

Abstract
Please cite this paper as: Getie‐Kebtie et al. (2012) Label‐free mass spectrometry‐based quantification of hemagglutinin and neuraminidase in influenza virus preparations and vaccines. Influenza and Other Respiratory Viruses 7(4), 521–530.
Background Influenza vaccination is the primary method for preventing influenza and its severe complications. An accurate rapid method to determine hemagglutinin (HA) concentration would facilitate reference antigen preparation and consequently expedite availability of seasonal as well as pandemic vaccines.
Objective The goal of this study was to develop a label‐free mass spectrometry (MS) based method that enables simultaneous identification and quantification of HA, neuraminidase (NA), and other viral proteins and protein contaminations in influenza vaccine or virus preparations.
Methods The method presented is based on LC/MSE analysis of vaccine or virus preparations tryptic digests spiked with a known amount of protein standard from which a universal response factor is generated and applied to calculate the concentration of proteins identified in the mixture.
Results We show that, with the use of an appropriate internal standard, the label‐free MS‐based protein quantification method is applicable for simultaneous identification and absolute quantification of HA and identification and relative quantification of other influenza proteins as well as protein impurities in influenza vaccines and virus preparations. We show that different subtype recombinant HA is preferred internal standard that provides the most accurate results in absolute quantification of HAs and other influenza proteins. We applied this method to measure the absolute quantity of HA as well as relative quantities of other viral proteins and impurities in preparations of whole virus and monovalent vaccine, providing data to demonstrate strain‐dependent differences in the amount of NA.
Conclusion The label‐free MS method presented here is ideally suited for timely preparation of reference material needed for potency testing of seasonal and pandemic vaccines.
Keywords: Hemagglutinin, influenza, mass spectrometry, neuraminidase, proteomics
Introduction
Although both HA and neuraminidase (NA) play an important role in the induction of protective immune responses, 1 , 2 HA is the predominant antigen in influenza vaccine. The HA content of the virus preparations can be calculated using total protein concentration and percent HA as measured by densitometry of a Coomassie‐stained SDS‐PAGE HA band(s). 3 This method of determining HA concentration is subject to inconsistency owing to potential co‐migration of proteins on SDS‐PAGE 4 and the fact that protein quantification methods have different sensitivities for different proteins. 5 A fast and more accurate alternative method to measure HA content would facilitate more rapid preparation of reference standards and support production and availability of seasonal as well as pandemic vaccines. To this effect, some physicochemical methods with a potential application for quantification of HA have recently been developed and evaluated, including reverse‐phase HPLC 6 and liquid chromatography–tandem mass spectrometry (LC‐MS/MS). 7
The liquid chromatography–tandem mass spectrometry (LC‐MS/MS) method for the absolute quantification of HA using isotope dilution in conjunction with multiple reaction monitoring (MRM) has been applied to quantify HA in purified virus preparations, monovalent bulk concentrates, or trivalent inactivated influenza vaccines. 7 This method quantifies targeted peptides released by proteolytic digestion of the sample as a stoichiometric representative of the analyte protein. A stable isotope‐labeled reference peptide is spiked into the sample as an internal standard (IS). Quantification of HA is achieved by comparing the peak area of the isotopically labeled reference peptide with that of the endogenous target peptide. This method allows simultaneous quantification of multiple proteins, provided labeled peptides are included for each specific target. However, this approach heavily depends on the availability of a library of labeled HA peptides that are pre‐selected to represent different types and subtypes of HA. In the event of an emerging pandemic influenza virus with a unique HA sequence, additional time and resources may be necessary to prepare a new set of labeled peptides.
Recently, a new type of label‐free quantification method known as LC/MSE was introduced for quadrupole time‐of‐flight (Q‐Tof) mass spectrometers. 8 For this method, alternating scans of low collision energy and elevated collision energy during LC/MS analysis are used to obtain both protein identity and quantity in a single experiment. Quantification is based on the experimental data showing that the average signal intensity measured by LC/MSE of the three most intense tryptic peptides for any given protein is constant at a given concentration, regardless of protein type and size. 8 As the signal intensity is proportional to concentration, the amount of any protein in the mixture can be estimated. LC/MSE provides substantial advantages for protein analysis over conventional LC/MS/MS approaches, as it utilizes parallel, multiplex fragmentation where all peptide precursors are simultaneously fragmented throughout the chromatographic separation process regardless of intensity. This allows data‐independent identification of lower abundance peptides and provides increased proteome coverage and dynamic range of protein quantification compared with data‐dependent LC/MS/MS. The method has been successfully applied for quantification of proteins in biological samples, 9 , 10 , 11 and our laboratory demonstrated its applicability for relative quantification of proteins separated by SDS gel electrophoresis. 12 Here we report the application of this method for absolute quantification of influenza proteins, including HA, NA, and protein impurities in a variety of influenza virus preparations.
Materials and methods
Materials
The following items were purchased from Sigma‐Aldrich (St. Louis, MO, USA): bovine liver catalase, acetonitrile, methanol, ammonium bicarbonate, iodoacetamide, and dithiothreitol (DTT). Formic acid was obtained from Fluka (Milwaukee, WI, USA). Sequencing‐grade modified porcine trypsin was obtained from Promega (Madison, WI, USA). RapiGest was purchased from Waters (Milford, MA, USA). Recombinant hemagglutinins (rHA) from A/Brisbane/59/2007, A/Solomon Islands/03/2006, A/New Caledonia/20/1999, A/Vietnam/1203/2004, and B/Brisbane/60/2008 were purchased from Protein Sciences Corporation (Meriden, CT, USA). Different lots of expired monovalent 2009 H1N1 vaccine (Novartis Vaccines and Diagnostics Limited, Cambridge, MA, USA) were kindly provided from the national pandemic vaccine stockpile by the Biomedical Advanced Research and Development Authority (BARDA).
