

Abstract
Background
Post-translational modification is considered an important biological mechanism with critical impact on the diversification of the proteome. Although a long list of such modifications has been studied, succinylation of lysine residues has recently attracted the interest of the scientific community. The experimental detection of succinylation sites is an expensive process, which consumes a lot of time and resources. Therefore, computational predictors of this covalent modification have emerged as a last resort to tackling lysine succinylation.
Results
In this paper, we propose a novel computational predictor called ‘Success’, which efficiently uses the structural and evolutionary information of amino acids for predicting succinylation sites. To do this, each lysine was described as a vector that combined the above information of surrounding amino acids. We then designed a support vector machine with a radial basis function kernel for discriminating between succinylated and non-succinylated residues. We finally compared the Success predictor with three state-of-the-art predictors in the literature. As a result, our proposed predictor showed a significant improvement over the compared predictors in statistical metrics, such as sensitivity (0.866), accuracy (0.838) and Matthews correlation coefficient (0.677) on a benchmark dataset.
Conclusions
The proposed predictor effectively uses the structural and evolutionary information of the amino acids surrounding a lysine. The bigram feature extraction approach, while retaining the same number of features, facilitates a better description of lysines. A support vector machine with a radial basis function kernel was used to discriminate between modified and unmodified lysines. The aforementioned aspects make the Success predictor outperform three state-of-the-art predictors in succinylation detection.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4336-8) contains supplementary material, which is available to authorized users.
Keywords: Post-translational modification, Lysine succinylation, Protein sequences, Amino acids, Prediction
Background
Once proteins are translated in the ribosome they undergo a series of chemical modifications known as post-translational modifications (PTMs). These PTMs play multiple biological roles, which influence cellular functions through complex post-translational networks [1, 2], by adding functional groups to specific residues in a protein. Among such PTMs are methylation [3], ubiquitination [4], acetylation [5] and phosphorylation [6]. Although a great number of PTMs have been extensively studied, a new modification coined succinylation [7, 8], has recently caught the interest of the research community. Succinylation was identified through mass spectrometry and sequence alignment [9], and reportedly contributes to the structure and function of proteins [8]. Succinylated enzymes are known to have essential roles in mitochondrial and fatty acid metabolism [10], whereas modified histones were shown to influence the function of chromatin [11].
Understanding how the succinylation mechanism works is of vital importance because of the involvement of this biological mark in cellular processes. Nevertheless, the detection of succinylation sites by traditional experimental techniques has proven to be expensive and time-consuming. In order to overcome these downsides, computational methods have emerged as a necessary detection approach. Some lysine succinylation predictors, which make use of the amino acid composition of proteins, are: iSuc-PseAAC which employs the position-specific propensity of peptides and the pseudo amino acid composition in order to train a support vector machine [12], iSuc-PseOpt [13] and pSuc-Lys [14] that incorporate the sequence-coupling effects into the composition of amino acids and include the k-nearest neighbor strategy for dealing with class imbalance (for prediction purposes, iSuc-PseOpt uses a random forest algorithm [13] whereas pSuc-Lys utilizes an ensemble of random forest classifiers [14]), SucPred which is a learning algorithm that only regards positive and unlabeled samples [15], SuccinSite that incorporates encoding schemes such as k-spaced amino acid pairs, binary scoring and amino acid index properties as input to a random forest classifier [16], and SuccFind which employs evolutionary information along with an improved feature strategy for optimization [17].
However, none of the above methods made use of a combination of structural and evolutionary information. The critical consideration of evolutionary features has been highlighted by a former study, which identified homologous succinylated proteins and conserved orthologs for them in several species [18]. In spite of all the efforts so far, the accurate detection of succinylated residues remains extremely limited. Therefore, new approaches, able to accurately discriminate between succinylated and non-succinylated lysine residues, are absolutely necessary. In this paper, we propose a novel computational predictor called ‘Success’, which efficiently uses structural features such as accessible surface area (ASA), backbone torsion angles and local structure conformations in addition to evolutionary information from the position-specific scoring matrix (PSSM) of proteins for predicting succinylation sites. We regarded the above characteristics due to their reportedly significance for lysine succinylation prediction. For instance, ASA has been previously employed to determine the surface density of succinylated amino groups in pharmacokinetic analyses [19], whereas lysine succinylation was reported to be evolutionarily conserved [8].
This study used a collection of 670 proteins, which contained annotated succinylated and non-succinylated lysines, from two PTM databases [20, 21]. When these sites were retrieved they amounted to 1782 and 18,344 succinylation and non-succinylation residues, respectively. For each lysine residue, we retrieved the sequence stretch comprising 15 amino acids upstream and downstream of it for feature extraction. The PSSM and the local structure with the highest probability were computed for each protein sequence. Both features were used for extracting the submatrix corresponding to each peptide sequence (15 residues, lysine residue, 15 residues) and subsequently converted to bigram probabilities [22]. Considering all the structural and evolutionary features, each lysine residue was defined by a 657-component vector. Because the numbers of succinylation and non-succinylation sites were hugely disproportionate, the resulting training matrix turned out to be unbalanced. For ameliorating this imbalance we employed a k-nearest neighbor strategy [13]. Subsequently, the remaining non-redundant sites were employed to train a support vector machine with a radial basis function kernel for prediction. We compared ‘Success’ with three benchmark predictors (iSuc-PseOpt [13], pSuc-Lys [14] and SuccinSite [16]). As a result, ‘Success’ showed a significant improvement in performance, being able to accurately predict succinylated lysines with 0.866 sensitivity, 0.838 accuracy and 0.677 Matthews correlation coefficient. To the best of our knowledge, these results have not been attained by any available predictor.
Methods
The proposed predictor combines structural and evolutionary information of amino acids with bigram profiles [22] for detecting succinylation and non-succinylation sites. The following sections describe the protein sequence dataset, the computed features, and the support vector machine employed for prediction.
Protein dataset
This study regarded a collection of 670 protein sequences, which were obtained from two PTM databases [20, 21]. The lysine residues of each protein sequence were previously annotated as succinylated or non-succinylated. Because of this, every sequence was analysed and its succinylation and non-succinylation residues were retrieved. As a result, 1782 positive lysines (succinylated) and 18,344 negative lysines (non-succinylated) were obtained.
