

Abstract
Background
Influenza A viruses circulating in pigs in Brazil are still not characterized, and only limited data are available about swine influenza epidemiology in the country. Therefore, we characterized the hemagglutinin (HA) and neuraminidase (NA) genes of influenza viruses isolated from Brazilian pigs. We also evaluated one case of probable swine‐to‐human transmission.
Methods
Twenty influenza viruses isolated from pigs during 2009–2010 in five Brazilian states (Minas Gerais, Sao Paulo, Parana, Rio Grande do Sul, and Mato Grosso) were used. One human isolate, from a technician who became ill after visiting a swineherd going through a respiratory disease outbreak, was also used in the study. Phylogenetic analysis for the HA and NA genes and hemagglutinin amino acid sequence alignment were performed.
Results
All isolates clustered with pandemic H1N1 2009 (pH1N1) viruses and appeared to have a common ancestor. Genetic diversity was higher in the HA than in the NA gene, and the amino acid substitution S203T in one of HA's antigenic sites was found in most of the samples. The human isolate was more related to swine isolates from the same herd visited by the technician than to other human isolates, suggesting swine‐to‐human transmission.
Conclusion
Our results show that pH1N1 was disseminated and the predominant subtype in Brazilian pigs in 2009–2010.
Keywords: Genetic characterization, hemagglutinin, influenza A virus, pandemic, swine
Introduction
Influenza A virus (IAV) infection in swine poses a threat to pig production and has important implications for the epidemiology and control of influenza infection in other species.1 IAVs have an antisense RNA genome that is divided into eight segments, which allows genetic reassortment between different viruses.2 The viral surface glycoproteins hemagglutinin (HA) and neuraminidase (NA) are the main targets of the host immune response, and they are important for virulence and host specificity.3 Three subtypes of swine influenza virus (SIV) are currently circulating in the swine population globally: H1N1, H3N2, and H1N2.1
Influenza A infection in pigs has been well characterized both in the United States and in Europe.4, 5 The classical SIV (cH1N1), which is similar to the human 1918 pandemic virus, remained antigenic and genetically conserved in the United States until the introduction of an H3N2 virus in 1998.5 The cocirculation of cH1N1 and the H3N2 virus led to reassortment and resulted in the introduction of the reassorted H3N2, H1N1,6 and H1N2 subtypes.7 Reassortant viruses currently endemic in North American swine have combinations of different HA and NA genes with a triple‐reassortant internal gene (TRIG) cassette formed by the human lineage PB1 gene, the avian lineage PA and PB2 genes, and the classical swine lineage M, NP, and NS genes.5 A cluster classification best depicts circulating H1 viruses; the α‐, β‐, and γ‐clusters are represented by cH1N1‐derived subtypes, and the δ‐cluster is represented by H1 strains carrying an HA gene that is most closely related to human seasonal viruses. Both N1 and N2 subtypes can be found in all four clusters.8 In Europe, an avian‐derived virus was the predominant H1N1 strain in swine until the introduction of a human H3N2 virus that led to a genetic reassortment in the mid‐1980s.4 The H1N2 subtype, with human lineage HA and NA genes and other genes from avian‐like H1N1 viruses, is also endemic in European pigs.9 Influenza A virus circulation in Brazilian pigs has been demonstrated by serological studies10, 11 and some viruses were isolated,12, 13 but the epidemiology of the infection is still unknown in the country.
After the introduction and spread of the swine‐origin pandemic H1N1 virus (pH1N1) in humans in 2009, a number of countries, including Brazil, reported infection in pigs.12, 14 The pandemic H1N1 has the M and NA genes from avian‐like H1N1 Eurasian SIV and the other six genes from the North American triple‐reassortant swine lineage.15
Although influenza A virus infection has already been detected in swine in Brazil, these circulating viruses have not been genetically characterized and subtyped in most cases.12, 13 Furthermore, the evolutionary and epidemiologic features of IAV strains circulating in pigs in Brazil are still unknown. Therefore, in this study, we genetically characterized the HA and NA genes of 20 influenza viruses isolated from pigs during 2009 and 2010 in five Brazilian states, which together account for more than 50% of the country's pig population.