Quantification of standards
Synthetic catalase peptides (referred to as Synth‐Catal in figure legends)
Ten tryptic peptides of bovine catalase (Table 1) that consistently demonstrated the strongest MS signals during repeated in‐solution digestion experiments (n = 10) were chosen as internal standards for quantification experiments. Peptides were synthesized by Fmoc chemistry, purified by HPLC, and analyzed by MALDI‐TOF by the CBER Facility for Biotechnology Resources.
Table 1.
List of synthetic catalase peptides used as internal standards
| Peptide sequence | Mol.Wt. |
|---|---|
| LGPNYLQIPVNCPYR | 1745·9 |
| VWPHGDYPLIPVGK | 1516·9 |
| DALLFPSFIHSQK | 1501·8 |
| GAGAFGYFEVTHDITR | 1739·8 |
| FSTVAGESGSADTVRDPR | 1850·9 |
| LNSLTVGPR | 955·6 |
| LFAYPDTHR | 1118·6 |
| LAHEDPDYGLR | 1284·6 |
| NFSDVHPEYGSR | 1406·6 |
| FNSANDDNVTQVR | 1478·7 |
Predigested catalase (referred to as PreD‐Catal in figure legends)
A 10 pmol/μl of catalase working solution was prepared from a 4 mg/ml stock solution in 50 mm ammonium bicarbonate buffer, pH 8·0. Catalase was denatured and reduced by boiling for 5 minutes in 10 mm DTT containing 0·1% RapiGest and then alkylated by incubation for 30 minutes at 60°C in 50 mm of iodoacetamide. After that, the protein was subjected to tryptic digest with trypsin to protein ratio 2:1. Tryptic digestion was stopped by formic acid (final concentration 1%).
Preparation of influenza whole virus
Influenza viruses A/California/07/2009X‐179A, A/Victoria/210/2009X‐187, A/Wisconsin/15/2009, and B/Brisbane/60/2008 were grown in 9‐ to 11‐day‐old embryonated chicken eggs for 60–72 hours at 33°C. The allantoic fluid was harvested, cell debris was removed by centrifugation (200 g, 10 minutes), and the virus was concentrated by ultracentrifugation (100 000 g, 2 hours). The virus pellet was resuspended in phosphate‐buffered saline, and aliquots were stored at 4°C.
Proteolytic digestion
To ensure homogenous preparations of the whole‐virus preparation, the original material was vortexed well and then sonicated three times (15 seconds pulses) prior to transfer of a 100‐μl aliquot. An equal volume of 0·1% RapiGest was then added, and the sample was incubated at room temperature for 30 minutes and vortexed before making a dilution to approximately 250 μg/ml. Ten microliter of the diluted whole‐virus preparation, recombinant HA (1 pmol/μl), or monovalent vaccine (undiluted) was mixed with 1 μl of the appropriate internal standard protein, that is, mixture of synthetic peptides of catalase, predigested catalase, intact catalase, or rHA from A/Vietnam/1203/2004 (10 pmol/μl), and 1 μl of the appropriate quality control protein (10 pmol/μl), that is, rHA from A/Brisbane/59/2007 or B/Brisbane/60/2008. Then 10 μl of 0·1% Rapigest in 50 mm ammonium bicarbonate containing 10 mm DTT was added, and the mixture was boiled for 5 minutes in a heating block. The sample was cooled to room temperature before alkylation with 50 mm iodoacetamide. The alkylation took place in the dark at 60°C for 30 minutes. Proteolytic digestion was performed by incubating the sample with modified trypsin (Promega) at ratios of 1:10 and 2:1 (trypsin to total protein, by weight) at 37°C for 4 hours. Tryptic digestion was stopped by adding formic acid to a final concentration of 1%. The final volume was adjusted to 100 μl by adding 0·1% formic acid (concentration of the internal standard in the final sample was 100 fmol/μl). Each sample was digested in triplicate (referred to as a biological replicate in Tables and Figure legends).
Data acquisition, processing, and quantification
Samples were analyzed by LC/MSE using a nanoACQUITY ultrapressure liquid chromatography (UPLC) and Synapt G2 mass spectrometer equipped with a nanolockspray ion source (Waters). proteinlynx global server v2.4 software (Waters) was used to process mass spectral raw data for subsequent protein identification and quantification. The average intensity value of the top three ionized tryptic peptides of the internal standard protein was used to convert the average intensity of top responsive peptides of analyte proteins to the corresponding absolute quantity of protein loaded on column. Further details of data acquisition, processing, and quantification are available as Data S1.
Results and discussion
Evaluation of assay variability
To demonstrate instrument performance consistency, we assessed the reproducibility of technical replicates used in label‐free MS‐based absolute quantification. Table 2 summarizes the results obtained from three biological replicates of a whole‐virus preparation of A/California/07/2009x179 that were run in triplicate for each biological sample. The results obtained demonstrate that the variation in measurement is insignificant (CV < 5%).
Table 2.
