

Abstract
Background
This work represents an extensive MD simulation / water-dynamics studies on a series of complexes of inhibitors (leupeptin, E-64, E-64-C, ZPACK) and plant cysteine proteases (actinidin, caricain, chymopapain, calotropin DI) of papain family to understand the various interactions, water binding mode, factors influencing it and the structural basis of differential inhibition.
Results
The tertiary structure of the enzyme-inhibitor complexes were built by visual interactive modeling and energy minimization followed by dynamic simulation of 120 ps in water environment. DASA study with and without the inhibitor revealed the potential subsite residues involved in inhibition. Though the interaction involving main chain atoms are similar, critical inspection of the complexes reveal significant differences in the side chain interactions in S2-P2 and S3-P3 pairs due to sequence differences in the equivalent positions of respective subsites leading to differential inhibition.
Conclusion
The key finding of the study is a conserved site of a water molecule near oxyanion hole of the enzyme active site, which is found in all the modeled complexes and in most crystal structures of papain family either native or complexed. Conserved water molecules at the ligand binding sites of these homologous proteins suggest the structural importance of the water, which changes the conventional definition of chemical geometry of inhibitor binding domain, its shape and complimentarity. The water mediated recognition of inhibitor to enzyme subsites (Pn...H2O....Sn) of leupeptin acetyl oxygen to caricain, chymopapain and calotropinDI is an additional information and offer valuable insight to potent inhibitor design.
Background
Recently the cysteine proteases of both the plant and animals have received a considerable attention because of their broad ranges of activities and critical role in the different intracellular / biological / pathological processes or disorders [1-7]. Uncontrolled proteolysis of certain proteases e.g. Cathepsins [8,9], Caspases [10,11], Cruzain [12,13] causes several pathological disorders. Therefore the development of potential inhibitors which can modulate or can moderately control the proteolytic activity has become a challenge in drug design. For combating that challenge, some stimulating investigation on the structural aspects of the complexes on those kind of cysteine protease with different organic inhibitors or ligands e.g. E-64, E-64-C, leupeptin etc have been done by x-ray methods, which revealed the 3D-interactional information about the substrate binding chemistry and the topological requisite for substrate-mimicking inhibitors. However, due to the lacking of detail inhibitor binding information, we are interested to carry out those studies by MD-simulation methods on plant cysteine protease-inhibitor complex of papain [14,15] superfamily.
In this regard, beside the common Sn-Pn (n = 1–3) interactional events of the inhibitor with the main chain of the active site residues in the respective complexes, the role of the water molecules can not be ignored as they are sometimes present as conserved at the ligand binding sites of homologous proteins. These structurally conserved water molecule can change or influence the shape and complimentarity of the inhibitor site, thus affect strategies for therapeutic design. So, in order to get valuable insight of the characteristics of the inhibitor, the modeled inhibitor-enzyme complex structures are analyzed in detail and compared with the crystallographic information. The model structures of the enzyme-inhibitor complexes are built for the present study using template of available x-ray structure of enzyme-inhibitor complexes from papain family. In each complex structure, either from a x-ray or model study, it is evident that binding and catalysis is a two fold mechanism and in each case the subsite (Sn-Pn) interactions [16] are the main features for consideration to understand their differences in specificity.
The chemical structures of the selected inhibitors E-64, E-64-C, leupeptin, ZPACK for the present study are shown in Fig. 1. The inhibitor, E-64, a trans epoxysuccinic acid attached to a modified dipeptide [(leucylamino)-4-guanidinobutane] [17,18] is a potent irreversible (covalent type) inhibitor for cysteine proteases in general and its binding modes with papain [19], actinidin [20], caricain [21] and other lysozomal cysteine proteases [7] have been reviewed. The other analogue, E-64-C [22], consisting of epoxysuccinyl, leucyl, and an isoamyl group, is also being studied for its mode of different subsite binding [23-25]. The efficacy of this kind of inhibitor, makes the epoxide and its derivatives potential candidates as drug for the suppression of elevated levels of cysteine protease activity associated with certain disease states [23].
Figure 1.

Chemical structures of Leupeptin, E-64-C, E-64 and ZPACK
Figure 3.

Stereoscopic view of average over the last 50 ps of MD simulation of the Actinidin (backbone only) with E-64-C (inhibitor, ball and stick) complex
Figure 4.

Stereoscopic view of average over the last 50 ps of MD simulation of the Caricain (backbone only) with Leupeptin (inhibitor, ball and stick) complex
Leupeptin is another biologically important peptidyl aldehyde (AC-leu-leu-Arginal), a naturally occurring protease inhibitor [26] of microbial origin. The arginal residue at the C-terminus of leupeptin is found to be essential for its inhibitory activity which inhibits cysteine proteases as well as serine proteases [27]. Aldehyde inhibitors such as leupeptin are promising lead compounds in the design of anticancer protease inhibitors [28,29]. They are mostly composed of amino acids and therefore have few metabolic degradation products that are toxic [27].
ZPACK (N-benzoyloxycarbonyl-L-phenylalanine-L-alanylchloromethyl ketone) [17,18], a substrate – like synthetic inhibitor [30], is specifically designed for cysteine proteases.
We have done model enzyme-inhibitor complexes for which no crystal structure is available with a view that, these structural study of the complexes by modeling along with the x-ray structures will afford further insight and complementary information in this area.