Materials and methods
Viral samples
Twenty virus isolates from swine lung tissue samples collected in Brazil were used in this study. The viruses were isolated from lung samples sent for routine diagnosis of respiratory disease to a private veterinary diagnostic laboratory during the years of 2009 and 2010. The lung samples were obtained during respiratory disease outbreaks in 13 swineherds located in five Brazilian states: Minas Gerais (Table 1: herds A‐F and I‐K), São Paulo (Table 1: Herd M), Paraná (Table 1: Herd L), Rio Grande do Sul (Table 1: Herd H), and Mato Grosso (Table 1: Herd G). All herds had animals showing acute respiratory signs, such as cough, fever, nasal discharge, and anorexia, mainly in nursery and grower pigs. Viruses were grown in MDCK cell culture and subsequently tested by HA, as described previously.16 Samples were numbered 1/2009–17/2009 and 18/2010–20/2010.
Table 1.
Complete characterization of viral samples studied
| Sample name | Herd | State | Host species | Year of collection | Accession number | |
|---|---|---|---|---|---|---|
| HA | NA | |||||
| A/swine/Brazil/1 | A | Minas Gerais | swine | 2009 | JQ666845 | JQ666866 |
| A/swine/Brazil/2 | swine | 2009 | JQ666846 | JQ666867 | ||
| A/swine/Brazil/3 | swine | 2009 | JQ666847 | JQ666868 | ||
| A/swine/Brazil/4 | B | Minas Gerais | swine | 2009 | JQ666848 | JQ666869 |
| A/swine/Brazil/5 | C | Minas Gerais | swine | 2009 | JQ666849 | JQ666870 |
| A/swine/Brazil/6 | swine | 2009 | JQ666850 | JQ666871 | ||
| A/swine/Brazil/7 | D | Minas Gerais | swine | 2009 | JQ666851 | JQ666872 |
| A/swine/Brazil/8 | E | Minas Gerais | swine | 2009 | JQ666852 | JQ666873 |
| A/swine/Brazil/9 | swine | 2009 | JQ666853 | JQ666874 | ||
| A/swine/Brazil/10 | swine | 2009 | JQ666854 | JQ666875 | ||
| A/swine/Brazil/11 | F | Minas Gerais | swine | 2009 | JQ666855 | JQ666876 |
| A/swine/Brazil/12 | G | MatoGrosso | swine | 2009 | JQ666856 | JQ666877 |
| A/swine/Brazil/13 | H | Rio Grande do Sul | swine | 2009 | JQ666857 | JQ666878 |
| A/swine/Brazil/14 | I | Minas Gerais | swine | 2009 | JQ666858 | JQ666879 |
| A/swine/Brazil/15 | J | Minas Gerais | swine | 2009 | JQ666859 | JQ666880 |
| A/swine/Brazil/16 | K | Minas Gerais | swine | 2009 | JQ666860 | JQ666881 |
| A/swine/Brazil/17 | swine | 2009 | JQ666861 | JQ666882 | ||
| A/swine/Brazil/18 | L | Paraná | swine | 2010 | JQ666862 | JQ666883 |
| A/swine/Brazil/19 | M | São Paulo | swine | 2010 | JQ666863 | JQ666884 |
| A/swine/Brazil/20 | swine | 2010 | JQ666864 | JQ666885 | ||
| A/Minas Gerais/21 | K | Minas Gerais | human | 2009 | JQ666865 | JQ666886 |
One human‐derived isolate, from a technician who showed influenza‐like respiratory symptoms after visiting a swineherd in Minas Gerais state (Table 1: Herd K) in which a respiratory disease outbreak was taking place, was also included in the study. Real‐time reverse transcription polymerase chain reaction (RT‐PCR) of the human nasal swab sample was performed by Brazilian health authorities according to the CDC protocol,17 confirming swine lineage influenza A/H1 infection. The human sample was named A/Minas Gerais/21/2010, and written authorization was given for the use of the sample and information.
The sample names, properties, state of origin, host species, year of collection, and GenBank accession numbers are shown in Table 1.
This study was approved by the Ethics Committee of Universidade Federal de Minas Gerais and followed the legal requirements set by the Genetic Heritage Management Council – CGEN of the Brazilian Ministry of the Environment.
RNA extraction and reverse transcription
Viral RNA was extracted from 140 μl cell supernatant using QIAamp viral RNA mini kit (QIAGEN Inc., Valencia, CA, USA) according to the manufacturer's specifications. Ten microliters of RNA were transcribed to cDNA at 42°C for 60 minutes using 0·2 μm of Uni12 primer (5′‐AGCAAAAGCAGG‐3′),16 100 U of M‐MLV reverse transcriptase (Promega, Madison, WI, USA), 5 μl of 5 × reaction buffer, 0·8 mm of each dNTP, 25 U of RNasin ribonuclease inhibitor, and water to a final volume of 25 μl.