Structural and evolutionary features
Each protein sequence was used for computing ten different characteristics related to ASA, backbone torsion angles, secondary structure and position-specific scoring matrix (PSSM). The first three types of characteristics were calculated by SPIDER2 [23], a newly developed tool that has reportedly provided reasonable outcomes when it comes to predicting the structural features of proteins [24–29]. For the computation of the PSSM, we used the PSI-BLAST program [30]. These characteristics are detailed in the following subsections.
Accessible surface area
ASA shows the approximate accessible area of each amino acid to a particular solvent in the 3D configuration of a protein [31, 32]. Because the ASA value of an amino acid depends on the protein configuration, considering the predicted values tends to be more informative than using an experimentally determined general value. ASA was computed for proteins with known 3D structures by the SPIDER2 tool [23]. As a result, for each amino acid in a protein sequence, we obtained one numeric ASA value.
Secondary structure
Secondary configuration provides useful information to understand the local 3D structure of proteins. This structure can be studied by looking at the amino acid contributions to local protein structures, namely, helix (ph), strand (pe) and coil (pc). Therefore, we run the SPIDER2 tool [23] on each protein sequence and predicted the contribution likelihood of every amino acid to the three aforementioned local structures. For each protein, we obtained three numerical vectors, each representing a different local structure. In addition, SPIDER2 returns the local structure with the highest likelihood as an L × 3 matrix, where L indicates the length of the protein sequence and the three columns are the contribution likelihoods to the three local structures (helix, strand and coil). Hereafter, this matrix will be referred to as SSpre.
Backbone torsion angles
While secondary structure regards the local configuration of amino acids of a protein [27], torsion angles complement the secondary structure feature by providing continuous information about the local structure of proteins. For instance, the backbone torsion angles ϕ and Ψ provide continuous information about the interaction of local amino acids along the protein backbone [33, 34]. More recent works have proposed two new angles based on the dihedral angles θ and τ [26]. To take the four angles into consideration, we run SPIDER2 [23] on each protein sequence, and attained four numerical vectors referred to as ϕ, Ψ, θ and τ hereafter.
Position-specific scoring matrix
The PSSM has been shown to provide useful evolutionary information about proteins [23–25, 35–37]. This matrix contains the substitution probability of each amino acid in a protein with all the amino acids of the genetic code. In order to compute such probabilities, we aligned each protein sequence to those in Protein Data Bank [38] with the PSI-BLAST algorithm [30]. PSI-BLAST was run on non-redundant proteins, with a threshold of 0.001 and three iterations. For each protein in our benchmark dataset, we thus obtained its respective PSSM which consisted of the linear probabilities of amino acids. The resulting PSSM will have a size of L × 20, where L is the protein length and the 20 columns represent the amino acids of the genetic code.
Lysine residues as feature vectors
Each lysine residue was described in terms of its 15 upstream and 15 downstream amino acids (Fig. 1a). The optimal residue window around a lysine has been widely explored [13, 39, 40]. Previous studies regarded different window sizes and concluded that the 15 amino acids upstream and downstream of a lysine provide useful information about succinylation sites. For this specific study, we also considered several residue windows and trained the predictor (see Additional file 1). Consequently, the same conclusions were drawn. In cases where a lysine was positioned close to either protein terminus, the gap of 15 (upstream or downstream) amino acids was filled by the mirror effect of amino acids [13] (Fig. 1b).
Fig. 1.
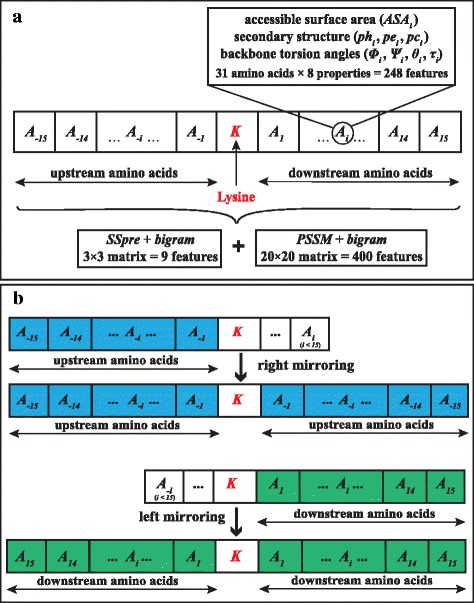
Description of a lysine residue (a) with enough amino acids to both sides, and (b) with missing upstream (left) and downstream (right) amino acids
Now let us consider the following peptide P,
| 1 |
which describes the lysine K, and comprises A−i (1 ≤ i ≤ 15) upstream and Ai (1 ≤ i ≤ 15) downstream amino acids. Thereby, each succinylated or non-succinylated lysine was represented by a peptide P consisting of 31 amino acids (including itself). The 31 amino acids are thus expressed by structural characteristics, such as ASA, ph, pe, pc, ϕ, Ψ, θ and τ. The evolutionary features of peptide P were represented by bigram profiles [22], extracted from the PSSM and denoted here as PSSM + bigram. Similarly, the secondary structure with the highest likelihood was also represented in terms of bigram profiles, extracted from SSpre and denoted as SSpre + bigram. The transformation PSSM + bigram returns a 20 × 20 matrix (or a 400-dimensional feature vector), whereas that of SSpre + bigram results in a 3 × 3 matrix (or a 9-dimensional feature vector). These two vectors were then used to obtain the corresponding information about each lysine in peptide P. We employed the bigram feature extraction technique because of its promising results in solving protein analysis problems [22, 41–48]. The bigram approach is independent of window sizes, which has advantage in this particular study. In other words, it returns 400- (for PSSM) and 9- (for SSpre) dimensional feature vectors regardless of the residue window size. Previous studies have shown that using a large residue window could provide necessary information for discriminating between lysines and their neighbouring amino acids [13]. Therefore, the bigram approach enables us to resize the residue window around a lysine without necessarily increasing the number of features.
