

Abstract
Semipermeable cell culture membranes are commonly used in multilayered microfluidic devices to mimic the basement membrane in vivo and to create compartmentalized microenvironments for physiological cell growth and differentiation. However, existing membranes are predominantly made up of synthetic polymers, providing limited capacity to recapitulate cellular interactions with native extracellular matrices that play a crucial role in the induction of physiological phenotypes. Here we describe a new type of cell culture membranes engineered from native extracellular matrix (ECM) materials that are thin, semipermeable, optically transparent, and amenable to integration into microfluidic cell culture devices. Facile and cost-effective fabrication of these membranes was achieved by controlled sequential steps of vitrification that transformed three-dimensional (3D) ECM hydrogels into structurally stable thin films. By modulating the composition of ECM, our technique provided a means to tune key membrane properties such as optical transparency, stiffness, and porosity. For microfluidic cell culture, we constructed a multilayered microdevice consisting of two parallel chambers separated by a thin membrane insert derived from different types of ECM. This study showed that our ECM membranes supported attachment and growth of various types of cells (epithelial, endothelial, and mesenchymal cells) under perfusion culture conditions. Our data also revealed the promotive effects of the membranes on adhesion-associated intracellular signaling that mediates cell-ECM interactions. Moreover, we demonstrated the use of these membranes for constructing compartmentalized microfluidic cell culture systems to induce physiological tissue differentiation or to replicate interfaces between different tissue types. Our approach provides a robust platform to produce and engineer biologically active cell culture substrates that serve as promising alternatives to conventional synthetic membrane inserts. This strategy may contribute to developing physiologically relevant in vitro cell culture models for a wide range of applications.
Graphical abstract
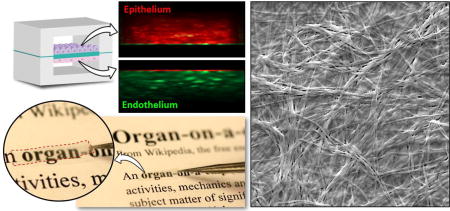
This paper presents a new type of cell culture membranes engineered from native extracellular matrix (ECM) materials that are thin, semipermeable, optically transparent, and amenable to integration into microfluidic cell culture devices.
Introduction
Microphysiological cell culture models, collectively known as organs-on-chips, are rapidly emerging as a novel platform to emulate the essential units of living organs for a wide variety of applications (1–3). By enabling new capabilities to present cultured cells with physiologically relevant structural, biochemical, and biomechanical cues, organ-on-a-chip models make it possible to mimic the native phenotype of various tissue types and their integrative behaviors that give rise to complex organ-level functions. Over the last decade, considerable success has been achieved in demonstrating the feasibility of leveraging this biomimetic microengineering strategy to model the functional units of various organs for basic and translational research (4–7).
Construction of these microphysiological models often requires perfusable microfluidic systems that consist of stacked layers of microfabricated cell culture chambers (8). This design provides a compartmentalized environment advantageous for co-culture of different cell types to replicate cellular heterogeneity and multilayered tissue structures found in virtually all organs. As a key component in this type of microdevices, semipermeable membranes containing nano- or microscopic pores are commonly used as cell culture substrates sandwiched between two adjacent chambers. In this configuration, the membranes provide a physical barrier to cell migration and enable the compartmentalization of different cell populations while permitting their exchange of soluble signaling molecules through the pores, recapitulating the role of the basement membrane in vivo (8, 9). This approach has been used extensively in microengineered cell culture models to reconstitute various types of tissue-tissue interfaces and to study their physiological functions in a range of contexts including immune responses (7), biomolecular transport (4), gas and fluid exchange (10), drug delivery (5), and nanoparticle absorption (11).