Reproducibility of technical replicates. Three biological replicates of the tryptic digest of the whole‐virus preparation of H1N1 influenza A virus, A/California/07/2009X‐179A, were prepared, and each was analyzed using nanoAcquity UPLC and Synapt G2 MS in triplicates. A mixture of synthetic catalase peptides was used as an internal standard
| Tech‐1 | Tech‐2 | Tech‐3 | Aver | %CV | |
|---|---|---|---|---|---|
| Biological replicate 1‐ amount of protein in ng | |||||
| HA | 26·7 | 27·4 | 28·0 | 27·3 | 2·4 |
| NA | 9·9 | 9·5 | 10·00 | 9·8 | 2·8 |
| M1 | 18·5 | 18·2 | 18·8 | 18·5 | 1·6 |
| NP | 10·5 | 9·9 | 10·3 | 10·2 | 2·6 |
| Biological replicate 2‐ amount of protein in ng | |||||
| HA | 32·5 | 31·3 | 32·5 | 32·1 | 2·1 |
| NA | 13·0 | 12·6 | 13·9 | 13·2 | 5·0 |
| M1 | 23·4 | 22·4 | 22·8 | 22·9 | 2·1 |
| NP | 13·5 | 12·3 | 13·1 | 13·0 | 4·5 |
| Biological replicate 3‐ amount of protein in ng | |||||
| HA | 38·4 | 36·5 | 35·8 | 36·9 | 3·7 |
| NA | 16·8 | 16·4 | 16·4 | 16·6 | 1·2 |
| M1 | 28·7 | 26·3 | 27·9 | 27·6 | 4·3 |
| NP | 15·9 | 14·9 | 14·7 | 15·2 | 4·4 |
HA, hemagglutinin; MS, mass spectrometry.
On the other hand, Table 3 shows that the variability among biological replicates is higher, with a CV in some instances close to 20%. Considering the high consistency of technical replicates, this indicates that sample preparation and assay conditions impact measurement precision. Consequently, we identified and studied the influence of three critical sample preparation steps that could potentially contribute to this variability, namely the homogeneity of the influenza virus preparations, the completeness of tryptic digestion, and the selection of the appropriate internal standard.
Table 3.
Reproducibility of biological replicates. See Table 2 for description
| Biol‐1 | Biol‐2 | Biol‐3 | Aver | %CV | |
|---|---|---|---|---|---|
| Biological replicates‐amount of protein in ng | |||||
| HA | 27·3 | 32·1 | 36·9 | 32·1 | 13·1 |
| NA | 9·8 | 13·2 | 16·6 | 13·2 | 22·4 |
| M1 | 18·5 | 22·9 | 27·6 | 23·0 | 17·4 |
| NP | 10·2 | 13·0 | 15·2 | 12·8 | 17·2 |
HA, hemagglutinin.
Optimization of sample preparation and digestion conditions
Upon storage of the whole‐virus preparation at 4°C, a loosely packed pellet was noticed at the bottom of the tube. Without homogenous distribution of the analyte proteins, aliquots withdrawn from such a sample will likely have variable quantities of the proteins. We therefore mixed the original samples vigorously by vortexing and dispersed aggregates by sonication prior to dispensing aliquots. The virus was also solubilized by addition of RapiGest detergent before removing material for dilution or digestion, as described in Materials and methods.
Completeness of tryptic digestion is a critical step in any proteomics‐based quantification, either label‐free or label‐based. An incomplete digestion of a protein leads to an underestimation of the true protein quantity. In most proteomics experiments, a trypsin to protein mass ratio of 1:10 or less is used. In a recent study, Norrgran et al. 13 reported that the optimum trypsin to protein ratio to achieve maximum digestion efficiency of HA is close to 2:1. In our experiments, we compared two tryptic digestion protocols: that is, trypsin to protein ratio of 1:10 (Low trypsin) and trypsin to protein ratio of 2:1 (High trypsin). Figure 1 illustrates the result of this comparison in terms of the protein concentration obtained by label‐free MS‐based quantification of the digests. For all proteins analyzed, a significantly higher concentration of protein was obtained with the high trypsin protocol as compared to the low trypsin protocol, indicative of greater degree of tryptic digestion. For that reason, the high trypsin protocol was used in our study.
Figure 1.
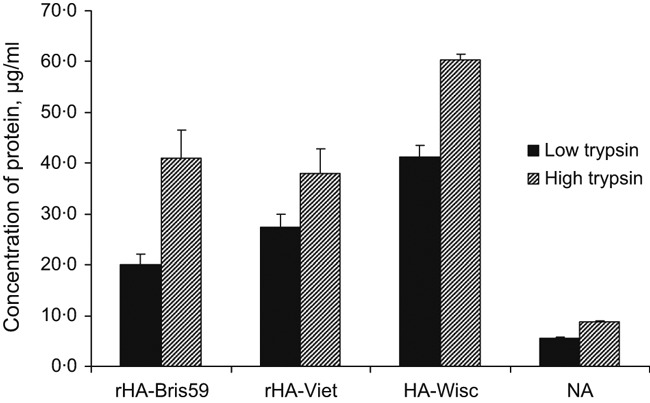
Comparison of low and high trypsin digestion protocols. Recombinant HAs from A/Brisbane/59/2007 and A/Vietnam/1203/2004 were spiked into a preparation of A/Wisconsin/15/2009 at concentrations of 63·2 and 64·4 μg/ml, respectively. A trypsin to protein ratio of 1:10 or 2:1 was used for the low or high trypsin digestion protocol, respectively. For absolute quantification, a mixture of synthetic peptides was used as an internal standard. Data shown are representative of the results of three biological replicates, each of which was obtained from three technical replicates.