HA and NA full‐length PCR amplification
Viral isolation was confirmed by real‐time RT‐PCR using primers for the matrix (M) gene, as described previously.17 Full‐length HA and NA genes were amplified using forward and reverse external primers published by18 (HAF1 and HAR1778; NAF1 and NAR1413), with 5 μl of 5 × reaction buffer, 1·25 U of GoTaq Hot Start DNA Polymerase (5 U/μl; Promega), 2 mm of MgCl2, 0·2 mm of each dNTP, 0·2 μm of each primer, 0·5 μg of cDNA, and nuclease‐free water to a final volume of 25 μl. The amplification reaction was performed under the following conditions: denaturation at 94°C for 5 minutes; 30 amplification cycles of denaturation at 94°C for 30 seconds, primer annealing at 60°C for 30 seconds, and extension at 72°C for 3 minutes; and a final extension at 72°C for 7 minutes.
Subsequently, the PCR products were submitted to separate nested PCR reactions using external–internal (HAF1 with HAR952 5′‐GRAGGCTGGTGTTTATAGCACC‐3′; NAF1 with NAR815 5′‐GGAGCATTCCTCRTAGTGRTAATTAGG‐3′) or internal–external (HAF809 5′‐CGAAGCAACTGGAAATCTAGTGG‐3′ with HAR1778; NAF646 5′‐TRAGAACACAAGAGTCWGAATGTG‐3′ with NAR1413) primer pairs. Each reaction consisted of 5 μl of 5 × reaction buffer, 1·25 U of GoTaq Hot Start DNA Polymerase (5 U/μl; Promega), 2 mm of MgCl2, 0·2 mm of each dNTP, 0·1 μm of each primer, 0·5 μg of cDNA, and nuclease‐free water to a final volume of 25 μl. The thermocycling conditions were: denaturation at 94°C for 5 minutes; 30 amplification cycles of denaturation at 94°C for 30 seconds, primer annealing at 60°C for 30 seconds, and extension at 72°C for 1 minute; and a final extension at 72°C for 7 minutes. Nested PCR products were visualized on a 1% agarose gel stained with ethidium bromide.
Sequencing and phylogenetic analysis
The HA and NA genes were sequenced bidirectionally from at least 2 distinct amplification products using a bigdye terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA), and reactions were resolved in ABI 3130 DNA Analyzer (Applied Biosystems). Sequences were edited with seqscape software v2.5 (Applied Biosystems) and aligned using ClustalW. The final sequences were deposited into GenBank under the accession numbers JQ666845‐JQ666886 and numbered from 1 to 17 for 2009 viruses and 18 to 20 for 2010 viruses (Table 1). The human and swine influenza virus sequences used for the phylogenetic analysis were obtained from the Influenza virus sequence Database (http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nphselect.cgi?go=database), comprising well‐known pandemic H1N1 sequences, deposited Brazilian sequences, and sequences with high homology to the isolates. The phylogenetic analysis for each gene was conducted using the neighbor‐joining evolutionary distance method19 with the Tamura–Nei model in mega 5.01.20 Bootstrap tests of 1000 replicates were used, and bootstrap values over 70 are indicated at the corresponding node.20 Nucleotide sequences translation into protein sequences and multiple sequence alignment were performed in mega 5.01. Networks were constructed using the neighbor‐net method, a distance‐based method in splitstree v4.8 software.21 In this type of dendrogram, each network is constructed to minimize the distances (or number of mutations) between haplotypes. Haplotype and nucleotide diversity analysis were conducted for samples 1–21 using dnasp software 5.1.22 Values were calculated as previously described.23, 24
Results
Influenza virus isolates
During 2009 and 2010, swine lung samples were collected from the south (Paraná and Rio Grande do Sul), central‐west (Mato Grosso), and southeast (Minas Gerais and São Paulo) regions of Brazil. The distribution of the sampled herds across the country is shown in Figure 1. All samples were collected during respiratory disease outbreaks from animals showing clinical signs, such as fever, coughing, nasal discharge, and anorexia. The diagnosis of influenza A infection was confirmed by virus isolation in cell culture and hemagglutination tests. In some cases, more than one animal/property were analyzed; thus, the virus isolates A/swine/Brazil/1 to 3/2009 came from the same property, as did A/swine/Brazil/5 and 6/2009, A/swine/Brazil/8 to 10/2009, A/swine/Brazil/16 and 17/2009, and A/swine/Brazil/19 and 20/2010.
