

Abstract
Objective
The DAVID gene functional classification tool requires adaptations for use in non-model species and there is little available information to guide selection of a kappa score. Our objective was to develop an R-script that allows custom gene identifiers and novel annotation information to be incorporated into analyses, then use such data to evaluate the number of differentially expressed genes (DEGs) in a comparison based on kappa score selection.
Results
Using an R-script we developed and multiple data sets ranging from 555 to 3340 annotated DEGs from a study in rainbow trout, we found the percentage of DEGs harbored within a module and the number of genes shared among multiple modules decreased with increasing kappa score regardless of the number of DEGs in the comparison. The number of genes in enriched modules peaked at a kappa score of 0.5 for the comparisons with 3340 and 1313 DEGs and 0.3 for 555 DEGs. The number of genes harbored within enriched modules generally decreased with increasing kappa score; however, this was affected by whether the largest modules were significantly enriched. Large non-enriched modules can be reanalyzed using a higher kappa score resulting in some of the genes clustering in smaller enriched modules.
Electronic supplementary material
The online version of this article (10.1186/s13104-018-3154-7) contains supplementary material, which is available to authorized users.
Keywords: Gene functional classification, kappa statistics, Fuzzy heuristic partition, Soft clustering
Introduction
Data analysis software program packages designed to conduct cluster analysis of genes derived from sequencing or microarray data is an essential step to identify functional modules and reveal biological insights [1]. Soft clustering that can assign one gene to multiple clusters has been extensively used in gene analysis [2]. Soft clustering can use gene expression values [3] as well as gene ontology (GO) and other gene or protein annotation sources [4, 5]. This approach takes advantage of the assumption that genes with similar annotation profiles have similar functions [5–7]. Challenges exist with the publicly available programs to conduct soft clustering when atypical annotation sources are required. Many programs are web based and the input format and data resources cannot be changed by users [4, 5, 8–12]. This is a particular problem when the annotation resources used in the program are not updated in a timely manner [13]. Many programs were designed for specific model organisms [5, 8, 14] and don’t provide the flexibility to analyze data derived from non-model organisms. Lastly, the selection of parameters and statistical tests are limited for some software [12, 14, 15].
The web based software DAVID has become one of the most frequently cited tools for gene functional analysis [13, 16]. This tool was initially designed for human, mouse, rat, and fruit fly genomes and has been adopted for use in other species [17, 18]; however, it cannot be used for minor species when custom gene identifiers and their novel transcription information need to be incorporated into the analysis [19]. Furthermore, there have been reports of the gene annotation databases at times being outdated [5, 13, 20]. When using the DAVID gene functional classification tool, the selection of the kappa score greatly affects how genes are clustered. Kappa statistics, which measures inter-rater agreement, has been shown to be a reliable measurement of the functional gene–gene relationships in DAVID’s algorithm when an appropriate kappa value is utilized [5]. The optimal kappa score for generation of modules with biologically significant relationships is dependent on the nature of the data set. Thus, selecting an appropriate kappa score for a specific data set is a critical step in the data analysis.
The present paper describes the implementation of the agglomeration algorithm behind the DAVID functional gene classification tool with use of custom gene identifiers and their novel transcription information for cluster analysis of differentially expressed genes (DEGs) from a study on rainbow trout. We wrote an R-script to allow a standalone version of the functional gene classification program with which one can directly apply the algorithm to any species using the latest updated resources, and without a limit on input gene identifiers. Using this program, we explore the impact of kappa statistics on clustering rainbow trout gene expression data for three comparisons with widely different numbers of DEGs.
Main text
Methods
The rainbow trout used in the study were about 2-years-old and from stocks maintained at the USDA National Center for Cool and Cold Water Aquaculture (NCCCWA, Kearneysville, WV). Fish were reared indoors under artificial ambient photoperiod, in continuous-flow treated spring water, at 13 ± 1 °C. Follicle enclosed oocytes from rainbow trout competent to undergo the resumption of meiosis in response to the maturation inducing hormone (MIH), 17α-20β-dihydroxy-4 pregnen-3-one, were incubated in vitro for 24 h with or without either MIH or salmon pituitary homogenate (SPH). Sample total RNA was isolated from follicles freshly collected from the fish (Fresh), follicles cultured without hormone treatment (Control), and follicles cultured with MIH or SPH treatment using Trizol reagent (Invitrogen, Carlsbad, CA) followed by lithium chloride precipitation. Libraries from twelve RNA samples, three replications per treatment, were constructed with TruSeq mRNA Preparation for GAIIx/HiSeq, and then sequenced in 6 lanes using the Illumina HiSeq 2000 platform. Bowtie2 was used with default settings to align raw sequencing reads to a rainbow trout transcriptome database [21] supplemented with an additional 72 gene sequences of interest selected from Gene Bank [22]. At the false discovery rate (FDR) < 0.05, both DESeq2 [23] and edgeR [24] identified 4239 DEGs for control vs MIH treatment (Control_MIH), 1691 DEGs for control vs freshly excised tissue (Control_Fresh), and 691 DEGs for control vs SPH treatment (Control_SPH) comparisons. Those DEGs were analyzed by Blast2GO software [25, 26], and 3340, 1313, and 555 DEGs were annotated for the three comparisons accordingly.
