | NR3C2 | 20 | 0.60 |
| Hexoprenaline97 | 3872806 | ADRB2 | 77 | 0.52 |
| Ketobemidone98 | 1600 | OPRD1 | 49 | 0.46 |
| OPRK1 | 45 | 0.48 | ||
| OPRM1 | 44 | 0.55 | ||
| Lercanidipine99 | 19685790 | CACNA1B | 0.49 | |
| CACNA1C | 107 | 0.70 | ||
| CACNA1D | 146 | 0.63 | ||
| Lexacalcitol100 | 4474609 | VDR | 144 | 0.62 |
| Meptazinol101 | 854 | OPRD1 | 44 | 0.48 |
| OPRK1 | 39 | 0.60 | ||
| OPRM1 | 38 | 0.55 | ||
| Metipranolol102 | 494 | ADRB1 | 27 | 0.45 |
| ADRB2 | 31 | 0.52 | ||
| Ormeloxifene103 | 5104028 | ESR1 | 86 | 0.51 |
| ESR2 | 58 | 0.44 | ||
| Paroxypropione104 | 1890 | ESR1 | 38 | 0.58 |
| ESR2 | 30 | 0.58 | ||
| Pipenzolate105 | 601314 | CHRM1 | 0.47 | |
| CHRM2 | 30 | 0.43 | ||
| CHRM3 | 57 | 0.53 | ||
| CHRM4 | 35 | 0.53 | ||
| CHRM5 | 40 | 0.53 | ||
| Pozanicline106 | 6562 | CHRNA2 | 33 | 0.57 |
| CHRNA4 | 0.57 | |||
| CHRNA10 | 53 | 0.55 | ||
| Propiverine107 | 1530934 | CHRM2 | 24 | 0.42 |
| CHRM3 | 50 | 0.57 | ||
| Revatropate108 | 4214265 | CHRM1 | 55 | 0.53 |
| CHRM2 | 33 | 0.53 | ||
| CHRM3 | 59 | 0.57 | ||
| GPM3 | 0.57 | |||
| Temazepam109 | 740 | GABA5 | 28 | 0.59 |
| Udenafil110 | 13916432 | PDE5A | 74 | 0.61 |
| Unoprostone111 | 8214703 | PTGER1 | 45 | 0.57 |
| PTGER2 | 30 | 0.40 | ||
| PTGER3 | 0.57 | |||
| PTGDR | 52 | 0.40 | ||
| PTGFR | 85 | 0.51 | ||
| Valategrast112 | 72190226 | ITGA4 | 60 | 0.32 |
| Verubulin113 | 35978229 | TUBB3 | 62 | 0.51 |
Among the 535 million predictions of protein−ligand affinity we expect numerous false positives and false negatives. These errors stem from three major classes of problem. (1) Issues with target annotation: annotated ligands may not be representative for a gene, such as curcumin (ZINC100067274), which is annotated for 32 genes and is probably artifactual for many of them.54 Annotated ligands may also be mis-annotations in ChEMBL, leading to false positives. For instance, nicotinamide (CHEMBL1140) is annotated for fatty-acid amide hydrolase 1 (FAAH), because it shares an abbreviation (NAM) with the actual ligand, N-arachidonylmaleimide.55 (2) Errors with the SEA method: We use ECFP4 fingerprints, which have little specificity for certain classes of molecules, such as peptides and sterols, which share many common features and thus are not well discriminated using this fingerprint. SEA also has high variance for small ligand sets and low sensitivity for large, diverse ligand sets. For instance, SEA fails to predict the well-known antihistamine drugs chlorcyclizine and propiomazine for histamine H1 receptor (HRH1), despite their having Tc values of 0.79 and 0.69, respectively, to the most similar HRH1 ligands. The pSEA values of 11 in each case have been diluted by the 9000 diverse ligands annotated to this target. A remedy might be to split targets with large number of ligands, perhaps by chemical clusters, mode of action, or binding site, if known. Note that Naïve Baysian classifiers can be trained to correctly predict these activities, as can be seen on ChEMBL’s ligand detail pages for these compounds. (3) No explicit model of promiscuity for SEA: We have made some progress here by stringent filtering of ligands we suspect are promiscuous (both PAINS and aggregator-like), but we fail to handle frequent hitters such as staurosporine (ZINC3814434, hits 365 targets in ChEMBL) and its ilk. Our current approach also performs poorly on sigma nonopioid intracellular receptor 1 (SIGMAR1) and cytochromes P450 3A4 (CYP3A4), because the ligands annotated to it are highly diverse. To remedy this problem for targets with many ligands, we could cluster by chemotype.
Genes Lacking Commercially Available Ligands
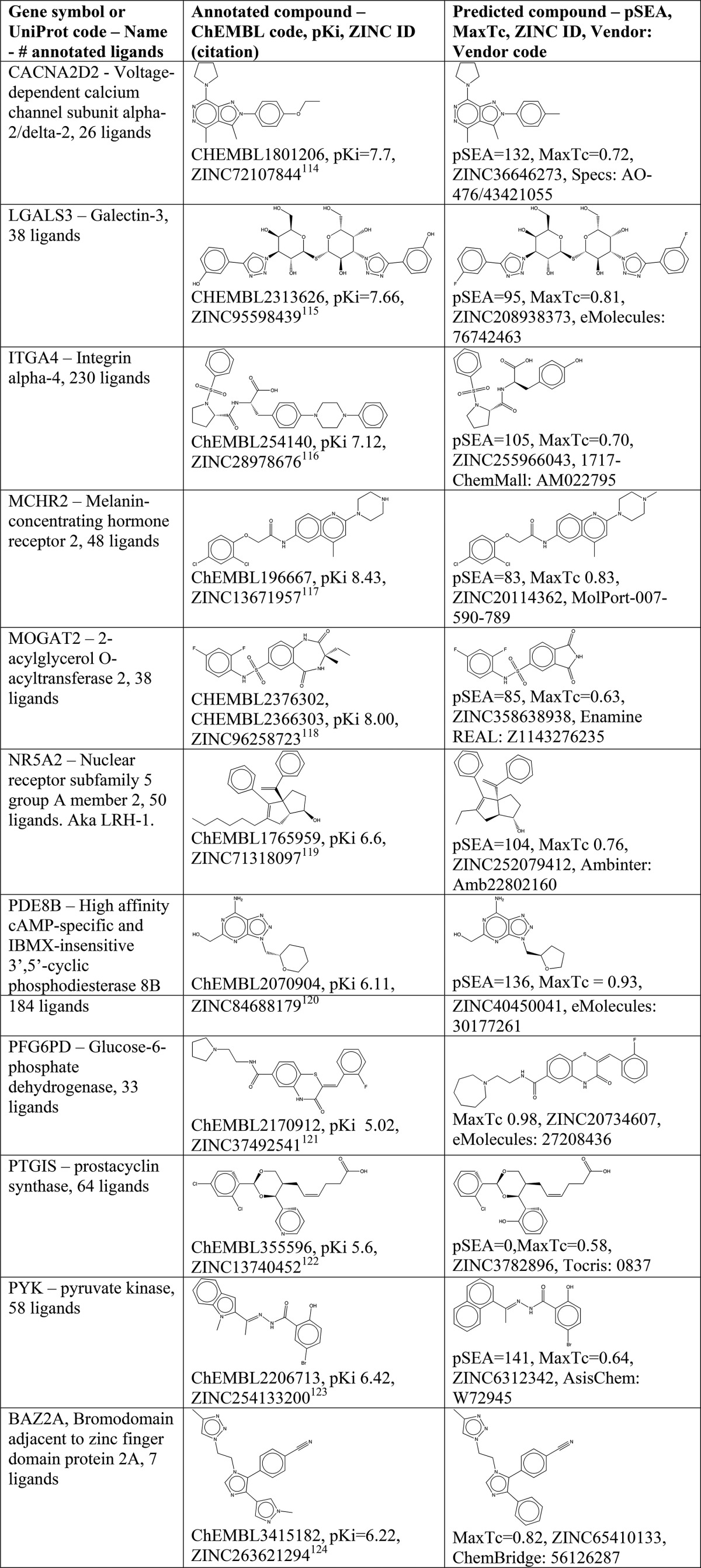
When a target has purchasable ligands, they can be used to rapidly probe its biological function without requiring synthetic chemistry expertise. Yet there are 69 targets with 20 or more annotated ligands in ChEMBL where none is readily purchasable (Table 2). To fill these holes in “target space”, we have identified purchasable compounds that are predicted to be active. In one example, voltage dependent calcium channel subunit alpha-2/delta-2 (CACNA2D2) has 26 ligands in ChEMBL, none of which is for sale, such as CHEMBL1801206 with a pKi of 7.7. The compound ZINC36664273, however, is sold by Specs as AO-476/43421055 and has a pSEA of 132 and a MaxTc of 0.72. Looking at these compounds side by side (Table 2) and without detailed experimental knowledge of this target, the Specs compound may be reasonable to try against this target. If successful, such compounds could become a purchasable control for these targets.
Table 2. Selected Plausible Predictions of Purchasable Compounds for Genes with No Purchasable Ligands in ChEMBL.
