

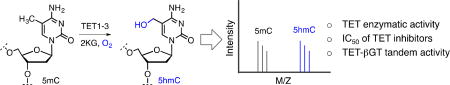
Abstract
Enzymatic methylation at carbon five on cytosine (5mC) in DNA is a hallmark of mammalian epigenetic programming and is critical to gene regulation during early embryonic development. It has recently been shown that dynamic erasure of 5mC by three members of the ten-eleven translocation (TET) family plays a key role in cellular differentiation. TET enzymes belong to Fe (II)- and 2-ketoglutarate (2KG) dependent dioxygenases that successively oxidize 5mC to 5-hydroxymethyl cytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxycytosine (5CaC), thus providing a chemical basis for the removal of 5mC which once was thought to be a permanent mark in mammalian genome. Since then a wide range of biochemical assays have been developed to characterize TET activity. Majority of these methods require multi-step processing to detect and quantify the TET-mediated oxidized products. In this study, we have developed a MALDI mass spectrometry based method that directly measures the TET activity with high sensitivity while eliminating the need for any intermediate processing steps. We applied this method to the measurement of enzymatic activity of TET1 and 3, Michaleis-Menten parameters (KM and kcat) of TET-2KG pairs and inhibitory concentration (IC50) of known small-molecule inhibitors of TETs. We further demonstrated the suitability of the assay to analyze chemoenzymatic labeling of 5hmC by β-glucosyltransferase, highlighting the potential for broad application of our method in deconvoluting the functions of novel DNA demethylases.
Keywords: DNA demethylation, TET enzymes, β-Glucosyltransferase, MALDI mass spectrometry, Michaelis-Menten parameters, TET inhibitors
Graphical abstract
Introduction
Chemical modifications on DNA and histones are primary epigenetic mechanisms that play critical role in gene regulation.1 Notable example includes 5-methylcytosine (5mC) in DNA, and acetylation and methylation of lysine ε-amine in the unstructured tail of nucleosomal histones.2 Ten-Eleven Translocation (TET)-mediated successive oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5CaC) is considered a landmark finding as it provides a biochemical basis for active DNA demethylation and is essential to early mammalian development (Fig. 1A).3,4,5 Humans have three TET isoforms (TET 1, 2 and 3), all of which are members of the Fe (II) and 2-ketoglutaric acid 1 (2KG)-dependent dioxygenases, a superfamily that is present in all forms of life (Fig. 1B).6 TET-mediated oxidation of 5mC is critical to cellular differentiation.7 Growing evidence also suggests that each of the TET oxidized products (5hmC, 5fC and 5CaC) can independently regulate gene expression, aside from playing a role as intermediates in the demethylation pathway.8,9,10 Overexpression and catalytically inactive mutations of TET enzymes are involved in the development of certain cancers as well.11
Fig. 1.

Ten-eleven translocation (TET) enzymes and their biochemistry. (A) Successive oxidation of 5mC (generated by DNMTs and SAM) to 5hmC, 5fC and 5CaC by TET enzymes and 2KG leads to active DNA demethylation. Decarboxylation of 5CaC to C is achieved primarily by Thymine DNA glycosylase (TDG) followed by base excision repair (BER) mechanism. Putative decarboxylase may also be involved in direct removal of the carboxylic group. (B) Domain structures of TET1-3 showing double stranded β helix (DSβH) fold, preceding cysteine (Cys)-rich domain and a CXXC domain in TET1 and 3. (C) Colored-coded domain structure of catalytic portion of the human TET2 enzyme. A low-complexity insert (aa 1481–1843) was replaced with a GS linker of 15 amino acids.