Comparison of internal standards for suitability in absolute protein quantification
Selection of an appropriate internal standard is critical for successful label‐free absolute protein quantification. Label‐based absolute quantification methods use a stable isotope‐labeled endogenous target peptide as a stoichiometric representative of the protein from which it is cleaved. The method presented here uses either a simulated tryptic digest (mixture of synthetic tryptic peptides) or a whole protein (pre‐digested or co‐digested with the sample) as internal standard (IS). The universal signal response factor (counts/mol of protein) obtained from the average MS signal response of the top three MS signals of tryptic peptides from the IS is then used to determine a corresponding absolute quantity of a protein of interest. 8 Theoretically, accurate quantification is achieved when both the internal standard and analyte proteins are digested to a similar proportion. To test this, we spiked an internal standard protein, catalase, into influenza whole‐virus preparations in three different forms, that is, intact, predigested, or as a mixture of selected synthetic tryptic peptides. In addition, an intact form of recombinant HA from H5N1 influenza virus (A/Vietnam/1203/2004) was added to determine whether a related protein would provide different results. A whole‐virus preparation of A/Wisconsin/15/2009 (H3N2) was used for this analysis. The rationale for using a mixture of catalase synthetic tryptic peptides that simulate a completely digested form of an IS was to control for incomplete enzymatic digestion that could result in an underestimation of the analyte protein quantity. A more accurate estimation of protein concentration is probable when whole protein is used as internal standard, because tryptic digestion of the standard and analyte protein is likely to be proportional. The IS can be spiked into the sample as an intact protein, or digested separately (pre‐digested) and then spiked into the sample digest. This approach allows normalization of tryptic digestion, assuming digestion efficiency of the analyte and IS is proportional. This proportionality is likely to be best achieved by using an IS that is analogous to the analyte protein. We used intact recombinant HA of a different subtype to the analyte as a structurally similar IS; for the A/Wisconsin/15/2009 (H3N2) strain, we used rHA from A/Vietnam/1203/2004 (H5N1).
Figure 2 shows results that compare concentrations of HA and other proteins in whole‐virus preparation of A/Wisconsin/15/2009 measured using each of the aforementioned internal standards. To identify the best approach for accurate determination of the proteins’ quantities, we ideally needed to have prior knowledge of the quantities of the proteins in the sample analyzed. For the influenza preparation, this is not practical; therefore, we spiked a known concentration of rHA from A/Brisbane/59/2007 (H1N1) into the sample as an internal performance control.
Figure 2.
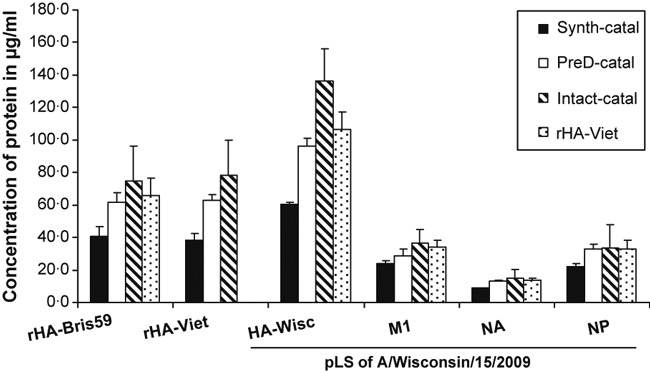
Comparison of different internal standards for suitability in absolute quantification of influenza virus proteins. A known quantity of recombinant hemagglutinin (HA) from A/Brisbane/59/2007 was spiked into a preparation of A/Wisconsin/15/2009 at a concentration of 63·2 μg/ml to assess the accuracy of the different approaches in absolute quantification of HA. Recombinant HA from A/Vietnam/1203/2004 was used as an internal standard at a concentration of 64·4 μg/ml. A known quantity of catalase in the form of intact, predigested, or mixture of synthetic peptides was spiked into the sample at the concentration of 60·1 μg/ml. Data shown are representative of the results of three biological replicates, each of which was obtained from three technical replicates.
It is apparent from Figure 2 that the measured concentrations of all proteins are noticeably different for different types of internal standards. It is highly likely that incomplete tryptic digestion contributes to this difference. For example, the concentrations of all proteins were significantly lower when quantified using synthetic catalase peptides as internal standard in comparison with using pre‐digested catalase or intact catalase. The measured concentration of rHA from A/Brisbane/59/2007 (spiked at a concentration of 63·2 μg/ml) using synthetic catalase peptides was 40·9 μg/ml, suggesting approximately 65% of the protein was digested. This implies that the internal standard protein with digestion efficiency close to 65% yields the most accurate estimate of rHA concentration. When intact catalase was used as an internal standard, the protein’s measured concentration was overestimated, that is, 74·8 μg/ml, indicating the digestion efficiency of catalase is well <65%. On the other hand, the use of intact rHA enabled accurate determination of the concentration of the protein with an error of 3·8%, suggesting close to 65% digestion efficiency for the internal standard rHA. The noticeable difference in concentrations measured using intact catalase and intact rHA as IS implies that the accuracy of this method depends not only on the form, that is, synthetic tryptic peptides, pre‐digested or intact, but also on the type of protein used as an IS.
