

Abstract
Objectives
Studies which consider the molecular mechanisms of degeneration and regeneration of cartilaginous tissues are seriously hampered by problematic ribonucleic acid (RNA) isolations due to low cell density and the dense, proteoglycan-rich extracellular matrix of cartilage. Proteoglycans tend to co-purify with RNA, they can absorb the full spectrum of UV light and they are potent inhibitors of polymerase chain reaction (PCR). Therefore, the objective of the present study is to compare and optimise different homogenisation methods and RNA isolation kits for an array of cartilaginous tissues.
Materials and Methods
Tissue samples such as the nucleus pulposus (NP), annulus fibrosus (AF), articular cartilage (AC) and meniscus, were collected from goats and homogenised by either the MagNA Lyser or Freezer Mill. RNA of duplicate samples was subsequently isolated by either TRIzol (benchmark), or the RNeasy Lipid Tissue, RNeasy Fibrous Tissue, or Aurum Total RNA Fatty and Fibrous Tissue kits. RNA yield, purity, and integrity were determined and gene expression levels of type II collagen and aggrecan were measured by real-time PCR.
Results
No differences between the two homogenisation methods were found. RNA isolation using the RNeasy Fibrous and Lipid kits resulted in the purest RNA (A260/A280 ratio), whereas TRIzol isolations resulted in RNA that is not as pure, and show a larger difference in gene expression of duplicate samples compared with both RNeasy kits. The Aurum kit showed low reproducibility.
Conclusion
For the extraction of high-quality RNA from cartilaginous structures, we suggest homogenisation of the samples by the MagNA Lyser. For AC, NP and AF we recommend the RNeasy Fibrous kit, whereas for the meniscus the RNeasy Lipid kit is advised.
Cite this article: M. Peeters, C. L. Huang, L. A. Vonk, Z. F. Lu, R. A. Bank, M. N. Helder, B. Zandieh Doulabi. Optimisation of high-quality total ribonucleic acid isolation from cartilaginous tissues for real-time polymerase chain reaction analysis. Bone Joint Res 2016;5:560–568. DOI: 10.1302/2046-3758.511.BJR-2016-0033.R3.
Keywords: Ribonucleic acid, Ribonucleic acid isolation, Nucleus pulposus, Annulus fibrosus, Articular cartilage, Meniscus, Real-time polymerase chain reaction
Article focus
To determine the optimal ribonucleic acid (RNA) isolation and tissue homogenisation method for four different cartilaginous tissues: nucleus pulposus, annulus fibrosus, articular cartilage and meniscus.
Key messages
Homogenisation of cartilaginous tissues is best achieved by use of the Magna-lyser (homogenisation of samples simultaneous and more cost-effective).
RNA isolation using the RNeasy Fibrous kit and the RNeasy Lipid kit result in the highest quality RNA for subsequent real time polymerase chain reaction.
Strengths and limitations
This is an extensive study comparing four different commonly used and commercially available methods for RNA isolation of different cartilaginous tissues.
Although a limited number of donors where used, internal checks (each tissue sample split over the various processing methods) were included to allow valid comparisons.
Introduction
Low back pain and osteoarthritis are health problems prevalent in Western society, with a high risk of resulting in disability and high socio-economic costs.1 Many recent studies have been performed to investigate either the aetiology of degeneration of cartilaginous tissues, e.g., articular cartilage (AC) and the intervertebral disc (IVD), or tissue regeneration. Both research fields will benefit from knowledge of changes in cellular behaviour, as can be obtained via real-time polymerase chain reaction (PCR) and microarray analysis.
Both real-time PCR and microarray analysis of cartilaginous tissues are seriously hampered by the problematic ribonucleic acid (RNA) isolation of these tissues.2-4 Low cell density (1% to 2%) and a dense, highly cross-linked extracellular matrix composed of primarily proteoglycans impede the RNA isolation process.4,5 Moreover, when analysing the aetiology of IVD degeneration or osteoarthritis, RNA degradation is increased in comparison with healthy tissues, and concomitantly a decrease in viable cells and an increase in cell death occurs.2,6 The large, negatively charged proteoglycans not only tend to co-purify with the RNA,7 but they can also absorb the full spectrum of ultraviolet (UV) light, interfering with the UV photometric quantification of nucleic acids and thereby may impede a solid and reliable RNA quantification. In addition, proteoglycans are potent inhibitors of polymerase chain reactions which make amplification of specific genes from the isolated RNA even more difficult.8
Therefore, most data on gene expression of cartilaginous tissues are based on RNA extracted from isolated and/or in vitro-cultured cells. Since chondrocytes, and nucleus pulposus (NP) and annulus fibrosus (AF) cells dedifferentiate during in vitro culture, gene expression analysis may not reflect the in vivo situation.9-11 A number of methods have been developed to isolate RNA from the AC,2,4,12 but only one study compares different methods for RNA extraction from intervertebral disc tissue.3 Changes in gene expression in IVD tissue after different interventions aimed at regeneration of the IVD were shown in various in vivo studies. However, most of these studies involve small animals, and the results found in small animal models cannot be translated directly to the human situation due to the different dimensions and physiology of the IVD and the presence of notochordal cells in these small animals.13,14 The large animal studies available tend to use different RNA isolation kits for different cartilaginous tissues but, more importantly, they fall short in showing the quality and quantity of the isolated RNA.3,15-17
To overcome this lack of data, we compared four different RNA isolation kits for four different cartilaginous tissues obtained from a large animal model, the goat (fibrous tissue meniscus (M), AF proteoglycan-rich AC and NP). Aside from the standard method for RNA isolation via guanidine thiocyanate (GITC) using TRIzol, we also investigated the use of the RNeasy Lipid Tissue kit (Qiagen, Venlo, The Netherlands), the RNeasy Fibrous Tissue kit and the Aurum Total RNA Fatty and Fibrous Tissue kit (Bio-Rad, Veenendaal, The Netherlands), all commercially available. The RNeasy Lipid Tissue kit and RNeasy Fibrous Tissue kit are silica gel-based purification kits, which are designed for optimal lysis of tissues rich in fat or fibre, respectively. Aurum total RNA Fatty and Fibrous kit is also designed for tissues that are difficult to disrupt such as fatty and fibrous tissues, and is based on filter-binding extraction but in a vacuum format using the Bio-Rad Aurum vacuum manifold setup. Moreover, two different methods of tissue homogenisation were used to examine the effect of homogenisation on the different RNA isolation kits: the MagNA Lyser (Roche Diagnostics, Almere, The Netherlands) and the Freezer Mill (Spex CertiPrep, Middlesex, United Kingdom ). From these studies we deduced the optimal RNA isolation and tissue homogenisation specified for each tissue.