Efforts to understand the functions of TET proteins in eukaryotic gene regulation have fueled the development of novel analytical methods to detect and quantify the enzymatic activity of TETs as well as their oxidized products both in vitro and in vivo.12,13 Several chemical approaches coupled with next-generation sequencing techniques have been developed for base-resolution mapping of TET products in cell- and tissue-specific manners.14,15 Such methods have led to a detailed understanding of TET mediated processes and their relevance in gene regulation. In parallel, efforts are underway for the biochemical and structural characterization of TET proteins in vitro.16,17 Representative methods for the analysis of modified nucleotides are (1) one- and two-dimensional thin-layer chromatography (1D- and 2D-TLC), (2) tandem liquid chromatography and mass spectrometry (LC-MS/MS), (3) Western blotting with cytosine modification-specific antibodies, and (4) chemoenzymatic labeling of the modified cytosines particularly 5hmC.13 Novel methods are also being developed for strand-specific quantitative analysis of TET activity.13
A common assay to characterize TET activity involves LC-MS/MS analysis of the oxidized oligonucleotide products. Following enzymatic oxidation, oligonucleotide product(s) are purified using nucleotide purification kits and subjected to enzymatic degradation to individual nucleotides prior to mass spectrometric assignment. Most of the current approaches involve multiple enzymatic reactions and purification steps that invariably lead to the loss of oxidized products 5hmC, 5fC and 5CaC) and errors in quantification. Herein we report the development and optimization of a Matrix-assisted laser desorption/ionization (MALDI) based mass spectrometric assay that facilitates direct and efficient measurement of enzymatic activity of TET and eliminates the need for subsequent enzymatic digestion and purification of the oxidized oligonucleotide products. Employing the optimized conditions, we further determine the kinetic constants of TET enzymes and IC50 of the selected small-molecule inhibitors of TETs. Finally, we demonstrate the utility of the assay to directly examine the subsequent chemoenzymatic functionalization of TET enzymatic products particularly 5hmC.
Materials and methods
Expression and purification of TET2 and TET3 in E. coli
The catalytic domain of TET2 is comprised of a Cys-rich domain and a DSβH (also known as jelly-roll motif) domain with a large low-complexity insert (Fig. 1C).3 It has been reported by Yanhui Xu group that this unstructured insert poses significant challenge for crystallization.16,17 By screening a series of deletion mutants, Xu group found TET2 (1099–1936 with residues 1481–1843 replaced by a 15-residue GS-linker GGGGSGGGGSGGGGS) to be the minimum catalytically active fragment suitable for bacterial expression (Fig. 1C). A 6×His tag was introduced at the N-terminal for the purpose of initial purification. A similar deletion mutant was also generated for TET3 (689–1596 with residues 1061–1500 replaced by a 13-residue GS-linker GGGSGGGGSGGGS). Both the expression constructs were obtained from Xu group at Fudan University.16,17
The plasmids were transformed into Escherichia coli strain BL21 star (DE3) competent cells (Invitrogen) using pET-28b kanamycin-resistant vector. A single colony was picked up and grown overnight at 37 °C in 10 mL of Luria-Bertani (LB) broth in presence of 50 µg/mL kanamycin. The culture was diluted 100-fold and allowed to grow at 37 °C to an optical density (OD600) of 0.8, and protein expression was induced overnight at 17 °C with 0.5 mM IPTG in an Innova 44® Incubator shaker (New Brunswick Scientific). Proteins were purified as follows: harvested cells were resuspended in 15 mL lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM β-mercaptoethanol, 10% glycerol, 25 mM imidazole, Lysozyme, DNase, and Roche protease inhibitor cocktail). The cells were lysed by pulsed sonication (Qsonica-Q700), and centrifuged at 13,000 rpm for 40 min at 4 °C. The soluble extracts were subjected to Ni-NTA agarose resin (Thermo) according to manufacturer’s instructions. After passing 20 volumes of washing buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM β-mercaptoethanol, 10% glycerol, and 25 mM imidazole), proteins were eluted with a buffer containing 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM β-mercaptoethanol, 10% glycerol, and 400 mM imidazole. Proteins were further purified by gel filtration chromatography (Superdex-200) using AKTA pure FPLC system (GE healthcare) with buffer containing 50 mM Tris-HCl pH 8.0, 150 mM NaCl, and 10% glycerol. Purified proteins were concentrated using Amicon Ultra-10k centrifugal filter device (Merck Millipore Ltd.). The protein concentration was determined using Bradford assay kit (BioRad Laboratories) with BSA as a standard. The concentrated proteins were stored at −80 °C before use.
Synthesis of DNA substrates
Oligonucleotides carrying 5mC and 5hmC were synthesized using standard DNA phosphoramidite monomers (Glen Research) under ultra-mild conditions using 1H-tetrazole as activator reagent in an EXPIDITE Nucleic Acid Synthesis System (PerSeptive Biosystems) with DMT-ON protocol. While 5mC phosphoramidite was purchased from Glen Research, 5hmC phosphoramidite was synthesized according to previously reported procedure.18,19 To ensure good coupling elongated (4 minutes and 30 seconds) coupling times were applied for the coupling of modified bases and for standard bases (2 minutes) normal coupling was applied. The crude oligonucleotide was cleaved from the beads and deprotected by incubating with ammonium hydroxide (33% v/v) at 25 °C for 24 hr. A preliminary purification and DMT deprotection were carried out using Poly PakII purification cartridge (Glen Research) according to the standard protocol provided by the manufacturer. Crude oligonucleotides were purified by HPLC using a C-18 column (Solvent A: 0.1 M TEAA, Solvent B: Acetonitrile; gradient: 0 min 5% B, 10 min 40% B, 15 min 100% B with a flow rate 4 mL/min). The fractions were collected and concentrated by SpeedVac concentrator followed by lyophilization, and re-dissolved in RNAase free water. The quality and purity of synthesized DNAs were established by high resolution MALDI-TOF-MS. For TET assay, the oligonucleotides were annealed in Duplex Buffer (100 mM Potassium Acetate, 30 mM HEPES, pH 7.5, Integrated DNA Technology) by heating the mixed equimolar concentrations of oligonucleotides to 94°C for 2 min and gradually cooling down to room temperature.