The features PSSM and SSpre were transformed into bigram profiles as described below. Let a PSSM of size L × 20 be Ps whose elements mpq represent the transitional probabilities of q-th amino acids at p-th locations in the protein sequence. Thereby, matrix Ps would be represented by a bigram profile [22] as
| 2 |
where 1 ≤ p ≤ 20 and 1 ≤ q ≤ 20.
Eq. (2) will consequently return a 20 × 20 bigram occurrence matrix B, which consists of all the bigram frequencies Bp, q (for p = 1, 2, …, 20 and q = 1, 2, …, 20). This bigram matrix B (or PSSM + bigram) was transformed into a feature vector as
| 3 |
for i = 1, …, 20 and j = 1, …, 20, and where superscript T denotes transpose.
In a similar way, the bigram matrix B′ for SSpre (or SSpre + bigram) can be described as
| 4 |
where 1 ≤ p ≤ 3 and 1 ≤ q ≤ 3, and where elements rp, q are the transition probabilities of each amino acid to the three local conformations (helix, strand and coil).
The bigram matrix B′ was also transformed into a feature vector as
| 5 |
for i = 1, …, 3 and j = 1, …, 3.
Support vector machine for classification
Support vector machine (SVM) is a well-known pattern classification scheme [49], which has been successfully used in regression and classification applications [50–55]. The ultimate goal of SVMs is to maximize the margin between hyperplanes, which represent linear boundaries between classes. To deal with non-linear boundaries, function kernels were consequently introduced [56]. These functions could be radial basis, polynomial or linear. In this work, we designed a SVM that makes use of a radial basis function kernel to find a margin between succinylated (yi = + 1) and non-succinylated (yi = − 1) lysine residues. If the feature vector of i-th lysine residue is defined as xi with class label yi (either succinylated or non-succinylated), then an unknown lysine residue x′ can be predicted by the following function,
| 6 |
where αi are adjustable weights, β represents a bias, n is the number of samples and K() indicates the radial basis function kernel. The SVM classifier was designed with the Weka tool (C = 1, tolerance = 0.001, ϵ = 10−12 and γ = 0.01) [57].
Results and discussion
Any computational approach, aimed at predicting succinylation sites, requires a critical assessment of its performance. The following sections explain the statistical metrics used for evaluation purposes as well as the comparison of the Success predictor and three state-of-the-art predictors.
Performance metrics
We have considered four well-defined metrics for assessing the performance of the Success predictor and other recently proposed predictors. These metrics include sensitivity, specificity, accuracy and Matthews correlation coefficient (MCC) [41, 58–62]. Sensitivity, which varies between 0 and 1, evaluates the correctness of succinylation site identification. A value of 0 indicates the inability of the predictor to detect succinylated lysines (true positive rate), whereas that of 1 depicts a predictor able to correctly identify all the succinylated lysines. Specificity assesses the capability of a predictor to recognize non-succinylation sites (true negative rate). It varies between 0 (completely incorrect classification) and 1 (completely correct classification). Accuracy measures the total number of correctly classified lysine residues, and ranges from 0 (the least accurate predictor) to 1 (the most accurate predictor). MCC indicates the classification quality of a predictor. A value of −1 indicates a completely negative correlation, whereas that of +1 means a highly positive correlation.
Now let us consider a benchmark dataset, which consists of K+ succinylated sites and K− non-succinylated sites. This can be further expressed as
| 7 |
| 8 |
where and are the succinylated residues correctly classified (true positives) as such, and incorrectly classified as non-succinylated sites (false negatives), respectively. Likewise, and are those non-succinylated sites correctly classified (true negatives) as such, and incorrectly classified as succinylated sites (false positives), respectively. The above statistical metrics could be defined as
| 9 |
| 10 |
| 11 |
| 12 |
Any method that performs the highest in all of these metrics would be the ideal predictor. However, an improved predictor should at least show a higher sensitivity when compared with other approaches. This is because a predictor with lower sensitivity is unable to correctly detect succinylated lysine residues, and hence inappropriate for tackling such prediction problems.
Cross-validation strategy
In order to accurately assess the performance of the Success predictor in each statistical metric, we utilized a cross-validation procedure. Two commonly used cross-validation approaches include n-fold cross-validation and jackknife [63, 64]. Although jackknife is regarded to be the least arbitrary approach and yields unique outcomes for a dataset [65], we implemented the n-fold cross-validation procedure which requires less processing time. This cross-validation strategy was conducted in five steps as follows:
Step 1. Randomly partition the initial dataset into n parts of roughly equal size.
Step 2. Retain n − 1 folds as training data and the remaining fold as validation data.
Step 3. Use the training samples for estimating the predictor parameters.
Step 4. Compute the four statistical metrics on the validation fold.
Step 5. Repeat Step 1 to Step 4 n times and calculate the average of each metric.
In this study, we carried out 6-, 8- and 10-fold cross-validations whose results are presented in the subsequent sections.
Dataset balancing
Our benchmark dataset comprised 670 protein sequences, which accounted for 1782 succinylation sites (positive set) and 18,344 non-succinylation sites (negative set). Such a difference between positive and negative sets indicates a huge imbalance between both classes. Although it is reasonable to assume that the number of non-succinylated lysines might be greater than that of succinylated lysines, this disproportion can severely bias any machine learning classifier.
A long list of methods, aimed at dealing with class imbalance, have been proposed. For instance, sampling methods balance the class distribution by subsampling the majority class, or by sampling with replacement the minority class [66]; ensemble methods consider the majority class in a supervised manner, or learn the characteristics of the original majority class in an unsupervised manner [67]; other methods remove noise and instances in the boundaries [68], or remove instances far away from the decision boundary from the majority class [69]. However, because the k-nearest neighbor strategy has been widely used in this scenario [12, 13], we also implemented it in order to establish a fair comparison with previous predictors. In doing so, we first considered a k = 10 (derived from a data ratio of 10:1), and eliminated any negative sample whose 10-nearest neighbours included at least one positive sample. We subsequently increased the k value until similar numbers of positive and negative samples were obtained. As a result, the number of negative samples was drastically reduced to 1872 samples. Both sets were then used to perform n-fold cross-validation, and assess the Success predictor against three benchmark predictors [13, 14, 16].