Materials and Methods
Tissue samples
Tissue samples from the AC, M, intervertebral disc AF and intervertebral disc NP were obtained from skeletally mature three- to five-year-old Dutch female dairy goats (n = 3) at autopsy. The research protocol was approved by the scientific board and the animal ethics committee of the VU University Medical Center, and is in accordance with national guidelines and regulations. After collection, samples were cut in half, immediately frozen in liquid nitrogen and stored at -80°C until further use. Tissue samples of less than 100 mg were used in this study. Additionally, a few samples were stored in RNA Later Solution (Life Technologies, Bleiswijk, The Netherlands) to test for optimal storage conditions of the tissues. Figure 1 describes the flow of the performed experiments (arrows) as well as the comparisons made in order to evaluate the different RNA extraction and homogenisation methods (numbers).
Fig. 1.
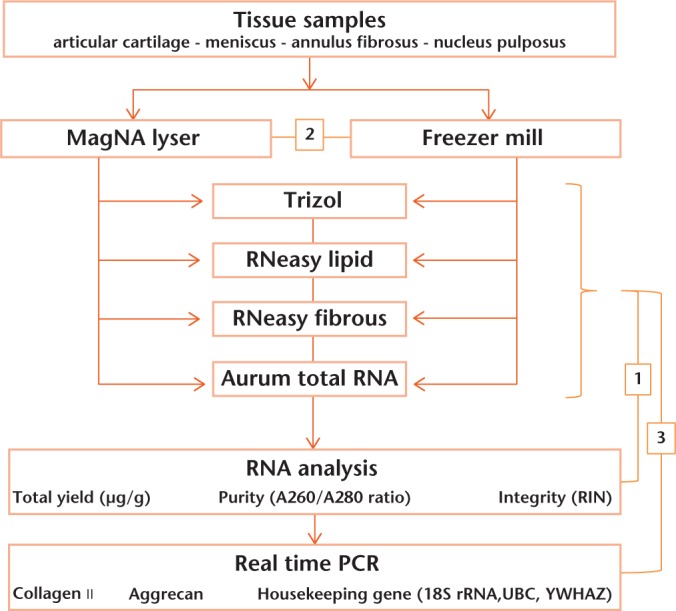
Overview of the experimental series. Arrows show the flow of the experiment: tissue samples were collected from goats, homogenised using two different methods, and ribonucleic acid (RNA) was isolated from each homogenate by four different techniques. RNA yield and purity was determined by NanoDrop. Real-time polymerase chain reaction (PCR) was used to determine the quality and quantity of mRNA transcripts for the gene expression of type II collagen and aggrecan. Numbers in boxes show the different comparisons between the different RNA isolation kits.(1: RNA yield and purity compared for the four different kits.2: Comparison of the two homogenisation methods, i.e. MagNA Lyser and Freezer Mill. 3: Gene expression analysis used to compare the different RNA isolation kits.) (18S rRNA, 18S ribosomal RNA; UBC, Ubiquitin C; YWHAZ, Tyrosine 3-monooxygenase; RIN, RNA integrity number.)
Tissue homogenisation
Each half of the obtained tissue samples was homogenised by either the MagNA Lyser or the Freezer Mill. The MagNA Lyser is a homogeniser that disrupts, and simultaneously homogenises tissue by rapid agitation of a 2 ml screw tube containing the tissue, 1.4 mm ceramic beads and a lysis buffer. Although tissue disruption and tissue homogenisation are two different processes, in this manuscript we will only use the term homogenisation for both processes. Tissues to be homogenised by the MagNA Lyser were divided into four parts and transferred into pre-cooled tubes, filled with ceramic beads, and the lysis buffers derived from the different RNA isolation kits which were added. Homogenisation occurred in four runs of 40 seconds each at 6500 rpm. In between the runs, samples were cooled at 4°C for two minutes. In the case that the samples were very viscous, they were placed on a roller mixer for approximately ten minutes. The Freezer Mill is an impact grinder that uses a steel impactor which moves back and forth between the metal ends of a vial by strong magnetic fields in liquid nitrogen. For the Freezer Mill, frozen samples were added to a pre-cooled grinding container. Tissue was ground in liquid nitrogen in three runs of two minutes each, at a frequency of 10 Hz. A cooling step of two minutes between each run was applied to achieve optimal brittleness. The homogenised tissue powder was divided into four groups and suspended in the lysis buffer provided by the different RNA isolation kits.