When rHA was used as the IS, we observe that degree of digestion similarly impacted measurement of catalase concentrations. To normalize concentrations by taking into consideration the extent of digestion, we used the measured concentrations of the three forms of catalase reported in Figure 3 (synthetic tryptic peptides, predigested catalase, and whole protein) to calculate correction factors. In all cases, the actual concentration of catalase spiked into the sample was expected to be 60·1 μg/ml. However, the measured concentration of catalase spiked into the sample as a mixture of synthetic peptides was 103·8 μg/ml when rHA was used as standard (Figure 3). This difference is, at least partly, attributed to the fact that the internal rHA standard was incompletely digested, leading to an overestimation of the catalase concentration. On the other hand, the calculated concentration of catalase spiked as intact protein (52·8 μg/ml) and as pre‐digested protein (57·3 μg/ml), although different, was much closer to the expected 60·1 μg/ml when rHA was used as the IS, suggesting some proportionality of tryptic digestion efficiency regardless of whether catalase was digested separately or in combination with other proteins.
Figure 3.
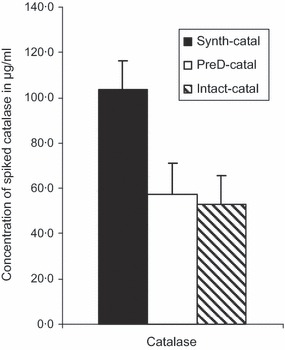
Quantification of the different forms of spiked catalase using recombinant hemagglutinins from A/Vietnam/1203/2004 as internal standard. A known quantity of catalase in the form of intact, predigested, or mixture of synthetic peptides was spiked into a preparation of A/Wisconsin/15/2009 at the concentration of 60·1 μg/ml. Correction factors determined from the ratio of the measured and expected concentrations of intact, predigest, and synthetic peptide forms of catalase were 0·88, 0·95, and 1·73, respectively. Data shown are representative of the results of three biological replicates, each of which was obtained from three technical replicates.
The results shown in Figure 2 were re‐calculated using the correction factors calculated from the data in Figure 3 (0·88, 0·95, and 1·73 for intact, predigested, and synthetic peptide forms of catalase, respectively), and the corrected concentrations are plotted in Figure 4. The differences in measured concentrations that were observed before correction (Figure 2) were noticeably reduced after correction, confirming the need to consider completeness of digestion and type of IS when using the label‐free MS‐based quantification method.
Figure 4.
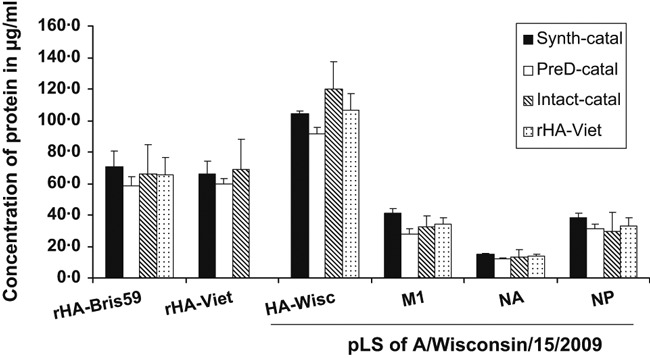
Concentrations of proteins in a preparation of A/Wisconsin/15/2009 and spiked recombinant hemagglutinins derived from the data shown in Figure 2 using the calculated correction factors for intact, pre‐digested, and synthetic tryptic peptide forms of catalase used as internal standards. The correction factors were 0·88, 0·95, and 1·73, respectively.
Quantification of HA in purified protein and whole‐virus preparations
To assess the accuracy of this label‐free MS method for absolute protein quantification, we analyzed solutions of purified rHA containing known quantities of protein.
Table 4 shows the results of absolute quantification of rHAs from three H1N1 influenza virus strains. In this analysis, rHA from the H5N1 influenza virus A/Vietnam/1203/2004 was used as an IS. The quantities of rHAs determined by the label‐free mass spectrometry (MS) method were compared with the expected quantities according to manufacturer’s specifications as well as quantities determined by the BCA protein assay. There was <10% difference between the manufacturer’s specifications and the label‐free MS measurements for all rHAs, except that of A/Brisbane/59/2007. Although a significant (37%) difference was observed between the label‐free MS measured quantities of rHA from A/Brisbane/59/2007 and the manufacturer’s specification, the concentration of protein determined by BCA method was not much different from that of the label‐free MS measurement (Table 4).
Table 4.
Quantification of rHAs from three H1N1 influenza virus strains using rHA from A/Vietnam/1203/2004 as an internal standard
| Influenza virus strain (H1N1) | Concentration of protein, fmol/μ | ||
|---|---|---|---|
| Manufacturer’s specification | Type of analysis | ||
| Label‐free MS (%CV) | BCA (%CV) | ||
| A/Solomon Islands/03/2006 | 100 | 96·1 (2·3) | 89·3 (0·1) |
| A/New Caledonia/20/1999 | 100 | 106·0 (3·7) | 88·9 (0·1) |
| A/Brisbane/59/2007 | 100 | 136·6 (15·8) | 128·9 (0·2) |
MS, mass spectrometry; rHA, recombinant hemagglutinins.
Data shown are representative of the results of three biological replicates, each of which was obtained from three technical replicates.