Success versus benchmark predictors
We compared the Success predictor with three recently proposed predictors, namely, iSuc-PseOpt [13], SuccinSite [16] and pSuc-Lys [14]. These predictors are available as active web servers to which any protein sequence can be uploaded for succinylation site identification. It is worth noting that many of our query proteins were utilized to train these predictors, and therefore the results could be somehow biased in their favour. Besides the performance of the three approaches (iSuc-PseOpt [13], SuccinSite [16] and pSuc-Lys [14]) was reported on the validation data (i.e., samples held-out for testing during n-fold cross-validation). For the Success predictor, this validation data was not used to estimate its training parameters, and thereby we could easily provide the resulting AUCs (area under the curve) for 6-, 8- and 10-fold cross-validations. However, because it was unknown which samples the three benchmark predictors used for training we were unable to report their respective AUCs.
The performance of all the predictors is summarized in Table 1. It can be clearly observed that the proposed predictor outperforms all the benchmark predictors in metrics such as sensitivity, accuracy and MCC. For instance, sensitivity was significantly improved by 40.8%, accuracy by 15%, and MCC by 43.7%. To the best of our knowledge, these promising results have not been achieved by any predictor in the literature. Although SuccinSite [16] showed a high specificity (0.902), its sensitivity was very poor (~0.3), which indicates that ~70% of succinylated lysine residues remained undetected. In addition, the Success predictor reached AUCs of 0.838 for 6-, 8- and 10-fold cross-validations. One of the reasons why the specificity of the proposed predictor turned out lower than that of benchmark methods is because of the extensive removal of negative instances. Those removed sites, though close to positive sites, appear to contain useful information. Nevertheless, this strategy proves to significantly improve sensitivity.
Table 1.
Performance of the Success predictor and three benchmark predictors
| Predictor | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| iSuc-PseOpt [13] | 0.615 | 0.779 | 0.699 | 0.400 |
| SuccinSite [16] | 0.302 | 0.902 | 0.609 | 0.256 |
| pSuc-Lys [14] | 0.587 | 0.864 | 0.729 | 0.471 |
| Success (6-fold cross-validation) | 0.861 | 0.815 | 0.838 | 0.677 |
| Success (8-fold cross-validation) | 0.866 | 0.809 | 0.837 | 0.676 |
| Success (10-fold cross-validation) | 0.864 | 0.811 | 0.837 | 0.676 |
The highest values are highlighted in bold
The above results clearly illustrate the capability of the Success predictor to accurately discriminate between succinylation and non-succinylation sites. Such a combination of evolutionary and structural information apparently provides accurate descriptions of succinylated lysines. Additionally, the transformation of PSSM and SSpre matrices by the bigram feature extraction technique contributes to effectively refine the information of the surrounding amino acids, and thereby capture the differences between each type of lysine. In order to substantiate the previous claim about the importance of the bigram approach, we also trained the proposed predictor without considering any bigram transformation. However, the highest statistical metrics were achieved when the bigram approach was taken into consideration (Table 2). Finally, the SVM classifier with a radial basis function kernel appears to find a maximal hyperplane separation when evolutionary and structural characteristics are employed.
Table 2.
Performance of the Success predictor without regarding the bigram feature extraction strategy
| cross-validation | |||
|---|---|---|---|
| 6-fold | 8-fold | 10-fold | |
| Sensitivity | 0.860 | 0.859 | 0.859 |
| Specificity | 0.813 | 0.813 | 0.809 |
| Accuracy | 0.836 | 0.835 | 0.834 |
| MCC | 0.673 | 0.672 | 0.669 |
| AUC | 0.836 | 0.836 | 0.834 |
Insights into succinylation prediction
We manually analyzed the proteins whose succinylation sites were predicted by the predictors: Success, iSuc-PseOpt [13], SuccinSite [16] and pSuc-Lys [14] (see Additional file 2). It turned out that the four predictors successfully detected all the succinylation sites of specific proteins. These proteins included succinate-CoA ligase subunit alpha (UniProtKB ID Q9WUM5) whose absence causes severe disorders with antenatal manifestations [70], serine hydroxymethyltransferase (UniProtKB ID B1XB26) which regulates the metabolic partitioning of methylenetetrahydrofolate [71], and glutamate dehydrogenase 1 (UniProtKB ID P00366) which is involved in the breakdown and synthesis of the neurotransmitter glutamate [72]. However, the Success predictor was the only one capable of detecting all the succinylation sites of proteins involved in apoptosis and cytoskeleton functions. Some of these proteins included elongation factor 1-alpha 1 (UniProtKB ID P10126) which regulates apoptosis and actin cytoskeleton, and acts as a mediator of lipotoxicity [73] as well as T-complex protein 1 subunit gamma (UniProtKB ID E9Q133) which contributes to assemble and fold cytoskeleton proteins [74]. In addition, the Success predictor correctly detected the only succinylation site, which went undetected by the three benchmark predictors, of other proteins. Two of these proteins are transketolase (UniProtKB ID P40142) that affects the NAPDH production in order to counteract oxidative stress [75], and RNA polymerase I-specific transcription initiation factor RRN3 (UniProtKB ID B2RS91) which acts as a connector between RNA polymerase I and transcription factors [76]. Nevertheless, such a unique succinylated lysine was only detected by iSuc-PseOpt [13], SuccinSite [16] and pSuc-Lys [14] predictors for proteins such as galectin (UniProtKB ID B1AQR8), which acts as an immunomodulatory and enhances transforming growth factor-b signaling [77]. For other proteins, their only succinylated lysine was discovered by all the predictors. These proteins included peptidyl-prolyl cis-trans isomerase FKBP3 (UniProtKB ID Q62446) which stimulates auto-ubiquitylation and proteasomal degradation [78], proline dehydrogenase (UniProtKB ID F6YFQ5) that causes DNA damage-induced senescence [79], and sulfite oxidase (UniProtKB ID Q8R086) which catalyses the oxidation of toxic sulfite to sulfate [80]. Finally, none of the predictors was able to detect the succinylation sites of other proteins. A few examples are lon protease homolog (UniProtKB ID Q8CGK3) which recognises and degrades unfolded proteins [81], caveolin (UniProtKB ID D3Z0J2) which is involved in vesicular transport, cholesterol homeostasis, signal transduction and tumor suppression [82], and kinesin-like protein (UniProtKB ID E9PWU7) that caps microtubules released from the centrosome during interphase [83].