The DEGs with mapped GO terms were input into an R-script with the DAVID gene functional classification algorithms [5] for grouping the genes into functionally related clusters. The script first calculates the kappa score to measure the degree of annotated gene pair co-occurrence, then searches for seeding genes, and then conducts functional clustering (Fig. 1). The R-script was tested with DAVID’s sample data and is available in Additional file 1: R_script for clustering.
Fig. 1.
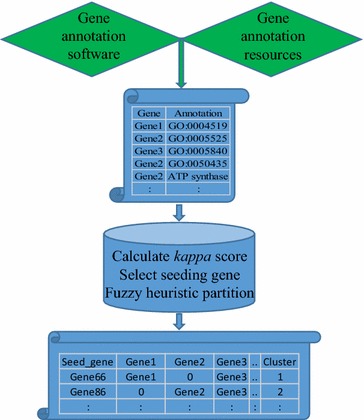
Flowchart of gene functional classification with DAVID’s algorithms in non-model organisms. Steps executed through R-script are in blue
Module enrichment scores were generated by calculating the geometric mean of the P-values which were derived from hypergeometric test on the input gene sets followed by negative log transformation of the geometric mean. The described rainbow trout transcriptome served as the reference genes used for the hypergeometric test. Pearson’s correlation coefficients were calculated using the R program.
Results
Number of modules and harbored genes
Cluster numbers were dynamically changed under different kappa scores for all comparisons (Fig. 2a). The number of modules for Control_MIH and Control_SPH increased as increasing kappa scores subdivided modules, but then decreased as fewer gene pairs met the increasing stringency. As would be expected, the peak number of modules was observed at a greater kappa score for Control_MIH with 3340 DEGs, than Control_SPH with 555 DEGs. The percentage of DEGs harbored in all modules decreased with increased kappa score and decreased at a greater rate as the number of DEGs in the comparison decreased (Fig. 2b).
Fig. 2.
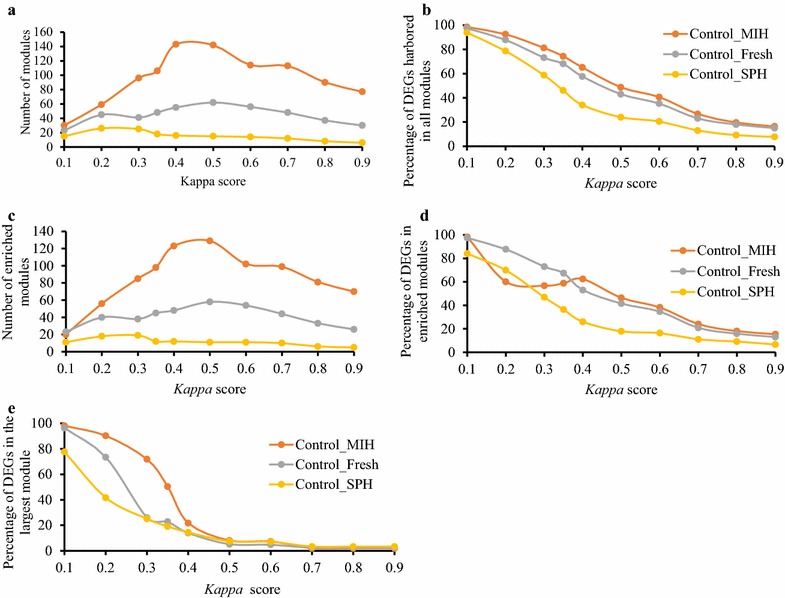
Changes in the number of modules (a), percentage of DEGs harbored in all modules (b), the number of gene set enriched modules (c), percentage of genes harbored in significantly enriched modules (d), and the percentage of differentially expressed genes harbored within the largest module (e) with kappa score
Genes shared by different modules
As an individual gene may be involved in multiple biological functions, it is reasonable that these multi-function genes are shared by multiple modules with each module composed of genes associated with a disparate function. In our data sets, the number of genes harbored in multiple modules decreased as kappa score increased (Table 1). The percent of genes clustered in multiple modules also decreased with increasing kappa score as the number of DEGs in the comparison decreased. Less than 10% of the genes clustered to multiple modules when the kappa score exceeded 0.5 for Control_MIH, 0.4 for Control_Fresh, and 0.3 for Control_SPH.