In vitro enzymatic assays
For in vitro enzymatic activity assays, 10 µM of various double-stranded DNA substrates (Fig. 2A) are incubated with TET2 (1099–1936 del-insert) and TET3 (689–1596 del-insert) proteins (10 µM) in buffer containing 50 mM HEPES (pH 8.0), 100 mM NaCl, 100 mM Fe(NH4)2(SO4)2, 2 mM ascorbate, 1 mM DTT, 1 mM ATP, and 1 mM 2-KG at 37 °C for 3 hr. The product DNA was purified using following methods:
Nucleotide clean up method: The oligonucleotides were purified using QIAquick Nucleotide Removal Kit (QIAGEN) following manufacturer’s instructions and denatured at 100 °C for 10 min. The oligonucleotides were further concentrated using speedvac concentrator for 10 min and analyzed by MALDI-TOF mass spectrometry (AB SCIEX Voyager DE Pro) by spotting 1 µL of sample and then mixed with 1 µL of 3-Hydroxypicolinic Acid (3-HPA) matrix on MALDI plate.
Cation-exchange resin method: The product DNA was desalted by adding 10–15 µL of AG® 50W-X8 Cation Exchange Resin (BioRad, Cat # 143–5441) directly into the biochemical mixture and agitated followed by incubation for 5 min at room temperature. The samples were centrifuged at 10,000 rpm for 2 min. The oxidized products were analyzed by MALDI-TOF mass spectrometry (AB SCIEX Voyager DE Pro) by spotting 1 µL of sample and then mixed with 1 µL of 3-Hydroxypicolinic Acid (3-HPA) matrix on MALDI plate.
Fig. 2.

Assay optimization and TET activity. (A) A two-step optimization involving substrate selection (I) and product purification (II) to measure TET activity. Among the series of DNA substrates that were examined initially, 8 nucleotide containing substrate 5 showed robust MALDI signal as shown in B. The initial purification included multi-step protocol involving passing through desalting column, washing and eluting the DNA followed by concentration using vacuum evaporator. The final condition involved desalting by adding AG® 50W-X8 cation exchange resin and directly spotting on to MALDI plate. The condition led to robust detection of 5hmC formed by TET2/3 on 8-mer DNA as shown in C.
Relative quantification of 5mC and 5hmC DNAs (standard curve)
For the standard curve, the percentage of 5mC and 5hmC containing synthetic DNAs was varied from 0 to 100 while keeping the total DNA concentration at 10 µM. After mixing, the samples were analyzed by MALDI-TOF mass spectrometry by spotting 1 µL of sample and then mixed with 1 µL of 3-Hydroxypicolinic Acid (3-HPA) matrix on a MALDI plate. The relative intensities of the synthesized DNAs were fitted to a straight line with nonlinear regression using GraphPad Prism.
Measurements of TET kinetic parameters
For kinetics experiments, 5 µM of 8-nt double-stranded DNA substrate (Fig. 2A) was incubated with TET2 (1099–1936 del-insert) and TET3 (689–1596 del-insert) proteins (5 µM) with varied 2KG concentrations (3 – 100 µM) in a buffer containing 50 mM HEPES (pH 8.0), 100 mM NaCl, 100 mM Fe(NH4)2(SO4)2, 2 mM ascorbate, 1 mM DTT, and 1 mM ATP at 37 °C. The samples were collected in different time intervals ranging from 10 – 90 min. Samples were desalted using AG® 50W-X8 Resin (BioRad, Cat # 143–5441). The TET products were analyzed through MALDI using 3-hydroxypicolinic acid (3-HPA) matrix. Only points within the linear range were used to calculate the slope for each 2KG concentration and the values were fitted to the Michaelis-Menten equation v0 = (Vmax[S])/(Km+[S]) to get the KM and kcat values. Experiments were performed in triplicates. The MALDI experiments were conducted in reflectron negative TOF mode and the ion intensities were normalized to the highest peak. The background noise was corrected for calculating signal/noise ratios using AB Sciex Data Explorer (4.0) and the apex peak intensities were measured for the kinetic analysis.