Dark Chemical Matter
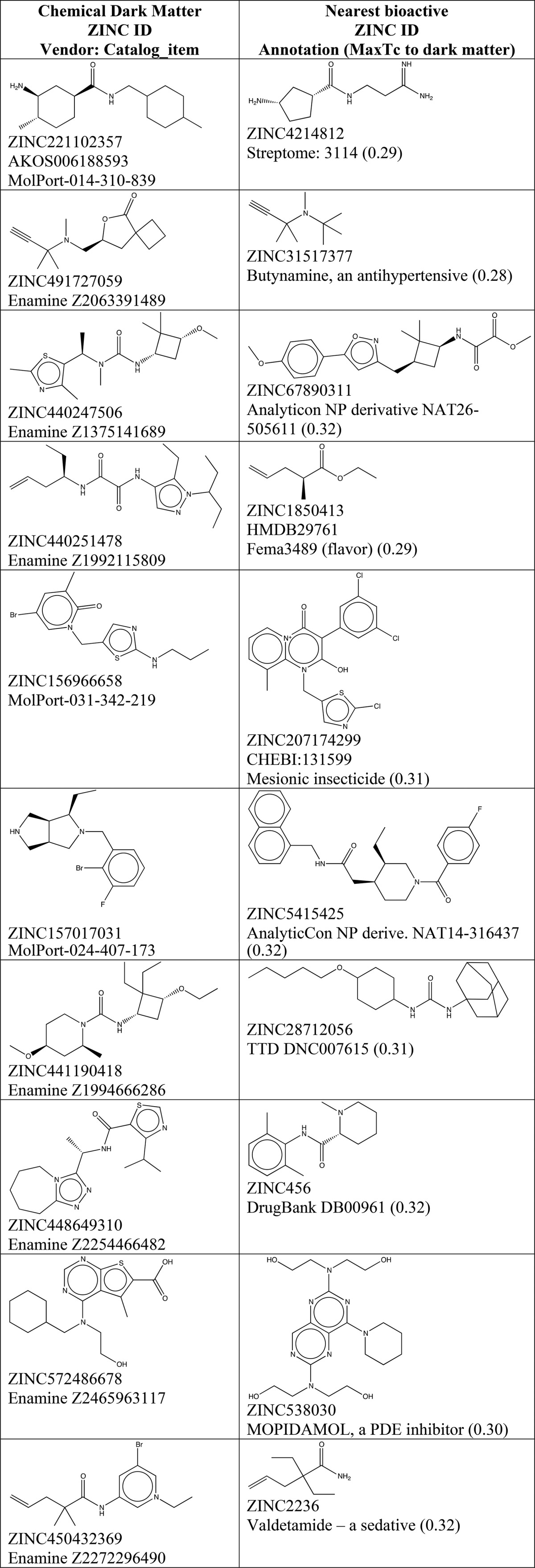
Intriguingly, 229 million purchasable compounds have no prediction at all by either pSEA ≥ 40 or Tanimoto similarity Tc ≥ 0.40. Some of these will have just missed our cutoffs, wherever the cutoffs may be drawn. A few will be known actives, or analogs of actives, that simply lack a direct binding annotation in ChEMBL. Still, these compounds are generally interesting because they do not much resemble any direct binding actives in ChEMBL. Should they be found to be active in an assay, they are more likely to have fewer off-targets, at least against well-studied targets, and are less likely to be encumbered by patents. A substantial body of literature explores the strengths and pitfalls of dark chemical matter.56−59 To illustrate what a user of this resource can expect to find in this underexploited yet commercially available space, we have highlighted ten compounds (Table 3). For each commercially available molecule, we show the nearest precedented bioactive from public sources available to ZINC, which may also include compounds not in ChEMBL. Dark chemical matter56−59 may be browsed online at zinc15.docking.org/substances/having/no-predictions and downloaded at scale by physical property tranches (https://files.docking.org/dark-matter/current), by vendor catalogs (e.g., for ChemBridge at https://files.docking.org/catalogs/50/chbr/chbr.predict.txt.gz) and by the genes they are predicted to bind (https://files.docking.org/genes/<genesymbol>/<genesymbol>.predictions.txt.gz).
Table 3. Compounds with No Predictions “Chemical Dark Matter”a.
To browse, use: https://zinc15.docking.org/substances/having/no-predictions. To download: https://files.docking.org/special/dark-matter. To browse annotated compounds similar to any compound (e.g., at least 0.30 similar to ZINC compound 14). https://zinc15.docking.org/substances/having/genes?ecfp4_fp-tanimoto-30=14 or 0.30 similar to SMILES https://zinc15.docking.org/substances/having/genes?ecfp4_fp-tanimoto-30=c1ccccc1NOCOCN. Also try https://zinc15.docking.org/substances/subsets/in-vitro?ecfp4_fp-tanimoto-30={zincorsmiles}. For similarity to natural products, try, https://zinc15.docking.org/substances/subsets/biogenic?ecfp4_fp-tanimoto-30=<zincorsmiles>. Please note: these queries are efficient if there are few matches, but will time out if too many hits are found. As a general rule, use tanimoto-50 first, which will be fast, and decrease progressively to −40 and then −30 only if no matches are found. This calculation is intensive, and we may limit usage if there are too many queries that return multiple thousands of hits to allow us to keep this service freely available.
A new research tool is now available within ZINC15 for public use. We demonstrate the use of these new tools in four use cases, which illustrate how to access predictions both interactively and via static downloads, below.
Use Case One
The user is interested in a well-studied target such as the serotonin 2A receptor (HTR2A) and seeks compounds to purchase that are likely to work but have not been reported active in ChEMBL21. The user first checks how many ligands are annotated active at 10 μM or better (5031, interactively at https://zinc15.docking.org/genes/HTR2A/substances or statically downloaded at https://files.docking.org/genes/current/HTR2A/HTR2A.smi). The user then queries how many commercially available ligands have SEA predictions at an exceptionally strong statistical significance, with pSEA = 80 (30 952 at https://zinc15.docking.org/genes/HTR2A/predictions/subsets/strong+purchasable). For instance, ZINC462039162 available from Enamine, catalog number Z1269906839, with a pSEA = 82 and MaxTc = 0.63 (https://zinc15.docking.org/substances/ZINC000462039162/predictions/table.html). Millions of other commercially available molecules can be obtained in a similar way. All predictions are downloaded immediately using https://files.docking.org/genes/current/HTR2A/HTR2A.predictions.txt.gz, from which compounds may be selected.
Use Case Two
The user wishes to obtain a screening library for projects involving several voltage-gated ions channels. The user wishes to find purchasable compounds that do not seem too similar, yet are more likely to be ligands than purely random compounds, i.e., having a high MaxTc between 0.65 and 0.70, corresponding to an expected precision of 0.35−0.40. The library should be downloaded in 2D for chemoinformatics and 3D for docking. In ZINC, there are 14 849 already annotated ligands for any such channel in ChEMBL21 at 10 μM or better (https://zinc15.docking.org/subclasses/vgic/substances). Of these, 1108 (7.5%) are purchasable and may be a good starting point for the library. A further 21 242 purchasable predicted ligands also are available, such as ZINC629100 (https://zinc15.docking.org/substances/ZINC000000629100/predictions/table.html), which is Tc 0.69 to the nearest annotated active CHEMBL1097858, active at pKi of 7.7. To obtain the first 1000 ZINC codes for these molecules, the user accesses: https://zinc15.docking.org/subclasses/vgic/predictions/subsets/purchasable.txt?maxtc-between=65+70&count=1000. To download 3D models of these compounds, please see Obtaining 3D Models, below. A second approach to download predicted compounds for voltage gated ion channels would be to first obtain the names of all the genes: https://zinc15.docking.org/subclasses/vgic/genes.txt:name. Then, the user would use this list to download the static predictions by gene. For example, for the sodium channel protein type 5 subunit alpha (SCN5A), the predictions are in https://files.docking.org/genes/SCN5A/SCN5A.predictions.txt.gz.
Use Case Three
The user would like to know all predictions for a particular vendor catalog. Vendors may be interested to know possible targets of their compounds for marketing purposes. Vendors may also wish to know which of their make-on-demand compounds might be prioritized for synthesis based on possible activity. Academic centers that screen vendor libraries may be interested in individual vendors because they have negotiated special pricing, or because the vendor makes plates available at a discount to facilitate the mechanics of screening. We have been precomputed searches to enable such investigations to save time. To access them, the user would complete the following steps:
-
1.
Browse to https://files.docking.org/catalogs to select the catalog of interest.
-
2.
Download the file of predictions. For instance, for ChemBridge, the code is chbr and the URL is https://files.docking.org/catalogs/50/chbr/chbr.predict.txt.gz. Each row contains the vendor code, ZINC ID, InChIKey, predicted gene, MaxTc, and pSEA: one molecule per row.
-
3.
Break the downloaded files into subsets using Unix command-line tools to filter by MaxTc, pSEA, and predicted gene.
To download these in 3D for docking, please see Obtaining 3D Models, below.
Use Case Four
The user wishes to download dark chemical matter screening libraries in 2D or 3D formats. To do so, the user browses to https://files.docking.org/dark-matter. The compounds have been binned into tranches by physical property using our standard scheme (http://wiki.docking.org/index.php/Physical_property_space). The 2D files are available as compressed text files organized by purchasability. Each row contains one molecule with its SMILES, ZINC ID, physical property tranche, purchasability, and reactivity. The 3D files will likewise be prepared in future but are meanwhile available as described in Obtaining 3D Models, below.
Obtaining 3D Models
To download 3D models for a set of molecules in bulk for one of the above use cases, here is a general approach that will work for any arbitrary set of ZINC IDs:
-
1.
Obtain the codes of the molecules to download using the previous use cases or otherwise and store the codes in zinc-codes.txt.
-
2.
Select mol2, db, or db2 file formats. mol2 may be converted to other formats as required. The latter two are used by the UCSF DOCK 3.x programs only.
-
3.
Download the script getfiles.csh from https://files.docking.org/catalogs/getfiles.csh.
-
4.
Edit the file by hand following the instructions within.
-
5.