Results and discussion
All the inhibitors selected for the present study (E-64, E-64-C, Leupeptin, ZPACK) can form the covalent bond with active site Cys 25 Sγ atom (with distance ~1.80 Å) of each cysteine proteases. This crucial interactional step which is mainly manifested in their inhibitory activity seems to remain conserved in all of our considered thiol proteases (papain, caricain, chymopapain, actinidin, calotropin DI) belonging to papain superfamily. The study reveals that each of the inhibitor molecule forms hydrogen bonds with the S1 subsite residues of enzyme molecule almost in a same fashion (Table I) as were observed in x-ray structures. The main residues that are involved in the conserved interactions are Cys 25 Sγ, Cys 25 amide nitrogen, the side chain of Gln 19 Nε2H, His 159 Nδ1H and in some cases carbonyl oxygen of Asp 158. The other residue Gly 66 offers its carbonyl oxygen and amide nitrogen to form hydrogen bond with the backbone of inhibitor molecule in most of the cases which are shown in Table I. It is observed that all these interactions help to orient the inhibitor molecule suitably in the active site cleft for the required nucleophilic attack by the active Cys 25 Sγ atom in the complex structure [31]. The S1-P1interactions of each of the inhibitor with the proteases papain, actinidin, caricain, chymopapain and calotropin DI are included in Table I. Both the MD simulation study and solved enzyme-inhibitor crystal structure of papain superfamily reveal, although S1-P1 interaction remains conserved / similar in all cases, however essential differences arise mainly at the S2-P2 and S3-P3 side chain interaction sites, depending on the amino acid residues constituting the respective sites of the proteases. The reaction propensities of the enzyme residues of the model enzyme-inhibitor complexes can be looked from DASA results (Fig. 6 – Fig. 9) which show distinctly the residues involved in electrostatic and hydrophobic interaction with the E-64, E-64-C, Leupeptin, ZPACK. The interaction pattern obtained from MD-simulation study indicates that the differential interaction in the S2 subsite is a dominant factor in defining the substrate specificity of cysteine proteases of papain family. This S2 subsite, which is generally hydrophobic in nature, is the best defined substrate binding site that deserved the term pocket and involves the residues both from -R and -L domain of enzyme [31]. Table II shows the involved residues of the selected proteases in S2 subsite region. Another residue (205, papain numbering) Ser for papain, Met (211) for actinidin, Gln (205) for calotropin DI at the back of S2 pocket is found to play a significant role in deciding the subtrate specificity of these enzymes at the P2 position of an inhibitor. The residues involved in S3 binding site (Table II) are generally found to be present on the enzyme surface and the interaction seems to prevail only in the side chain region with no main chain interaction.
Table I.
Significant atomic distances (Å) of different catalytic site residues of cysteine proteases with selected inhibitors at S1 - P1 site
| E-64 & Protease | Papain Caricain | Chymo | Actinidin | Calotropin | |
| [19] | [21] | papain | [20] | DI | |
| O5.....66(Gly):N | 2.88 | 2.78 | - | 2.92 | 3.37 |
| N10.....66(Gly):O | 3.04 | 2.94 | 3.23 | 2.91 | 3.49 |
| N6.....158(Asp):O | 3.27 | 3.58 | 3.15 | 3.60 | |
| O18.....159(His):ND1 | 2.90 | 2.88 | 2.76 | 2.80 | 3.10 |
| O17.....19(Gln):NE2 | 2.87 | 2.74 | 2.88 | 2.91 | 3.22 |
| O17.....25(Cys):N | 2.95 | 3.01 | 3.05 | 2.94 | 3.54 |
| O17.....25(Cys):SG | 3.36 | 3.33 | 3.24 | 2.81 | |
| E-64-C & Protease | Papain | Caricain | Chymo | Actinidin | Calotropin |
| [33] | Papain | DI | |||
| O5...66(Gly):N | 3.16 | 2.88 | 3.28 | 3.05 | 3.21 |
| N10...66(Gly):O | 3.46 | - | 3.30 | 3.64 | - |
| O17...19(Gln):NE2 | 2.90 | 3.35 | 3.12 | 3.25 | 3.18 |
| O17...25(Cys):N | 2.75 | 3.20 | 3.11 | 2.36 | 2.84 |
| O17...25(Cys):SG | 2.61 | 3.35 | 3.01 | 3.08 | 2.79 |
| O18...159(His):ND1 | 3.76 | 3.38 | 3.32 | 3.56 | 3.39 |
| N6...158(Asp):O | 2.99 | 3.17 | 3.27 | 3.43 | 3.40 |
| Leupeptin & Protease | Papain | Caricain | Chymo | Actinidin | Calotropin |
| [34] | Papain | DI | |||
| 2 Leu:N...66(Gly):O | 3.05 | 3.14 | 3.50 | 3.24 | 3.29 |
| 2 Leu:O...66(Gly):N | 2.78 | 3.17 | 3.13 | 3.09 | 3.07 |
| 3 Arg:N...25(Cys):SG | 3.01 | 3.06 | 3.26 | 3.22 | 3.02 |
| 3 Arg:N...158(Asp):O | 3.03 | 2.69 | 3.09 | 2.80 | 2.93 |
| 3 Arg:O...19(Gln):NE2 | 2.88 | 3.17 | 3.21 | 3.17 | 3.18 |
| 3 Arg:O...25(Cys):SG | 2.62 | 2.65 | 2.66 | 2.71 | 2.56 |
| 3 Arg:O...25(Cys):N | 3.00 | 3.28 | 3.50 | 3.16 | 3.46 |
| ZPACK & Protease | Papain | Caricain | Chymo | Actinidin | Calotropin |
| [30] | papain | DI | |||
| N11...66(Gly):O | 2.84 | 2.98 | - | 2.85 | 3.61 |
| O8...66(Gly):N | 2.96 | 3.10 | 3.29 | 3.10 | 2.85 |
| O3...25(Cys):N | 3.61 | 3.25 | 3.40 | 2.85 | 3.84 |
| O3...25(Cys):SG | 3.30 | 3.45 | 3.18 | 3.05 | 3.28 |
| O3...19(Gln):NE2 | 3.56 | 3.36 | 3.60 | 3.36 | 3.20 |
Footnote: The numbers in square brackets are reference numbers In the table, the distances are typed as normal for the modeled complexes and x-ray crystallographic values are in italics. Residue numbers are according to papain sequence
Figure 6.