These results indicate that although the Success predictor detected a large number of succinylation sites in comparison to the other predictors, all the predictors should be used in a complementary way for more complete outcomes.
Conclusions
This study proposes a new computational predictor called ‘Success’, which is aimed at detecting succinylation sites of modified proteins. The proposed method makes an optimum use of the structural and evolutionary information of amino acids around lysine residues. Features such as PSSM and SSpre were transformed into frequency vectors using the bigram feature extraction approach, which proved effective to describe each type of lysine. The studied characteristics were appropriate for the SVM classifier with a radial basis function kernel to find the maximal separation between modified and unmodified lysine residues.
Additional files
Performance of the Success predictor using 6-, 8- and 10-fold cross-validation. (DOCX 148 kb)
Numbers of succinylation sites detected by each predictor. (XLS 119 kb)
Acknowledgements
Yosvany López and Alok Sharma thank the members of Tsunoda Laboratory for their constructive comments and suggestions.
Funding
Publication of this article was funded by JSPS KAKENHI Grant Number 15F15385, and partly supported by JST CREST Grant Number JPMJCR1412, Japan.
Availability of data and materials
The training matrix and SVM classifier script can be found at https://github.com/YosvanyLopez/Success.
About this supplement
This article has been published as part of BMC Genomics Volume 19 Supplement 1, 2018: 16th International Conference on Bioinformatics (InCoB 2017): Genomics. The full contents of the supplement are available online at https://bmcgenomics.biomedcentral.com/articles/supplements/volume-19-supplement-1.
Abbreviations
- ASA
Accessible surface area
- AUC
Area under the curve
- MCC
Matthews correlation coefficient
- PSSM
Position-specific scoring matrix
- PTM
Post-translational modification
- SVM
Support vector machine
Authors’ contributions
YL and AS conducted the classification part of the project and drafted the manuscript. AD and SPL extracted the structural and evolutionary information of proteins and improved the draft. GT and ABS analyzed the outcomes of the benchmark predictors for comparison purposes. TT supervised the entire project. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4336-8) contains supplementary material, which is available to authorized users.
Contributor Information
Yosvany López, Email: yosvany.lopez.alvarez@gmail.com.
Alok Sharma, Email: alok.sharma@griffith.edu.au.
Abdollah Dehzangi, Email: abdollah.dehzangi@griffithuni.edu.au.
Sunil Pranit Lal, Email: lal.sunil@gmail.com.
Ghazaleh Taherzadeh, Email: ghazaleh.taherzadeh@griffithuni.edu.au.
Abdul Sattar, Email: a.sattar@griffith.edu.au.
Tatsuhiko Tsunoda, Email: tsunoda.mesm@mri.tmd.ac.jp.
References
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed. 2005;44(45):7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Xu Y, Chou K-C. Recent progress in predicting posttranslational modification sites in proteins. Curr Top Med Chem. 2016;16(6):591–603. doi: 10.2174/1568026615666150819110421. [DOI] [PubMed] [Google Scholar]
- 3.Qiu W-R, Xiao X, Lin W-Z, Chou K-C. iMethyl-PseAAC: identification of protein Methylation sites via a pseudo amino acid composition approach. Biomed Res Int. 2014;2014:947416. doi: 10.1155/2014/947416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiu W-R, Xiao X, Lin W-Z, Chou K-C. iUbiq-Lys: prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a gray system model. J Biomol Struct Dyn. 2015;33(8):1731–1742. doi: 10.1080/07391102.2014.968875. [DOI] [PubMed] [Google Scholar]
- 5.Hou T, Zheng G, Zhang P, Jia J, Li J, Xie L, Wei C, Li Y. LAceP: lysine Acetylation site prediction using logistic regression classifiers. PLoS One. 2014;9(2):e89575. doi: 10.1371/journal.pone.0089575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ubersax JA, Ferrell JE. Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8(7):530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 7.Weinert BT, Schölz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C. Lysine Succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with Acetylation. Cell Rep. 2013;4(4):842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. 2011;7(1):58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen ON. Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr Opin Chem Biol. 2004;8(1):33–41. doi: 10.1016/j.cbpa.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 10.Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BMM, Skinner ME, et al. SIRT5-mediated lysine Desuccinylation impacts diverse metabolic pathways. Mol Cell. 2013;50(6):919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, Zhao Y. Lysine Succinylation and lysine Malonylation in Histones. Mol Cell Proteomics. 2012;11(5):100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Ding Y-X, Ding J, Lei Y-H, Wu L-Y, Deng N-Y. iSuc-PseAAC: predicting lysine succinylation in proteins by incorporating peptide position-specific propensity. Sci Rep. 2015;5:10184. doi: 10.1038/srep10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia J, Liu Z, Xiao X, Liu B, Chou K-C. iSuc-PseOpt: identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal Biochem. 2016;497:48–56. doi: 10.1016/j.ab.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Jia J, Liu Z, Xiao X, Liu B, Chou K-C. pSuc-Lys: predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J Theor Biol. 2016;394:223–230. doi: 10.1016/j.jtbi.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 15.Zhao X, Ning Q, Chai H, Ma Z. Accurate in silico identification of protein succinylation sites using an iterative semi-supervised learning technique. J Theor Biol. 2015;374:60–65. doi: 10.1016/j.jtbi.2015.03.029. [DOI] [PubMed] [Google Scholar]