Run the script, with the list of ZINC codes in the same directory. The 3D files will be downloaded.
Please note that 3D models are currently available for about 120 million of the 400 million compounds in ZINC. We are continually building and rebuilding them, prioritizing the popular lead-like and fragment-like areas best suited to docking. If a 3D model is not available, the molecule detail page contains a “Request Generation” button in the 3D representations section. If a 3D model does not exist, it is either because it fails to build or because it is still on our action list.
Discussion
Four major results emerge in this work. First, using ZINC and ChEMBL, we predict molecular target activities for 171 million commercially available compounds at 2629 targets and store them in an accessible database. Second, we create an interface to search, access, and download the predictions (https://zinc15.docking.org and https://files.docking.org). Predictions can be accessed individually or downloaded in bulk, and are available in a range of formats ready for both docking and chemoinformatics, or for purchase. To demonstrate the utility of these predictions, we perform a retrospective 5-fold cross-validation of the ChEMBL bioactivity data set. Further, we identify likely targets of drugs known in the literature where direct binding annotations are not available in ChEMBL. Finally, this new tool allows us to quantify predicted target biases of purchasable chemical space. Target bias predicted by this model is substantial—some genes are represented by millions of purchasable compounds, others have very few. Nearly 60% of purchasable compounds in ZINC have no prediction at all, allowing us to offer purchasable “dark chemical matter”. We take up each of these results in turn.
We predict targets for over 40% of the 400 million compounds currently for sale in ZINC. The number is admittedly arbitrary, as we were obliged to choose pSEA and Tanimoto similarity cutoffs. Knowing that this approach would produce false positives and false negatives, we attempted to strike a useful balance, and equip the user to apply further constraints. Many compounds with MaxTc as low as 0.40 to the nearest active may not bind the predicted target−previous work suggests 18% precision might be a good estimate44 and this is consistent with the results we found in Figure 1 (blue circle). Likewise, those with a pSEA near our chosen threshold of pSEA = 40 may not be active against the predicted target. Should such chemically novel predictions be confirmed experimentally, they may represent new starting points for optimization and could lead to new biology. If the user wishes higher confidence hits, more stringent cutoffs in pSEA or MaxTc are easily applied. We refer the reader to the set of thresholds examined in our cross-validation of the ChEMBL bioactivity data set (Supporting Information Figure S1) for guidance in choosing pSEA and MaxTc values to optimize the desired output. For the highest rates of precision at an acceptable recall, we recommend threshold values at pSEA ≥ 80 and MaxTc ≥ 80 (Figure 1, pink circle), noting this may reduce the number of novel compound−target associations that pass the cutoff.
For those wishing to buy a compound that works, the user might only consider the most similar compounds, having high Tc to a precedented bioactive. For those seeking chemical novelty against a target, where testing 10 or even 50 more novel compounds to find new chemical matter is acceptable, more novel compounds may be sought. Users of virtual screening methods such as docking may want particularly novel (low MaxTc) compounds, because their screening method makes an independent assessment of each prediction. Some will prefer to pursue the most novel—and potentially most interesting—the purchasable chemical dark matter, those compounds that do not seem similar to any of the annotated compounds used to make these predictions. Whatever the appetite for risk, investigators are empowered by these tools to select predictions that are right for their project.
Interfacing the prediction database through ZINC allows predictions to be searched, grouped, filtered, compared, and downloaded using the extensive ZINC machinery. Thus 3D models of predicted compounds may be accessed for molecular docking screens, while SMILES strings or molecular properties may be downloaded for ligand-based methods. Predicted compounds for any of 2629 genes may be accessed and downloaded in any of eight formats. Results may be filtered by prediction statistics (pSEA, MaxTc), molecular properties (e.g., molecular weight, calculated logP, polar surface area, fraction sp3) and purchasability (in stock, make-on-demand, or by vendor). Both 2D and 3D results can be organized by gene (e.g., ADRB2, SRC), minor class (e.g., GPCR Class B, voltage-gated ion channel), major class (e.g., transcription factor or membrane protein), Kingdom (bacterial, eukaryotic, viral), vendor, and physical property tranche. Attributes of predictions may be downloaded in tabular form for analysis. A REST API, exemplified in this work, described previously9 and documented online,60 allows automated queries and machine-readable results, so that this database may be incorporated into third-party software applications.
We examined drugs and investigational compounds without an established molecular target annotation in ChEMBL to assess the relevance of the predictions. The 50 we highlighted exemplify typical results that can be expected using our approach for the millions of molecules that have never been assayed (Table 1). Whereas an exhaustive analysis is impractical, this result supports the view that our predictions are often consistent with experimentally observed binding.
A global picture of target bias in commercially available libraries emerges. Of the 535 million compound−target predictions, over 500 000 predictions on 400 000 compounds have a MaxTc better than 0.60 (ECFP4) to a ligand annotated for that target; a level of similarity that suggests 35% precision.44 A further 1.6 million predictions on 1.4 million compounds with MaxTc between 50 and 59 are also strong candidates for experimental testing. Many of these two million compounds could have been predicted by pairwise Tanimoto similarity alone, without the help of SEA. The pSEA adds most value below MaxTc 0.50, where it provides a global similarity measure to the set of annotated ligands as a group instead of a single pairwise one. This becomes even more acute below MaxTc of 0.40, where we only retain predictions with pSEA ≥ 40 as the Tanimoto coefficient alone becomes too untrustworthy, with precision falling rapidly below 10%.
Our analysis provides additional resources. We have predicted compounds for 69 targets61 for which none of the 20 or more actives is commercially available (Table 2). If confirmed experimentally, these genes could now be represented in screening panels of commercially available compounds, and these new ligands used as controls or perhaps even starting points for design. For each of 2629 genes, a range of commercially available compounds from high-confidence, having high MaxTc, to more-novel-yet-intriguing at lower MaxTc are now available. For the most studied targets, there is a deep bench of predictions running into the millions of compounds each. Massive biases for some targets, such as the dopamine D2 (DRD2) and beta-2 adrenergic (ADRB2) receptors for instance, echoes our earlier work62 that commercial libraries are heavily biased toward long-studied, important biological targets. Correspondingly, less-well-studied targets with few ligands often have sparse representation in commercial libraries, which can occur when the known actives are natural products or their derivatives. We have also assembled a database of “dark chemical matter”, 229 million purchasable compounds that received no target prediction and that generally do not resemble known bioactives, which is available from our website in 2D and 3D formats. If these compounds were active in a screen, they would likely represent new starting points for optimization.
Our approach has other liabilities. Our cutoffs in MaxTc and pSEA inevitably exclude sensible predictions. Some classes of compounds such as sterols, peptides, and nucleotides suffer from higher mis-prediction rates, a subject of continuing research. pKa and explicit charge are poorly treated in our current protocol based on stereochemistry-naïve ECFP4 fingerprints, making amide nitrogens and basic amines too much alike, for instance, leading to some obviously wrong predictions. Massive turnover in the chemical marketplace means stored predictions may lag the appearance of new compounds in ZINC. ChEMBL contains artifacts and errors, which this approach can magnify. The SEA and MaxTc approaches quantify whole-molecule similarities and are thereby naïve of critical chemical moieties (often called warheads).
Notwithstanding these limitations, our database of predicted biological activities for purchasable chemical space is a pragmatic tool that should be useful to a broad audience. It affords both a retail view—buy this compound for this target—as well as a wholesale one—this target is well represented, and here are some compounds for it. Our predictions can be rapidly tested because the compounds are purchasable. We intend to continue to update the database as purchasable chemical space evolves and ChEMBL is enhanced. This database is provided in the hope that it will be useful, but you must use it at your own risk.
Methods
Library Preparation
We used CHEMBL21 compounds annotated for targets better than 10 μM and grouped by Uniprot gene symbol across eukaryotes, as previously described in ZINC15.9 Thus in this scheme, DRD2_HUMAN, DRD2_RAT, and DRD2_MOUSE are all grouped into a single gene annotation DRD2, and predictions are made against the unified collection for the gene and not the individual orthologs. In situations where the target is composed of several gene products, as in some ion channels for instance, we used the ChEMBL name. When no gene has been formally assigned by Uniprot, we use the Uniprot accession code itself as the gene name, as in ZINC15.
SEA Reference Library Construction
We grouped ligands by affinity. We computed an affinity bin as the negative log of the molar affinity, which is variously expressed as Ki, IC50, and EC50 among others in ChEMBL21 and which we refer to as pKi in this work for simplicity. Thus in this scheme, bin 6 contains all compounds with 1 μM affinity or better. Lower affinity bins were inclusive of compounds from all higher affinity bins. We built three SEA libraries as follows. In the first library, we only proceed if there are at least five distinct compounds active against a single gene, we only accept activities of 1 μM or better. We found 1382 such genes, which we defined as being well described by their ligands. In the second library, we only predict for those single gene targets that did not qualify for the first pass, accepting activities as weak as 10 μM, and as few as one good ligand. We found 1347 of these less-well-described genes. The third library was an attempt to overcome a statistical weakness, which diluted the signal of genes having many diverse ligands. We clustered ligands to describe individual chemotypes of 302 genes having 300 ligands or more each. For each library we computed a statistical background for SEA based on the 410 624 annotated compounds. We computed the pSEA based on an extreme value distribution and the maximum Tanimoto similarity of the prediction to the annotated compounds (MaxTc). Throughout we suppressed from the libraries compounds with PAINS patterns or similarity to a precedented aggregator by 0.70 (ECFP4) having an affinity worse than 100 nM.48 This was likely too conservative, but earlier, more permissive attempts at this library often suffered from excessive erroneous predictions, likely owing to these fraught compounds.