Despite widespread use in microfluidic culture, however, an existing selection of commercially available or custom-designed semipermeable membranes suffer from several limitations. Most notably, the vast majority of cell culture membranes in use today are made of synthetic polymers, such as polyester, polycarbonate, or poly(dimethylsiloxane) (PDMS), that significantly differ from the native ECM. The ECM represents the key insoluble component of the cellular microenvironment and serves as anchorage substrates for adherent cells by engaging ECM ligand-specific cell surface receptors (12, 13). To mimic this critical aspect of cell-ECM interactions, synthetic membranes can be modified by absorptive coating or covalent bonding of ECM proteins on the surface to support cell attachment (8, 14). However, the bulk material remains foreign and fails to mimic the biochemical composition of the basement membrane that provides instructive cues for expression of physiological cellular phenotypes (15). These polymeric membranes also lack the ability to recapitulate the fibrous architecture and physical properties (e.g. stiffness) of native matrices that profoundly influence the structure and function of cells (16). These inherent limitations often become the source of discrepancies between microphysiological models and their in vivo counterparts. The lack of optical transparency is another common problem in certain types of synthetic membranes (e.g., electrospun substrates, microporous Transwell inserts) that imposes constraints on imaging and analysis of cells in membrane-containing microfluidic devices. In addition, the fabrication of porous membranes demands specialized and expensive manufacturing techniques such as track etching (17), electrospinning (18), and chemical etching (19). This requirement presents a major practical challenge for routine production and optimization of cell culture membranes necessary for rapid-prototyping microphysiological systems in a research laboratory environment.
In an effort to address these problems, here we describe a simple and cost-effective strategy to generate semipermeable cell culture membranes derived from native ECM proteins that can be easily integrated into microfabricated devices. This technique utilizes natural evaporation-driven dehydration and vitrification of ECM hydrogel scaffolds to form thin ECM films without requiring specialized equipment or infrastructure. The resulting membranes are fibrous, clear, permeable, and mechanically stable enough to retain their structural integrity during bonding and assembly of multilayered microfluidic devices. Using collagen hydrogel and Matrigel as representative materials, we demonstrate new capabilities to tune the properties of ECM membranes and to modulate attachment and organization of different types of adherent cells. Furthermore, we show the proof-of-principle for using these membranes in compartmentalized microdevices to engineer living human barrier tissues that resemble various types of tissue-tissue interfaces in vivo. Our naturally derived membranes offer new opportunities to overcome the major limitations of conventional semipermeable membranes and to improve the physiological relevance and predictive capacity of microfluidic cell culture models.
Methods
Membrane Fabrication
The production of ECM membranes was accomplished by the multi-step fabrication process depicted in Figure 1. To begin, a predetermined volume of ECM hydrogel precursor solution was distributed evenly on a flat PDMS slab and incubated at 37°C for 1 hour to allow for gelation (Fig. 1A). Subsequently, the hydrogel was dried in a sterile environment at room temperature overnight. During this process, an evaporative loss of water content from the gel caused a drastic volume reduction and eventually led to the formation of a thin sheet on the PDMS surface that showed a pink color and crystallized residues (Fig. 1B). The ECM film was then rehydrated in pure distilled deionized (DDI) water for 4 hours to remove salts, phenol red, and other impurities (Fig. 1C). Following gentle aspiration of water, the film underwent another drying cycle to create a thin ECM membrane supported by an underlying PDMS substrate (Fig. 1D). Finally, the membrane was peeled from the PDMS pedestal using fine forceps (Fig. 1E) and cut to desired size and shape for use in microfluidic devices. To yield membranes strips of uniform thickness as measured, 1–2 mm were trimmed off the edges of the ECM membranes after the peeling step to remove sloped boundary regions. The uniform thickness was verified by measuring the thickness of each membrane at multiple locations using the image analysis technique described in the next section.
Figure 1. Fabrication of ECM-derived membrane inserts for microfluidic cell culture.

A. ECM hydrogel cast on PDMS surface. B. Dehydration of ECM hydrogel to yield a dried ECM film. C. Rehydration of dried ECM film to remove salts and other impurities followed by transglutaminase cross-linking (for fabrication of COL+MAT). D. Dehydration of purified and cross-linked ECM film to yield ECM membranes used as microfluidic cell culture inserts. E. Peeling of ECM membrane from underlying PDMS surface using forceps, followed by manual trimming with scissors if necessary. F. Microfluidic channel slabs fabricated by soft lithography are stamped with uncured PDMS to facilitate bonding of ECM membrane inserts over microfluidic channels. G. An ECM membrane is placed over the lower channel using forceps. H. The upper channel slab stamped with uncured PDMS is bonded to the lower channel slab to create an enclosed three-layer channel system. The cross-sectional view of the fully assembled device is shown in I and J. I. Cells are seeded on the ECM-derived membrane inserts in microfluidic devices. J. During perfusion culture, the seeded cells proliferate on the membrane surface to form stable, confluent monolayers in microdevices.