RNA isolation
After tissue homogenisation, samples were subjected to RNA isolation using either TRIzol (Life Technologies), the RNeasy Lipid Tissue kit, the RNeasy Fibrous Tissue kit, or the Aurum Total RNA Fatty and Fibrous Tissue kit. Kits are further referred to as TRIzol, Lipid, Fibrous and Aurum, respectively. The homogenised tissue was separated into two equal parts and RNA was isolated with the same kit but independently, i.e. n = 2 per RNA isolation method per homogenisation method performed for cartilaginous material obtained from three different goats. RNA isolation occurred according to the manufacturer’s protocol. For TRIzol isolations, the proposed additional steps for RNA isolation from tissues rich in proteoglycans were tested: high salt precipitation; extra centrifuging step; and precipitation using lithium chloride (LiCl, 8M) instead of propanol. Minor modifications and suggested improvements for the RNeasy Fibrous kit include using 20 μl instead of 10 µl of proteinase K for NP tissues as pilot studies showed increased RNA quality and quantity. Furthermore, 70% ethanol (EtOH), isopropanol and phenol:chloroform:isoamyl alcohol were tried, in addition to 100% EtOH. Both the Lipid and Fibrous kits were used in centrifuge form, whereas the Aurum kit was used in vacuum form. DNase I treatment (Qiagen) was performed according to the manufacturer’s protocol for all kits except TRIzol. Additionally, RNA clean-up sets, MinElute (Qiagen) and RNA clean-up kit (Invitek, Glasgow, United Kingdom) were used after RNA isolation via TRIzol.
RNA analysis
The quantity and quality of the isolated RNA were determined by NanoDrop ND-1000 (NanoDrop Technologies, Inc. Wilmington, New Jersey). The RNA concentration was determined by measuring the absorbance at 260 nm, and purity was assessed by determining the A260/A280 ratio, which should be between 1.8 and 2.0, and the A260/A230 ratio. Additionally, RNA integrity of specific samples was verified by calculating the RNA integrity number (RIN) from the 28S/18S ratios, using the Agilent RNA 6000 Pico Chips (Agilent Technologies, Amstelveen, The Netherlands) according to the manufacturer’s instructions.
Complementary DNA synthesis
First strand complementary (c)DNA synthesis was performed using 100 ng total RNA as input, in a 25 μl reverse transcription reaction mix consisting of 5 U Transcriptor Reverse Transcriptase, 0.08 U random primers, 10 U Protector RNase Inhibitor, and 1 mM of each dNTP in 1 × Transcriptor reverse transcription (RT) buffer (all Roche Diagnostics). Reactions were incubated at 42°C for 45 minutes, followed by an inactivation of the transcriptase enzyme at 80°C for five minutes.
Real-time PCR
Real-time PCR reactions were performed using the SYBR Green reaction kit according to the manufacturer’s protocol (Roche Diagnostics) in a LightCycler 480 System (Roche Diagnostics). The LightCycler reactions were prepared in 20 μl total volume with 7 μl PCR-H2O, 0.5 μl forward primer (0.2 μM), 0.5 μl reverse primer (0.2 μM), 10 μl LightCycler Mastermix (LightCycler 480 SYBR Green I Master; Roche Diagnostics), to which 2 μl of ten times diluted cDNA was added as a PCR template. Controls in the real-time PCR reaction included RT reactions without the reverse transcriptase (control for DNA carryover) and RT reactions without template (control for reagent contamination). Primers (Life Technologies) used for real-time PCR are listed in Table I. Primers were designed from sequences available in the data banks, based on homology in conserved domains comparing human, mouse, rat, dog and/or cow. Primers were previously used by Vonk et al.18,19 The amplified PCR fragment extended over at least one exon border, except for 18S. The 18S ribosomal RNA (18S rRNA), Ubiquitin C (UBC) and Tyrosine 3-monooxygenase (YWHAZ) were used as housekeeping genes. The stability of the housekeeping genes was checked by the geNorm software.20 Gene expression levels were normalised for the normalisation factor calculated with the equation . Expression of the target genes, type II collagen (Col2A1) and aggrecan (ACAN), was quantified with the fit point method in the LightCycler software, crossing points from a standard curve of six serial dilutions ranging from 2 pg to 10 fg, of DNA, derived from each gene, were assessed. Relative gene expression is shown as the ratio between absolute expression of the gene of interest and the absolute normalisation factor for housekeeping gene expression of the same sample. PCR efficiency (E) was automatically calculated using the fit point method, as provided by LightCycler (E = 10−1/slope). Gene expression data were used only if the PCR efficiency was calculated as being between 1.85 and 2.0 (Table I).
Table I.