To determine the dynamic range of the label‐free quantification method, a serial dilution of rHA from B/Brisbane/60/2008 was prepared ranging from 1·57 to 50·00 μg/ml. The same amounts of rHAs from A/Brisbane/59/2007 and A/Vietnam/1203/2004 were spiked to each dilution as performance control and internal standard proteins, respectively. The concentrations of rHAs from B/Brisbane/60/2008 and A/Brisbane/59/2007 at each dilution were determined using the label‐free MS‐based quantification method. The results of this experiment are presented in Figure 5. We observed an excellent linear correlation (R 2 = 0·997) between the expected and determined concentrations of rHA from B/Brisbane/60/2008 within the concentration range studied. The slope of the graph (1·18) approaches 1, indicating that the expected and determined concentrations of rHA are not significantly different at all dilutions. The concentration of the quality control rHA was constant with standard deviation of 0·3, and the average concentration (5·2 μg/ml) was close to the expected value (6·3 μg/ml) throughout all dilutions, confirming the accuracy of the quantification method.
Figure 5.
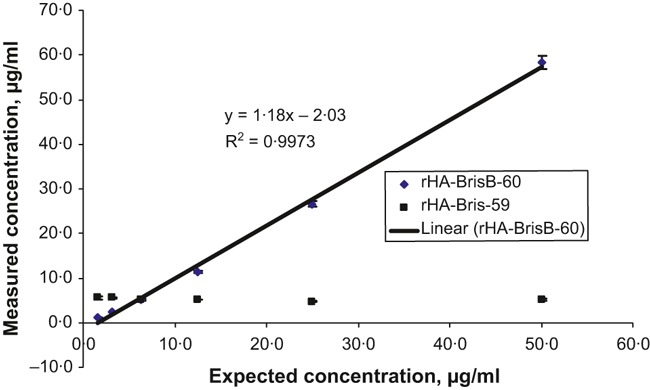
Quantification of recombinant hemagglutinins (rHA) from serial dilutions of the protein at concentrations ranging from 1·57 to 50·00 μg/ml. This concentration range brackets the concentration of hemagglutinin in different whole‐virus preparations and vaccines of influenza. Recombinant hemagglutinins from A/Brisbane/59/07 was spiked as a quality control protein at a concentration of 6·3 μg/ml. Recombinant hemagglutinins from A/Vietnam/1203/2004 was used as an internal standard protein. Data shown are representative of the results of three biological replicates, each of which was obtained from three technical replicates.
To assess the applicability of the method in the quantification of HA in different whole‐virus preparations, we determined the HA content of several H1N1, H3N2, and B virus suspensions. In this case, we spiked a known quantity of a second rHA (in addition to rHA used as IS) into each preparation, to monitor the accuracy of the native HA quantification. We assumed that if the quantity of the spiked rHA is determined accurately, the measured quantity of the native HA should be accurate too. Our results (Table 5) show that the performance control rHA was accurately measured with a maximum error of 10·7%.
Table 5.
Viral protein concentration (μg/ml) in various whole‐virus preparations. rHA from A/Brisbane/59/07 was spiked into A/Wisconsin/15/09, A/Victoria/210/09, and B/Brisbane/60/08 at a concentration of 63·0 μg/ml, and rHA from B/Brisbane/60/08 was spiked into A/California/07/09X‐179A at a concentration of 60·5 μg/ml. rHA from A/Vietnam/1203/2004 was used as an internal standard
| Subtype | Average of three biological replicates (%CV) | |||||
|---|---|---|---|---|---|---|
| Strain | HA | NA | M1 | NP | % Error (rHA)* | |
| H1N1 | A/California/07/09X‐179A | 37·1 (11·1) | 11·7 (6·9) | 18·9 (19·5) | 36·0 (21·3) | 10·7 |
| H3N2 | A/Wisconsin/15/09 | 79·4 (5·4) | 13·4 (7·5) | 39·1 (10·4) | 55·9 (15·9) | 5·6 |
| A/Victoria/210/09X‐187 | 23·8 (6·7) | 2·2 (13·6) | 13·6 (12·5) | 17·2 (24·4) | 8·7 | |
| B | B/Brisbane/60/2008 | 81·0 (4·6) | 4·5 (4·4) | 45·0 (6·7) | 35·0 (5·4) | 0·2 |
HA, hemagglutinin; rHA, recombinant hemagglutinins.
Data shown are representative of the results of three biological replicates, each of which was derived from three technical replicates.
*Percent error represents the percent difference of the measured and expected amount of the rHA added into the sample for quality control of the measurement efficiency.
Quantification of influenza proteins in the 2009 H1N1 influenza vaccine
To assess the applicability of the method in the quantification of influenza proteins in commercial influenza vaccines, we analyzed three lots of a monovalent 2009 H1N1 influenza vaccine. The results of this study are summarized in Table 6. The data demonstrate excellent consistency in HA content in all three lots. Assuming consistency in the manufacturing process, the consistency in HA content implies the practical applicability of the quantification method. This was further demonstrated by the accurate measurement of the spiked performance control recombinant HA from B/Brisbane/60/08.
Table 6.
Absolute quantification of proteins from three lots of the 2009 H1N1 vaccine (Novartis). rHA from A/Vietnam/1203/2004 was used as an internal standard. A known quantity of rHA from B/Brisbane/60/08 was spiked for quality control at a concentration of 60·5 μg/ml
| Protein | Protein concentration, μg/ml (%CV) | ||
|---|---|---|---|
| Lot 1 | Lot 2 | Lot 3 | |
| HA | 25·5 (15·7) | 25·6 (5·8) | 25·5 (6·3) |
| NA | 9·5 (9·4) | 9·4 (5·3) | 9·0 (5·5) |
| NP | 3·5 (14·3) | 3·1 (6·4) | 1·6 (31·2) |
| Spiked rHA‐BrisB | 64·0 (6·2) | 64·5 (6·9) | 70·5 (7·8) |
HA, hemagglutinin; rHA, recombinant hemagglutinins.