Table 1.
Changes in the number of genes shared among different numbers of modules with kappa score
| Comparison | Number of modules | Number of genes shared among modules | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| K = 0.1 | K = 0.2 | K = 0.3 | K = 0.35 | K = 0.4 | K = 0.5 | K = 0.6 | K = 0.7 | K = 0.8 | K = 0.9 | ||
| Control_MIH | 1 | 861 | 1145 | 985 | 1128 | 1539 | 1443 | 1229 | 883 | 650 | 548 |
| 2 | 1511 | 1384 | 1167 | 847 | 486 | 158 | 110 | 2 | 0 | 0 | |
| 3 | 746 | 436 | 415 | 343 | 110 | 23 | 17 | 4 | 0 | 0 | |
| 4 | 146 | 105 | 117 | 131 | 23 | 2 | 0 | 0 | 0 | 0 | |
| 5 | 24 | 20 | 16 | 27 | 16 | 1 | 0 | 0 | 0 | 0 | |
| 6 | 1 | 0 | 4 | 5 | 2 | 0 | 0 | 0 | 0 | 0 | |
| 7 | 0 | 0 | 9 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Control_Fresh | 1 | 431 | 391 | 585 | 704 | 657 | 536 | 444 | 302 | 236 | 197 |
| 2 | 653 | 439 | 264 | 147 | 83 | 25 | 15 | 0 | 0 | 0 | |
| 3 | 172 | 235 | 91 | 34 | 11 | 4 | 4 | 0 | 0 | 0 | |
| 4 | 22 | 70 | 17 | 9 | 6 | 0 | 0 | 0 | 0 | 0 | |
| 5 | 1 | 14 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 6 | 0 | 5 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Control_SPH | 1 | 201 | 282 | 281 | 236 | 184 | 132 | 113 | 71 | 51 | 43 |
| 2 | 224 | 101 | 39 | 20 | 5 | 1 | 1 | 1 | 0 | 0 | |
| 3 | 72 | 44 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 4 | 19 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 5 | 5 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
Number of enriched modules
Another important factor in gene functional classification is the enrichment score of modules, which helps to identify the most biologically relevant gene clusters. Nevertheless, some modules with an enrichment score of less than 1.3 (P < 0.05) could be potentially interesting [15]. The number of enriched modules combining all three GO categories peaked at a kappa score of about 0.5 for Control_MIH and Control_Fresh, but peaked earlier at 0.3 for Control_SPH which had the least number of DEGs (Fig. 2c). However, the total number of genes harbored among enriched modules generally decreased drastically with increasing kappa score (Fig. 2d). The Pearson’s correlations between the kappa scores and number of genes harbored in the enriched modules were − 0.947, − 0.983, and − 0.914 for Control_MIH, Control_Fresh, and Control_SPH respectively, and were highly significant (P < 0.001).
Cluster size
Implementing the fuzzy heuristic multiple-linkage partition in DAVID often resulted in one extremely large cluster of DEGs when low kappa scores were applied (Additional file 2: Table S1, module 1). The percentages of the DEGs in the largest module decreased dramatically with increased kappa score, and decreased more rapidly as the number of DEGs in the comparisons decreased (Fig. 2e). When using the data for all three GO categories, the enrichment scores of the largest modules were significant for all comparisons for all kappa scores except kappa scores below 0.4 for Control_MIH (Additional file 2: Table S2). Enrichment scores for the largest module increased consistently with kappa score for Control_MIH, but peaked at mid kappa scores for Control_Fresh and Control_SPH. This pattern held for Control_MIH when looking at data for the GO categories individually, but the patterns were less consistent among GO categories for Control_Fresh and Control_SPH (Additional file 2: Table S3).