DASA study of inhibitor-enzyme complexes
Figure 9.

DASA study of inhibitor-enzyme complexes
Table II.
Residues involved in S2 and S3 sites of different protease systems
| Name of the Protease | Residues involved in the S2 | Residues involed in the S3 |
| sites | sites | |
| Papain [14,15] | Val-133, Val-157, Ala-160, | Tyr-61, Tyr-67 |
| Ser-205, Asp-158, Pro-68 | ||
| Actinidin [35] | Ala-136, Ile-160, Met-211, | Asn-61, Tyr-69 |
| Ala-163, Asp-161, Ile70 | ||
| Caricain [40] | Val-133, Val-157, Ser-209, | His-61, Tyr-67 |
| Ala-160, Asp-158, Pro-68 | ||
| Chymopapain [36] | Leu-133, Leu-157, Ala-160, | Tyr-61, Tyr-67 |
| Asp-158, Gln-68 | ||
| CalotropinDI [41] | Gly-130,Gln-134,Val-154, | Tyr-61, Tyr-67 |
| Ala-160,Gln-205,Asp-158, | ||
| Tyr-68 |
Probable interaction of the inhibitors with the active site residues ofActinidin, Chymopapain, Caricain and Calotropin DI
Binding mode of E-64 and E-64-C to proteases (Caricain, Chymopapain, Actinidin, Calotropin DI)
All the main chain hydrogen bonding are similar to those found in papain complexes (Table I). The P1 carbonyl oxygens of E-64 and E-64-C forms hydrogen bonds with Cys-25 N, Sγ and Gln 19 Nε2H. The P2 main chain keto O and amide NH (Table III) make two hydrogen bonds with NH and O of Gly-66 respectively in most of the cases forming one residue stretch parallel β sheet structure. Although the interaction at the S1 subsite of enzymes are similar in nature for both of the inhibitors, significant differences arise at the P2 position while making interaction to S2 pocket of the enzymes. P2 residue (Leu) of E-64 extends inside into the pocket (Fig. 10) whereas corresponding P2 residue of E-64-C is located just at the entry of the S2 hydrophobic pocket (Fig. 11) interacting weakly. This point also gets support from DASA study (Fig. 6 – Fig. 9) for E-64-C-protease complexes where the peak for one of the leading residue at the S2 pocket is either very low or absent : Gly-130 (for calotropin DI), Ile-133 (for chymopapain), Val-133 (for caricain), Ala-136 (for actinidin). Another significant feature arising from MD simulation study that also get support from DASA study, is the residues of proteases especially Asp-158 in model complexes with E-64, E-64-C show a considerable decrease of their accessibilities suggesting that the Sn (n = 1–3) subsites of the modeled complexes are perfectly shielded from the solvent phase by binding with the inhibitors. The large difference in accessibility may occur due to many interaction with the inhibitor molecule. There is a second mode of binding for E-64-C to protease where isoamylamide and leu group of E-64-C involved in hydrophobic interaction with S2 and S3 site (contrary to S3 and S2 sites)of proteases respectively, but it does not change any basic interaction characters at these sites. In both cases the attack on the active site thiol group of the cysteine proteases occur from the backside of the inhibitor resulting in opening of the epoxide ring and at the same time the formation of a covalent bond between inhibitor and enzyme [19,20,32,33].
Table III.
Residues involved in various subsites of the selected inhibitors
Figure 10.

Stereoscopic view of probable binding mode of E-64 with the active site residues of Calotropin DI
Figure 11.

Stereoscopic view of probable binding mode of E-64-C with the active site residues of Actinidin
Binding mode of ZPACK and Leupeptin to proteases (Caricain, Chymopapain, Actinidin, Calotropin DI)
Both ZPACK (N-benzoyloxycarbonyl-L-phenylalanyl choromethyl ketone) and leupeptin form hydrogen bonds with the proteases in model complexes in usual fashion (Table I). But the important feature of β sheet nature of hydrogen bonding pair formed between the P2 moiety of ZPACK (phe) and Leupeptin (Leu) (Table III) with protease Gly-66 is antiparallel, different from E-64/E-64-C-protease complexes. The bulky P2 sidechain of ZPACK, which is Phe, is comfortably accommodated in the space available making hydrophobic interaction with all the residues Leu-133, Leu-157 (for chymopapain), Ala-136, Ile-160, Met-211 (for actinidin), Val-133 and Val-157 (for caricain), Gly-130, Val-154, Gln-205 (for calotropin DI). The P2 moiety (Table III) extends itself far inside the S2 pocket and thus away from carbonyl oxygen atom of Asp 158 (Fig. 12) which makes P2 backbone unable to form hydrogen bond with this residue. DASA study done on the model complexes with ZPACK shows the peak for Asp-158 for respective enzymes notably less prominent compared to other complexes. Thus it can be argued that the absence of this particular H-bond makes this protease-inhibitor complex less stable compared to other protease-inhibitor complexes. The residues involved in S2 subsite interaction for ZPACK-protease model are also same and responsible for preference of substrate splitting in leupeptin-protease complexes (Fig. 13). P2 moiety of leupeptin and E-64, despite being inside the S2 pocket, orients its backbone in such a way that they are able to form the additional H-bond (Table I) with carbonyl oxygen atom of Asp-158 giving extra stability to the respective complexes. There is another pocket on the left of the cleft called S3 subsite defined by the segment 61, 67 (papain numbering). The CBZ (benzoyloxycarbonyl) group of ZPACK simulating its P3 residue in the present complex makes aromatic-aromatic interaction with phenyl ring of S3 subsite with the shortest distance from aromatic to aromatic group > 4Å (Fig. 12), whereas the P3 moiety (Leu) of Leupeptin extends its sidechain towards the surface of S3 site interacting with its constituent residues (Fig. 13) of proteases.