Measurements of inhibitory concentration (IC50) of TET2 inhibitors
The assay mixture containing 5 µM TET2 (1099–1936 del-insert) was incubated with varying concentrations of NOG (N-Oxalylglycine, Cat # O9390, Sigma) (0.05mM – 5mM) and 2HG (D-2-hydroxyglutaric acid, Cat # sc-227739 and L-2-hydroxyglutaric acid, Cat # sc-361834, Santa cruz biotechnology) (0.5mM – 30mM) in a buffer containing 50 mM HEPES (pH 8.0), 100 mM NaCl, 100 mM Fe(NH4)2(SO4)2, 2 mM ascorbate, 1 mM DTT, 1 mM ATP for 5 min on ice. Demethylation was initiated by adding 100 µM 2-KG and 10 µM 8-nt double stranded 5mC DNA and further incubated at 37 °C for 3 hr. The product DNA was denatured at 100 °C for 10 min and desalted using cation exchange resin method described above. The concentrated oligonucleotides were analyzed by MALDI-TOF mass spectrometry and the values were fitted to the 4-parameter non-linear regression algorithm (Y=Bottom + (Top - Bottom)/(1+10^((LogIC50 - X)*Hill slope))) of the GraphPad Prism software. X: log of dose or concentration; Y: response, decreasing as X increases; Top and bottom: upper and lower values of a given curve; logIC50: same log units as X; HillSlope: Slope factor or Hill slope, unitless.
Expression and purification of β-GT in E. coli
The N-terminal 6×His-tagged T4 β-GT (β-glucosyltransferase) plasmid was a kind gift of Prof. Heinrich Leonhardt at the Ludwig Maximilians University, Munich.20 The pET28b kanamycin-resistant vector was used to transform E. Coli BL21 codon plus (DE3) RIPL competent cells (Invitrogen). A single colony was picked up and grown overnight at 37 °C in 10 mL of Luria-Bertani (LB) broth in presence of 50 µg/mL kanamycin and 35 µg/mL chloramphenicol. The culture was diluted 100-fold and allowed to grow at 37 °C to an optical density (OD600) of 0.8, and protein expression was induced overnight at 18 °C with 0.5 mM IPTG in an Innova 44® Incubator shaker (New Brunswick Scientific). Protein purification protocol was followed as described above.
β-glucosyltransferase (β-GT)-mediated glucosylation of 5hmC
For 5hmC glucosylation, first a demethylase assay was carried out using TET2 (1099–1936 del-insert) on 8-nt 5mC containing double stranded DNA substrate to obtain 5hmC DNA. A 50 µL of demethylase assay containing 10 µM TET2, 10 µM 8-nt 5mC DNA and 1 mM 2-KG was incubated at 37 °C for 3 hr. in buffer containing 50 mM HEPES (pH 8.0), 100 mM NaCl, 100 mM Fe(NH4)2(SO4)2, 2 mM ascorbate, 1 mM DTT and 1 mM ATP. After 3 hr of incubation, 25 µL of assay mixture was withdrawn to confirm 5hmC formation. 5 µM T4-β-GT, 100 µM UDP-glucose (MP Biomedicals, LLC. Cat # 101208) were added to the remaining 25 µL of assay and the glucosylation reaction carried out at 25 °C for 1 hr. The product DNA was denatured at 100 °C for 10 min and added cation exchange resin method described above. The desalted oligonucleotide products were analyzed by MALDI-TOF mass spectrometry.