When the number of DEGs harbored in the largest module is high and the DEGs in the module is not significantly enriched, such as in the combined GO category data for Control_MIH kappa scores 0.1–0.35, with 3279–1684 genes, respectively (Additional file 2: Table S2); the total number of genes harbored among enriched modules can be reduced relative to higher kappa scores (Fig. 2d). Thus, the ability to identify interactions of those genes which are not found in enriched modules with our other DEGs, is reduced in the analysis. One strategy to generate significantly enriched gene clusters for genes within these large modules is to break down the large module into smaller sub-modules by using a higher kappa score. We tested this at kappa score of 0.35 for Control_MIH. Using kappa score 0.35 for the complete data set (3340 DEGs), the largest module contains 1684 DEGs (see Additional file 2: Table S2), among which 449 DEGs are not found in other modules that were significantly enriched. However, at kappa score 0.6, this large module yielded 74 sub-modules of which 18 contained a total of 250 of the genes that were not previously incorporated into any enriched module. Seventeen of these 18 sub-modules were significantly enriched and contained 246 of the DEGs not incorporated into enriched modules in the initial analysis using kappa 0.35.
Discussion
Our R-script provides a flexible way to conduct gene functional cluster analysis for model and non-model organisms with DAVID’s algorithms [5] (Fig. 1). When using this program, there is no restriction to input annotated gene identifiers. In addition, users can prepare a flat matrix by using any software or laboratory experiment to get desired information from any gene resource.
Clustering of data sets into modules in which the genes have a functional relationship is highly dependent on the kappa score used in the analysis [5, 15]. In general, as the kappa score is increased the number of genes in the largest modules decreases. As the number of genes in a module decreases, the shared function of those genes becomes more specific; however, modules with few genes can only provide insight into the interactions of those limited numbers of genes. Thus, an investigator must choose a stringency or kappa score that is appropriate for their data set. Some papers report a kappa score of 0.35 [17, 27] as suggested in the DAVID program, but many papers either don’t mention the kappa score [18, 28–35] or report using scores above 0.35; e.g. 0.45 [36], 0.5 [37–39], 0.80 [40], or even 0.85 [41]. Few publications provide information on how or why kappa scores were selected.
In our RNA-seq data analysis, the number of modules in the comparison with the greatest number of DEGs (Control_MIH) was observed at a much higher kappa value than for the comparison with the least number of DEGs (Control_SPH) (Fig. 2a). As mentioned, the number of modules increases as increasing kappa scores subdivides modules with large gene sets, but then the number of modules decreases as more gene pairs fail to meet the increasing stringency. As expected, the more DEGs in the comparisons the greater the likelihood of more modules with large gene sets at low kappa scores (Additional file 2: Table S1). Regardless of the number of DEGs in a comparison, the percentage of genes harbored in all modules decreased as kappa score increased presumably as more gene pairs failed to meet stringency (Fig. 2b). Similar patterns were observed in terms of how kappa score affected the number of enriched modules and the percentage of genes harbored within enriched modules (Fig. 2c, d).
Limitations
Although we used multiple data sets ranging from 555 to 3340 annotated genes in rainbow trout to test the impact of kappa score on functional gene cluster analysis, the results only serve as a guide for the impacts of changes in data set size. Actual results will likely be impacted by differences in the size and diversity of the transcriptome among tissues, reference genome annotation, and species.
Additional files
Additional file 1. R_script for clustering.
Additional file 2: Table S1. Changes in the number of genes harbored in each module with kappa score. Table S2. The enrichment score for the largest module estimated by using three GO categories. Table S3. Changes in the enrichment score for the largest module with kappa score, estimated for each GO category.
Authors’ contributions
GMW and HM designed the study and wrote the paper. HM wrote the R script and conducted data analysis. GG helped in data analysis. All authors read and approved the final manuscript.
Acknowledgements
The authors acknowledge invaluable technical and animal caretaking contributions from Jill Birkett and Josh Kretzer. Mention of trade names is solely to provide accurate information and does not imply endorsement by the USDA. The USDA is an equal opportunity employer and provider.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The raw sequencing data used in the manuscript was deposited in NCBI Sequence Read Archive database (SRA Accession: SRP108755).
Consent for publication
Not applicable.
Ethics approval and consent to participate
All experiments were conducted under a protocol approved by the USDA/ARS National Center for Cool and Cold Water Aquaculture Institutional Animal Care and Use Committee (protocol #50).
Funding
This work was supported by USDA-ARS CRIS Project 8082-31000-012.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- DEGs
differentially expressed genes
- MIH
maturation inducing hormone
- SPH
salmon pituitary homogenate
- GO
gene ontology
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13104-018-3154-7) contains supplementary material, which is available to authorized users.