Primer sequences used for PCR
| Gene | Oligonucleotide Sequence | Annealing temperature (°C) | Product size (bp) | PCR efficiency | |
|---|---|---|---|---|---|
| 18S | Forward | 5′ GTAACCCGTTGAACCCCATT 3′ | 57 | 151 | 1.91 |
| Reverse | 5′ CCATCCAATCGGTAGTAGCG 3′ | ||||
| UBC | Forward | 5′ GCGGTGAACGCCGATGATTAT 3′ | 56 | 202 | 1.86 |
| Reverse | 5′ TTTGCCTTGACATTCTCGATGG 3′ | ||||
| YWHAZ | Forward | 5′ GATGAAGCCATTGCTGAACTTG 3′ | 56 | 229 | 1.97 |
| Reverse | 5′ CTATTTGTGGGACAGCATGGA 3′ | ||||
| COL2A1 | Forward | 5′ AGGGCCAGGATGTCCGGCA 3′ | 56 | 195 | 2.0 |
| Reverse | 5′ GGGTCCCAGGTTCTCCATCT 3′ | ||||
| ACAN | Forward | 5′ CAACTACCCGGCCATCC 3′ | 57 | 160 | 2.0 |
| Reverse | 5′ GATGGCTCTGTAATGGAACAC 3′ | ||||
UBC, ubiquitin C; YWHAZ, Tyrosine 3-monooxygenase; COL2A1, collagen type II; ACAN, aggrecan; PCR, polymerase chain reaction
Statistical analysis
The normality of the data for the different groups was checked and a Wilcoxon rank-sum test was used to determine significant differences in gene expression. SPSS Statistics software version 20 was used (IBM Corp., Armonk, New York). To assess RNA quality and be able to compare the different homogenisation methods and RNA isolation kits, the variance in gene expression of duplicate samples was calculated, assuming that the fold difference for perfect duplicates is one (Fig. 1; orange blocks 2 and 3). After tissue homogenisation, samples were split and processed independently, i.e. n = 2 per RNA isolation method per homogenisation method performed for cartilaginous material obtained from three different goats.
Results
RNA Isolation
Table II shows the average weight of the tissue samples, RNA yield and quality (A260/A280 and A260/A230 ratio) determined for four different cartilaginous tissues, homogenised by either the MagNA Lyser (Magna) or Freezer Mill (Freezer). RNA extraction was executed using four different kits (TRIzol (TRIzol), RNeasy Lipid Tissue kit (Lipid), RNeasy Fibrous Tissue kit (Fibrous), and Aurum Total RNA Fatty and Fibrous Tissue kit (Aurum)). RNA isolation with all four kits for all cartilaginous tissues resulted in quantifiable amounts of RNA. Yields obtained with the Lipid and Fibrous kits were lower for all tissues and homogenisation methods, compared with TRIzol and the Aurum kit. RNA yields obtained with the Aurum kit show high standard deviations (sd) in comparison with the other three kits, especially for the yields of the Aurum samples homogenised by the MagNA Lyser. For all four tissues, higher A260/ A280 ratios were found for the Lipid and Fibrous kits compared with TRIzol and Aurum RNA isolations, suggesting that although the RNA yield for these kits is lower, the obtained RNA is less contaminated. The high sds and low purity of the RNA isolated with the Aurum kit indicate that the kit has low reproducibility for RNA isolation of cartilaginous tissues. The additional steps as suggested by the manufacturer to isolate pure RNA from material containing a very high level of proteoglycans for TRIzol and the Fibrous kit, did all not improve RNA quality (data not shown). In addition, the modifications prior to isolation, i.e., tissue storage in an RNA Later Solution, and RNA clean-up kits after isolation, also did not improve the quality of the obtained RNA (data not shown). Moreover, storage of the tissue samples in the RNA Later solution deteriorated the homogenisation process as the tissues became very rigid.
Table II.
Comparison of ribonucleic acid (RNA) yield and purity isolated using two different homogenisation methods and four different RNA isolation methods for four different types of cartilaginous tissues. RNA yield, purity of A articular cartilage (AC), B meniscus (M), C nucleus pulposus (NP), and D annulus fibrosus (AF) as measured by NanoDrop. Data are presented as mean and standard deviation (sd), n = 3 (goats), n = 2 (per goat per tissue per homogenisation method per RNA isolation method)
| A | RNA isolation kit | Homogenisation method | Tissue weight (mg) | Purity |
Yield (µg RNA/g tissue) | |
|---|---|---|---|---|---|---|
| A260/280 | A260/A230 | |||||
| AC | TRIzol | Magna | 70 (23.66) | 1.28 (0.11) | 0.27 (0.09) | 14.39 (12.26) |
| Spex | 56.67 (25.82) | 1.49 (0.08) | 0.22 (0.1) | 12.79 (5.06) | ||
| Lipid | Magna | 66.67 (2.58) | 1.93 (0.15) | 0.34 (0.09) | 3.33 (1.27) | |