Figure 12.

Stereoscopic view of probable binding mode of ZPACK with the active site residues of Chymopapain
Figure 13.

Stereoscopic view of probable binding mode of Leupeptin with the active site residues of Caricain
Role of water in inhibitor binding to the respective proteases
Water molecules have endowed several important role in the recognition and stabilization of the interaction between the ligand and its site. Table IV(A,B,C) gives a generalized view of interaction with the water molecules in active site cleft of the proteases. Extensive MD simulation / water dynamics study done on a series of enzyme-inhibitor complexes from papain plant family demonstrates some unique features of interesting nature. Conformational and dynamic properties of the model complexes are compared among themselves and with respect to x-ray structure. Solvent networks found in the x-ray structure were reproduced by the simulation which was unbiased with respect to the crystalline hydration structure. These networks seem to play an important role in the stability of the complexes, evidence of this is found in the structure of the ligand water interaction of the active site. We have identified water molecules, by studying a series of MD simulated complexes, in the active site cleft that may be essential in binding of inhibitor in the cleft.
Table IV (A).
Water involved in the interaction inside active site cleft of the enzyme-inhibitor complexes
| Papain + Leupeptin[34] | Distances in Å | Subsites of inhibitor | |
| in interaction | |||
| Leup:3Arg:NE | 419:OH2 | 3.28* | P1 |
| Leup:3Arg:NH1 | 437:OH2 | 2.67* | P1 |
| Caricain + Leupeptin | |||
| Leup:3Arg:NE | 439:OH2 | 3.21* | P1 |
| Leup:3Arg:NE | 411:OH2 | 2.85* | P1 |
| Leup:3Arg:NH1 | 1365:OH2 | 2.92* | P1 |
| Leup:3Arg:NH2 | 439:OH2 | 2.85 | P1 |
| Leup:3Arg:O | 1313:OH2 | 2.63** | P1 |
| Leup:Ac:O | 1293:OH2 | 2.86 | P3 |
| Caricain:157:Val:O | 1293:OH2 | 2.78 | |
| Chymopapain + Leupeptin | |||
| Leup:3Arg:NE | 1288:OH2 | 3.05* | P1 |
| Leup:3Arg:NH1 | 1324:OH2 | 2.99* | P1 |
| Leup:3Arg:NH2 | 1288:OH2 | 3.01 | P1 |
| Leup:3Arg:NH2 | 1397:OH2 | 3.19 | P1 |
| Leup:3Arg:O | 1505:OH2 | 2.98** | P1 |
| 1324:OH2 | 1402:OH2 | 2.74 | |
| Leup:1Leu:N | 1402:OH2 | 3.32 | P2 |
| Leup:Ac:O | 1437:OH2 | 3.13 | P3 |
| Chymo:66:Gly:O | 1437:OH2 | 2.74 | |
| Actinidin + Leupeptin | |||
| Leup:3Arg:NH1 | 1249:OH2 | 2.83* | P1 |
| Leup:3Arg:NH1 | 1303:OH2 | 2.86* | P1 |
| Act:158:O | 1303:OH2 | 2.72 | |
| Leup:3Arg:NH2 | 1249:OH2 | 3.01 | P1 |
| Leup:3Arg:NH2 | 1593:OH2 | 3.11 | P1 |
| Leup:3Arg:O | 186:OH2 | 3.26** | P1 |
| Leup:Ac:O | 1391:OH2 | 3.28 | P3 |
| Calotropin DI + Leupeptin | |||
| Leup:3Arg:NE | 1370:OH2 | 3.23* | P1 |
| Leup:3Arg:NH1 | 1370:OH2 | 3.17* | P1 |
| Leup:3Arg:NH1 | 1553:OH2 | 3.49* | P1 |
| Leup:3Arg:NH1 | 1435:OH2 | 3.37 | P1 |
| Leup:3Arg:NH2 | 437:OH2 | 2.56 | P1 |
| Leup:3Arg:O | 1320:OH2 | 3.33** | P1 |
| Leu :Ac :O | 1595 :O1 | 3.00 | P3 |
| 1595 :OH2 | CaloDI:67:Tyr :OH | 3.49 | |
| Leup:Ac:O | CaloDI:67:TYR:OH | 3.22 | P3 |
Table IV (B).