Database Loading
Predictions were loaded into ZINC. To minimize ligands whose charge differed sharply from precedent, we computed the mean and the standard deviation of the average microspecies charge using ChemAxon’s CXCALC program for each gene. When loading each prediction, if the charge of a 3D representation at pH 7.4 (reference model) was available, we suppressed loading if the charge on the molecule fell outside 1.5 standard deviations from the mean charge for ligands annotated to that gene. This remains an area of ongoing research. The result was to suppress predictions that we likely would have thrown out on inspection, in a scalable if incomplete and imperfect way.
ChEMBL Cross-Validation
We evaluated the predictive performance of SEA+TC using ChEMBL’s bioactivity data set (version 21). Receiver operating characteristic curves were generated from independent 5-fold cross-validation runs for each method examined (SEA, SEA+TC, NBC). For SEA and NBC cross-validation sets, each point on the curve represents the average true-positive rate (TPR) and false-positive rate (FPR) from all 5 folds. TPRs and FPRs along the curve were determined by stepping a decision threshold across the range of possible SEA p-values (0.0−1.0), for all predicted compound-target interactions. To examine the sensitivity of these results to how well the target is described by ligands, we ran the analysis using targets with a minimum of 5 ligands and also with 50 ligands.
For SEA+TC cross-validation sets, TPRs and FPRs along the curve were determined by two separate decision thresholds; one for the SEA p-value and another for the maximum Tanimoto coefficient (MaxTc). As ROC curves evaluate a binary classifier using a single discrimination threshold, assessing performance by simultaneously stepping across both metrics was not ideal. To account for this, we generated ROC curves by stepping across all possible values of MaxTc, while holding the pSEA decision threshold constant (Figure 1). Predicted compound−target associations are therefore positive if their pSEA or MaxTc passes either of the respective cutoffs. A consequence of this bivariate thresholding is that the static pSEA threshold prevents the TPR and FPR from ever reaching zero. To highlight this, the distance between a fully stratified classifier (TPR = 0; FPR = 0) and the minimum point at which both decision thresholds begin to affect performance is shown in dashed lines (Figure 1). Performance metrics for a range of pSEA decision thresholds are shown in Supporting Information Figure S1A and B. Complementary curves stepping across pSEA while holding a separate MaxTc decision threshold constant are shown in Supporting Information Figure S1C and D.
Interface
We added support for SEA predictions to the user interface on the Molecule Detail, Target Detail and Gene Detail pages of ZINC. The interface classifies each gene by one of 15 major target classes (e.g., membrane receptor, ion channel, transporter) and by one of 42 subclasses (e.g., Class A GPCR, voltage gated ion channel, etc) whose pages also allow access to the SEA predictions. The results are downloadable in eight formats: SMILES, mol2, SDF, pdbqt, json, xml, txt, and xls. The predictions may be accessed visually via a web browser or programmatically using an application program interface, both located at https://zinc15.docking.org/predictions/home. Static files are accessible via https://files.docking.org/predictions, https://files.docking.org/genes, https://files.docking.org/catalogs, and https://files.docking.org/dark-matter.
Caveats
Vendors often advertise stereochemically ambiguous molecular descriptions and thus the number of compounds and predictions strongly depends on how these are treated. Since ZINC is a 3D focused database, we are obliged to commit to a 3D representation. Where there is ambiguity, we enumerate up to a maximum of four possible stereoisomers (R/S and E/Z) and readily admit that this inflates the numbers in this work.
Acknowledgments
This work was supported by GM71896 (to B. K. Shoichet and J.J.I.), GM093456 (to M.J.K.), and the Paul G. Allen Family Foundation (to M.J.K.). We thank Greg Landrum, Novartis, and the RDKit community for RDKit, ChemAxon (chemaxon.com) for licenses for JChem and Marvin, and OpenEye Scientific Software (eyesopen.com) for software licenses for OEChem. We thank SeaChange Pharmaceuticals (seachangepharma.com) for improvements to the SEAware software. We thank Dr. Matthew O’Meara for reading the manuscript. We are grateful the anonymous reviewers who made helpful suggestions that substantially improved the manuscript. While ZINC itself remains noncommercial, the contribution of a single company, Enamine Ltd (Kyiv Ukraine, http://enamine.net), offering over 80% of the compounds currently offered for sale, is acknowledged.
Glossary
Abbreviations
- ChEMBL
EBI medicinal chemistry database
- FPR
false positive rate
- GPCR
G protein-coupled receptor
- NBC
Naïve-Bayesian classifier
- PR
precision-recall
- PRC
PR curves
- AUPRC
area under the PR curve
- ROC
receiver operating characteristic
- AUROC
area under the ROC curve
- SEA
Similarity Ensemble Approach
- pSEA
negative log SEA p-value
- SEA+TC
combinatorial approach combining SEA and Tanimoto similarity as described in the text
- Tc
Tanimoto coefficient
- MaxTc
Maximum Tc
- TPR
true-positive rate
- ZINC
the ZINC Is Not Commercial chemistry database.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.7b00316.
Three figures as follows: (S1) Performance metrics for a range of pSEA decision thresholds. (S2) Complementary curves stepping across pSEA while holding a separate MaxTc decision threshold constant. (S3) Scatterplot of predicted ligands per gene vs annotated ligands per gene, both axes on a log scale (PDF)
Author Contributions
⊥ J.J.I. and G.G. contributed equally to this work
The authors declare no competing financial interest.
Supplementary Material
References
- Levchenko K.; Datsenko O. P.; Serhiichuk O.; Tolmachev A.; Iaroshenko V. O.; Mykhailiuk P. K. Copper-Catalyzed O-Difluoromethylation of Functionalized Aliphatic Alcohols: Access to Complex Organic Molecules with an Ocf2h Group. J. Org. Chem. 2016, 81, 5803–5813. 10.1021/acs.joc.6b00628. [DOI] [PubMed] [Google Scholar]
- Tolmachev A.; Bogolubsky A. V.; Pipko S. E.; Grishchenko A. V.; Ushakov D. V.; Zhemera A. V.; Viniychuk O. O.; Konovets A. I.; Zaporozhets O. A.; Mykhailiuk P. K.; Moroz Y. S. Expanding Synthesizable Space of Disubstituted 1,2,4-Oxadiazoles. ACS Comb. Sci. 2016, 18, 616–624. 10.1021/acscombsci.6b00103. [DOI] [PubMed] [Google Scholar]
- Bogolubsky A. V.; Moroz Y. S.; Mykhailiuk P. K.; Ostapchuk E. N.; Rudnichenko A. V.; Dmytriv Y. V.; Bondar A. N.; Zaporozhets O. A.; Pipko S. E.; Doroschuk R. A.; Babichenko L. N.; Konovets A. I.; Tolmachev A. One-Pot Parallel Synthesis of Alkyl Sulfides, Sulfoxides, and Sulfones. ACS Comb. Sci. 2015, 17, 348–354. 10.1021/acscombsci.5b00024. [DOI] [PubMed] [Google Scholar]
- Bogolubsky A. V.; Moroz Y. S.; Mykhailiuk P. K.; Pipko S. E.; Zhemera A. V.; Konovets A. I.; Stepaniuk O. O.; Myronchuk I. S.; Dmytriv Y. V.; Doroschuk R. A.; Zaporozhets O. A.; Tolmachev A. 2,2,2-Trifluoroethyl Chlorooxoacetate−Universal Reagent for One-Pot Parallel Synthesis of N(1)-Aryl-N(2)-Alkyl-Substituted Oxamides. ACS Comb. Sci. 2015, 17, 615–622. 10.1021/acscombsci.5b00091. [DOI] [PubMed] [Google Scholar]
- Druzhenko T.; Denisenko O.; Kheylik Y.; Zozulya S.; Shishkina S. S.; Tolmachev A.; Mykhailiuk P. K. Design, Synthesis, and Characterization of So2-Containing Azabicyclo[3.N.1]Alkanes: Promising Building Blocks for Drug Discovery. Org. Lett. 2015, 17, 1922–1925. 10.1021/acs.orglett.5b00608. [DOI] [PubMed] [Google Scholar]
- Bogolubsky A. V.; Moroz Y. S.; Mykhailiuk P. K.; Granat D. S.; Pipko S. E.; Konovets A. I.; Doroschuk R.; Tolmachev A. Bis(2,2,2-Trifluoroethyl) Carbonate as a Condensing Agent in One-Pot Parallel Synthesis of Unsymmetrical Aliphatic Ureas. ACS Comb. Sci. 2014, 16, 303–308. 10.1021/co500025f. [DOI] [PubMed] [Google Scholar]
- Bogolubsky A. V.; Moroz Y. S.; Mykhailiuk P. K.; Panov D. M.; Pipko S. E.; Konovets A. I.; Tolmachev A. A One-Pot Parallel Reductive Amination of Aldehydes with Heteroaromatic Amines. ACS Comb. Sci. 2014, 16, 375–380. 10.1021/co5000568. [DOI] [PubMed] [Google Scholar]
- Bogolubsky A. V.; Moroz Y. S.; Mykhailiuk P. K.; Pipko S. E.; Konovets A. I.; Sadkova I. V.; Tolmachev A. Sulfonyl Fluorides as Alternative to Sulfonyl Chlorides in Parallel Synthesis of Aliphatic Sulfonamides. ACS Comb. Sci. 2014, 16, 192–197. 10.1021/co400164z. [DOI] [PubMed] [Google Scholar]
- Sterling T.; Irwin J. J. Zinc 15-Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teague S. J.; Davis A. M.; Leeson P. D.; Oprea T. I. The Design of Leadlike Combinatorial Libraries. Angew. Chem., Int. Ed. 1999, 38, 3743–3748. . [DOI] [PubMed] [Google Scholar]
- Carr R. A.; Congreve M.; Murray C. W.; Rees D. C. Fragment-Based Lead Discovery: Leads by Design. Drug Discovery Today 2005, 10, 987–992. 10.1016/S1359-6446(05)03511-7. [DOI] [PubMed] [Google Scholar]
- Bento A. P.; Gaulton A.; Hersey A.; Bellis L. J.; Chambers J.; Davies M.; Kruger F. A.; Light Y.; Mak L.; McGlinchey S.; Nowotka M.; Papadatos G.; Santos R.; Overington J. P. The Chembl Bioactivity Database: An Update. Nucleic Acids Res. 2014, 42, D1083–1090. 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZINC Annotated Catalogs . https://zinc15.docking.org/catalogs/subsets/annotated (accessed April 10, 2017).