Figure 1.
States from which influenza viruses were isolated. Herds from Minas Gerais and Sao Paulo states in the southeast region, Parana and Rio Grande do Sul states in the south region, and Mato Grosso in the central‐west region of Brazil are shown. N, number of properties analyzed per state.
After visiting a commercial swineherd in Minas Gerais state, Brazil (Table 1: herd K), during a respiratory disease outbreak, a technician became ill with respiratory symptoms similar to the ones observed in the pigs, which started 3 days after the visit and lasted for 1 week. The technician had no contact with any other swineherd for at least 7 days before and after that visit and had no contact with humans showing respiratory symptoms. The outbreak began in August 2009, when sows and grower pigs showed respiratory clinical signs as described above, which lasted for 1 week. A nasal swab sample was collected from the technician, and influenza H1 virus infection was confirmed by real‐time RT‐PCR using the CDC protocol,17 followed by virus isolation (Table 1: sample A/Minas Gerais/21/2010). Viral isolates from 2 pigs from the same herd had also been obtained (Table 1: samples A/swine/Brazil/16/2009 and A/swine/Brazil/17/2009).
Phylogenetic analysis
For each virus isolate, the HA (1658nt, 32–1689) and NA (1363nt, 25–1387) genes were sequenced. Phylogenetic trees based on the HA and NA genes of the influenza viruses isolated here are shown in Figure 2a,b respectively, along with other influenza virus reference sequences from GenBank. Both HA and NA genes from all twenty swine isolates and from the human isolate showed high identity (approximately 99%) and clustered with gene sequences from the pandemic H1N1 virus introduced in the human population in 2009. For both genes, H1N1 and H1N2 seasonal swine and human influenza viruses formed a distinct branch from the Brazilian 2009–2010 isolates.
Figure 2.
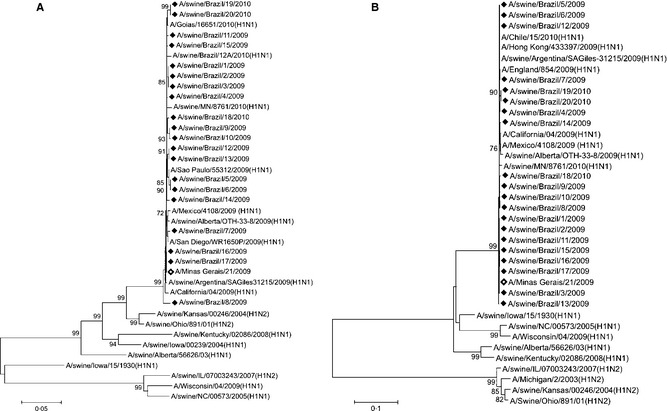
Phylogenetic trees of swine and human Brazilian isolates. Neighbor‐joining tree of (a) hemagglutinin (HA ‐ 1638nt) and (b) neuraminidase (NA ‐ 1358nt). The HA and NA gene sequences of human and swine pandemic H1N1 and seasonal H1N1 and H1N2 viruses were included in the analysis. Closed diamonds = Brazilian pandemic influenza viruses isolated from swine in this study; open diamond = Brazilian pandemic influenza virus isolated from human in this study; A/swine/Brazil/12A/2010: pandemic influenza virus previously isolated from pigs in Brazil.
During influenza outbreaks, active viruses may be genetically related to several others from different regions. To take this population‐level phenomenon into account, we constructed networks that can illustrate the multiple connections between the viral isolates. Networks based on the neighbor‐net method were built with HA and NA sequences from the isolates described in this study and from pandemic H1N1 viruses isolated from humans and swine around the world (Figure 3). Most of the haplotypes were unique, especially for the HA gene (haplotypic diversity = 0·952 ± 0·028; nucleotide diversity = 0·0049 ± 0·0008), which presented higher diversity than the NA gene (haplotypic diversity = 0·962 ± 0·026; nucleotide diversity = 0·0034 ± 0·0006). The networks suggest a common origin for all virus isolates, irrespective of region or host. Furthermore, most of the human and swine isolates appear to have originated from a human‐derived ancestral strain (Figure 3). Nonetheless, identical haplotypes were found in some cases, mainly among isolates from the same properties (Figure 3a: isolates 5–6 and 16–17; Figure 3b: isolates 1–3, 8–10, and 19–20). Notably, the human isolate 21/2009 HA sequence is identical to those of isolates 16/2009 and 17/2009. Its NA sequence is also more related to those swine samples than to other samples in the network (Figure 3).