Water involved in the interaction inside active site cleft of the enzyme-inhibitor complexes
| Papain + E-64-C[32,33] | Distances in Å | Subsites of inhibitor | |
| in interaction | |||
| E-64-C (I):O18 | 232:OH2 | 2.98** | P1 |
| E-64-C(II):O18 | 389:OH2 | 2.60** | P1 |
| Caricain + E-64-C | |||
| E-64-C:O18 | 1468:OH2 | 3.47** | P1 |
| E-64-C:O17 | 1520:OH2 | 3.46 | P1 |
| Chymopapain + E-64-C | |||
| E-64-C:O18 | 1464:OH2 | 2.69** | P1 |
| E-64-C:O9 | 1317:OH2 | 3.20 | P2 |
| Actinidin + E-64-C | |||
| E-64-C:O18 | 1489:OH2 | 2.85** | P1 |
| E-64-C:O9 | 1152:OH2 | 3.32 | P2 |
| E-64-C:O1 | 1454:OH2 | 3.21 | P1 |
| CalotropinDI + E-64-C | |||
| E-64-C:O18 | 445:OH2 | 3.09** | P1 |
| E-64-C:O18 | 441:OH2 | 3.29** | P1 |
| E-64-C:O17 | 455:OH2 | 3.21 | P1 |
| E-64-C:O9 | 1299:OH2 | 2.84 | P2 |
| E-64-C:O5 | 1406:OH2 | 3.41 | P1 |
| 1299:OH2 | 1406:OH2 | 3.16 | |
| Papain + E-64[19] | |||
| E-64:O18 | 217:OH2 | 2.67** | P1 |
| E-64:N17 | 216:OH2 | 2.45* | P3 |
| Actinidin + E-64[20] | |||
| E-64:O18 | 582H:OH2 | 2.81** | P1 |
| E-64:N17 | 257H:OH2 | 3.23* | P3 |
| E-64:N15 | 54H:OH2 | 3.01 | |
| E-64:N17 | 54:OH2 | 3.57 | P3 |
| E-64:N15 | 54:OH2 | 3.01 | P3 |
| Caricain + E-64[21] | |||
| E-64:O18 | 310H:O | 2.98** | P1 |
| Chymopapain + E-64 | |||
| E-64:O18 | 302:OH2 | 3.21** | P1 |
| E-64:O9 | 1357:OH2 | 2.85 | P2 |
| E-64:N18 | 1128:OH2 | 3.14 | P3 |
| CalotropinDI + E-64 | |||
| E-64:O18 | 196:OH2 | 2.91** | P1 |
| E-64:O17 | 196:OH2 | 3.39 | P1 |
| E-64:O9 | 1370:OH2 | 2.94 | P2 |
| E-64:N15 | 1310:OH2 | 3.43* | P3 |
| E-64:N18 | 1625:OH2 | 3.25 | P3 |
| E-64:N18 | 1363:OH2 | 3.06 | P3 |
| E-64:N17 | 1310:OH2 | 3.22* | P3 |
Table IV (C).
Water involved in the interaction inside active site cleft of the enzyme-inhibitor complexes
| Papain + ZPACK[30] | Distances in Å | Subsites of inhibitor | |
| in interaction | |||
| (No water mediated interaction | |||
| was reported) | |||
| Caricain + ZPACK | |||
| ZPACK:O13 | 981:OH2 | 3.01 | P3 |
| ZPACK:O3 | 396:OH2 | 3.40 | P1 |
| ZPACK:O3 | 402:OH2 | 2.25** | P1 |
| Chymopapain + ZPACK | |||
| ZPACK:O13 | 341:OH2 | 3.14 | P3 |
| ZPACK:O14 | 377:OH2 | 3.07 | P3 |
| ZPACK:N11 | 377:OH2 | 3.42' | P2 |
| ZPACK:O3 | 290:OH2 | 3.28** | P1 |
| Actinidin + ZPACK | |||
| ZPACK:O13 | 345:O1 | 2.84 | P3 |
| ZPACK:N6 | 345:OH2 | 2.47 | P1 |
| ZPACK:O3 | 366:OH2 | 2.82** | P1 |
| Calotropin DI + ZPACK | |||
| ZPACK:O13 | 1279:OH2 | 3.20 | P3 |
| ZPACK:O13 | 1346:OH2 | 3.31 | P3 |
| ZPACK:O3 | 1300:OH2 | 3.09 | P1 |
Foot notes: Names in Italics are x-ray structures Names in normal font are modelled structures * denotes the interactions found both in x-ray and modeled structure ** denotes water found near oxyanion hole
In all Leupeptin-protease model complexes we found P1 side chain (Arg) of leupeptin is extensively involved in water interaction network (Table IV(A,B,C)). These water interactions though less in number, are also found to be present in x-ray structure of papain-leupeptin complex [34]. These interactions support the fact that P1 side chain of inhibitor, leupeptin, makes few interactions with S1 site to avoid steric hindrance with carbonyl group of Gly-23 of protease molecule and extend straight up out of the cleft towards solvent [8] making a number of interactions. Moreover the keto oxygen (O3) of ZPACK, epoxy and carbonyl oxygens of E-64, E-64-C seem to stabilize through strong and weak H-bond with water molecules within the groove of the enzymes. The amide nitrogen (N11) of ZPACK and keto oxygen (O9) of E-64, E-64-C are observed to form H-bonds with water sites thus assisting the chemical potentiality of P2 site. MD simulations also characterize some other important stereochemically potential interaction sites of inhibitors in complexation with different proteases associated with P3 site of E-64, leupeptin, ZPACK and involved in solvation network within the active site. Guanidinobutane group of Arg of E-64, keto oxygen (O13, O14) of CBZ of ZPACK and acetyl keto oxygen of leupeptin involved in water interaction at S3 site with each protease in model complexes (Table IV(A,B,C)). The water molecules easily access the surface region of S3 site of enzyme to interact with the polar group involved in P3 site of those inhibitor molecules. Solved x-ray structure of complexes from papain family also shows this solvation network of P3 site of inhibitor only for papain-E-64 [19] and actinidin-E-64 [20] complexes but few in number. The inhibitor bonded water molecular bridging H-bonds of leupeptin acetyl oxygen directly to protein S2 / S3 subsite residues at amide oxygen of Val-157 (in caricain) and Gly-66 (in chymopapain) and at side chain Tyr-67-OH group of calotropinDI are more conducive allowing the ligand binding by Pn...H2O...Sn type of interaction which are evidenced and are supported from our studies. Thus the hydration dynamic study on series of modeled complexes helps us to understand the role of water involved in stabilizing the inhibitor molecules in S2 / S3 subsite of proteases, where polar group involved in P3 site of inhibitor may add extra stability to enzyme-inhibitor complexes by water mediating interactions of ligand and enzyme residues involved in this segment.