- 16.Hasan MM, Yang S, Zhou Y, Mollah MNH. SuccinSite: a computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol BioSyst. 2016;12(3):786–795. doi: 10.1039/C5MB00853K. [DOI] [PubMed] [Google Scholar]
- 17.Xu H-D, Shi S-P, Wen P-P, Qiu J-D. SuccFind: a novel succinylation sites online prediction tool via enhanced characteristic strategy. Bioinformatics. 2015;31(23):3748–3750. doi: 10.1093/bioinformatics/btv439. [DOI] [PubMed] [Google Scholar]
- 18.Zhen S, Deng X, Wang J, Zhu G, Cao H, Yuan L, Yan Y. First comprehensive proteome analyses of lysine Acetylation and Succinylation in seedling leaves of Brachypodium distachyon L. Sci Rep. 2016;6:31576. doi: 10.1038/srep31576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamasaki Y, Sumimoto K, Nishikawa M, Yamashita F, Yamaoka K, Hashida M, Takakura Y. Pharmacokinetic analysis of in vivo disposition of Succinylated proteins targeted to liver Nonparenchymal cells via scavenger receptors: importance of molecular size and negative charge density for in vivo recognition by receptors. J Pharmacol Exp Ther. 2002;301(2):467–477. doi: 10.1124/jpet.301.2.467. [DOI] [PubMed] [Google Scholar]
- 20.Liu Z, Wang Y, Gao T, Pan Z, Cheng H, Yang Q, Cheng Z, Guo A, Ren J, Xue Y. CPLM: a database of protein lysine modifications. Nucleic Acids Res. 2014;42(Database issue):D531–D536. doi: 10.1093/nar/gkt1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z, Cao J, Gao X, Zhou Y, Wen L, Yang X, Yao X, Ren J, Xue Y. CPLA 1.0: an integrated database of protein lysine acetylation. Nucleic Acids Res. 2011;39(Database issue):D1029–D1034. doi: 10.1093/nar/gkq939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma A, Lyons J, Dehzangi A, Paliwal KK. A feature extraction technique using bi-gram probabilities of position specific scoring matrix for protein fold recognition. J Theor Biol. 2013;320:41–46. doi: 10.1016/j.jtbi.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 23.Heffernan R, Paliwal K, Lyons J, Dehzangi A, Sharma A, Wang J, Sattar A, Yang Y, Zhou Y. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci Rep. 2015;5:11476. doi: 10.1038/srep11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faraggi E, Zhang T, Yang Y, Kurgan L, Zhou Y. SPINE X: improving protein secondary structure prediction by multistep learning coupled with prediction of solvent accessible surface area and backbone torsion angles. J Comput Chem. 2012;33(3):259–267. doi: 10.1002/jcc.21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 26.Lyons J, Dehzangi A, Heffernan R, Sharma A, Paliwal K, Sattar A, Zhou Y, Yang Y. Predicting backbone Cα angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network. J Comput Chem. 2014;35(28):2040–2046. doi: 10.1002/jcc.23718. [DOI] [PubMed] [Google Scholar]
- 27.Faraggi E, Yang Y, Zhang S, Zhou Y. Predicting continuous local structure and the effect of its substitution for secondary structure in fragment-free protein structure prediction. Structure. 2009;17(11):1515–1527. doi: 10.1016/j.str.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heffernan R, Dehzangi A, Lyons J, Paliwal K, Sharma A, Wang J, Sattar A, Zhou Y, Yang Y. Highly accurate sequence-based prediction of half-sphere exposures of amino acid residues in proteins. Bioinformatics. 2016;32(6):843–849. doi: 10.1093/bioinformatics/btv665. [DOI] [PubMed] [Google Scholar]
- 29.Yang Y, Heffernan R, Paliwal K, Lyons J, Dehzangi A, Sharma A, Wang J, Sattar A, Zhou Y: SPIDER2: A Package to Predict Secondary Structure, Accessible Surface Area, and Main-Chain Torsional Angles by Deep Neural Networks. In: Prediction of Protein Secondary Structure. Edited by Zhou Y, Kloczkowski A, Faraggi E, Yang Y, vol. 1484: Springer New York; 2016: 55-63. [DOI] [PubMed]
- 30.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lins L, Thomas A, Brasseur R. Analysis of accessible surface of residues in proteins. Protein Sci. 2003;12(7):1406–1417. doi: 10.1110/ps.0304803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan B-B, Yang F, Ye Y, Wu Q, Li C, Huber T, Su X-C. 3D structure determination of a protein in living cells using paramagnetic NMR spectroscopy. Chem Commun. 2016;52(67):10237–10240. doi: 10.1039/C6CC05490K. [DOI] [PubMed] [Google Scholar]
- 33.Dor O, Zhou Y. Real-SPINE: an integrated system of neural networks for real-value prediction of protein structural properties. Proteins: Structure, Function, and Bioinformatics. 2007;68(1):76–81. doi: 10.1002/prot.21408. [DOI] [PubMed] [Google Scholar]
- 34.Xue B, Dor O, Faraggi E, Zhou Y. Real-value prediction of backbone torsion angles. Proteins: Structure, Function, and Bioinformatics. 2008;72:427–433. doi: 10.1002/prot.21940. [DOI] [PubMed] [Google Scholar]
- 35.Dehzangi A, Paliwal K, Lyons J, Sharma A, Sattar A. Proposing a highly accurate protein structural class predictor using segmentation-based features. BMC Genomics. 2014;15(Suppl 1):S2. doi: 10.1186/1471-2164-15-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taherzadeh G, Yang Y, Zhang T, Liew AW-C, Zhou Y. Sequence-based prediction of protein–peptide binding sites using support vector machine. J Comput Chem. 2016;37(13):1223–1229. doi: 10.1002/jcc.24314. [DOI] [PubMed] [Google Scholar]
- 37.Taherzadeh G, Zhou Y, Liew AW-C, Yang Y. Sequence-based prediction of protein-carbohydrate binding sites using support vector machines. J Chem Inf Model. 2016;56(10):2115–2122. doi: 10.1021/acs.jcim.6b00320. [DOI] [PubMed] [Google Scholar]