Figure 3.
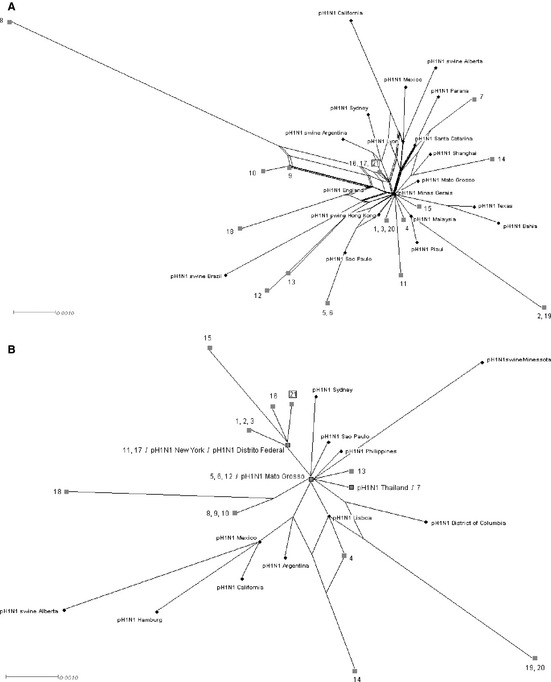
Neighbor‐net dendrogram of (a) hemagglutinin (HA) and (b) neuraminidase (NA) genes of swine and human Brazilian isolates, indicated by gray filled squares. The HA and NA gene sequences of human and swine pandemic H1N1 viruses isolated previously were included in the analysis and are indicated by black circles. Numbers 1–20 indicate swine isolates; the human isolate is indicated as number 21 and highlighted with an open square; gray squares with black border = Brazilian isolates identical to other reference sequences; pH1N1: reference strains for human pandemic H1N1; pH1N1swine: reference strains for swine pandemic H1N1.
Receptor‐binding and antigenic site analysis
We analyzed the viral hemagglutinin subunit 1 protein (HA1) sequences to evaluate the amino acid changes capable of affecting receptor‐binding specificity and antigenicity. The HA1 amino acid sequence alignment is shown in Figure 4. The H1 amino acid numbering of the mature peptide was used.25
Figure 4.
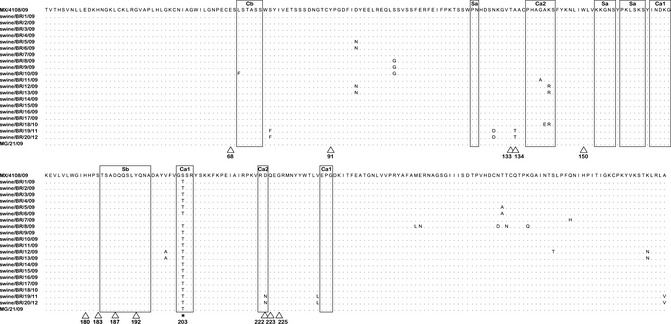
Hemagglutinin 1 (HA1) protein sequence alignment of H1N1 pandemic swine and human Brazilian isolates. Sequences were aligned and numbered using mature HA1. Dots represent amino acids similar to the consensus A/Mexico/4108/2009 (GenBank accession no. GQ162170). The antigenic sites (Sa, Sb, Ca1, Ca2, and Cb) are shown as large rectangles. The amino acid residues at the receptor‐binding site are indicated by a triangle. * = mutation common to all but one isolate.
All 2009–2010 Brazilian swine and human isolates contained residues of the swine lineage in the receptor‐binding pockets, with no substitutions in those sites relative to the A/Mexico/4108/09 strain, except for the substitutions A134T and D222N in isolates A/swine/Brazil/19/2010 and A/swine/Brazil/20/2010. A number of different amino acid substitutions at the HA1 antigenic sites of the Brazilian isolates were detected, all located at Ca1, Ca2, or Cb sites. Interestingly, the amino acid substitution S203T in HA1 and both the V106I and N248D mutations in NA (data not shown) were observed in all but two 2009–2010 Brazilian pandemic H1N1 isolates. The isolate A/swine/Brazil/7/2009 had the Q293H substitution in HA1 and both the V106I and N248D mutations in NA, while isolate A/swine/Brazil/14/2009 contained the S203T substitution in HA1 but only the V106I substitution in NA.