The other key feature and the most important one that has emerged out from the study is the participation of a key water molecule (marked by ** in Table IV(A,B,C)) in the vicinity of oxyanion hole within the active site of proteases which seemingly play a vital role in inhibitor binding. This particular water molecule found near the region of oxyanion hole, comprising of Cys 25 NH and Gln 19 Nε2H, forms hydrogen bond with the carbonyl / carboxyl oxygen of P1 moiety of inhibitor at a distance of ~2.63 Å to 3.47 Å [O18 of E-64 and E-64-C, O3 of ZPACK and mainchainO of Arg of leupeptin] (Fig. 10 – Fig. 13). This observation when extended to the solved x-ray structure of papain family either native or complexed [15,19-21,33,35,36], we found that this particular water is also present in several of these cases (PDB codes are 1PPN, 2ACT, 2AEC, 1MEG, 1GEC, 1YAL, 1PPP, 1PE6). This important observation lead us to suggest that the water molecule present in the solved structure and model complexes may be involved in stabilizing the inhibitor molecule during nucleophilic attack of cysteine protease. And perhaps this water which behaves like a conserved water, is involved in acylation / deacylation step during catalysis by proteases in papain family. Table IV(A,B,C) highlights that for each inhibitor the interactions are not random, but rather highly specific, usually involving specific residues / regions of the ligand. These water molecules contribute directly to the stability of the complex by holding themselves in the right position through network of hydrogen bonds. These water networks are probably crucial for the stability of protein-ligand complexes and may be important for any site directed drug design strategies. Thus our observation demonstrates that incorporation of water molecules into the system improves interpretation and predictive ability of the models and are in good agreements with the x-ray structural analysis of some complexes and offers valuable insights into new characteristics of the ligands which may be exploited for design of more potent inhibitors.
Material and Method
The different inhibitors (Fig. 1) were docked to the active site cleft of the selected proteases from the papain family using chemical intuition and viable interaction strategy. The position and orientation of these inhibitors in the active site cleft was primarily based on the knowledge of x-ray structures [19-21,30-34,37-39] of different cysteine proteases with the inhibitors.
Each of the modeled structures was subjected to constrained energy minimization to relieve residual strain maintaining the integrity of the model. The modeling and energy minimization was done with the InsightII software package (Molecular Simulations Inc., San Diego, CA, version 95.0) on a Silicon Graphics Indigo workstation. The minimization was done with the Discover Platform of the InsightII program with a consistent valence force field (CVFF). The algorithms utilized for minimization were typically the conjugate gradient options in vacuum and were chosen to be distance dependent. The covalently linked ligand was minimized while the enzyme backbone was kept rigid using the Discover option constraint. During energy minimization, in addition to the usual energy terms (bond, angle, dihedral, improper dihedral, electrostatic and van der Waals), distance constraints were also introduced initially. Different selected inhibitors were docked into the caricain [21,40], chymopapain [36], actinidin [35], calotropinDI [41] environments lead by the information of x-ray crystallographically solved complex structures of proteases from the papain family. The nonbonded interactions included van der Waal's and electrostatic forms which used the Lenard-Jones and Coulomb functions respectively. The cut off radius for nonbonded interactions was 30 Å.
Molecular dynamics (MD) simulations of the modeled enzyme-inhibitor complex structure
Each of the energy minimized structure was solvated by a 10 Å layer of water molecules using SOAK option of insight II (Molecular Simulations Inc., San Diego, CA, version 95.0). During molecular dynamic simulations and minimization, a distance dependent dielectric constant of 1.0 was used. The parameter used was the consistent valence forcefield (CVFF). Prior to simulations, the co-ordinates of all the enzyme-inhibitor atoms were fixed and solvent molecules were minimized using steepest decent method in all constrained minimizations for 1000 iterations. Finally, the conjugate gradient minimizations were performed with all atoms free to move until maximum derivative was < 0.5 Kcal/mol/Å. The minimized co-ordinates of the whole system were used as a starting point for NVT (constant volume and temperature) at 300K to generate possible stable conformation. After 20 ps equilibrium stage, the simulations were carried out using velocity verlet algorithm for another 100 ps. Only the amino acid residues and solvent within a 12 Å radius of the inhibitor was permitted to move during simulation; the remainder of protein was fixed throughout the simulation. Surrounding the sphere of moving residue and solvent was a layer of waters whose positions were fixed in order to ensure that none of the moving solvent molecules escaped from the vicinity of the active site simulation. The average structures over the last 50 ps of each kind of MD simulations were taken to find out probable interactions of the inhibitors with the active site residues (Fig 2 – Fig. 5).
Figure 2.

Stereoscopic view of average over the last 50 ps of MD simulation of the Calotropin DI (backbone only) with E-64 (inhibitor, ball and stick) complex
Figure 5.

Stereoscopic view of average over the last 50 ps of MD simulation of the Chymopapain (backbone only) with ZPACK (inhibitor, ball and stick) complex
All the optimized structures were finally analyzed by PROCHECK [42].