Results and discussion
Design and optimization of a MALDI-based assay for measurement of TET activity
LC-MS based methods have been widely employed to measure the activity of TET enzymes on various DNA substrates.13 Two recent studies have demonstrated catalytic activity of bacterially expressed TET2 and 3 on a 58 nucleotide-containing palindromic DNA with a single 5mC modification.16,17 We synthesized both the strands of the 58-mer DNA substrate 2 and examined for TET-mediated 5hmC formation (Fig. 2A). However, when we attempted to examine the product formation directly by MALDI mass spectrometry, the oxidized products could not be detected, likely due to the extremely poor signal-to-noise (S/N) ratio. Subsequent desalting of the enzymatic products using various nucleotide purification kits did not improve S/N ratio on the MALDI spectrum. We reasoned that a shorter DNA sequence, due to efficient ionization, might help in obtaining stronger peak(s) with improved S/N ratio. TET2-DNA crystal structure shows that critical interactions between the enzyme and its substrate map only to a few nucleotides adjacent to the 5mC base, suggesting that TET2 can potentially oxidize a short DNA substrate. Consistently, it has been reported that TET proteins also act on DNA substrates containing six nucleotides.21 Using a solid phase DNA synthesis technique, we prepared three palindromic DNA sequences 3, 4, and 5 each containing 22, 14 and 8 nucleotides, respectively, based on the original 58-mer DNA substrate (Fig. 2A). The integrity of the purified DNA substrates was confirmed by MALDI spectrometry (Fig. S1–3). Following TET2-mediated oxidation, the products were purified using nucleotide removal kit and subjected to MALDI based detection. We clearly observed the formation of 5hmC on all three short DNA substrates (Fig. S1–3). No oxidation of 5mC occurred in absence of TET2 enzyme. Although we did not observe any preference for the length of the three DNA substrates used (22-mer/14-mer/8-mer), as the extent of product formation was essentially identical for all the three substrates, we decided to use the shortest 8-mer DNA 5 for subsequent experiments.
After demonstrating that the enzymatic activity of TET2 can be monitored using our MALDI based assay, we focused on further streamlining the detection process (Fig. 2A). In our initial efforts, it was necessary to pass the enzymatic products through a nucleotide cleaning kit prior to concentrating the eluent to detect 5hmC formation, thus introducing lengthy steps. The purpose of using a nucleotide purification column is to eliminate salts present as assay components to improve signal strength in the mass spectrum. We reasoned that addition of cation exchange resin directly to the assay upon completion of the enzymatic oxidation would eliminate the need for desalting columns. To test our rationale, we added pre-washed AG® 50W-X8 cation exchange resin directly to the assay at specified times.22 Resin-treated sample was then spotted on the MALDI plate and subjected to mass spectrometric analysis without passing through the column. TET oxidized products were detected with significantly improved signal-to-noise ratio in the MALDI spectra (Fig. 2B,C). The assay was also used to examine the activity of bacterially expressed TET3 enzyme (Fig. S4). Overall, we have developed an extremely simple and straightforward MALDI-based method to determine the activity of TET enzymes that does not require multiple enzymatic digestions, liquid chromatographic optimization and separation of the digested products prior to analysis.
Measurement of Michaelis-Menten parameters for TET enzymes
Straightforward and direct measurement of TET oxidized products by sensitive mass spectrometric method is suitable for measuring kinetic parameters of TET enzymes. Although kcat and KM for various DNA substrates of TET2 have been recently determined,17 similar kinetic studies have not been reported for the cofactor 2KG 1. To precisely measure the amount of 5hmC formation compared to the substrate 5mC, we first sought to determine the difference, if any, between the relative ionization potential of the substrate (5mC) and that of the product (5hmC) DNAs. Towards this end, we synthesized the 5hmC-containing DNA, corresponding to the enzymatic product of TET2, using solid phase technique. The crude DNA was purified and confirmed by MALDI. The synthesized 5mC and 5hmC DNA were mixed in appropriate ratios and their relative ion intensities were determined by MALDI mass spectrometry. The observed signals of the synthesized DNAs corresponded linearly to their relative amounts present in the mixture as evident from the straight line of a slope of 1.0 ± 0.01 (Fig. 3A,B,S5). Our data clearly suggest that the substrate (5mC) and product (5hmC) DNA molecules ionize in mass spectrometer with equal efficiency and the intensities of the observed peaks correspond well to their relative abundance.
Fig. 3.

Measurement of Michaelis-Menten parameters for 2KG-TET activity. (A) MALDI mass spectrometric signals of 1:1 mixture of synthesized 5mC and 5hmC containing 8-mer DNAs. (B) Relative intensities (%) of MALDI mass spectrometric signals for various mixtures of synthesized 5mC and 5hmC containing 8-mer DNAs. Slope 1.0±0.01 suggests similar ionization potential of the two DNAs. (C) Dose- and time-response activity of 2KG towards TET2 enzyme. (D) Michaelis-Menten parameters of TET2-2KG pair. (E) Dose- and time-response activity of 2KG towards TET3 enzyme. (F) Michaelis-Menten parameters of TET3-2KG pair.