| Spex | 61.67 (5.16) | 1.84 (0.17) | 0.46 (0.46) | 3.92 (1.32) | ||
| Fibrous | Magna | 56.17 (9.97) | 1.94 (0.18) | 0.40 (0.05) | 3.65 (1.09) | |
| Spex | 63.33 (10.33) | 1.79 (0.1) | 0.67 (0.29) | 6.81 (3.68) | ||
| Aurum | Magna | 72 (26.78) | 1.39 (0.11) | 0.23 (0.09) | 153.6 (303.6) | |
| Spex | 86.67 (18.07) | 1.46 (0.14) | 0.26 (0.12) | 7.78 (7.21) | ||
| B | RNA isolation kit | Homogenisation method | Tissue weight (mg) | Purity |
Yield (µg RNA/g tissue) | |
| A260/280 | A260/A230 | |||||
| M | TRIzol | Magna | 85.00 (13.42) | 1.41 (0.16) | 0.33 (0.21) | 12.9 (15.5) |
| Spex | 73.33 (12.91) | 1.38 (0.17) | 0.19 (0.16) | 26.34 (28.68) | ||
| Lipid | Magna | 71.67 (5.16) | 1.85 (0.16) | 0.53 (0.41) | 3.66 (2.66) | |
| Spex | 51.67 (2.58) | 1.73 (0.07) | 0.69 (0.31) | 9.65 (1.69) | ||
| Fibrous | Magna | 71.00 (21.04) | 1.95 (0.18) | 0.47 (0.24) | 4.50 (3.58) | |
| Spex | 58.33 (2.58) | 1.66 (0.17) | 0.30 (0.29) | 4.14 (3.15) | ||
| Aurum | Magna | 88.33 (5.50) | 1.44 (0.10) | 0.49 (0.32) | 114.9 (174) | |
| Spex | 80.00 (15.49) | 1.29 (0.03) | 0.19 (0.14) | 36.39 (80.13) | ||
| C | RNA isolation kit | Homogenisation method | Tissue weight (mg) | Purity |
Yield (µg RNA/g tissue) | |
| A260/280 | A260/A230 | |||||
| AF | TRIzol | Magna | 78.33 (18.07) | 1.32 (0.22) | 0.24 (0.2) | 12.97 (6.14) |
| Spex | 83.33 (5.16) | 1.24 (0.16) | 0.12 (0.04) | 6.17 (3.95) | ||
| Lipid | Magna | 70.00 (0.00) | 1.88 (0.20) | 0.24 (0.10) | 2.16 (0.67) | |
| Spex | 68.33 (5.16) | 1.73 (0.20) | 0.44 (0.42) | 3.94 (2.00) | ||
| Fibrous | Magna | 95.17 (31.28) | 1.94 (0.20) | 0.36 (0.17) | 2.42 (0.81) | |
| Spex | 51.67 (2.58) | 1.78 (0.05) | 0.19 (0.12) | 2.65 (0.18) | ||
| Aurum | Magna | 83.83 (6.01) | 1.45 (0.09) | 0.47 (0.36) | 113.2 (79.18) | |
| Spex | 66.67 (18.07) | 1.32 (0.10) | 0.26 (0.13) | 23.6 (32.32) | ||
| D | RNA isolation kit | Homogenisation method | Tissue weight (mg) | Purity |
Yield (µg RNA/g tissue) | |
| A260/280 | A260/A230 | |||||
| NP | TRIzol | Magna | 75 (19.49) | 0.98 (0.13) | 0.13 (0.04) | 26.06 (9.8) |
| Spex | 83.33 (5.16) | 1.04 (0.08) | 0.15 (0.04 | 5.44 (2.99) | ||
| Lipid | Magna | 68.33 (14.38) | 1.49 (0.12) | 0.22 (0.20) | 3.22 (1.67) | |
| Spex | 65.00 (15.49) | 1.67 (0.13) | 0.47 (0.34) | 2.46 (0.28) | ||
| Fibrous | Magna | 86.67 (29.44) | 1.65 (0.20) | 0.46 (0.18) | 2.86 (0.83) | |
| Spex | 43.33 (12.91) | 1.76 (0.15) | 0.13 (0.08) | 4.03 (0.59) | ||
| Aurum | Magna | 64.83 (23.16) | 1.52 (0.26) | 0.8 (0.7) | 102.8 (143.9) | |
| Spex | 53.33 (20.66) | 1.36 (0.21) | 0.26 (0.11) | 43.66 (87.12) | ||
As protein contaminations and solvent carryovers could interfere with the UV photometric quantification of nucleic acids, RNA concentration and integrity of randomly selected samples was also determined with the Agilent NanoChip in parallel with the NanoDrop. The mean RIN value for all tissues was 4.13 (sd 0.64). For all tissues, homogenisation with the MagNA Lyser resulted in better RIN values compared with the Freezer Mill (mean 4.45, sd 0.57 and 3.80, sd 0.48, respectively). Clear peaks in both the 18S and 28S bands were observed for all four tissues (see supplementary material).
Gene expression analysis to validate RNA quality
The high concentration of proteoglycans in the cartilaginous tissues not only causes problems in the RNA isolation process but can also inhibit real-time PCR reactions, therefore quantitative real-time PCR analysis was performed to validate the quality and purity of the isolated RNA by analysing collagen type II (COL2A1) and aggrecan (ACAN) gene expression. For NP, AF and AC, expression of COL2A1 and ACAN was measured after RNA isolation with both the Lipid and the Fibrous kits, whereas for some samples isolated by TRIzol or the Aurum kit no gene expression could be observed (Table III). For meniscus tissue, only the Lipid kit resulted in quantifiable gene expression of all samples. RNA quality was validated by comparing the variance in gene expression between duplicate samples, assuming that the fold difference for perfect duplicates is one. Tissue samples were split into two after homogenisation; RNA isolation and gene expression analysis were performed in a similar manner but independently for each half. Since the expression of COL2A1 was highly variable between all four cartilaginous tissues, we only present the data of ACAN expression.
Table III.