Solvent accessibility study
The difference accessible surface area (DASA) study of a protein with and without inhibitor reveals the potential interactional sites of the protein. In order to estimate the fit of contact between the protein and the inhibitor, the difference accessible surface area (DASA) was calculated using DASA = [ASA (enzyme) – ASA (complex)] where ASA represents the accessible surface area given by Lee and Richard [43] (1971). ASA (complex) and ASA (enzyme) are the accessible surface area of each residue in a protease structure with and without the inhibitor respectively. The DASA study, performed on each of the modeled complexes using Biosym (MSI, San Diego, CA, InsightII package, 95.0 version), shows distinctly the protease residues involved in electrostatic and hydrophobic interaction with the inhibitors. Difference plots of solvent accessibilities to indicate the interacting residues of different numbers of papain-family cysteine proteases are shown in Fig. (6,7,8,9).
Figure 7.

DASA study of inhibitor-enzyme complexes
Figure 8.
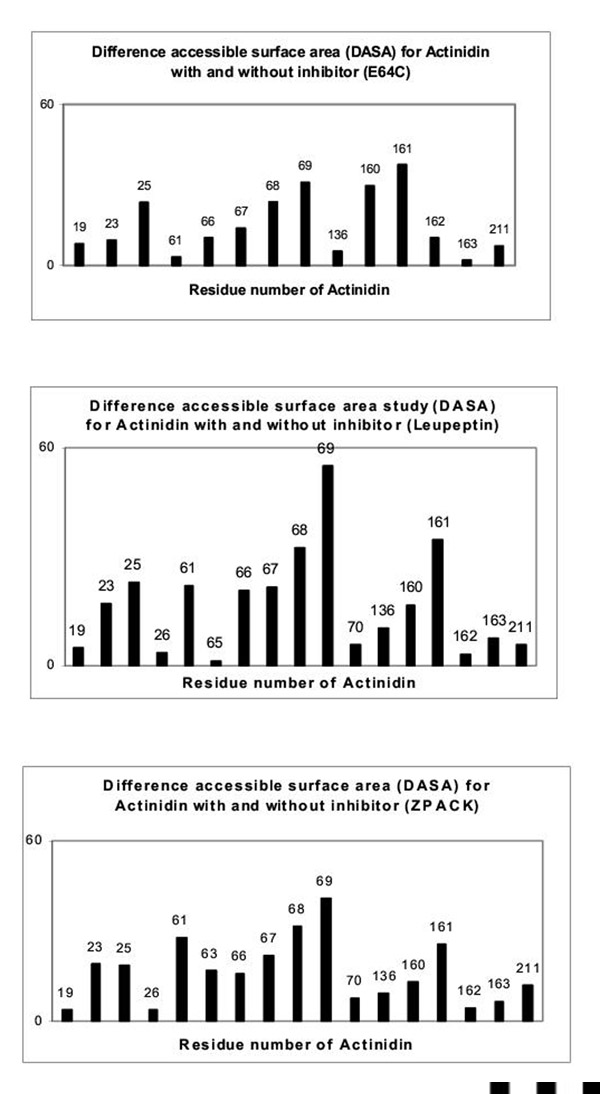
DASA study of inhibitor-enzyme complexes
Acknowledgments
Acknowledgement
We are thankful to the members of the Biophysics Department, Bose Institute. Prof. N. K. Sinha for his assistance and Prof. W. Saenger for providing us with Calotropin DI co-ordinate at 1.9 Å in 1992 through personal communication.
Contributor Information
Suparna Bhattacharya, Email: Suparna99@yahoo.com.
Sreya Ghosh, Email: ashoke@boseinst.ernet.in.
Sibani Chakraborty, Email: ashoke@boseinst.ernet.in.
Asim K Bera, Email: ashoke@boseinst.ernet.in.
Bishnu P Mukhopadhayay, Email: ashoke@boseinst.ernet.in.
Indrani Dey, Email: ashoke@boseinst.ernet.in.
Asok Banerjee, Email: ashoke@boseinst.ernet.in.
References
- Barrett AJ. Proteinases in Mammalian Cells and Tissues, Elsevier / North-Holland, Amsterdam, 1977.
- Delaisse JM, Eeckhout Y, Vaes G. Biochem Biophys Res Commun. 1984;125:441–447. doi: 10.1016/0006-291x(84)90560-6. [DOI] [PubMed] [Google Scholar]
- Delaisse JM, Ledent P, Vaes G. Biochem J. 1991;279:167–274. doi: 10.1042/bj2790167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denhardt D, Greenberg AH, Egan SE, Hamilton RT, Wright JA. Oncogene. 1987;2:55–59. [PubMed] [Google Scholar]
- Neurath H. Science. 1984;224:350–357. doi: 10.1126/science.6369538. [DOI] [PubMed] [Google Scholar]
- Page A, Warburton MJ, Chambers TJ, Hayman AR. Biochem Ser Trans. 1991. p. 286S. [DOI] [PubMed]
- Turk B, Turk V, Turk D. Biol Chem. 1997;378:141–150. [PubMed] [Google Scholar]
- McGrath ME. Annu Rev Biophys Biomol Struct. 1999;28:181–204. doi: 10.1146/annurev.biophys.28.1.181. [DOI] [PubMed] [Google Scholar]
- Turk B, Turk D, Turk V. Biochim Biophys Acta. 2000;1477:98–111. doi: 10.1016/S0167-4838(99)00263-0. [DOI] [PubMed] [Google Scholar]
- Los M, Stroh C, Janicke Ru, Engles IH, Schulze-Osthof K. Trends Immunol. 2001. pp. 356–62. [DOI] [PubMed]
- Nunez G, Benedic MA, Inohara N, Hu Y. Oncogene. 1998;17:3237–45. doi: 10.1038/sj/onc/1202581. [DOI] [PubMed] [Google Scholar]
- Cazzulo JJ, Stoka V, Turk V. Biol Chem. 1997;378:1–10. doi: 10.1515/bchm.1997.378.1.1. [DOI] [PubMed] [Google Scholar]
- McGrath ME, Eakin AE, Engel JC, McKerrow JH, Craik CS, Fletterick RJ. J Mol Biol. 1995;247:251–259. doi: 10.1006/jmbi.1994.0137. [DOI] [PubMed] [Google Scholar]
- Drenth J, Jansonius JN, Koekoek R, Wolthers BG. Adv Protein Chem. 1971;25:79–115. doi: 10.1016/s0065-3233(08)60279-x. [DOI] [PubMed] [Google Scholar]
- Kamphuis IG, Kalk KH, Swarte MBA, Drenth J. J Mol Biol. 1984;179:233–256. doi: 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- Schechter I, Berger A. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- Hanada K, Tamai M, Yamagishi M, Ohmura S, Sawada J, Tanaka I. Agric Biol Chem. 1978. pp. 523–528.