Contributor Information
Hao Ma, Email: hao.ma@ars.usda.gov.
Guangtu Gao, Email: guangtu.gao@ars.usda.gov.
Gregory M. Weber, Email: greg.weber@ars.usda.gov
References
- 1.D’haeseleer P. How does gene expression clustering work? Nat Biotechnol. 2005;23(12):1499–1501. doi: 10.1038/nbt1205-1499. [DOI] [PubMed] [Google Scholar]
- 2.Kumar L, Futschik MA. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2(1):5–7. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Futschik ME, Carlisle B. Noise-robust soft clustering of gene expression time-course data. J Bioinform Comput Biol. 2005;3(4):965–988. doi: 10.1142/S0219720005001375. [DOI] [PubMed] [Google Scholar]
- 4.Ramirez F, Lawyer G, Albrecht M. Novel search method for the discovery of functional relationships. Bioinformatics. 2012;28(2):269–276. doi: 10.1093/bioinformatics/btr631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan W. Incorporating gene functions as priors in model-based clustering of microarray gene expression data. Bioinformatics. 2006;22(7):795–801. doi: 10.1093/bioinformatics/btl011. [DOI] [PubMed] [Google Scholar]
- 7.Krejnik M, Klema J. Empirical evidence of the applicability of functional clustering through gene expression classification. IEEE ACM Trans Comput Biol. 2012;9(3):788–798. doi: 10.1109/TCBB.2012.23. [DOI] [PubMed] [Google Scholar]
- 8.Yu GC, Li F, Qin YD, Bo XC, Wu YB, Wang SQ. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26(7):976–978. doi: 10.1093/bioinformatics/btq064. [DOI] [PubMed] [Google Scholar]
- 9.Beissbarth T, Speed TP. GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics. 2004;20(9):1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 10.Hu Q, Wang ZG, Zhang ZG. FSim: a novel functional similarity search algorithm and tool for discovering functionally related gene products. Biomed Res Int. 2014;2014:509149. doi: 10.1155/2014/509149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin D, Brun C, Remy E, Mouren P, Thieffry D, Jacq B. GOToolBox: functional analysis of gene datasets based on gene ontology. Genome Biol. 2004;5(12):R101. doi: 10.1186/gb-2004-5-12-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khatri P, Draghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21(18):3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wadi L, Meyer M, Weiser J, Stein LD, Reimand J. Impact of outdated gene annotations on pathway enrichment analysis. Nat Methods. 2016;13(9):705–706. doi: 10.1038/nmeth.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 16.Fresno C, Fernandez EA. RDAVIDWebService: a versatile R interface to DAVID. Bioinformatics. 2013;29(21):2810–2811. doi: 10.1093/bioinformatics/btt487. [DOI] [PubMed] [Google Scholar]
- 17.Zambonelli P, Zappaterra M, Soglia F, Petracci M, Sirri F, Cavani C, Davoli R. Detection of differentially expressed genes in broiler pectoralis major muscle affected by White Striping—wooden breast myopathies. Poultry Sci. 2016;95(12):2771–2785. doi: 10.3382/ps/pew268. [DOI] [PubMed] [Google Scholar]
- 18.Libro S, Slatko BE, Foster JM. Characterization of innate immunity genes in the parasitic nematode Brugia malayi. Symbiosis. 2016;68(1–3):145–155. doi: 10.1007/s13199-015-0374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szczesniak MW, Gaffney DJ, Elo LL, Zhang X, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13. doi: 10.1186/s13059-016-0881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(9):R60. doi: 10.1186/gb-2003-4-9-r60. [DOI] [PubMed] [Google Scholar]
- 21.Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noel B, Bento P, Da Silva C, Labadie K, Alberti A, et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun. 2014;5:3657. doi: 10.1038/ncomms4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.https://www.ncbi.nlm.nih.gov/gene.