- 38.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dehzangi A, López Y, Lal SP, Taherzadeh G, Michaelson J, Sattar A, Tsunoda T, Sharma A. PSSM-Suc: accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction. J Theor Biol. 2017;425:97–102. doi: 10.1016/j.jtbi.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 40.López Y, Dehzangi A, Lal SP, Taherzadeh G, Michaelson J, Sattar A, Tsunoda T, Sharma A. SucStruct: prediction of succinylated lysine residues by using structural properties of amino acids. Anal Biochem. 2017;527:24–32. doi: 10.1016/j.ab.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 41.Dehzangi A, Heffernan R, Sharma A, Lyons J, Paliwal K, Sattar A. Gram-positive and gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into Chou's general PseAAC. J Theor Biol. 2015;364:284–294. doi: 10.1016/j.jtbi.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 42.Paliwal KK, Sharma A, Lyons J, Dehzangi A. A tri-gram based feature extraction technique using linear probabilities of position specific scoring matrix for protein fold recognition. IEEE Transactions on NanoBioscience. 2014;13(1):44–50. doi: 10.1109/TNB.2013.2296050. [DOI] [PubMed] [Google Scholar]
- 43.Dehzangi A, Sohrabi S, Heffernan R, Sharma A, Lyons J, Paliwal K, Sattar A. Gram-positive and gram-negative subcellular localization using rotation forest and physicochemical-based features. BMC Bioinformatics. 2015;16(Suppl 4):S1. doi: 10.1186/1471-2105-16-S4-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma R, Dehzangi A, Lyons J, Paliwal K, Tsunoda T, Sharma A. Predict gram-positive and gram-negative subcellular localization via incorporating evolutionary information and physicochemical features into Chou's general PseAAC. IEEE Transactions on NanoBioscience. 2015;14(8):915–926. doi: 10.1109/TNB.2015.2500186. [DOI] [PubMed] [Google Scholar]
- 45.Nanni L, Brahnam S, Lumini A. Prediction of protein structure classes by incorporating different protein descriptors into general Chou’s pseudo amino acid composition. J Theor Biol. 2014;360:109–116. doi: 10.1016/j.jtbi.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 46.Wei L, Liao M, Gao X, Zou Q. Enhanced protein fold prediction method through a novel feature extraction technique. IEEE Transactions on NanoBioscience. 2015;14(6):649–659. doi: 10.1109/TNB.2015.2450233. [DOI] [PubMed] [Google Scholar]
- 47.Hayat M, Tahir M, Khan SA. Prediction of protein structure classes using hybrid space of multi-profile Bayes and bi-gram probability feature spaces. J Theor Biol. 2014;346:8–15. doi: 10.1016/j.jtbi.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 48.Zakeri P, Jeuris B, Vandebril R, Moreau Y. Protein fold recognition using geometric kernel data fusion. Bioinformatics. 2014;30(13):1850–1857. doi: 10.1093/bioinformatics/btu118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vapnik VN. The nature of statistical learning theory. New York: Springer; 1995. [Google Scholar]
- 50.Ben-Hur A, Horn D, Siegelmann HT, Vapnik V. Support vector clustering. J Mach Learn Res. 2001;2:125–137. [Google Scholar]
- 51.Lyons J, Biswas N, Sharma A, Dehzangi A, Paliwal KK. Protein fold recognition by alignment of amino acid residues using kernelized dynamic time warping. J Theor Biol. 2014;354:137–145. doi: 10.1016/j.jtbi.2014.03.033. [DOI] [PubMed] [Google Scholar]
- 52.Lyons J, Dehzangi A, Heffernan R, Yang Y, Zhou Y, Sharma A, Paliwal K. Advancing the accuracy of protein fold recognition by utilizing profiles from hidden Markov models. IEEE Trans Nanobioscience. 2015;14(7):761–772. doi: 10.1109/TNB.2015.2457906. [DOI] [PubMed] [Google Scholar]
- 53.Lyons J, Paliwal KK, Dehzangi A, Heffernan R, Tsunoda T, Sharma A. Protein fold recognition using HMM-HMM alignment and dynamic programming. J Theor Biol. 2016;393:67–74. doi: 10.1016/j.jtbi.2015.12.018. [DOI] [PubMed] [Google Scholar]
- 54.Suykens JAK, Vandewalle J. Least squares support vector machine classifiers. Neural Process Lett. 1999;9(3):293–300. doi: 10.1023/A:1018628609742. [DOI] [Google Scholar]
- 55.Tong S, Koller D. Support vector machine active learning with applications to text classification. J Mach Learn Res. 2002;2:45–66. [Google Scholar]
- 56.Bishop CM. Pattern recognition and machine learning. New York: Springer; 2006. [Google Scholar]
- 57.Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH. The WEKA data mining software: an update. SIGKDD Explorations. 2009;11(1):10–18. doi: 10.1145/1656274.1656278. [DOI] [Google Scholar]
- 58.Chen W, Feng P, Ding H, Lin H, Chou K-C. iRNA-methyl: identifying N6-methyladenosine sites using pseudo nucleotide composition. Anal Biochem. 2015;490:26–33. doi: 10.1016/j.ab.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 59.Ding H, Deng E-Z, Yuan L-F, Liu L, Lin H, Chen W, Chou K-C. iCTX-type: a sequence-based predictor for identifying the types of Conotoxins in targeting ion channels. Biomed Res Int. 2014;2014:286419. doi: 10.1155/2014/286419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu B, Fang L, Wang S, Wang X, Li H, Chou K-C. Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. J Theor Biol. 2015;385:153–159. doi: 10.1016/j.jtbi.2015.08.025. [DOI] [PubMed] [Google Scholar]
- 61.Liu Z, Xiao X, Qiu W-R, Chou K-C. iDNA-methyl: identifying DNA methylation sites via pseudo trinucleotide composition. Anal Biochem. 2015;474:69–77. doi: 10.1016/j.ab.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 62.Xiao X, Min J-L, Lin W-Z, Liu Z, Cheng X, Chou K-C. iDrug-target: predicting the interactions between drug compounds and target proteins in cellular networking via benchmark dataset optimization approach. J Biomol Struct Dyn. 2015;33(10):2221–2233. doi: 10.1080/07391102.2014.998710. [DOI] [PubMed] [Google Scholar]
- 63.Alpaydin E: Introduction to Machine Learning, Third edn: The MIT Press; 2014.