Data shown are representative of the results of three biological replicates, each of which was obtained from three technical replicates.
Determination of HA/NA ratios of different influenza virus preparations
Despite evidence that NA induces protective immunity, 14 the amount of NA in influenza vaccines is not quantified, partly because of a lack of an appropriate analytical method, 15 but also because its concentration is likely to be dependent on the NA content of the vaccine seed virus. The method we describe generates data that allow quantification of HA, NA, as well as any other viral and non‐viral protein that is at a reasonable concentration.
Our results show that there is a significant difference in the HA/NA ratio for different influenza viruses (Table 7). The ratio was different not only for different subtypes, but also for different strains within a subtype. For example, the ratio of the two H3N2 influenza viruses A/Wisconsin/15/2009 and A/Victoria/210/2009 was ∼6:1 and 12:1, respectively. Similarly, the ratios of the two H1N1 viruses, A/Brisbane/59/2007 and A/California/7/2009, were ∼5:1 and 3:1. The proportion of NA in this 2009 H1N1 pandemic virus is much greater than observed for other viruses. In contrast, the amount of NA in B/Brisbane/60/2008 was far less than expected, with a HA/NA ratio of 18:1. While there was no significant difference in the ratio between the three lots of the 2009 H1N1 vaccines studied, the small difference the ratio between the vaccine and whole‐virus preparation of A/California/07/09X‐179A may result from the use of a different vaccine seed virus for vaccine manufacture or may indicate enrichment of the HA during the manufacturing process.
Table 7.
Ratios of hemagglutinin to neuraminidase across different influenza viruses
| Preparation | Strain | Subtype | Ratio of HA/NA (%CV) |
|---|---|---|---|
| Whole virus | A/Wisconsin/15/09 | H3N2 | 5·9 (5·1) |
| Whole virus | A/Victoria/210/09X‐187 | H3N2 | 10·8 (7·4) |
| Whole virus | B/Brisbane/60/08 | B | 18·0 (10·0) |
| Whole virus | A/Brisbane/59/07 | H1N1 | 4·5 (4·4) |
| Whole virus | A/California/07/09X‐179A | H1N1 | 3·1 (12·9) |
| 2009 monovalent vaccine ‐Lot 1 | A/California/07/09X‐179A | H1N1 | 2·7 (7·4) |
| 2009 monovalent vaccine‐Lot 2 | A/California/07/09X‐179A | H1N1 | 2·7 (7·4) |
| 2009 monovalent vaccine‐Lot 3 | A/California/07/09X‐179A | H1N1 | 2·8 (7·1) |
HA, hemagglutinin.
Determination of the quantities of other influenza virus proteins and contaminant proteins
Unlike other MS‐based absolute protein quantification methods that target specific proteins in a complex mixture of proteins, the label‐free MS method presented here offers a unique opportunity for quantification of all proteins identified in the sample. While most of the other MS‐based quantitative methods require an initial LC/MS run to confirm the presence of the protein to be quantified, the label‐free method described here allows performing identification and quantification of proteins in a single run. As a result, this method provides comprehensive characterization of the sample not only in terms of the quantity of the protein of interest, but also in terms of identification and quantification of other influenza proteins present in a sample as well as other protein impurities. For example, individual proteins that originate from chicken eggs that are used in vaccine production can be identified and quantified (Table 8). The European Pharmacopoeia specifies that the amount of ovalbumin in inactivated whole‐virion influenza vaccine per human dose should be ≤1 μg and that the concentration of proteins other than HA in the vaccine should not be more than six times the total HA content of the vaccine. 16 While the concentration of ovalbumin in some of our whole‐virus preparations was >1 μg/ml, there was no detectable amount of ovalbumin in each of the three lots of subunit monovalent 2009 H1N1 vaccine. The label‐free MS quantification method could provide full characterization and quantification of vaccine components in a single analysis, and therefore may be a beneficial method to use as an in‐process or quality control test.
Table 8.
List of impurities identified and quantified from whole‐virus preparations of three different influenza virus strains. rHA from A/Vietnam/1203/2004 was used as an IS
| Strain | Contaminant protein | Accession number | Concentration in sample, μg/ml (%CV) |
|---|---|---|---|
| A/California/07/2009X‐179A (H1N1) | Alpha fetoprotein | P84407 | 5·0 (16·0) |
| Apovitellenin 1 | P02659 | 2·3 (4·3) | |
| Hemoglobin alpha | P01994 | 1·8 (16·6) | |
| Hemoglobin beta | P02112 | 7·1 (9·9) | |
| Hemoglobin epsilon | P02128 | 2·9 (13·8) | |
| Hemoglobin pi | P02007 | 2·8 (7·1) | |
| Hemoglobin rho | P02127 | 2·9 (13·8) | |
| Myosin 9 | P14105 | 6·0 (13·3) | |
| Ovalbumin | P01012 | 6·2 (4·8) | |
| A/Wisconsin/15/2008 (H3N2) | Annexin A2 | P17785 | 2·3 (8·7) |
| Apolipoprotein A | P08250 | 3·1 (9·6) | |
| Hemoglobin beta | P02112 | 1·8 (27·7) | |
| Histone H4 | P70081 | 0·8 (25·0) | |
| Ovalbumin | P01012 | 4·1 (22·0) | |
| B/Brisbane/60/2008 | Actin cytoplasmic type 5 | P53478 | 34·0 (8·8) |
| Annexin A2 | P17785 | 4·2 (7·1) | |
| Tubulin alpha 1 | P02552 | 2·5 (20·0) | |
| Tubulin beta | 7 P09244 | 1·4 (14·3) |
rHA, recombinant hemagglutinins.