- Gillet V. J. New Directions in Library Design and Analysis. Curr. Opin. Chem. Biol. 2008, 12, 372–378. 10.1016/j.cbpa.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Bender A.; Glen R. C. Molecular Similarity: A Key Technique in Molecular Informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. 10.1039/b409813g. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Nicholls A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- Steindl T. M.; Schuster D.; Wolber G.; Laggner C.; Langer T. High-Throughput Structure-Based Pharmacophore Modelling as a Basis for Successful Parallel Virtual Screening. J. Comput.-Aided Mol. Des. 2007, 20, 703–715. 10.1007/s10822-006-9066-y. [DOI] [PubMed] [Google Scholar]
- Button A. L.; Hiss J. A.; Schneider P.; Schneider G. Scoring of De Novo Designed Chemical Entities by Macromolecular Target Prediction. Mol. Inf. 2017, 36, 1600110. 10.1002/minf.201600110. [DOI] [PubMed] [Google Scholar]
- Schneider G.; Schneider P. Macromolecular Target Prediction by Self-Organizing Feature Maps. Expert Opin. Drug Discovery 2017, 12, 271. 10.1080/17460441.2017.1274727. [DOI] [PubMed] [Google Scholar]
- Fu G.; Nan X.; Liu H.; Patel R. Y.; Daga P. R.; Chen Y.; Wilkins D. E.; Doerksen R. J. Implementation of Multiple-Instance Learning in Drug Activity Prediction. BMC Bioinformatics.. BMC Bioinf. 2012, 13 (Suppl 15), S3. 10.1186/1471-2105-13-S15-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azencott C. A.; Ksikes A.; Swamidass S. J.; Chen J. H.; Ralaivola L.; Baldi P. One- to Four-Dimensional Kernels for Virtual Screening and the Prediction of Physical, Chemical, and Biological Properties. J. Chem. Inf. Model. 2007, 47, 965–974. 10.1021/ci600397p. [DOI] [PubMed] [Google Scholar]
- Cereto-Massague A.; Ojeda M. J.; Valls C.; Mulero M.; Pujadas G.; Garcia-Vallve S. Tools for in Silico Target Fishing. Methods 2015, 71, 98–103. 10.1016/j.ymeth.2014.09.006. [DOI] [PubMed] [Google Scholar]
- Basak S. C. Mathematical Descriptors for the Prediction of Property, Bioactivity, and Toxicity of Chemicals from Their Structure: A Chemical-Cum-Biochemical Approach. Curr. Comput.-Aided Drug Des. 2013, 9, 449–462. 10.2174/15734099113096660041. [DOI] [PubMed] [Google Scholar]
- Yu P.; Wild D. J. Fast Rule-Based Bioactivity Prediction Using Associative Classification Mining. J. Cheminf. 2012, 4, 29. 10.1186/1758-2946-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugaya N. Training Based on Ligand Efficiency Improves Prediction of Bioactivities of Ligands and Drug Target Proteins in a Machine Learning Approach. J. Chem. Inf. Model. 2013, 53, 2525–2537. 10.1021/ci400240u. [DOI] [PubMed] [Google Scholar]
- Murrell D. S.; Cortes-Ciriano I.; van Westen G. J.; Stott I. P.; Bender A.; Malliavin T. E.; Glen R. C. Chemically Aware Model Builder (Camb): An R Package for Property and Bioactivity Modelling of Small Molecules. J. Cheminf. 2015, 7, 45. 10.1186/s13321-015-0086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal A.; Ahn Y. Y.; Wild D. J. Optimizing Drug-Target Interaction Prediction Based on Random Walk on Heterogeneous Networks. J. Cheminf. 2015, 7, 40. 10.1186/s13321-015-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskar M.; Zeller G.; Zhao X. M.; van Noort V.; Bork P. Drug Discovery in the Age of Systems Biology: The Rise of Computational Approaches for Data Integration. Curr. Opin. Biotechnol. 2012, 23, 609–616. 10.1016/j.copbio.2011.11.010. [DOI] [PubMed] [Google Scholar]
- Peragovics A.; Simon Z.; Tombor L.; Jelinek B.; Hari P.; Czobor P.; Malnasi-Csizmadia A. Virtual Affinity Fingerprints for Target Fishing: A New Application of Drug Profile Matching. J. Chem. Inf. Model. 2013, 53, 103–113. 10.1021/ci3004489. [DOI] [PubMed] [Google Scholar]
- Chen X.; Yan C. C.; Zhang X.; Zhang X.; Dai F.; Yin J.; Zhang Y. Drug-Target Interaction Prediction: Databases, Web Servers and Computational Models. Briefings Bioinf. 2016, 17, 696–712. 10.1093/bib/bbv066. [DOI] [PubMed] [Google Scholar]
- Wang L.; Ma C.; Wipf P.; Liu H.; Su W.; Xie X. Q. Targethunter: An in Silico Target Identification Tool for Predicting Therapeutic Potential of Small Organic Molecules Based on Chemogenomic Database. AAPS J. 2013, 15, 395–406. 10.1208/s12248-012-9449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Liang Y.; Xu J. Toward Automated Biochemotype Annotation for Large Compound Libraries. Mol. Diversity 2006, 10, 495–509. 10.1007/s11030-006-9047-z. [DOI] [PubMed] [Google Scholar]
- Nguyen H. P.; Koutsoukas A.; Mohd Fauzi F.; Drakakis G.; Maciejewski M.; Glen R. C.; Bender A. Diversity Selection of Compounds Based on ’Protein Affinity Fingerprints’ Improves Sampling of Bioactive Chemical Space. Chem. Biol. Drug Des. 2013, 82, 252–266. 10.1111/cbdd.12155. [DOI] [PubMed] [Google Scholar]
- Huang T.; Mi H.; Lin C. Y.; Zhao L.; Zhong L. L.; Liu F. B.; Zhang G.; Lu A. P.; Bian Z. X. Most: Most-Similar Ligand Based Approach to Target Prediction. BMC Bioinf. 2017, 18, 165. 10.1186/s12859-017-1586-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann S. L.; Brown S. P.; Muchmore S. W.; Patel H.; Merta P.; Locklear J.; Hajduk P. J. A Unified, Probabilistic Framework for Structure- and Ligand-Based Virtual Screening. J. Med. Chem. 2011, 54, 1223–1232. 10.1021/jm1013677. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Dornhofer A. A.; Langer T. Efficient Overlay of Small Organic Molecules Using 3d Pharmacophores. J. Comput.-Aided Mol. Des. 2007, 20, 773–788. 10.1007/s10822-006-9078-7. [DOI] [PubMed] [Google Scholar]
- Keiser M. J.; Roth B. L.; Armbruster B. N.; Ernsberger P.; Irwin J. J.; Shoichet B. K. Relating Protein Pharmacology by Ligand Chemistry. Nat. Biotechnol. 2007, 25, 197–206. 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- Keiser M. J.; Setola V.; Irwin J. J.; Laggner C.; Abbas A. I.; Hufeisen S. J.; Jensen N. H.; Kuijer M. B.; Matos R. C.; Tran T. B.; Whaley R.; Glennon R. A.; Hert J.; Thomas K. L.; Edwards D. D.; Shoichet B. K.; Roth B. L. Predicting New Molecular Targets for Known Drugs. Nature 2009, 462, 175–181. 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGraw A. J.; Keiser M. J.; Ochocki J. D.; Shoichet B. K.; Distefano M. D. Prediction and Evaluation of Protein Farnesyltransferase Inhibition by Commercial Drugs. J. Med. Chem. 2010, 53, 2464–2471. 10.1021/jm901613f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lounkine E.; Keiser M. J.; Whitebread S.; Mikhailov D.; Hamon J.; Jenkins J. L.; Lavan P.; Weber E.; Doak A. K.; Cote S.; Shoichet B. K.; Urban L. Large-Scale Prediction and Testing of Drug Activity on Side-Effect Targets. Nature 2012, 486, 361–367. 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggner C.; Kokel D.; Setola V.; Tolia A.; Lin H.; Irwin J. J.; Keiser M. J.; Cheung C. Y.; Minor D. L. Jr.; Roth B. L.; Peterson R. T.; Shoichet B. K. Chemical Informatics and Target Identification in a Zebrafish Phenotypic Screen. Nat. Chem. Biol. 2011, 8, 144–146. 10.1038/nchembio.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux G. A.; Keiser M. J.; Sassano M. F.; Laggner C.; Mayer F.; Bainton R. J.; Werb Z.; Roth B. L.; Shoichet B. K.; Ashrafi K. In Silico Molecular Comparisons of C. Elegans and Mammalian Pharmacology Identify Distinct Targets That Regulate Feeding. PLoS Biol. 2013, 11, e1001712. 10.1371/journal.pbio.1001712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto T. T.IBM Internal Report; 1957.