Overview of successful analysis of gene expression
| RNA isolation kit | Homogenisation method | Articular cartilage |
Meniscus |
Annulus fibrosus |
Nucleus pulposus |
||||
|---|---|---|---|---|---|---|---|---|---|
| COL2A1 | ACAN | COL2A1 | ACAN | COL2A1 | ACAN | COL2A1 | ACAN | ||
| TRIzol | MagNA | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 2/6 | 2/6 |
| Freezer | 6/6 | 6/6 | 4/6 | 5/6 | 5/6 | 5/6 | 4/6 | 4/6 | |
| Lipid | MagNA | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 |
| Freezer | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | |
| Fibrous | MagNA | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 |
| Freezer | 6/6 | 6/6 | 2/6 | 2/6 | 6/6 | 6/6 | 6/6 | 6/6 | |
| Aurum | MagNA | 5/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 3/6 | 3/6 |
| Freezer | 6/6 | 6/6 | 2/6 | 2/6 | 5/6 | 5/6 | 5/6 | 5/6 | |
Real-time polymerase chain reaction analysis was performed for all samples to validate the quality of the isolated RNA. Numbers indicate the total of quantifiable samples. Samples were obtained from three goats. Per goat, two duplicate samples were run independently for both homogenisation methods and for the four different methods of RNA extraction
RNA, ribonucleic acid; COL2A1, collagen type II; ACAN, aggrecan; MagNA, MagNA Lyser; Freezer, Freezer Mill
Comparison of tissue homogenisation methods
By determining the fold differences in gene expression between duplicate samples, we compared the two different homogenisation methods for each isolation kit. For the Aurum RNA isolation kit, large variations in gene expression between duplicate samples, up to > 1500-fold differences, were observed. We decided to exclude results obtained with the Aurum kit from further analysis due to these large variations, the relative high number of samples where gene expression could not be determined (Table III), and the abnormal RNA yields and A260/A280 ratios (Table II).
Large differences between duplicates were observed for the samples isolated with TRIzol, regardless of homogenisation method, especially for meniscus and AF tissue as can be observed for aggrecan expression in Figure 2. Only for articular cartilage did RNA isolation with TRIzol result in similar gene expression for the duplicate samples. For the Lipid and Fibrous kits, homogenisation method does not influence gene expression analysis as no significant differences could be detected between duplicate samples (Fig. 2a).

Graph showing the comparison of tissue homogenisation methods. The variances in aggrecan gene expression between two duplicate samples for the four cartilaginous tissues isolated with the same kit homogenised by either the MagNA Lyser (dark orange) or Freezer Mill (second bar from left): a) for the RNeasy Lipid (left hand bars × 2) and Fibrous (right hand bars × 2) kits; and b) for TRIzol. Data are presented as mean and standard error. Note the linear scale for graph (a) and the logarithmic scale for graph (b). As the aggrecan gene expression of NP tissue after RNA isolation with TRIzol could only be determined for the duplicates of the samples of one goat for both homogenisation methods, no error bars are shown. (AC, articular cartilage; M, meniscus; AF, annulus fibrosus; NP, nucleus pulposus.)
Comparison and validation of the different RNA isolation kits
Since no differences between the two homogenisation methods for the Lipid and Fibrous kits were observed, data from samples isolated by either one of these kits but homogenised by different methods were pooled. In order to compare these two kits with the frequently used RNA isolation method TRIzol, we also pooled the data from the two homogenisation methods, despite the observed differences (Fig. 2b).
Reproducible gene expression was found for all tissues isolated with the Lipid and Fibrous kits and no differences between the two kits were observed (Fig. 3a), whereas large variations were observed for the TRIzol RNA isolations (Fig. 3b). Although for the meniscus no significant difference between the Lipid and Fibrous kits was found, Figure 3b shows less difference between duplicates for the Lipid kit.

Comparison of ribonucleic acid (RNA) isolation kits analysed by real-time polymerase chain reaction. Results of RNA isolation with the different kits and for the different cartilaginous tissues are compared by analysing the differences in aggrecan gene expression between duplicate samples: a) for the RNeasy Lipid (dark orange) and Fibrous (light orange) kits; and b) for TRIzol. Tissues homogenised with the same method are pooled. Data are presented as mean and standard error of the mean. Note the linear scale for graph (a) and the logarithmic scale for graph (b). (AC, articular cartilage; M, meniscus; AF, annulus fibrosus; NP, nucleus pulposus.)
Discussion
The extraction of high-quality RNA is a critical step to evaluate regenerative strategies for cartilaginous tissues. However, RNA extraction from these tissues has been difficult due to the low cell density and the dense, highly cross-linked extracellular matrix rich in proteoglycans. In this study, we compared different RNA isolation methods in order to determine the optimal extraction procedure for the obtainment of high-quality RNA from different cartilaginous tissues, i.e. the more fibrous tissue M and AF and the proteoglycan-rich AC and NP. For meniscus tissue, the RNeasy Lipid Tissue kit showed the best results whereas for the other tissues (NP, AF and AC), optimal RNA isolation and subsequent real-time PCR analysis was achieved with both the RNeasy Fibrous and RNeasy Lipid kits.
Efficient tissue homogenisation is an important step for obtaining high-quality RNA for real-time PCR analysis from cartilaginous tissues due to the relatively small amounts of extractable RNA and generally limited quantities of tissue available for analysis. Since more than 95% of the tissues at hand consist of extracellular matrix, and the tissue samples are small and hard, homogenisation with a mortar and pestle is difficult. In this study two homogenisation methods, MagNA Lyser and Freezer Mill, were used. We observed no differences in RNA quality and yield and gene expression results obtained by real-time PCR between the two homogenisation methods for the RNeasy Lipid and RNeasy Fibrous kits. As the MagNA Lyser can be used to homogenise 16 samples at the same time and the Freezer Mill can only process one sample and requires large quantities of liquid nitrogen, we suggest that the MagNA Lyser is the best method for the homogenisation of cartilaginous tissues. Furthermore, homogenisation with MagNa Lyser occurs in the lysis buffer derived from the RNA isolation kit; in this way, RNA degradation is prevented compared with tissue powder obtained after homogenisation with the Freezer Mill. In contrast to our results, Ruettger et al2 observed a negative effect of homogenisation by milling the tissue before RNA isolation via TRIzol and the RNeasy kits.