None of the 2009–2010 Brazilian pandemic H1N1 isolates had the H275Y or N295S substitutions in NA (data not shown), which are known to confer resistance to oseltamivir.26
Discussion
Pigs have receptors for both human and avian influenza viruses in their respiratory tracts and thus play an important role for interspecies transmission. They are considered a “mixing vessel” in which reassortment can occur, producing novel viruses to which the human population is naive.27 In Brazil, aside from a case report of pandemic H1N1 2009 virus infecting pigs in the south region,12 no detailed genetic or epidemiologic characterization has been reported. Therefore, this is the most comprehensive study on the molecular characterization of influenza viruses infecting pigs in different states in Brazil.
Our results indicate that influenza virus was widely spread in swine in Brazil during 2009–2010 and is associated with clinical illness in pigs of various ages. Isolates were sampled from pigs showing influenza‐like signs similar to those observed during seasonal outbreaks and in previous reports of pandemic infection.28 All of the Brazilian swine influenza isolates described here clustered with the pandemic H1N1 influenza viruses. Thus, pandemic H1N1 virus circulated in pigs at least during 2009 and 2010, and it continued to circulate in the Brazilian swine population even when viral activity in humans was low.
Host immune pressure is thought to be the main selective force driving amino acid substitutions, which may lead to antigenic drift, and HA is the main target of neutralizing antibodies.29 As expected, genetic diversity in the HA gene of swine isolates was higher than in the NA gene, although all isolates clustered within the H1N1 pandemic clade. Several samples that came from the same herd were identical, indicating within‐herd viral dissemination. These findings also support the hypothesis that immune pressure for viral mutation in pigs might be lower due to the constant introduction of naive animals into a herd.30 However, some amino acid substitutions were found at antigenic sites in Brazilian swine isolates, mainly at Ca and Cb antigenic sites, but the effect on the antigenicity of these viruses is unknown.
The constructed networks were used to provide insights upon the several possible ancestral relationships among haplotypes. Both the HA and NA dendrograms show that the pH1N1 isolates from distinct parts of the world are related to each other with a relatively small distance and share a single common origin, as expected for a pandemic outbreak.31 The observation that most of the human‐ and swine‐derived isolates appear to have originated from human‐derived isolates is also consistent with a higher frequency of human‐to‐human and human‐to‐swine transmission compared with zoonotic swine‐to‐human transmission.15 Indeed, reports on the 2009 pandemic outbreak suggest that the virus evolved silently in the swine host until its introduction to the human population,32 after which it disseminated rapidly among humans and frequently spilled over from humans to pigs.14 Although in line with other reports, the relationships displayed in the dendrograms should be interpreted carefully. The analysis displayed here does not account for the likely possibility that an heterogeneous mixture of viruses at the nucleotide level is produced within an infected host, as only the majority or consensus sequences are generated based on Sanger sequencing methods. Therefore, more detailed studies are needed to confirm the relationships described here.
Nonetheless, the data presented here also provide evidence of a possible swine‐to‐human influenza virus transmission. A technician became ill after entering a herd during a confirmed influenza outbreak. The onset of the illness was consistent with the influenza incubation period, suggesting that the visited swineherd was the most probable source of infection. Furthermore, both HA and NA genes from the human (21/2009) case show the same origin and have higher genetic relatedness to the animal samples from the same herd (16/2009 and 17/2009) than to any other human‐ or pig‐derived isolate.
Seven distinct clades of pandemic H1N1 viruses were identified globally circulating in the beginning of the pandemic.33 The HA substitution S203T combined with NA V106I and N248D found in 18 of the 20 swine isolates and in the human isolate described here suggests that these Brazilian isolates might be members of clade 7 of pH1N1.33 Isolate 7/2009 could be clustered with clade 6 because it did not show the HA substitution. Interestingly, isolate 14/2009, which did not have the N248D NA mutation, cannot be clustered within any of the seven clades. However, a detailed amino acid analysis of all eight viral genes is necessary to confirm these assumptions. It has been demonstrated that pandemic H1N1 2009 evolved and shifted from an initial mixed clade pattern to a clade 7–predominant pattern.34 The subsequent selection and evolution of clade 7 resulted in the circulation of variants D222G/N or E.35 Mutation D222G in the hemagglutinin protein has been correlated with the clinical onset of disease, and it was frequently found in severe/fatal cases of the pandemic influenza in humans.36 Likewise, the substitution D222N, which was observed in isolates 19/2010 and 20/2010, is more frequent among fatal cases of the disease in humans.37 However, the animals from which those viruses were isolated did not show signs of severe disease.