Production of ECM membranes with different compositions and thickness
In our study, we generated three types of ECM membranes composed of i) collagen Type I (COL), ii) collagen Type I and Matrigel (COL-MAT), or iii) collagen Type 1 and alginate (COL-ALG). For production of pure COL membranes, rat tail collagen Type I (Corning) solution was prepared at 2 mg/ml according to the manufacturer’s protocols. 400µl of this solution was used to carry out the multi-step fabrication procedure described above. To generate COL-MAT membranes, Matrigel (~10 mg/ml as supplied by Corning) was mixed with 2 mg/ml collagen solution at volume ratios of 1:4, 1:1, and 4:1. The remainder of the fabrication process was identical with the addition of an incubation step with 10 mU/ml transglutaminase in 1× PBS solution for 2 hours at 37 °C prior to rehydration in DDI water to cross-link Matrigel components to the collagen type I matrix. COL-ALG membranes were fabricated using collagen (2 mg/ml) and alginate (10 mg/ml) solutions mixed at ratios of 2:1, 1:1, and 1:2 (v/v). The same fabrication steps were followed with the exception that the membranes were soaked in DDI water for 2 hours at room temperature and another 2 hours at 37 °C to remove alginate used as a sacrificial material to increase the porosity of the resultant membranes.
To change the thickness of our ECM membranes, we used sequential layering techniques to generate membranes consisting of stacked layers of COL membranes. The number of COL layers ranged from 2 to 4 to vary the thickness of the resultant membrane. After the first layer was formed using the aforementioned protocol, it was wetted with 10 mU/ml transglutaminase solution and carefully overlaid with the second layer to prevent air bubble formation between the layers. The layered COL membranes were incubated for 2 hours in transglutaminase solution at 37 °C, and this step was followed by 3 washes in 1× DPBS. These steps were repeated for additional layers. To measure membrane thickness, one edge of the membrane was sandwiched between sets of glass slides while the center of the membrane remained freely suspended. For each membrane, a z-stack acquisition of 100 µm was performed using a long working distance inverted microscope (Zeiss) at the center of the membrane. Using the ZEN software (Zeiss), an orthogonal projection was created from the z-stack and further processed using the maximum intensity projection along the z-axis. The final image exhibited each pixel at its maximum intensity over the entire image stack. Image analysis was carried out using ImageJ to quantify membrane thickness.
Microdevice fabrication
Microchannels used in our study were fabricated using conventional soft lithography (4, 5). In brief, a prepolymer of poly(dimethylsiloxane) (PDMS) (Sylgard, Dow Corning) was mixed with curing agent at 10:1 (w/w) and degassed in a desiccator to remove air bubbles. The mixture was then casted on a photographically prepared silicon master and cured at 65°C for at least 2 hours. After curing, the PDMS slab was peeled from the mold and cut into desired size.
To construct multilayered microfluidic devices, we fabricated upper and lower microchannels with a rectangular cross-section having the width and height of 500 µm and 100 µm, respectively. A 1 mm biopsy punch was used to create fluidic access ports in these channels. To assemble a three-layer device, the lower channel slab was gently dipped into a thin layer of uncured PDMS prepared by spin-coating of 10:3 PDMS on a petri-dish at 2500 rpm for 5 minutes. When the slab was removed, the PDMS film was transferred onto the surface containing the microchannel features (Fig. 1F). Next, an ECM membrane was placed over the lower channel on the PDMS-stamped surface (Fig. 1G) and cured at room temperature overnight. After curing, the upper channel slab was coated with uncured PDMS using the same stamping technique and immediately bonded to the membrane-containing lower PDMS slab (Fig. 1H). The assembled device was left at room temperature overnight to ensure complete bonding.