- Muchmore S. W.; Debe D. A.; Metz J. T.; Brown S. P.; Martin Y. C.; Hajduk P. J. Application of Belief Theory to Similarity Data Fusion for Use in Analog Searching and Lead Hopping. J. Chem. Inf. Model. 2008, 48, 941–948. 10.1021/ci7004498. [DOI] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Koutsoukas A.; Lowe R.; Kalantarmotamedi Y.; Mussa H. Y.; Klaffke W.; Mitchell J. B.; Glen R. C.; Bender A. In Silico Target Predictions: Defining a Benchmarking Data Set and Comparison of Performance of the Multiclass Naive Bayes and Parzen-Rosenblatt Window. J. Chem. Inf. Model. 2013, 53, 1957–1966. 10.1021/ci300435j. [DOI] [PubMed] [Google Scholar]
- Gregori-Puigjane E.; Setola V.; Hert J.; Crews B. A.; Irwin J. J.; Lounkine E.; Marnett L.; Roth B. L.; Shoichet B. K. Identifying Mechanism-of-Action Targets for Drugs and Probes. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11178–11183. 10.1073/pnas.1204524109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Duan D.; Torosyan H.; Doak A. K.; Ziebart K. T.; Sterling T.; Tumanian G.; Shoichet B. K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 7076–7087. 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59, 4103–4120. 10.1021/acs.jmedchem.5b02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich C.; Bertozzi C.; Georg G. I.; Kiessling L.; Lindsley C.; Liotta D.; Merz K. M. Jr.; Schepartz A.; Wang S. The Ecstasy and Agony of Assay Interference Compounds. J. Med. Chem. 2017, 60, 2165–2168. 10.1021/acs.jmedchem.7b00229. [DOI] [PubMed] [Google Scholar]
- ZINC Genes Having Ligands of 10um or Better, after the Treatment for Pains and Aggregator Analogs Described in the Methods. https://zinc15.docking.org/genes/?num_substances-gt=0&num_predictions-gt=0 (accessed May 27, 2017).
- Inui J.; Imamura H. Beta-Adrenoceptor Blocking and Electrophysiological Effects of Bufetolol in the Guinea Pig Atria. Eur. J. Pharmacol. 1977, 41, 251–260. 10.1016/0014-2999(77)90318-1. [DOI] [PubMed] [Google Scholar]
- Masumiya H.; Tanaka Y.; Tanaka H.; Shigenobu K. Inhibition of T-Type and L-Type Ca(2+) Currents by Aranidipine, a Novel Dihydropyridine Ca(2+) Antagonist. Pharmacology 2000, 61, 57–61. 10.1159/000028381. [DOI] [PubMed] [Google Scholar]
- Nelson K. M.; Dahlin J. L.; Bisson J.; Graham J.; Pauli G. F.; Walters M. A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. 10.1021/acs.jmedchem.6b00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saario S. M.; Poso A.; Juvonen R. O.; Jarvinen T.; Salo-Ahen O. M. Fatty Acid Amide Hydrolase Inhibitors from Virtual Screening of the Endocannabinoid System. J. Med. Chem. 2006, 49, 4650–4656. 10.1021/jm060394q. [DOI] [PubMed] [Google Scholar]
- Bray N. Lead Identification: Shedding Light on Dark Chemical Matter. Nat. Rev. Drug Discovery 2015, 14, 817. 10.1038/nrd4787. [DOI] [PubMed] [Google Scholar]
- Macarron R. Chemical Libraries: How Dark Is Hts Dark Matter?. Nat. Chem. Biol. 2015, 11, 904–905. 10.1038/nchembio.1937. [DOI] [PubMed] [Google Scholar]
- Muegge I.; Mukherjee P. Performance of Dark Chemical Matter in High Throughput Screening. J. Med. Chem. 2016, 59, 9806–9813. 10.1021/acs.jmedchem.6b01038. [DOI] [PubMed] [Google Scholar]
- Wassermann A. M.; Lounkine E.; Hoepfner D.; Le Goff G.; King F. J.; Studer C.; Peltier J. M.; Grippo M. L.; Prindle V.; Tao J.; Schuffenhauer A.; Wallace I. M.; Chen S.; Krastel P.; Cobos-Correa A.; Parker C. N.; Davies J. W.; Glick M. Dark Chemical Matter as a Promising Starting Point for Drug Lead Discovery. Nat. Chem. Biol. 2015, 11, 958–966. 10.1038/nchembio.1936. [DOI] [PubMed] [Google Scholar]
- ZINC Zinc15 Resources Wiki Page . http://wiki.docking.org/index.php/ZINC15:Resources (accessed Oct 12, 2015).
- ZINC Genes Having 20 or More Ligands Where None Is for Sale, and Predictions. https://zinc15.docking.org/genes/?num_purchasable=0&num_predictions-gt=0&num_substances-gt=20 (acccessed May 27, 2017).
- Hert J.; Irwin J. J.; Laggner C.; Keiser M. J.; Shoichet B. K. Quantifying Biogenic Bias in Screening Libraries. Nat. Chem. Biol. 2009, 5, 479–483. 10.1038/nchembio.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Pina A. E.; McKnight W.; Dicay M.; Castaneda-Hernandez G.; Wallace J. L. Mechanisms Underlying the Anti-Inflammatory Activity and Gastric Safety of Acemetacin. Br. J. Pharmacol. 2007, 152, 930–938. 10.1038/sj.bjp.0707451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mross K.; Richly H.; Schleucher N.; Korfee S.; Tewes M.; Scheulen M. E.; Seeber S.; Beinert T.; Schweigert M.; Sauer U.; Unger C.; Behringer D.; Brendel E.; Haase C. G.; Voliotis D.; Strumberg D. A Phase I Clinical and Pharmacokinetic Study of the Camptothecin Glycoconjugate, Bay 38−3441, as a Daily Infusion in Patients with Advanced Solid Tumors. Ann. Oncol. 2004, 15, 1284–1294. 10.1093/annonc/mdh313. [DOI] [PubMed] [Google Scholar]
- Hofmann T. G.; Hehner S. P.; Bacher S.; Droge W.; Schmitz M. L. Various Glucocorticoids Differ in Their Ability to Induce Gene Expression, Apoptosis and to Repress Nf-Kappab-Dependent Transcription. FEBS Lett. 1998, 441, 441–446. 10.1016/S0014-5793(98)01609-3. [DOI] [PubMed] [Google Scholar]
- Raguenes-Nicol C.; Russo-Marie F.; Domage G.; Diab N.; Solito E.; Dray F.; Mace J. L.; Streichenberger G. Anti-Inflammatory Mechanism of Alminoprofen: Action on the Phospholipid Metabolism Pathway. Biochem. Pharmacol. 1999, 57, 433–443. 10.1016/S0006-2952(98)00312-8. [DOI] [PubMed] [Google Scholar]
- Schoemaker H.; Claustre Y.; Fage D.; Rouquier L.; Chergui K.; Curet O.; Oblin A.; Gonon F.; Carter C.; Benavides J.; Scatton B. Neurochemical Characteristics of Amisulpride, an Atypical Dopamine D2/D3 Receptor Antagonist with Both Presynaptic and Limbic Selectivity. J. Pharmacol Exp Ther. 1997, 280, 83–97. [PubMed] [Google Scholar]
- McLean R. C.; Baird S. W.; Becker L. C.; Townsend S. N.; Gerstenblith G.; Kass D. A.; Tomaselli G. F.; Schulman S. P. Response to Catecholamine Stimulation of Polymorphisms of the Beta-1 and Beta-2 Adrenergic Receptors. Am. J. Cardiol. 2012, 110, 1001–1007. 10.1016/j.amjcard.2012.05.029. [DOI] [PubMed] [Google Scholar]
- Tsukagoshi S. Pharmacokinetics of Azasetron (Serotone), a Selective 5-Ht3 Receptor Antagonist. Gan To Kagaku Ryoho. 1999, 26, 1001–1008. [PubMed] [Google Scholar]
- Oizumi K.; Nishino H.; Koike H.; Sada T.; Miyamoto M.; Kimura T. Antihypertensive Effects of Cs-905, a Novel Dihydropyridine Ca++ Channel Blocker. Jpn. J. Pharmacol. 1989, 51, 57–64. 10.1254/jjp.51.57. [DOI] [PubMed] [Google Scholar]
- Yamamoto M.; Shimizu M. Effects of a New Trh Analogue, Ym-14673 on the Central Nervous System. Naunyn-Schmiedeberg's Arch. Pharmacol. 1987, 336, 561–565. 10.1007/BF00169314. [DOI] [PubMed] [Google Scholar]
- Cambau E.; Matrat S.; Pan X. S.; Roth Dit Bettoni R.; Corbel C.; Aubry A.; Lascols C.; Driot J. Y.; Fisher L. M. Target Specificity of the New Fluoroquinolone Besifloxacin in Streptococcus Pneumoniae, Staphylococcus Aureus and Escherichia Coli. J. Antimicrob. Chemother. 2009, 63, 443–450. 10.1093/jac/dkn528. [DOI] [PubMed] [Google Scholar]
- Frishman W. H.; Goldberg R. J.; Benfield P. Bevantolol. A Preliminary Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Efficacy in Hypertension and Angina Pectoris. Drugs 1988, 35, 1–21. 10.2165/00003495-198835010-00001. [DOI] [PubMed] [Google Scholar]