The genetic and antigenic characterization of influenza viruses in swine populations worldwide is critical for understanding the epidemiology of the infection in that species and allowing the selection of ideal vaccine strains. Monitoring of Brazilian swineherds is necessary for establishing better control measures for swine influenza and reducing the risk of the introduction of novel strains to the susceptible human population that can lead to a pandemic. Furthermore, vaccination against swine influenza is not practiced in Brazilian herds; hence, pigs in Brazil may not have protective immunity. Together, our results suggest that the pandemic H1N1 subtype has become established in Brazilian swine populations and may become endemic in the country.
Acknowledgement
The authors wish to thank Dr. José Eustáquio Cavalcante and IPEVE Laboratory for providing samples. This work was financially supported by Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ), Instituto Nacional de Ciência e Tecnologia (INCT‐Pecuária), and Pró‐Reitoria de Pesquisa da UFMG. DSR, JKPR, RMCG, ZIPL, and RCL are CNPq fellowship recipients.
Rajão et al (2012) Genetic characterization of influenza virus circulating in Brazilian pigs during 2009 and 2010 reveals a high prevalence of the pandemic H1N1 subtype. Influenza and Other Respiratory Viruses. 7(5), 783–790
References
- 1. Olsen CW, Brown IH, Easterday BC, Van Reeth K. Swine influenza; in Straw BE, Zimmerman JJ, D'Allaire S, Taylor DJ. (eds): Diseases of Swine. 9th edn Ames: Blackwell Publishing, 2006; 469–482. [Google Scholar]
- 2. Palese P, Shaw ML. Orthomyxoviridae: the viruses and their replication; in Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (eds): Fields Virology. 5th edn Philadelphia: Lippincott Williams & Wilkins, 2007; 1647–1689. [Google Scholar]
- 3. Nicholls JM, Chan RW, Russell RJ, Air GM, Peiris JS. Evolving complexities of influenza virus and its receptors. Trends Microbiol 2008; 16:149–157. [DOI] [PubMed] [Google Scholar]
- 4. Van Reeth K. Avian and swine influenza viruses: our current understanding of the zoonotic risk. Vet Res 2007; 38:243–260. [DOI] [PubMed] [Google Scholar]
- 5. Vincent AL, Ma W, Lager KM, Janke BH, Richt JA. Swine influenza viruses: a North American perspective; in Maramorosch K, Shatkin AJ, Murphy FA. (eds): Advances in Virus Research. Burlington: Academic Press, 2008; 127–154. [DOI] [PubMed] [Google Scholar]
- 6. Webby RJ, Swenson SL, Krauss SL, Gerrish PJ, Goyal SM, Webster RG. Evolution of swine H3N2 influenza viruses in the United States. J Virol 2000; 74:8243–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Choi YK, Goyal SM, Joo HS. Prevalence of swine influenza virus subtypes on swine farms in the United States. Arch Virol 2002; 147:1209–1220. [DOI] [PubMed] [Google Scholar]
- 8. Vincent AL, Ma W, Lager KM, Gramer MR, Richt JA, Janke BH. Characterization of a newly emerged genetic cluster of H1N1 and H1N2 swine influenza virus in the United States. Virus Genes 2009; 39:176–185. [DOI] [PubMed] [Google Scholar]
- 9. Lam T, Hon C, Wang Z, Hui RK, Zeng F, Leung FC. Evolutionary analyses of European H1N2 swine influenza A virus by placing timestamps on the multiple reassortment events. Virus Res 2008; 131:271–278. [DOI] [PubMed] [Google Scholar]
- 10. Caron LF, Joineau MEG, Santin E, Richartz RRTB, Patricio MAC, Soccol VT. Seroprevalence of H3N2 influenza A virus in pigs from Paraná (South Brazil): interference of the animal management and climatic conditions. Virus Rev Res 2010; 15:1–11. [Google Scholar]
- 11. Rajão DS, Alves F, Del Puerto H et al Serological evidence of swine influenza in Brazil. Influenza Other Respi Viruses 2012; doi: 10.1111/j.1750‐2659.2012.00366.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schaefer R, Zanella JRC, Brentano L et al Isolation and characterization of a pandemic H1N1 influenza virus in pigs in Brazil. Pesq Vet Bras 2011; 31:761–767. [Google Scholar]
- 13. Mancini DAP, Cunha EMS, Mendonça RMZ et al Evidence of swine respiratory infection by influenza viruses in Brazil. Virus Rev Res 2006; 11:39–43. [Google Scholar]