We next investigated the steady-state kinetics of TET2 enzyme by systematically varying the concentration of cofactor 2KG in the optimized assay (Fig. 3C,D,S6). The DNA substrate was kept at a fixed concentration of 10 µM, well above its KM value. The concentration of TET2 was kept low at 2 µM to avoid the formation of higher oxidized products. Under these conditions, we determined an apparent KM of 15.7 µM for 2KG and the catalytic turnover (kcat) of 0.2 min−1 for wild type TET2 enzyme. To the best of our knowledge, this is the first measurement of KM of the cofactor 2KG for a TET enzyme. Following similar condition, we also determined the Michaelis-Menten parameters for TET3, another biologically important member of the TET family enzymes (Fig. 3E,F,S7). Consistent with TET2, we measured an apparent KM of 24.2 µM for the cofactor 2KG and kcat of 0.12 min−1. Thus overall catalytic efficiency (kcat/KM) of TET2 is ~2.6 fold higher than that of TET3 under the assay conditions. The improved catalytic efficiency of TET2 is consistent with its function as predominant TET enzyme in 5mC oxidation pathway in mammals. Humans have more than 60 2KG-dependent enzymes including histone, DNA and RNA demethylases, and prolyl hydroxylases.23 These enzymes are involved in wide-ranging biological processes involving gene regulation, hypoxic signaling, collagen biosynthesis and fatty acid metabolism to name a few. KM values for the cofactor 2KG for many of these enzymes have been previously measured and are typically in the low micromolar range as is the case for TET2 and 3 as determined in the current study, suggesting a similar cofactor-dependency of this family of enzymes.24 Overall, our assay provides a direct MALDI-based quantitative platform for the measurement of kinetic parameters of TET-2KG enzyme-cofactor pairs.
Evaluation of small-molecule inhibitors of TET2
Specific small-molecule inhibitors for TET enzymes are yet to be reported. N-oxalylglycine (NOG) is shown to inhibit a range of 2KG-dependent dioxygenases in a cofactor-competitive manner due mainly to its structural similarity with 2KG.25 Recently, 2-hydroxyglutarate (2HG), an oncometabolite produced by tumor-associated mutants of metabolic enzymes isocitrate dehydrogenase 1 and 2 (IDH1 and 2), has also been shown to inhibit TET proteins in 2KG competitive manner.26 Mutations of IDH1 and IDH2 are commonly found in a diverse range of cancers or solid tumors, including gliomas and acute myeloid leukemia with concomitant increase in 2HG level (up to 30 mM).27 These cancers are also associated with decreased levels of global 5hmC, suggesting that inhibition of TET activity by 2HG is critical for hematopoietic transformation. However, systematic measurement of the inhibitory effect of NOG and 2HG on human TET proteins has not been reported.
To determine the IC50 of NOG and 2HG directly by our MALDI-based assay, we optimized the assay conditions further. Given the cofactor-competitive nature of the inhibition, we further modified the assay by varying the concentration of 2KG and found 80 µM to be an optimal concentration for TET activity. In addition to NOG, we also examined both the enantiomers of 2HG (D-2HG and L-2HG). All the compounds displayed dose-dependent inhibition of TET2 enzyme. Under our assay condition, the IC50 for NOG was measured to be 149.5 ± 7.6 µM (Fig. 4A, S8). NOG has been reported to inhibit Naegleria Tet1, a homologue to mammalian Tet, with an IC50 of 49 ± 6 µM in a fluorescence polarization-based competitive binding assay that does not require the cofactor 2KG.28,29 In our assay, however, the inhibitory activity of NOG reflects its ability to modulate the catalytic potential of TET2 enzyme as it measures the formation of oxidized products.
Fig. 4.

Small-molecule inhibitors of TET2. Inhibitory activity (IC50) of NOG (A) and 2HG (B) towards TET2.
Using the dose-response inhibition studies, we determined IC50 values of 5.3 ± 0.3 M and 12.4 ± 2.4 µM for D-2HG and L-2HG, respectively (Fig. 4B, S9–10). Interestingly, in cancer cells, mutants IDH1 and 2 furnish the oncometabolite D-2HG and not its enantiomer L-2HG. Given that up to 35 mM of D-2HG accumulates in glioblastoma cells carrying IDH1 and IDH2 mutations,27 full inhibition of TET enzymes by D-2HG is highly likely in spite of its 5.3 mM IC50. The higher inhibitory activity of NOG over 2HG towards TET enzymes is very likely due to the presence of 2-keto acid moiety (much like natural cofactor 2KG) as well as the internal –NH– group (absent in 2HG) which can participate in strong H-bonding with the TET enzymes.