- Hanada K, Tamai M, Ohmura S, Sawada J, Tanaka I. Agric Biol Chem. 1978. pp. 529–536.
- Varughese KI, Armed FR, Cary PR, Hansman S, Huber CP, Storer AC. Biochemistry. 1989;28:1330–1332. doi: 10.1021/bi00429a058. [DOI] [PubMed] [Google Scholar]
- Varughese KI, Ying S, Cromwell D, Hasnain S, Xuong N. Biochemistry. 1992;31:5172–5176. doi: 10.1021/bi00137a012. [DOI] [PubMed] [Google Scholar]
- Katerelos NA, Taylor MAJ, Scott M, Goodenough PW, Pickersgill RW. FEBS Lett. 1996;392:35–39. doi: 10.1016/0014-5793(96)00697-7. [DOI] [PubMed] [Google Scholar]
- Waxman L. In Methods of Enzymology (L Lorand, Ed), Academic Press, New York, 1981. pp. 664–680. [DOI] [PubMed]
- Hanada K, Tamai M, Morimoto S, Adachi T, Oguma K, Ohmura S, Ohzeki M. Peptide Chemistry 1979 (Yonehara, H, ed) Protein Research Foundation, Osaka, Japan. 1980.
- Tamai M, Adachi T, Oguma K, Morimoto S, Hanada K, Ohmura S, Ohzeki M. Agric Biol Chem. 1981. pp. 675–679.
- Tamai M, Hanada K, Adachi T, Oguma K, Kashwagi K, Omura S, Ohzeki M. J Biochem. 1981;90:255–257. doi: 10.1093/oxfordjournals.jbchem.a133458. [DOI] [PubMed] [Google Scholar]
- Aoyogi T, Umezawa H. In: Proteases and Biological Control. E. Reich, D.B. Rifkin and E. Shaw, editor. Cold Spring Harbor Laboratory; 1975. pp. 429–454. [Google Scholar]
- Kurnov IV, Harrison RW. Protein Sci. 1996;5:752–758. doi: 10.1002/pro.5560050420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brtko J, Knopp J, Baker ME. Mol Cell Endocrinol. 1993;93:81–86. doi: 10.1016/0303-7207(93)90142-7. [DOI] [PubMed] [Google Scholar]
- Kennedy AR. Cancer Res. 1994. pp. 1899–1915.
- Drenth J, Kalk KH, Swen HM. Biochemistry. 1976;15:3731–3738. doi: 10.1021/bi00662a014. [DOI] [PubMed] [Google Scholar]
- Turk D, Guncar G, Pdobnik M, Turk B. JBiol Chem. 1998. pp. 137–147. [DOI] [PubMed]
- Kim MJ, Yamamoto D, Matsumoto B, Inoue M, Ishida T, Mizuno H, Sumiya S, Kitamura K. Biochem J. 1992;287:797–803. doi: 10.1042/bj2870797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto D, Matsumoto K, Ohishi H, Ishida T, Inoue M, Kitamura K, Mizuno H. J Biol Chem. 1991;266:14771–14777. doi: 10.2210/pdb1pe6/pdb. [DOI] [PubMed] [Google Scholar]
- Schroder E, Phillips C, Garman E, Harlos K, Crawford C. FEBS Lett. 1993;315:38–42. doi: 10.1016/0014-5793(93)81128-M. [DOI] [PubMed] [Google Scholar]
- Baker EN. J Mol Biol. 1980;141:441–484. doi: 10.1016/0022-2836(80)90255-7. [DOI] [PubMed] [Google Scholar]
- Maes D, Bouckaert J, Poortmans F, Wyns L, Looze Y. Biochemistry. 1996;35:16292–16298. doi: 10.1021/bi961491w. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Murata M, Sumiya S, Kitamura K, Ishida T. Biochim Biophys Acta. 1994;1208:268–276. doi: 10.1016/0167-4838(94)90113-9. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Murata M, Sumiya S, Mizuno K, Kitamura K, Ishida T. Biochim Biophys Acta. 1998;1383:93–100. doi: 10.1016/S0167-4838(97)00187-8. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Mizuno H, Kitamura K, Tse WC, Huber CP, Ishida T. Biopolymers. 1999;51:99–107. doi: 10.1002/(SICI)1097-0282(1999)51:1<99::AID-BIP11>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Pickersgill RW, Rizkallah P, Harries GW, Goodenough PW. Acta Crystallogr. 1991. pp. 766–771.
- Heinemann U, Pal GP, Hilgenfeld R, Saenger W. J Mol Biol. 1982;161:591–606. doi: 10.1016/0022-2836(82)90410-7. b) Saenger, W. 1992 Private Communication. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Cryst. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Lee B, Richards FM. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]