- Corcostegui R.; Labeaga L.; Innerarity A.; Berisa A.; Orjales A. Preclinical Pharmacology of Bilastine, a New Selective Histamine H1 Receptor Antagonist: Receptor Selectivity and in Vitro Antihistaminic Activity. Drugs R&D 2005, 6, 371–384. 10.2165/00126839-200506060-00005. [DOI] [PubMed] [Google Scholar]
- Bertrand F.; Lehmann O.; Galani R.; Lazarus C.; Jeltsch H.; Cassel J. C. Effects of Mdl 73005 on Water-Maze Performances and Locomotor Activity in Scopolamine-Treated Rats. Pharmacol., Biochem. Behav. 2001, 68, 647–660. 10.1016/S0091-3057(01)00448-8. [DOI] [PubMed] [Google Scholar]
- Hara H.; Ichikawa M.; Oku H.; Shimazawa M.; Araie M. Bunazosin, a Selective Alpha1-Adrenoceptor Antagonist, as an Anti-Glaucoma Drug: Effects on Ocular Circulation and Retinal Neuronal Damage. Cardiovasc. Drug Rev. 2005, 23, 43–56. 10.1111/j.1527-3466.2005.tb00156.x. [DOI] [PubMed] [Google Scholar]
- Malinowska B.; Kiec-Kononowicz K.; Flau K.; Godlewski G.; Kozlowska H.; Kathmann M.; Schlicker E. Atypical Cardiostimulant Beta-Adrenoceptor in the Rat Heart: Stereoselective Antagonism by Bupranolol but Lack of Effect by Some Bupranolol Analogues. Br. J. Pharmacol. 2003, 139, 1548–1554. 10.1038/sj.bjp.0705390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houin G.; Barre J.; Jeanniot J. P.; Ledudal P.; Cautreels W.; Tillement J. P. Pharmacokinetics of Butofilolol (Cafide) after Repeated Oral Administration in Man. Int. J. Clin Pharmacol Res. 1984, 4, 175–183. [PubMed] [Google Scholar]
- Holick M. F.; DeLuca H. F.; Avioli L. V. Isolation and Identification of 25-Hydroxycholecalciferol from Human Plasma. Arch. Intern. Med. 1972, 129, 56–61. 10.1001/archinte.1972.00320010060005. [DOI] [PubMed] [Google Scholar]
- Shibuya T.; Field R.; Watanabe Y.; Sato K.; Salafsky B. Structure-Affinity Relationships between Several New Benzodiazepine Derivatives and 3h-Diazepam Receptor Sites. Jpn. J. Pharmacol. 1984, 34, 435–440. 10.1254/jjp.34.435. [DOI] [PubMed] [Google Scholar]
- Fulton B.; Markham A. Mycophenolate Mofetil. A Review of Its Pharmacodynamic and Pharmacokinetic Properties and Clinical Efficacy in Renal Transplantation. Drugs 1996, 51, 278–298. 10.2165/00003495-199651020-00007. [DOI] [PubMed] [Google Scholar]
- Fischli W.; Clozel J. P.; Breu V.; Buchmann S.; Mathews S.; Stadler H.; Vieira E.; Wostl W. Ciprokiren (Ro 44−9375). A Renin Inhibitor with Increasing Effects on Chronic Treatment. Hypertension 1994, 24, 163–169. 10.1161/01.HYP.24.2.163. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Skolnick P. Triple Uptake Inhibitors: Therapeutic Potential in Depression and Beyond. Expert Opin. Invest. Drugs 2007, 16, 1365–1377. 10.1517/13543784.16.9.1365. [DOI] [PubMed] [Google Scholar]
- Ward D. A.; Abney K.; Oliver J. W. The Effects of Topical Ocular Application of 0.25% Demecarium Bromide on Serum Acetylcholinesterase Levels in Normal Dogs. Vet. Ophthalmol. 2003, 6, 23–25. 10.1046/j.1463-5224.2003.00257.x. [DOI] [PubMed] [Google Scholar]
- Gorrill M. J.; Marshall J. R. Pharmacology of Estrogens and Estrogen-Induced Effects on Nonreproductive Organs and Systems. J. Reprod Med. 1986, 31, 842–847. [PubMed] [Google Scholar]
- Dietz M.; Mohr P.; Kuhn B.; Maerki H. P.; Hartman P.; Ruf A.; Benz J.; Grether U.; Wright M. B. Comparative Molecular Profiling of the Pparalpha/Gamma Activator Aleglitazar: Ppar Selectivity, Activity and Interaction with Cofactors. ChemMedChem 2012, 7, 1101–1111. 10.1002/cmdc.201100598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H.; Shigenobu K. Efonidipine Hydrochloride: A Dual Blocker of L- and T-Type Ca(2+) Channels. Cardiovasc. Drug Rev. 2002, 20, 81–92. 10.1111/j.1527-3466.2002.tb00084.x. [DOI] [PubMed] [Google Scholar]
- Nabeshima T.; Matsuno K.; Kamei H.; Kameyama T. The Interaction of Eptazocine, a Novel Analgesic, with Opioid Receptors. Res. Commun. Chem. Pathol. Pharmacol. 1985, 48, 173–181. [PubMed] [Google Scholar]
- Zarbin M. A.; Palacios J. M.; Wamsley J. K.; Kuhar M. J. Axonal Transport of Beta-Adrenergic Receptors. Antero- and Retrogradely Transported Receptors Differ in Agonist Affinity and Nucleotide Sensitivity. Mol. Pharmacol. 1983, 24, 341–348. [PubMed] [Google Scholar]
- Aasmundstad T. A.; Xu B. Q.; Johansson I.; Ripel A.; Bjorneboe A.; Christophersen A. S.; Bodd E.; Morland J. Biotransformation and Pharmacokinetics of Ethylmorphine after a Single Oral Dose. Br. J. Clin. Pharmacol. 1995, 39, 611–620. 10.1111/j.1365-2125.1995.tb05720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer I. R.; Rider L.; Rodrigues L. U.; Gijon M. A.; Pac C. T.; Romero L.; Cimic A.; Sirintrapun S. J.; Glode L. M.; Eckel R. H.; Cramer S. D. Lipid Catabolism Via Cpt1 as a Therapeutic Target for Prostate Cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. 10.1158/1535-7163.MCT-14-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune M. E.; Katwala S. P.; Milicic I.; Witte D. G.; Kerwin J. F. Jr.; Meyer M. D.; Hancock A. A.; Williams M. Effect of Fiduxosin, an Antagonist Selective for Alpha(1a)- and Alpha(1d)-Adrenoceptors, on Intraurethral and Arterial Pressure Responses in Conscious Dogs. J. Pharmacol. Exp. Ther. 2002, 300, 487–494. 10.1124/jpet.300.2.487. [DOI] [PubMed] [Google Scholar]
- Sutherland R.; Croydon E. A.; Rolinson G. N. Flucloxacillin, a New Isoxazolyl Penicillin, Compared with Oxacillin, Cloxacillin, and Dicloxacillin. Br Med. J. 1970, 4, 455–460. 10.1136/bmj.4.5733.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodzki M.; Rutkowski R.; Jatczak M.; Kisiel M.; Czyzewska M. M.; Mozrzymas J. W. Comparison of Kinetic and Pharmacological Profiles of Recombinant Alpha1gamma2l and Alpha1beta2gamma2l Gabaa Receptors - a Clue to the Role of Intersubunit Interactions. Eur. J. Pharmacol. 2016, 784, 81–89. 10.1016/j.ejphar.2016.05.015. [DOI] [PubMed] [Google Scholar]
- Louca Jounger S.; Christidis N.; Hedenberg-Magnusson B.; List T.; Svensson P.; Schalling M.; Ernberg M. Influence of Polymorphisms in the Htr3a and Htr3b Genes on Experimental Pain and the Effect of the 5-Ht3 Antagonist Granisetron. PLoS One 2016, 11, e0168703. 10.1371/journal.pone.0168703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halobetasol Propionate: A Trihalogenated Ultrapotent Topical Corticosteroid. J. Am. Acad. Dermatol. 1991, 25, 1137−1186. [PubMed] [Google Scholar]
- Pinder R. M.; Brogden R. N.; Speight T. M.; Avery G. S. Hexoprenaline: A Review of Its Pharmacological Properties and Therapeutic Efficacy with Particular Reference to Asthma. Drugs 1977, 14, 1–28. 10.2165/00003495-197714010-00001. [DOI] [PubMed] [Google Scholar]
- Christensen C. B. The Opioid Receptor Binding Profiles of Ketobemidone and Morphine. Pharmacol. Toxicol. 1993, 73, 344–345. 10.1111/j.1600-0773.1993.tb01363.x. [DOI] [PubMed] [Google Scholar]
- Klotz U. Interaction Potential of Lercanidipine, a New Vasoselective Dihydropyridine Calcium Antagonist. Arzneim. Forsch. 2002, 52, 155–161. 10.1055/s-0031-1299873. [DOI] [PubMed] [Google Scholar]
- Dilworth F. J.; Williams G. R.; Kissmeyer A. M.; Nielsen J. L.; Binderup E.; Calverley M. J.; Makin H. L.; Jones G. The Vitamin D Analog, Kh1060, Is Rapidly Degraded Both in Vivo and in Vitro Via Several Pathways: Principal Metabolites Generated Retain Significant Biological Activity. Endocrinology 1997, 138, 5485–5496. 10.1210/endo.138.12.5594. [DOI] [PubMed] [Google Scholar]
- Holmes B.; Ward A. Meptazinol. A Review of Its Pharmacodynamic and Pharmacokinetic Properties and Therapeutic Efficacy. Drugs 1985, 30, 285–312. 10.2165/00003495-198530040-00001. [DOI] [PubMed] [Google Scholar]