RNA isolation with the RNeasy Lipid and Fibrous kits showed less protein contamination compared with TRIzol and the Aurum kit. For TRIzol, the combination of a high salt precipitation and an additional centrifugation step did not improve RNA quality. Lee et al3 also showed that the addition of a high salt solution was not sufficient to increase the RNA quality extracted from bovine NP as contaminating precipitates were still observed. However, in their hands, using the RNeasy method for RNA extraction from bovine NP tissue did not result in increased RNA quality and yield compared with TRIzol isolations. The tissue weight of the samples were comparable with those of the current study. The use of a double amount of proteinase K during isolation with the fibrous kit for NP tissue in order to remove residual proteoglycans resulted in a clear improvement of RNA quality, which is consistent with the findings from Mallein-Gerin and Gouttenoire.21 Our findings that storage of tissue samples in RNAlater solution did not improve the RNA extraction are in accordance with the study by Ruettger et al2 who also advised not to store articular cartilage in RNAlater solution as the tissue becomes extremely rigid and difficult to homogenise.
As the proteoglycans not only impede RNA isolation but may also inhibit the polymerase chain reactions, we analysed type II Collagen and ACAN gene expression using real-time PCR. Based on these results, we excluded the Aurum kit due to large variations in duplicate samples, the relatively large number of samples where no quantifiable gene expression could be observed, and the large variations found in RNA quality and quantity compared with the other kits. In line with the results found for RNA yield and quality, gene expression analysis after RNA extraction with either the RNeasy Fibrous or RNeasy Lipid kits was also found to be more stable and reproducible compared with TRIzol. For meniscus tissue, however, the RNeasy Lipid kit showed the best results. RNA extraction using TRIzol is not recommended for cartilaginous tissues, which is in accordance with the recommendations by Ruettger et al2 who suggested using TRIzol for RNA isolation of cultured chondrocytes and the rather costly, time-consuming RNAqueous kit22 for isolation of articular cartilage. On the other hand, Lee et al3 excluded RNA isolation with an RNeasy kit and further optimised RNA isolation using TRIzol, however, it must be noted that this study focuses on optimising tissue homogenisation via cryosectioning, presumably due to the low RNA yield obtained with the RNeasy kit.
RIN values of the isolated RNA in this study are relatively low (approximately 4.13 sd 0.64), and, according to guidelines, of insufficient quality for further analysis (< 6).23 However, successful real-time PCR analysis of samples with a low RIN value showed that the isolated RNA is of high enough quality for real-time PCR. Moreover, the RIN values of several duplicate samples are contradictory compared with the observed gene expression. For example, in two different NP samples, the RIN value of one duplicate was not quantifiable (sample 1: duplicate values of 3.8 and not applicable (N/A), and sample 2: duplicate values of 4.6 and N/A), whereas real-time PCR results showed barely any difference between duplicates (1.06 and 1.08). Conversely, comparable RIN values (sample 1:5.6 and sample 2:5.5) for duplicates resulted in a 6.95-fold difference in gene expression for articular cartilage.
In addition to the A260/A280 ratio, the A260/A230 ratio is also commonly checked as a marker for phenol and salt contaminations. The goal of our study was to isolate RNA from cartilaginous tissues for subsequent real-time PCR analysis as both the isolation and PCR analysis are hampered by the high concentration of proteoglycans present in the tissue. The A260/A230 ratios we found in our study are low (0.34, sd 0.2) but similar to the ratios found by other studies for nucleus pulposus and annulus fibrosus tissue.2,3 Moreover, in our study only the RNA isolation using TRIzol has a potential for phenol and salt contamination, whereas all A260/A230 ratios were low irrespective of RNA isolation method. Most importantly, real-time PCR was possible despite the low A260/A230 ratios, showing that the isolated RNA is of good enough quality.
In conclusion, for AC, AF and NP tissue no differences between the RNeasy Fibrous and RNeasy Lipid kits were observed with respect to RNA quality and yield, and gene expression analysis. Since the RNeasy Fibrous kit does not involve toxic chemicals such as phenol and chloroform, we propose this kit for the isolation of high-quality RNA for these tissues. For meniscus tissue the RNeasy Lipid kit is recommended.
This study is the first to compare RNA extraction of four different cartilaginous tissues with four different commercially available RNA isolation kits. We have shown that high-quality RNA can be isolated from small samples of cartilaginous tissue, and is suitable for the analysis of gene expression by quantitative real-time PCR.
Footnotes
Author Contribution: M. Peeters: Analysis and interpretation of data, Drafting of manuscript.
C. L. Huang: Study conception and design, Acquisition of data, Analysis and interpretation of data, Drafting of manuscript.
L. A. Vonk: Acquisition of data, Editing manuscript.
Z. F. Lu: Acquisition of data, Editing manuscript.
R. A. Bank: Editing manuscript.
M. N. Helder: Study conception and design, Analysis and interpretation of data, Editing manuscript.
B. Zandieh Doulabi: Study conception and design, Acquisition of data, Analysis and interpretation of data, Editing manuscript.