Cell culture
Human umbilical vein endothelial cells (HUVEC), both normal (Lonza) and GFP-expressing (Angio-Proteomie) were cultured in EGM-2 medium (Lonza). Murine pericytes genetically modified to express a Tomato Red-labeled form of the pericyte marker Gli-1 (gift from Dr. Benjamin Humphreys, Washington University, St. Louis) and human lung adenocarcinoma cells (A549, ATCC) were cultured in standard 10% FBS containing DMEM medium. Human bronchial epithelial cells (BEAS-2b, ATCC) were maintained in bronchial epithelial growth medium (BEGM, Lonza). Normal human lung fibroblasts (NHLFs, Lonza) were cultured in FGM-2 medium (Lonza).
Microfluidic cell culture
Microfluidic cell culture was conducted in the three-layer microfluidic system described above. Prior to cell seeding, the microchannels were incubated with cell culture medium used for each cell type at 37 °C for at least 2 hours. For devices containing fibronectin-coated polyester membranes, 40 µg/mL fibronectin solution was introduced into the channels pretreated with corona point discharge and incubated at 37 °C and 5% CO2 for 30 minutes prior to incubation with cell culture medium. Next, cells suspended in culture medium at approximately 10 million cells/ml were injected into the upper channel (Fig. 1I) and allowed to settle and attach to the membrane surface under static conditions at 37 °C and 5% CO2 for 2 hours. Following microscopic examination to confirm cell attachment, the microchannels were gently flushed to remove non-adherent cells and then connected to syringe pumps (Braintree Scientific) that generated a flow of culture medium at volumetric flow rates of 70–100 µl/hour. Cells were cultured for 24–72 hours with continuous medium flow as needed to establish confluent monolayers (Fig. 1J) and for over 7 days in select experiments (Fig. S5). Cell viability was assessed by fluorescence microscopy imaging of cells labeled with calcein-AM and ethidium bromide homodimer according to standard protocols (Live/Dead kit, Invitrogen).
Scanning electron microscopy (SEM)
Membrane samples for SEM were fixed at 4°C overnight in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer at pH 7.4. After several washing steps, the membranes were post-fixed in 2.0% osmium tetroxide for 1 hour, washed again in buffer, and dehydrated in a graded ethanol series. Subsequently, the samples were taken through a graded hexamethyldisilazane (HMDS) series and air dried prior to mounting and sputter-coating with gold/palladium. SEM images of the membranes were taken by an FEI Quanta FEG 250 scanning electron microscope. The obtained scanning electron micrographs were analyzed using the Analyze Particles function of ImageJ to measure the size distribution of membrane pores.
Atomic Force Microscopy – Nanoindentation
An Asylum Atomic Force Microscope (AFM) was used to characterize the mechanical properties of hydrated/wetted COL, COL-MAT and polyester membranes. A chromium-gold coated cantilever with a spring constant of 44.03 pN/nm and a pyramid indenter was used to extract force-indentation curves. The Young’s modulus was calculated from the force-indentation data using AtomicJ software.
Analysis of membrane permeability and transparency
Optical transparency of ECM membranes was quantified in the wavelength range of 350–700 nm using a standard spectrophotometer (Infinite M200, TECAN). To analyze permeability, we created ECM membrane-containing three-layer microfluidic devices using the fabrication method detailed above. In this system, membrane permeability was evaluated by i) loading the upper microchannel with a 20 kDa FITC-dextran solution (0.2 mM, Sigma-Aldrich), ii) collecting the outflow from the lower channel over 3 hours, and iii) measuring fluorescence intensity of the collected samples using a fluorimetric plate reader (Infinite M200, TECAN). During these experiments, flows in the upper and lower channels were driven in the same direction at 100 µl/hour for 3 hours. For comparison between different types of ECM membranes, data obtained in this study were normalized to the average permeability of pure collagen (COL) membranes.