Analysis of enzymatic functionalization of TET-oxidized products
Analysis of genomic distribution and base resolution sequencing of 5hmC in mammalian genome have been facilitated by various post-oxidative modifications. Recently He and Leonhardt groups have independently shown that 5hmC in DNA can be glycosylated by T4 phage β-glucosyltransferase (β-GT) using uridine diphosphoglucose (UDP-glucose).30,20 The He group further demonstrated that 5hmC functionalized with azide-containing UDP-glucose by β-GT can undergo ‘click’ reaction with alkyne moiety for various downstream applications particularly single molecule real time (SMRT)-based next-generation sequencing.30
To demonstrate that our MALDI based assay can be employed to directly analyze the post-oxidative functionalization of 5hmC, we coupled the β-GT mediated enzymatic glucosylation in tandem (Fig. 5A). The TET-modified 5hmC DNA was directly incubated with β-GT and UDP-glucose without any purification of the TET product and monitored using MALDI mass spectrometry (Fig. 5B). Significant amounts of glucosylated 5hmC (g5hmC) DNA was observed within an hour. However, the amount of g5hmC reached a plateau and did not increase further over time. This is most likely due to the product inhibition of β-GT by UDP as observed by the Pradhan group in a scintillation assay using UDP-[3H]glucose.31 Although, the scintillation assay accurately characterizes the catalytic activity of β-GT, the assay requires multiple steps following enzymatic modification such as spotting/ drying of the reaction mixtures on DE81 membrane, extensive washing, placing in scintillation vials and counting of tritium incorporation. In contrast, using the MALDI mass spectrometry assay described here, we can directly monitor the functionalization of 5hmC without any need for intermediate processing.
Fig. 5.

Functionalization of TET2 oxidized product. (A) scheme showing glucosylation of 5hmC by β-GT using UDP-glucose. (B) MALDI spectra showing direct monitoring of conversion of 5mC to 5hmC by TET2 and subsequent glucosylation to 5ghmC by β-GT.
Conclusions
We developed an efficient MALDI mass spectrometry based method to detect and quantify the activity of TET dioxygenases. The assay involves direct measurement of 5hmC produced by TET enzymes on a double stranded short palindromic DNA of eight nucleotides. We demonstrated multiple applications of the optimized assay. These include measurement of apparent KM and kcat for TET2 and TET3 towards the cofactor 2KG as well as determination of IC50 of various small-molecule inhibitors of TET proteins. We also showed that the assay is highly suitable for determining post-oxidative functionalization, such as 5hmC glucosylation by β-GT for downstream applications. The highlight of the assay developed here is the sensitivity and simplicity to analyze TET products, unlike the previous methods that require multiple rounds of enzymatic degradation, tedious purification of the subsequent products and hazardous detection methods. More importantly, direct mass spectroscopic measurement eliminates the obvious need for a validation step by a secondary assay, thus making the overall process scalable. Moreover, with the recent technological advancement in automated data collection (e.g. Bruker-ultrafleXtreme™ MALDI-TOF/TOF spectrometer), it is highly likely that screening a large number of samples would be viable in a medium throughput manner. The assay developed here has the potential for broad applicability as it can be extended to a range of 2KG-dependent human dioxygenases including histone demethylases and RNA demethylases for their biochemical characterization in a rapid and scalable manner.
Supplementary Material
Highlights.
Active demethylation of 5mC by TET enzymes is one of the key mechanisms of epigenetic regulation in vertebrates.
MALDI-based assay is developed to characterize catalytic activity of TET enzymes
The method is employed to determine kinetic parameters of TETs and IC50 of TET inhibitors
The assay is characterized by its minimum sample preparation, rapid and straightforward analysis
Acknowledgments
We thank the University of Pittsburgh and the National Institute of Health (1R01GM123234-01) for financial support; Profs. Y. Xu and H. Leonhardt for TET and β-GT constructs, respectively; Dr. D. Chakraborty and members of our laboratory for critical reading and editing of the manuscript. Support for MALDI-TOF MS instrumentation was provided by a grant from the National Science Foundation (CHE-1625002).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A. Supplementary data
Supplementary data related to this article is available.