- Brooks A. M.; Gillies W. E. Ocular Beta-Blockers in Glaucoma Management. Clinical Pharmacological Aspects. Drugs Aging 1992, 2, 208–221. 10.2165/00002512-199202030-00005. [DOI] [PubMed] [Google Scholar]
- Shelly W.; Draper M. W.; Krishnan V.; Wong M.; Jaffe R. B. Selective Estrogen Receptor Modulators: An Update on Recent Clinical Findings. Obstet Gynecol Surv. 2008, 63, 163–181. 10.1097/OGX.0b013e31816400d7. [DOI] [PubMed] [Google Scholar]
- Gustavo R. P. Anti-Gonadotropic Action of Possipione. Quad Clin Ostet Ginecol. 1958, 13, 307–315. [PubMed] [Google Scholar]
- Attwood D. Aggregation of Antiacetylcholine Drugs in Aqueous Solution: Monomer Concentrations in Non-Micellar Drug Systems. J. Pharm. Pharmacol. 1976, 28, 762–765. 10.1111/j.2042-7158.1976.tb04043.x. [DOI] [PubMed] [Google Scholar]
- Marks M. J.; Wageman C. R.; Grady S. R.; Gopalakrishnan M.; Briggs C. A. Selectivity of Abt-089 for Alpha4beta2* and Alpha6beta2* Nicotinic Acetylcholine Receptors in Brain. Biochem. Pharmacol. 2009, 78, 795–802. 10.1016/j.bcp.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraki Y. Comparative Functional Selectivity of Imidafenacin and Propiverine, Antimuscarinic Agents, for the Urinary Bladder over Colon in Conscious Rats. Naunyn-Schmiedeberg's Arch. Pharmacol. 2015, 388, 1171–1178. 10.1007/s00210-015-1155-9. [DOI] [PubMed] [Google Scholar]
- McGorum B. C.; Nicholas D. R.; Foster A. P.; Shaw D. J.; Pirie R. S. Bronchodilator Activity of the Selective Muscarinic Antagonist Revatropate in Horses with Heaves. Vet. J. 2013, 195, 80–85. 10.1016/j.tvjl.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Greenblatt D. J. Pharmacology of Benzodiazepine Hypnotics. J. Clin Psychiatry. 1992, 53 (Suppl), 7–13. [PubMed] [Google Scholar]
- Kang S. G.; Kim J. J. Udenafil: Efficacy and Tolerability in the Management of Erectile Dysfunction. Ther. Adv. Urol. 2013, 5, 101–110. 10.1177/1756287212470019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe S.; Watabe H.; Takaseki S.; Aihara M.; Yoshitomi T. The Effects of Prostaglandin Analogues on Intracellular Ca2+ in Ciliary Arteries of Wild-Type and Prostanoid Receptor-Deficient Mice. J. Ocul. Pharmacol. Ther. 2013, 29, 55–60. 10.1089/jop.2011.0197. [DOI] [PubMed] [Google Scholar]
- Halland N.; Blum H.; Buning C.; Kohlmann M.; Lindenschmidt A. Small Macrocycles as Highly Active Integrin Alpha2beta1 Antagonists. ACS Med. Chem. Lett. 2014, 5, 193–198. 10.1021/ml4004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopus M.; Smiyun G.; Miller H.; Oroudjev E.; Wilson L.; Jordan M. A. Mechanism of Action of Ixabepilone and Its Interactions with the Betaiii-Tubulin Isotype. Cancer Chemother. Pharmacol. 2015, 76, 1013–1024. 10.1007/s00280-015-2863-z. [DOI] [PubMed] [Google Scholar]
- Myatt J. W.; Healy M. P.; Bravi G. S.; Billinton A.; Johnson C. N.; Matthews K. L.; Jandu K. S.; Meng W.; Hersey A.; Livermore D. G.; Douault C. B.; Witherington J.; Bit R. A.; Rowedder J. E.; Brown J. D.; Clayton N. M. Pyrazolopyridazine Alpha-2-Delta-1 Ligands for the Treatment of Neuropathic Pain. Bioorg. Med. Chem. Lett. 2010, 20, 4683–4688. 10.1016/j.bmcl.2010.05.026. [DOI] [PubMed] [Google Scholar]
- van Hattum H.; Branderhorst H. M.; Moret E. E.; Nilsson U. J.; Leffler H.; Pieters R. J. Tuning the Preference of Thiodigalactoside- and Lactosamine-Based Ligands to Galectin-3 over Galectin-1. J. Med. Chem. 2013, 56, 1350–1354. 10.1021/jm301677r. [DOI] [PubMed] [Google Scholar]
- Saku O.; Ohta K.; Arai E.; Nomoto Y.; Miura H.; Nakamura H.; Fuse E.; Nakasato Y. Synthetic Study of Vla-4/Vcam-1 Inhibitors: Synthesis and Structure-Activity Relationship of Piperazinylphenylalanine Derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 1053–1057. 10.1016/j.bmcl.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Ulven T.; Frimurer T. M.; Receveur J. M.; Little P. B.; Rist O.; Norregaard P. K.; Hogberg T. 6-Acylamino-2-Aminoquinolines as Potent Melanin-Concentrating Hormone 1 Receptor Antagonists. Identification, Structure-Activity Relationship, and Investigation of Binding Mode. J. Med. Chem. 2005, 48, 5684–5697. 10.1021/jm050103y. [DOI] [PubMed] [Google Scholar]
- Scott J. S.; Berry D. J.; Brown H. S.; Buckett L.; Clarke D. S.; Goldberg K.; Hudson J. A.; Leach A. G.; MacFaul P. A.; Raubo P.; Robb G. Achieving Improved Permeability by Hydrogen Bond Donor Modulation in a Series of Mgat2 Inhibitors. MedChemComm 2013, 4, 1305–1311. 10.1039/c3md00156c. [DOI] [Google Scholar]
- Whitby R. J.; Stec J.; Blind R. D.; Dixon S.; Leesnitzer L. M.; Orband-Miller L. A.; Williams S. P.; Willson T. M.; Xu R.; Zuercher W. J.; Cai F.; Ingraham H. A. Small Molecule Agonists of the Orphan Nuclear Receptors Steroidogenic Factor-1 (Sf-1, Nr5a1) and Liver Receptor Homologue-1 (Lrh-1, Nr5a2). J. Med. Chem. 2011, 54, 2266–2281. 10.1021/jm1014296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNinno M. P.; Wright S. W.; Etienne J. B.; Olson T. V.; Rocke B. N.; Corbett J. W.; Kung D. W.; DiRico K. J.; Andrews K. M.; Millham M. L.; Parker J. C.; Esler W.; van Volkenburg M.; Boyer D. D.; Houseknecht K. L.; Doran S. D. Discovery of Triazolopyrimidine-Based Pde8b Inhibitors: Exceptionally Ligand-Efficient and Lipophilic Ligand-Efficient Compounds for the Treatment of Diabetes. Bioorg. Med. Chem. Lett. 2012, 22, 5721–5726. 10.1016/j.bmcl.2012.06.079. [DOI] [PubMed] [Google Scholar]
- Preuss J.; Maloney P.; Peddibhotla S.; Hedrick M. P.; Hershberger P.; Gosalia P.; Milewski M.; Li Y. L.; Sugarman E.; Hood B.; Suyama E.; Nguyen K.; Vasile S.; Sergienko E.; Mangravita-Novo A.; Vicchiarelli M.; McAnally D.; Smith L. H.; Roth G. P.; Diwan J.; Chung T. D.; Jortzik E.; Rahlfs S.; Becker K.; Pinkerton A. B.; Bode L. Discovery of a Plasmodium Falciparum Glucose-6-Phosphate Dehydrogenase 6-Phosphogluconolactonase Inhibitor (R,Z)-N-((1-Ethylpyrrolidin-2-Yl)Methyl)-2-(2-Fluorobenzylidene)-3-Oxo-3,4-Dihydr O-2h-Benzo[B][1,4]Thiazine-6-Carboxamide (Ml276) That Reduces Parasite Growth in Vitro. J. Med. Chem. 2012, 55, 7262–7272. 10.1021/jm300833h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faull A. W.; Brewster A. G.; Brown G. R.; Smithers M. J.; Jackson R. Dual-Acting Thromboxane Receptor Antagonist/Synthase Inhibitors: Synthesis and Biological Properties of [2-Substituted-4-(3-Pyridyl)-1,3-Dioxan-5-Yl] Alkenoic Acids. J. Med. Chem. 1995, 38, 686–694. 10.1021/jm00004a014. [DOI] [PubMed] [Google Scholar]
- Kumar N. S.; Amandoron E. A.; Cherkasov A.; Finlay B. B.; Gong H.; Jackson L.; Kaur S.; Lian T.; Moreau A.; Labriere C.; Reiner N. E.; See R. H.; Strynadka N. C.; Thorson L.; Wong E. W.; Worrall L.; Zoraghi R.; Young R. N. Optimization and Structure-Activity Relationships of a Series of Potent Inhibitors of Methicillin-Resistant Staphylococcus Aureus (Mrsa) Pyruvate Kinase as Novel Antimicrobial Agents. Bioorg. Med. Chem. 2012, 20, 7069–7082. 10.1016/j.bmc.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Drouin L.; McGrath S.; Vidler L. R.; Chaikuad A.; Monteiro O.; Tallant C.; Philpott M.; Rogers C.; Fedorov O.; Liu M.; Akhtar W.; Hayes A.; Raynaud F.; Muller S.; Knapp S.; Hoelder S. Structure Enabled Design of Baz2-Icr, a Chemical Probe Targeting the Bromodomains of Baz2a and Baz2b. J. Med. Chem. 2015, 58, 2553–2559. 10.1021/jm501963e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.