Analysis of membrane surface adsorption
Absorption of biomolecules on membrane surfaces was measured by treating bare COL membranes, pericyte-seeded COL membranes, and Transwell membranes with 1 mg/ml fluorescein-conjugated bovine serum albumin (FITC-BSA) for 2 hours at 37 °C. This step was followed by two washes with PBS for 5 minutes prior to detection and measurement of FITC fluorescence. The average fluorescence intensity from at least 13 micrographs per group was measured as the mean grey value using binarized images in ImageJ.
Immunofluorescence
After completion of cell culture experiments, devices were disconnected from the syringe pumps, and the channels were washed gently 3 times by perfusing 1× PBS. Cells on the membrane surface were then fixed by introducing 4% paraformaldehyde (Affymetrix) into both the upper and lower channels and incubating at room temperature for 15–20 minutes. The channels were then washed 3 times with 1× PBS and stored in a humid, refrigerated environment prior to antibody labeling and fluorescent microscopy. Following cell permeabilization and blocking with 0.1% Triton-X and 3% bovine serum albumin (BSA, Sigma) in 1× PBS for 30 minutes, the cells were incubated with primary antibodies against FAK-Y397 (Cell Signaling), pan-laminin (Sigma-Aldrich), and alpha-6 integrin (Abcam) diluted at 1:50 in 1% BSA-containing 1× PBS solution for 2 hours at room temperature. Subsequently, the cells were washed at least 5 times by gently flowing 1× PBS and then treated for 30–45 minutes with appropriate secondary antibodies diluted at 1:500 in 1× PBS containing 1% BSA. Actin cytoskeleton was labeled using Alexa488-conjugated phalloidin (Life Technologies) at a concentration of 1 µg/ml in 1× PBS, either added alone or mixed with the secondary antibodies. Immunofluorescence imaging was carried out using an inverted microscope with long working distance objectives (Zeiss). To quantify FAK activation on various membranes, we acquired 10 randomly positioned high magnification fields per membrane type and used ImageJ software (NIH) to manually select at least 40 individual cells with clearly discernable borders as regions of interest to measure averaged fluorescence intensity. These data were presented as fluorescence intensity on a per cell basis, normalized to the signal obtained from cells cultured on untreated polyester Transwell™ membranes.
Statistical Analysis
Results were reported as the mean ± standard deviation. Statistical significance of variance across groups was assessed by ANOVA with two-tailed Student’s t-test for individual comparisons using GraphPad software (Instat).
Results and discussion
Production of ECM-derived biomimetic membranes
The basement membrane is composed of two structurally distinct layers (15, 20). The first layer is the basal lamina made up of cell adhesion molecules and anchoring filaments that adhere to the basolateral side of cells comprising epithelium, vascular endothelium, peripheral nerve axons, adipose tissue, and muscle (15). This ultrathin (< 100 nm) layer is connected to 3D networks of ECM fibers known as the reticular lamina (20). This specialized zone serves to anchor the basal lamina to the underlying connective tissue and plays a critical role in compartmentalization of different tissue types (20, 21). As the main ECM component of the reticular lamina, collagen forms striated fibrils that are assembled in a hierarchical manner to provide structural support to the basement membrane (21). Inspired by this important role of collagen as a major structural protein, we used commercially available collagen type I as a base material for developing a simple and cost-effective method to generate ECM-derived cell culture membranes.
As shown in Figure 2A, the sequential process of collagen hydrogel dehydration resulted in the formation of completely dried planar sheets within 48 hours that could be peeled, trimmed to desired dimensions, and easily handled using fine forceps. With 400 µl of collagen hydrogel uniformly spread over an area of 200 mm2 (10 mm × 20 mm), the average thickness of the resulting films was measured to be 20 µm (Fig. S1). Membrane thickness was adjustable by changing the initial volume of collagen hydrogel (data not shown) and/or sequentially repeating the same rehydration cycle to deposit additional membrane layers (see Methods) (Fig. S1). Importantly, scanning electron microscopy revealed that the collagen (COL) membranes consisted of randomly oriented fibrils organized into dense 3D networks (Fig. 2B), mimicking the fibrous architecture of the basement membrane in vivo. The individual fibers comprising the meshwork also exhibited the characteristic banding pattern of native fibrillar collagen (inset, Fig. 2B). Furthermore, these membranes contained nanoscopic pores over the entire surface that were clearly visible in the scanning electron micrographs (inset, Fig. 2B).