References
- 1.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu X, Zhao BS, He C. TET family proteins: oxidation activity, interacting molecules, and functions in diseases. Chem. Rev. 2015;115:2225–2239. doi: 10.1021/cr500470n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song CX, He C. Potential functional roles of DNA demethylation intermediates. Trends Biochem. Sci. 2013;38:480–484. doi: 10.1016/j.tibs.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachman M, Uribe-Lewis S, Yang X, Burgess HE, Iurlaro M, Reik W, Murrell A, Balasubramanian S. 5-Formylcytosine can be a stable DNA modification in mammals. Nat. Chem. Biol. 2015;11:555–557. doi: 10.1038/nchembio.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Munzel M, Wagner M, Muller M, Khan F, Eberl HC, Mensinga A, Brinkman AB, Lephikov K, Muller U, Walter J, Boelens R, van Ingen H, Leonhardt H, Carell T, Vermeulen M. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, Liu XS, Aravind L, Agarwal S, Maciejewski JP, Rao A. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen L, Zhang Y. Enzymatic analysis of Tet proteins: key enzymes in the metabolism of DNA methylation. Methods Enzymol. 2012;512:93–105. doi: 10.1016/B978-0-12-391940-3.00005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu MY, DeNizio JE, Kohli RM. Quantification of Oxidized 5-Methylcytosine Bases and TET Enzyme Activity. Methods Enzymol. 2016;573:365–385. doi: 10.1016/bs.mie.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Booth MJ, Raiber EA, Balasubramanian S. Chemical methods for decoding cytosine modifications in DNA. Chem. Rev. 2015;115:2240–2254. doi: 10.1021/cr5002904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song CX, Yi C, He C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat. Biotech. 2012;30:1107–1116. doi: 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, Shi YG, Zhu J, Wang P, Xu Y. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155:1545–1555. doi: 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 17.Hu L, Lu J, Cheng J, Rao Q, Li Z, Hou H, Lou Z, Zhang L, Li W, Gong W, Liu M, Sun C, Yin X, Li J, Tan X, Wang P, Wang Y, Fang D, Cui Q, Yang P, He C, Jiang H, Luo C, Xu Y. Structural insight into substrate preference for TET-mediated oxidation. Nature. 2015;527:118–122. doi: 10.1038/nature15713. [DOI] [PubMed] [Google Scholar]
- 18.Schroder AS, Steinbacher J, Steigenberger B, Gnerlich FA, Schiesser S, Pfaffeneder T, Carell T. Synthesis of a DNA promoter segment containing all four epigenetic nucleosides: 5-methyl-5-hydroxymethyl-5-formyl-, and 5-carboxy-2'-deoxycytidine. Angew. Chem. Int. Ed. Eng. 2014;53:315–318. doi: 10.1002/anie.201308469. [DOI] [PubMed] [Google Scholar]
- 19.Dai Q, Song CX, Pan T, He C. Syntheses of two 5-hydroxymethyl-2'-deoxycytidine phosphoramidites with TBDMS as the 5-hydroxymethyl protecting group and their incorporation into DNA. J. Org. Chem. 2011;76:4182–4188. doi: 10.1021/jo200566d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szwagierczak A, Bultmann, Schmidt CS, Spada F, Leonhardt H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010;38:e181. doi: 10.1093/nar/gkq684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kizaki S, Sugiyama H. CGmCGCG is a versatile substrate with which to evaluate Tet protein activity. Org. Biomol. Chem. 2014;12:104–107. doi: 10.1039/c3ob41823e. [DOI] [PubMed] [Google Scholar]
- 22.Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, Smemo S, Dai Q, Bailey K, Nobrega MA, Han KL, Cui Q, He C. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nature Commun. 2013;4:1798. doi: 10.1038/ncomms2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol. 2008;4:152–156. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- 24.Breski M, Dey D, Obringer S, Sudhamalla B, Islam K. Engineering Biological C-H Functionalization Leads to Allele-Specific Regulation of Histone Demethylases. J. Am. Chem. Soc. 2016;138:13505–13508. doi: 10.1021/jacs.6b08653. [DOI] [PubMed] [Google Scholar]
- 25.Hamada S, Kim TD, Suzuki T, Itoh Y, Tsumoto H, Nakagawa H, Janknecht R, Miyata N. Synthesis and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorg. Med. Chem. Lett. 2009;19:2852–1855. doi: 10.1016/j.bmcl.2009.03.098. [DOI] [PubMed] [Google Scholar]
- 26.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Develop. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hashimoto H, Pais JE, Zhang X, Saleh L, Fu ZQ, Dai N, Correa IR, Jr, Zheng Y, Cheng X. Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature. 2014;506:391–395. doi: 10.1038/nature12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marholz LJ, Wang W, Zheng Y, Wang X. A Fluorescence Polarization Biophysical Assay for the Naegleria DNA Hydroxylase Tet1. ACS Med. Chem. Lett. 2016;7:167–171. doi: 10.1021/acsmedchemlett.5b00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, Turner SW, He C, Korlach J. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Methods. 2011;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terragni J, Bitinaite J, Zheng Y, Pradhan S. Biochemical characterization of recombinant beta-glucosyltransferase and analysis of global 5-hydroxymethylcytosine in unique genomes. Biochem. 2012;51:1009–1019. doi: 10.1021/bi2014739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.