- 64.Chou K-C, Shen H-B. Cell-PLoc: a package of web servers for predicting subcellular localization of proteins in various organisms. Nat Protoc. 2008;3(2):153–162. doi: 10.1038/nprot.2007.494. [DOI] [PubMed] [Google Scholar]
- 65.Hajisharifi Z, Piryaiee M, Beigi MM, Behbahani M, Mohabatkar H. Predicting anticancer peptides with Chou's pseudo amino acid composition and investigating their mutagenicity via Ames test. J Theor Biol. 2014;341:34–40. doi: 10.1016/j.jtbi.2013.08.037. [DOI] [PubMed] [Google Scholar]
- 66.Japkowicz N, Stephen S. The class imbalance problem: a systematic study. Intelligent Data Analysis. 2002;6(5):429–449. [Google Scholar]
- 67.Liu X-Y, Wu J, Zhou Z-H. Exploratory Undersampling for class-imbalance learning. IEEE Transactions on Systems, Man, and Cybernetics, Part B (Cybernetics) 2009;39(2):539–550. doi: 10.1109/TSMCB.2008.2007853. [DOI] [PubMed] [Google Scholar]
- 68.Tomek I. Two modifications of CNN. IEEE Transaction on Systems Man and Communications. 1976;6:769–772. [Google Scholar]
- 69.Hart P. The condensed nearest neighbor rule. IEEE Trans Inf Theory. 1968;14(3):515–516. doi: 10.1109/TIT.1968.1054155. [DOI] [Google Scholar]
- 70.Rouzier C, Guédard-Méreuze SL, Fragaki K, Serre V, Miro J, Tuffery-Giraud S, Chaussenot A, Bannwarth S, Caruba C, Ostergaard E, et al. The severity of phenotype linked to SUCLG1 mutations could be correlated with residual amount of SUCLG1 protein. J Med Genet. 2010;47(10):670–676. doi: 10.1136/jmg.2009.073445. [DOI] [PubMed] [Google Scholar]
- 71.MacFarlane AJ, Liu X, Perry CA, Flodby P, Allen RH, Stabler SP, Stover PJ. Cytoplasmic serine Hydroxymethyltransferase regulates the metabolic partitioning of Methylenetetrahydrofolate but is not essential in mice. J Biol Chem. 2008;283(38):25846–25853. doi: 10.1074/jbc.M802671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frigerio F, Karaca M, Roo MD, Mlynárik V, Skytt DM, Carobbio S, Pajęcka K, Waagepetersen HS, Gruetter R, Muller D, et al. Deletion of glutamate dehydrogenase 1 (Glud1) in the central nervous system affects glutamate handling without altering synaptic transmission. J Neurochem. 2012;123(3):342–348. doi: 10.1111/j.1471-4159.2012.07933.x. [DOI] [PubMed] [Google Scholar]
- 73.Stoianov AM, Robson DL, Hetherington AM, Sawyez CG, Borradaile NM. Elongation factor 1A-1 is a mediator of Hepatocyte lipotoxicity partly through its canonical function in protein synthesis. PLoS One. 2015;10(6):e0131269. doi: 10.1371/journal.pone.0131269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhaskar, Kumari N, Goyal N. Cloning, characterization and sub-cellular localization of gamma subunit of T-complex protein-1 (chaperonin) from Leishmania donovani. Biochem Biophys Res Commun. 2012;429(1-2):70–74. doi: 10.1016/j.bbrc.2012.10.090. [DOI] [PubMed] [Google Scholar]
- 75.Xu IM-J, Lai RK-H, Lin S-H, Tse AP-W, Chiu DK-C, Koh H-Y, Law C-T, Wong C-M, Cai Z, Wong CC-L, et al. Transketolase counteracts oxidative stress to drive cancer development. Proc Natl Acad Sci U S A. 2016;113(6):E725–E734. doi: 10.1073/pnas.1508779113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stepanchick A, Zhi H, Cavanaugh AH, Rothblum K, Schneider DA, Rothblum LI. DNA binding by the ribosomal DNA transcription factor Rrn3 is essential for ribosomal DNA transcription. J Biol Chem. 2013;288:9135–9144. doi: 10.1074/jbc.M112.444265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ikeda M, Katoh S, Shimizu H, Hasegawa A, Ohashi-Doi K, Oka M. Beneficial effects of Galectin-9 on allergen-specific sublingual immunotherapy in a Dermatophagoides farinae-induced mouse model of chronic asthma. Allergol Int. 2017;66(2017):432–439. doi: 10.1016/j.alit.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 78.Ochocka AM, Kampanis P, Nicol S, Allende-Vega N, Cox M, Marcar L, Milne D, Fuller-Pace F, Meek D. FKBP25, a novel regulator of the p53 pathway, induces the degradation of MDM2 and activation of p53. FEBS Lett. 2009;583(2009):621–626. doi: 10.1016/j.febslet.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 79.Nagano T, Nakashima A, Onishi K, Kawai K, Awai Y, Kinugasa M, Iwasaki T, Kikkawa U, Kamada S. Proline dehydrogenase promotes senescence through the generation of reactive oxygen species. J Cell Sci. 2017;130:1413–1420. doi: 10.1242/jcs.196469. [DOI] [PubMed] [Google Scholar]
- 80.Belaidi AA, Röper J, Arjune S, Krizowski S, Trifunovic A, Schwarz G: Oxygen reactivity of mammalian sulfite oxidase provides a concept for the treatment of sulfite oxidase deficiency. Biochem J 2015, 469(2):211-221. [DOI] [PubMed]
- 81.Bezawork-Geleta A, Brodie EJ, Dougan DA, Truscott KN. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci Rep. 2015;5:17397. doi: 10.1038/srep17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Williams TM, Lisanti MP. The caveolin proteins. Genome Biol. 2004;5:214. doi: 10.1186/gb-2004-5-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nachbar J, Lázaro-Diéguez F, Prekeris R, Cohen D, Müsch A. KIFC3 promotes mitotic progression and integrity of the central spindle in cytokinesis. Cell Cycle. 2014;13(3):426–433. doi: 10.4161/cc.27266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Performance of the Success predictor using 6-, 8- and 10-fold cross-validation. (DOCX 148 kb)
Numbers of succinylation sites detected by each predictor. (XLS 119 kb)
Data Availability Statement
The training matrix and SVM classifier script can be found at https://github.com/YosvanyLopez/Success.