Figure 2. Gross appearance, surface ultrastructure, and composition of ECM-derived membranes.

A. Digital photo of a COL-MAT membrane held by forceps demonstrating mechanical integrity and transparency. B. Scanning electron microscopy (SEM) visualization of collagen type I (COL) membrane surface ultrastructure, scale bar = 10 µm. Inset: Characteristic banding pattern visible in larger fibrils. C. SEM visualization of collagen type I and Matrigel composite (COL-MAT) membranes surface ultrastructure, scale bar = 10 µm. D. Immunofluorescence staining of laminin protein (green) in COL membranes demonstrates an expected absence of laminin protein. Scale bar = 200 µm. E. Immunofluorescence staining for laminin protein (green) in COL-MAT membranes shows robust incorporation of laminin within the fibrous microarchitecture (inset). Scale bar = 200 µm. F. SEM visualization of Transwell membrane surface ultrastructure showing 400 nm pores and smooth culture surfaces, scale bar = 10 µm.
Based on these results, we then examined the feasibility of using our technique to create biomimetic membranes that recapitulate not only the structure of the basement membrane but also its ECM composition. The primary structural components of the basement membrane are laminin and collagen type IV which self-assemble into 3D networks with tissue-specific mixtures of proteoglycans and specialized glycoproteins such as entactin (22, 23). To integrate these native constituents into our membranes, we formed composite hydrogels by mixing collagen with Matrigel, a reconstituted basement membrane-like material composed of approximately 60% laminin, 30% collagen IV, 8% entactin, proteoglycans, and various growth factors (see Methods for product information). Since Matrigel components do not covalently link to collagen type I during hydrogel polymerization, we used transglutaminase after the rehydration step (Fig. 1C) to cross-link Matrigel components to the polymerized collagen type I matrix. As was the case with the COL membranes, our protocol generated planar collagen-Matrigel (COL-MAT) membranes with similar thickness and structural integrity that consisted of densely packed ECM fibers (Fig. 2C). Successful integration of Matrigel components was evidenced by immunofluorescence detection of laminin in COL-MAT membranes (Fig. 2E). The biomimetic structure and composition of our ECM membranes were in stark contrast to the structure of commercially available Transwell™ cell culture membranes that showed highly artificial and smooth surfaces with randomly distributed nanoscopic pores (Fig. 2F).
Taken together, these results illustrate that our method allows for a simple strategy to produce thin, porous membranes that closely approximate the structural organization and composition of the ECM in the native basement membrane.
Engineering the properties of ECM membranes
The ability to vary the properties of cell culture membranes makes it possible to engineer the insoluble cellular microenvironment that has a major influence on growth, differentiation, and maintenance of cells. Such capabilities are also beneficial for modeling biomolecular transport and exchange of soluble factors between different tissue compartments. Moreover, material characteristics of membrane inserts become an important consideration for cell imaging and analysis commonly required for in vitro studies. By leveraging the flexibility to vary the type and composition of starting hydrogel materials, we demonstrated the ability of our fabrication technique to modulate the following properties of ECM membranes.
Optical transparency
Optical transparency is an important property of membrane inserts desirable for microscopic imaging and analysis. Although ECM hydrogels underwent dehydration and transformation during our fabrication procedure, their initial optical clarity was retained relatively well, resulting in the formation of thin films whose transparency was superior to that of existing cell culture membranes. For instance, our analysis showed that the COL membranes absorbed less light across the visual spectrum compared to Transwell polyester (PE) membranes with 400 nm pores that are marketed as optically clear (Fig. 3A). When Matrigel was added to the collagen base, the resulting COL-MAT membranes appeared considerably more transparent to the naked eye (Fig. 3B). This observation was supported by the spectrophotometric data that the light absorbance of the COL-MAT membranes was significantly lower than that of COL and clear Transwell PE membranes (Fig. 3A).
Figure 3. Tunable biophysical properties of engineered ECM-derived membranes.