- 14. Sreta D, Tantawet S, Ayudhya SNN et al Pandemic (H1N1) 2009 virus on commercial swine farm, Thailand. Emerg Infect Dis 2010; 16:1587–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garten RJ, Davis CT, Russell CA et al Antigenic and genetic characteristics of swine‐origin 2009 A (H1N1) influenza viruses circulating in humans. Science 2009; 325:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. World Health Organization . WHO Manual on Animal Influenza Diagnosis and Surveillance, 2nd edn Geneva: World Health Organization, 2002. (document WHO/CDS/CSR/NCS/2002.5) [Google Scholar]
- 17. World Health Organization . CDC protocol of real time RT‐PCR for swine influenza A (H1N1). Geneva: World Health Organization, 2009. [Google Scholar]
- 18. Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full‐length amplification of all influenza A viruses. Arch Virol 2001; 146:2275–2289. [DOI] [PubMed] [Google Scholar]
- 19. Saitou N, Nei M. The neighbor‐joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987; 4:406–425. [DOI] [PubMed] [Google Scholar]
- 20. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood evolutionary distance and maximum parsimony methods. Mol Biol Evol 2011; 28:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 2006; 23:254–267. [DOI] [PubMed] [Google Scholar]
- 22. Librado P, Rozas J. DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009; 25:1451–1452. [DOI] [PubMed] [Google Scholar]
- 23. Jukes TH, Cantor CR. Evolution of protein molecules; in Munro HN. (eds): Mammalian Protein Metabolism. 3rd edn New York: Academic Press, 1969; 121–132. [Google Scholar]
- 24. Lynch M, Crease TJ. The analysis of population survey data on DNA sequence variation. Mol Biol Evol 1990; 7:377–394. [DOI] [PubMed] [Google Scholar]
- 25. Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 1982; 31:417–427. [DOI] [PubMed] [Google Scholar]
- 26. Le QM, Kiso M, Someya K et al Avian flu: isolation of drug‐resistant H5N1 virus. Nature 2005; 437:1108. [DOI] [PubMed] [Google Scholar]
- 27. Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev 1992; 56:152–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brookes SM, Núñez A, Choudhury B et al Replication pathogenesis and transmission of pandemic (H1N1) 2009 virus in non‐immune pigs. PLoS ONE 2010; 5:00–00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 2000; 69:531–569. [DOI] [PubMed] [Google Scholar]
- 30. Brown IH. The epidemiology and evolution of influenza viruses in pigs. Vet Microbiol 2000; 74:29–46. [DOI] [PubMed] [Google Scholar]
- 31. Rambaut A, Holmes EC. The early molecular epidemiology of the swine‐origin A/H1N1 human influenza pandemic. PLoS Curr 2009; 1: RRN1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Solovyov A, Greenbaum B, Palacios G, Lipkin WI, Rabadan R. Host dependent evolutionary patterns and the origin of 2009 H1N1 pandemic influenza. PLoS Curr 2010; 2:RRN1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nelson M, Spiro D, Wentworth D et al The early diversification of influenza A/H1N1pdm. PLoS Curr 2009; 1:RRN1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Valli MB, Meschi S, Selleri M et al Evolutionary pattern of pandemic influenza (H1N1) 2009 virus in the late phases of the 2009 pandemic. PLoS Curr 2010; 2:RRN1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller RS, Maclean AR, Gunson RN, Carman WF. Occurrence of haemagglutinin mutation D222G in pandemic influenza A(H1H1) infected patients in the West of Scotland United Kingdom 2009‐10. Euro Surveill 2010; 15:19546. [PubMed] [Google Scholar]
- 36. Kilander A, Rykkvin R, Dudman SG, Hungnes O. Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome Norway 2009‐2010. Euro Surveill 2010; 15:19498–19501. [DOI] [PubMed] [Google Scholar]
- 37. Houng HH, Garner J, Zhou Y et al Emergent 2009 influenza A(H1N1) viruses containing HA D222N mutation associated with severe clinical outcomes in the Americas. J Clinical Virol 2012; 53:12–15. [DOI] [PubMed] [Google Scholar]