Data shown are representative of the results of three biological replicates, each of which was obtained from three technical replicates.
As stated previously, accurate and absolute quantification of a protein with our method requires spiking of a corresponding internal standard protein that is structurally similar to the analyte protein. As we did not have a source of recombinant NA and/or other influenza virus proteins, the measured quantities of NA and other proteins reported in this work should be viewed as relative values.
Conclusion
We have shown that, with the utilization of an appropriate internal standard, the label‐free MS‐based protein quantification method developed in our laboratory can be used for simultaneous identification and absolute quantification of HA and identification and relative quantification of other influenza proteins as well as protein impurities in influenza vaccines and virus preparations. The addition of known concentrations of intact rHA of a different subtype to the mixture provided the most accurate results in absolute quantification of HAs and therefore is preferred as internal standard. Importantly, this method uses well‐characterized standards that are readily available and is therefore ideal for rapid determination of the HA concentration in primary standards, reducing the time needed to prepare reference material for potency testing of seasonal and pandemic vaccines.
Disclaimer
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
Supporting information
Data S1. Methods.
Supporting info item
Acknowledgements
This project was supported by funding provided by the CBER (Panflu funds) and by the Biomedical Advanced Research and Development Authority. We appreciate technical help from Kevin Yang and Schafer Eichelberger and critical reading of the manuscript by Jerry Weir and John Cipollo.
References
- 1. Webster RG, Bean WJ Jr. Genetics of influenza virus. Annu Rev Genet 1978; 12:415–431. [DOI] [PubMed] [Google Scholar]
- 2. Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 2000; 69:531–569. [DOI] [PubMed] [Google Scholar]
- 3. Who Expert Committee On Biological Standardization. Available at http://www.who.int/biologicals/expert_committee/BS2011.2183_Flu_vax_ERL_calibration_protocol.pdf, 17 to 21 October 2011. (Accessed 17 January 2012).
- 4. Harvey R, Wheeler JX, Wallis CL, Robertson JS, Engelhardt OG. Quantitation of haemagglutinin in H5N1 influenza viruses reveals low haemagglutinin content of vaccine virus NIBRG‐14 (H5N1). Vaccine 2008; 26:6550–6554. [DOI] [PubMed] [Google Scholar]
- 5. Olson BJ, Markwell J. Assays for determination of protein concentration. Curr Protoc Protein Sci 2007; Chapter 3:Unit 3.4; pp 3.4.1–3.4.29. [DOI] [PubMed] [Google Scholar]
- 6. Kapteyn JC, Saidi MD, Dijkstra R et al. Haemagglutinin quantification and identification of influenza A&B strains propagated in PER.C6 cells: a novel RP‐HPLC method. Vaccine 2006; 24:3137–3144. [DOI] [PubMed] [Google Scholar]
- 7. Williams TL, Luna L, Guo Z et al. Quantification of influenza virus hemagglutinins in complex mixtures using isotope dilution tandem mass spectrometry. Vaccine 2008; 26:2510–2520. [DOI] [PubMed] [Google Scholar]
- 8. Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics 2006; 5:144–156. [DOI] [PubMed] [Google Scholar]
- 9. Bostanci N, Heywood W, Mills K et al. Application of label‐free absolute quantitative proteomics in human gingival crevicular fluid by LC/MS E (gingival exudatome). J Proteome Res 2010; 9:2191–2199. [DOI] [PubMed] [Google Scholar]
- 10. Cheng FY, Blackburn K, Lin YM, Goshe MB, Williamson JD. Absolute protein quantification by LC/MS(E) for global analysis of salicylic acid‐induced plant protein secretion responses. J Proteome Res 2009; 8:82–93. [DOI] [PubMed] [Google Scholar]
- 11. Farnsworth A, Cyr TD, Li C, Wang J, Li X. Antigenic stability of H1N1 pandemic vaccines correlates with vaccine strain. Vaccine 2011; 29:1529–1533. [DOI] [PubMed] [Google Scholar]
- 12. Getie‐Kebtie M, Lazarev A, Eichelberger M, Alterman M. Label‐free mass spectrometry‐based relative quantification of proteins separated by one‐dimensional gel electrophoresis. Anal Biochem 2011; 409:202–212. [DOI] [PubMed] [Google Scholar]
- 13. Norrgran J, Williams TL, Woolfitt AR et al. Optimization of digestion parameters for protein quantification. Anal Biochem 2009; 393:48–55. [DOI] [PubMed] [Google Scholar]
- 14. Sylte MJ, Suarez DL. Influenza neuraminidase as a vaccine antigen. Curr Top Microbiol Immunol 2009; 333:227–241. [DOI] [PubMed] [Google Scholar]
- 15. Bright RA, Neuzil KM, Pervikov Y, Palkonyay L. WHO meeting on the role of neuraminidase in inducing protective immunity against influenza infection, Vilamoura, Portugal, September 14, 2008. Vaccine 2009; 27:6366–6369. [DOI] [PubMed] [Google Scholar]
- 16.European Pharmacopoeia 5th Ed. Main Volume 5.0, European Directorate For The Quality Of Medic, 2004; 677.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Methods.
Supporting info item