A. Plot of membrane absorbance from 350–700 nm. The ECM-derived membranes exhibits superior optical transparency compared to traditional transparent cell culture inserts such as Transwell polyester membranes. B. Digital photograph of COL-MAT membrane demonstrating its optical clarity. This membrane was trimmed to the approximate size used for device bonding and held over printed text using forceps. C. Plot of relative membrane permeability representing measurements of 20kDa FITC-dextran transport across COL, COL-MAT, COL-ALG, and PE membrane inserts over a period of 6 hours under continuous parallel flow perfusion at a flow rate of 100 µl/hr. ** and ns represent P < 0.01 and not significant, respectively. D. SEM visualization of collagen type I-alginate (COL-ALG) membrane surface ultrastructure demonstrating larger pores and fenestrations (arrows) created by using alginate as a water-soluble sacrificial material, scale bar = 2 µm. E. Atomic force microscopy (AFM) nanoindentation measurement of the elastic modulus for hydrated COL, 80:20 COL-MAT, 50:50 COL-MAT, 20:80 COL-MAT, and Transwell PE membranes. * represents P < 0.05.
Permeability
Exchange of macromolecules, such as growth factors and cytokines, between adjacent tissue compartments is essential for complex multicellular interactions that play a critical role in diverse physiological and pathophysiological processes. Biomolecular transport necessary for these types of interactions requires that the basement membrane be sufficiently permeable to large molecules. To find out whether our ECM membranes mimic this important feature of the native basement membrane, we used 20kDa FITC-dextran as a representative macromolecule and measured its transport across bare COL membranes embedded between two microfluidic channels under flow conditions (see Methods for details).
Fluorescence measurement of outflow collected from the microchannels indicated that the COL membranes allowed translocation of dextran molecules due to externally imposed concentration gradients. Both COL and COL-MAT membranes, however, were significantly less permeable than Transwell PE membranes with 400 nm pores (Fig. 3C), presumably due to their dense fiber architecture (Fig. 2B). To increase membrane permeability, we then devised a new technique in which water-soluble alginate (ALG) was added to the collagen base and used as a sacrificial material that was dissolved away during the rehydration of initially dried films. SEM visualization of the collagen membranes produced by this method (COL-ALG membranes) showed markedly increased bundling of collagen fibrils and more clearly visible fenestrations throughout the surface, suggesting increased membrane porosity (arrows, Fig. 3D). Our quantitative analyses of the scanning electron micrographs confirmed that the average size of membrane pores in the COL-ALG membranes (700 nm) was significantly larger than that in the COL membranes (250 nm) (Fig. S2). Consistent with these microscopic findings, the permeability of the COL-ALG membranes to 20 kDa FITC-dextran was measured to be higher than that of COL membranes and Transwell PE membranes by a factor of 8 and 1.2, respectively (Fig. 3C).
Another important factor that impacts the permeability of our ECM membranes is adsorption of biological molecules on the membrane surface. Our assay using FITC-BSA (see Methods) showed that surface adsorption on the COL membranes was significantly greater than that on Transwell polyester membranes (Fig. S3). Binding and sequestration of biological molecules are a critical function of the native ECM that is often challenging to recapitulate using synthetic cell culture membranes. Therefore, this unique property may be exploited to further enhance the biological activity of our ECM membranes in a controllable fashion.
Young’s modulus
The stiffness of the basement membrane varies significantly depending on the mechanical microenvironment of associated tissues (24). During early embryonic development, for example, the basement membrane is more elastic to accommodate rapid growth and expansion of developing organs, whereas it becomes stiffer at later stages to provide mechanical stability (24, 25). Studies have also shown that aging and various types of diseases (e.g., diabetes) are often accompanied by significant stiffening of the basement membrane and its adjacent tissue (26). Recapitulating these physiologically relevant mechanical alterations of the native basement membrane in vitro entails the ability to adjust the stiffness of cell culture membranes in a predetermined manner. To demonstrate this capability, we varied the matrix composition of our ECM membranes and used nanoindentation AFM as a quantitative way to measure their Young’s moduli.