M. Peeters and C. L. Huang contributed equally to this work
ICMJE conflict of interest: None declared
Supplementary material
Electropherograms showing clear peaks in both the 18S and 28S bands for nucleus pulposus tissues can be found alongside the online version of this paper at http://www.bjr.boneandjoint.org.uk/
Funding Statement
None declared
References
- 1. Hiligsmann M, Cooper C, Arden N, et al. Health economics in the field of osteoarthritis: an expert’s consensus paper from the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO). Semin Arthritis Rheum 2013;43:303-313. [DOI] [PubMed] [Google Scholar]
- 2. Ruettger A, Neumann S, Wiederanders B, Huber R. Comparison of different methods for preparation and characterization of total RNA from cartilage samples to uncover osteoarthritis in vivo. BMC Res Notes 2010;3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee JTY, Cheung KMC, Leung VYL. Extraction of RNA from tough tissues with high proteoglycan content by cryosection, second phase separation and high salt precipitation. J Biol Methods 2015;2:20. [Google Scholar]
- 4. Gehrsitz A, McKenna LA, Söder S, Kirchner T, Aigner T. Isolation of RNA from small human articular cartilage specimens allows quantifcation of mRNA expression levels in local articular cartilage defects. J Orthop Res 2001;19:478-481. [DOI] [PubMed] [Google Scholar]
- 5. Urban MR, Fairbank JC, Bibby SR, Urban JP. Intervertebral disc composition in neuromuscular scoliosis: changes in cell density and glycosaminoglycan concentration at the curve apex. Spine (Phila Pa 1976) 2001;26:610-617. [DOI] [PubMed] [Google Scholar]
- 6. Zhao CQ, Wang LM, Jiang LS, Dai LY. The cell biology of intervertebral disc aging and degeneration. Ageing Res Rev 2007;6:247-261. [DOI] [PubMed] [Google Scholar]
- 7. Carney SL, Muir H. The structure and function of cartilage proteoglycans. Physiol Rev 1988;68:858-910. [DOI] [PubMed] [Google Scholar]
- 8. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987;162:156-159. [DOI] [PubMed] [Google Scholar]
- 9. Kluba T, Niemeyer T, Gaissmaier C, Gründer T. Human anulus fibrosis and nucleus pulposus cells of the intervertebral disc: effect of degeneration and culture system on cell phenotype. Spine (Phila Pa 1976) 2005;30:2743-2748. [DOI] [PubMed] [Google Scholar]
- 10. Hayman DM, Blumberg TJ, Scott CC, Athanasiou KA. The effects of isolation on chondrocyte gene expression. Tissue Eng 2006;12:2573-2581. [DOI] [PubMed] [Google Scholar]
- 11. Eleswarapu SV, Leipzig ND, Athanasiou KA. Gene expression of single articular chondrocytes. Cell Tissue Res 2007;327:43-54. [DOI] [PubMed] [Google Scholar]
- 12. Adams ME, Huang DQ, Yao LY, Sandell LJ. Extraction and isolation of mRNA from adult articular cartilage. Anal Biochem 1992;202:89-95. [DOI] [PubMed] [Google Scholar]
- 13. Smit TH. The use of a quadruped as an in vivo model for the study of the spine - biomechanical considerations. Eur Spine J 2002;11:137-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoogendoorn RJW, Helder MN, Smit TH, Wuisman PIJM. Notochordal Cells in Mature Caprine Intervertebral Discs. Eur Cell Mater 2005;10:59. [Google Scholar]
- 15. Henriksson HB, Svanvik T, Jonsson M, et al. Transplantation of human mesenchymal stems cells into intervertebral discs in a xenogeneic porcine model. Spine (Phila Pa 1976) 2009;34:141-148. [DOI] [PubMed] [Google Scholar]
- 16. Paul CPL, Zuiderbaan HA, Zandieh Doulabi B, et al. Simulated-physiological loading conditions preserve biological and mechanical properties of caprine lumbar intervertebral discs in ex vivo culture. PLoS One 2012;7:e33147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Capossela S, Schläfli P, Bertolo A, et al. Degenerated human intervertebral discs contain autoantibodies against extracellular matrix proteins. Eur Cell Mater 2014;27:251-263. [DOI] [PubMed] [Google Scholar]
- 18. Vonk LA, Kroeze RJ, Doulabi BZ, et al. Caprine articular, meniscus and intervertebral disc cartilage: an integral analysis of collagen network and chondrocytes. Matrix Biol 2010;29:209-218. [DOI] [PubMed] [Google Scholar]
- 19. Vonk LA, Doulabi BZ, Huang C, et al. Collagen-induced expression of collagenase-3 by primary chondrocytes is mediated by integrin &α;1 and discoidin domain receptor 2: a protein kinase C-dependent pathway. Rheumatology (Oxford) 2011;50:463-472. [DOI] [PubMed] [Google Scholar]
- 20. No authors listed. geNorm. https://genorm.cmgg.be/ (date last accessed 07 October 2016).[[bibmisc]]
- 21. Mallein-Gerin F, Gouttenoire J. RNA extraction from cartilage. Methods Mol Med 2004;100:101-104. [DOI] [PubMed] [Google Scholar]
- 22. Geyer M, Grässel S, Straub RH, et al. Differential transcriptome analysis of intraarticular lesional vs intact cartilage reveals new candidate genes in osteoarthritis pathophysiology. Osteoarthritis Cartilage 2009;17:328-335. [DOI] [PubMed] [Google Scholar]
- 23. Schroeder A, Mueller O, Stocker S, et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 2006;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]