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenhouse DG, Murphy A, Mignatti P, Zavadil J, Galloway AC, Balsam LB. Mitral valve prolapse is associated with altered extracellular matrix gene expression patterns. Gene. 2016;586(1):56–61. doi: 10.1016/j.gene.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Akan I, Love DC, Harwood KR, Bond MR, Hanover JA. Drosophila O-GlcNAcase deletion globally perturbs chromatin O-GlcNAcylation. J Biol Chem. 2016;291(19):9906–9919. doi: 10.1074/jbc.M115.704783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yanez LZ, Han JN, Behr BB, Pera RAR, Camarillo DB. Human oocyte developmental potential is predicted by mechanical properties within hours after fertilization. Nat Commun. 2016;7:10809. doi: 10.1038/ncomms10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen ZH, Zhao F, Qi YD, Hu LF, Li DJ, Yin C, Su PH, Zhang Y, Ma JH, Qian J, et al. Simulated microgravity alters the expression of cytoskeleton- and ATP-binding-related genes in MLO-Y4 osteocytes. Acta Astronaut. 2016;129:186–192. doi: 10.1016/j.actaastro.2016.09.017. [DOI] [Google Scholar]
- 31.Mahmood K, Hojland DH, Asp T, Kristensen M. Transcriptome analysis of an insecticide resistant housefly strain: insights about SNPs and regulatory elements in cytochrome P450 genes. PloS ONE. 2016;11(3):e0151434. doi: 10.1371/journal.pone.0151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang LQ, Kumar S, Dahiya S, Wang F, Wu J, Newick K, Han RX, Samanta A, Beier UH, Akimova T, et al. Ubiquitin-specific protease-7 inhibition impairs Tip60-dependent Foxp3+ T-regulatory cell function and promotes antitumor immunity. Ebiomedicine. 2016;13:99–112. doi: 10.1016/j.ebiom.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li FB, Li YP, Zhang KW, Li Y, He P, Liu YJ, Yuan HY, Lu HH, Liu JX, Che ST, et al. FBLN4 as candidate gene associated with long-term and short-term survival with primary glioblastoma. Oncotargets Ther. 2017;10:387–395. doi: 10.2147/OTT.S117165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yim J, Rabkin SW. Differences in gene expression and gene associations in epicardial fat compared to subcutaneous fat. Horm Metab Res. 2017;49:327–337. doi: 10.1055/s-0042-119202. [DOI] [PubMed] [Google Scholar]
- 35.Omatsu-Kanbe M, Nozuchi N, Nishino Y, Mukaisho K, Sugihara H, Matsuura H. Identification of cardiac progenitors that survive in the ischemic human heart after ventricular myocyte death. Sci Rep-Uk. 2017;7:41318. doi: 10.1038/srep41318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hicks SD, Ignacio C, Gentile K, Middleton FA. Salivary miRNA profiles identify children with autism spectrum disorder, correlate with adaptive behavior, and implicate ASD candidate genes involved in neurodevelopment. BMC Pediatr. 2016;16:52. doi: 10.1186/s12887-016-0586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pettai K, Milani L, Tammiste A, Vosa U, Kolde R, Eller T, Nutt D, Metspalu A, Maron E. Whole-genome expression analysis reveals genes associated with treatment response to escitalopram in major depression. Eur Neuropsychopharm. 2016;26(9):1475–1483. doi: 10.1016/j.euroneuro.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 38.Sans JD, Krishnan S, Aggison LK, Adams HL, Shrikant MM, Lopez-Giraldez F, Petersen SL. Microarray analysis of neonatal rat anteroventral periventricular transcriptomes identifies the proapoptotic Cugbp2 gene as sex-specific and regulated by estradiol. Neuroscience. 2015;303:312–322. doi: 10.1016/j.neuroscience.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 39.Fukuda K, Sugihara E, Ohta S, Izuhara K, Funakoshi T, Amagai M, Saya H. Periostin is a key niche component for wound metastasis of melanoma. PloS ONE. 2015;10(6):e0129704. doi: 10.1371/journal.pone.0129704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia J, Rocke DM, Perry G, Ray M. Differential network analyses of Alzheimer’s disease identify early events in Alzheimer’s disease pathology. Int J Alzheimer’s Dis. 2014;2014:721453. doi: 10.1155/2014/721453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai XF, Li Y, Bai ZH, Tang XQ. Molecular portraits revealing the heterogeneity of breast tumor subtypes defined using immunohistochemistry markers. Sci Rep-Uk. 2015;5:14499. doi: 10.1038/srep14499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. R_script for clustering.
Additional file 2: Table S1. Changes in the number of genes harbored in each module with kappa score. Table S2. The enrichment score for the largest module estimated by using three GO categories. Table S3. Changes in the enrichment score for the largest module with kappa score, estimated for each GO category.
Data Availability Statement
The raw sequencing data used in the manuscript was deposited in NCBI Sequence Read Archive database (SRA Accession: SRP108755).