From qualitative visual examination, the dry ECM membranes prepared by our technique were observed to be rigid and resistant to excessive bending and twisting, regardless of composition. When hydrated, however, the membranes became softer and more compliant, exhibiting transition to a water-holding gelatinous appearance (Fig. S4). As plotted in Figure 3E, the wetted COL membranes had a mean Young’s modulus of approximately 660 kPa. When collagen was blended with Matrigel at a volume ratio of 80:20 (collagen: Matrigel), the stiffness of the resultant COL-MAT membranes decreased significantly, yielding a Young’s modulus of 549 kPa. Increasing the volume fraction of Matrigel to 50% led to further reduction of the Young’s modulus down to 429 kPa. Considering that Matrigel is comprised largely of globular basement membrane proteins, the observed changes in membrane stiffness are likely due to the reduction in fibrous collagen type I, rendering the composite membranes more compliant. Although little data have been reported on the mechanical properties of native basement membranes alone, our results are comparable to the physiological ranges of basement membrane stiffness measured in several types of human tissue (27), including the lens capsule (0.3–2.4 MPa) (28), retina (1 MPa) (27), cochlea (37–135 kPa) (29), and blood vessels (1–3 MPa) (30). In contrast, hydrated Transwell PE membranes were found to be more than 2 orders of magnitude stiffer, as demonstrated by their mean Young’s modulus of 180 MPa (Fig. 3E). These data illustrate tunable stiffness of our ECM membranes and their potential as promising alternatives to conventional membrane inserts for modeling physiologically relevant biophysical microenvironments.
Incorporation of ECM membranes into microfluidic culture
Next, we investigated the utility of our membranes as cell culture substrates in microfluidic systems. For this study, we embedded ECM membranes between two parallel microchannels to construct perfusable multilayer cell culture devices (Fig. 4A). Our bonding technique based on adhesive layers of uncured PDMS (Figs.1F–1H) permitted seamless integration of ECM membranes into the cell culture chambers without compromising structural integrity. Despite their small thickness, the membranes remained intact and flat across the channel width during and after assembly (Fig. 4B). Hydration of the membranes with culture media prior to cell seeding led to enhanced optical clarity but no undesirable structural changes were observed (data not shown).
Figure 4. Microfluidic cell culture using ECM-derived membranes.

A. Digital photograph of a microfluidic cell culture device comprised of upper (blue) and lower (red) microchannels separated by a COL-MAT membrane (shown with dotted line). Injection of red and blue food coloring dyes demonstrates patency of device bonding and partitioning function of the membrane. B. A cross-sectional view of the device along the line a-a shown in A. The ECM membrane indicated with a white arrow was immunostained to visualize type I collagen. The dotted lines show the channel walls. Channel width = 500 microns. C. Phase contrast micrograph of a confluent human bronchial epithelial cell (BEAS-2b) monolayer formed on a COL-membrane in a three-layer microdevice. The cells were cultured for 72 hours under flow conditions at a flow rate of 100 µl/hr. Scale bar = 200 µm. D. A confluent monolayer of BEAS-2b cells stained with red CellTracker dye. Scale bar = 200 µm. E. Human umbilical vein endothelial cells (HUVEC) grown to confluence on the surface of COL-MAT membrane visualized by fluorescence imaging of constitutive green fluorescent protein (GFP, green) expression following 48 hours of microfluidic perfusion culture at 100 µl/hour. Scale bar = 200 µm. F. Normal human lung fibroblasts (NHLFs) growing on the COL-MAT membrane surface. Red CellTracker dye was used to visualize the cells. Scale bar = 100 µm. G. Immunofluorescence staining of alpha smooth muscle actin (α-SMA) in NHLFs cultured in the microdevice. Scale bar = 100 µm.
To examine cell adhesion and growth on our ECM membranes, we used human bronchial epithelial cells (BEAS-2b) as a representative cell population. When the cells were seeded into our devices containing COL membranes, they attached to the membrane surface and began to adhere within 30 minutes in the absence of flow. Cell adhesion in these devices did not require pre-treatment of channel surfaces with ECM solutions – a common technique for achieving cell attachment to synthetic substrates in conventional microfluidic systems. Under perfusion culture conditions, the cells continued to proliferate over a period of 2–3 days until they formed a confluent monolayer (Fig. 4C, 4D). The same growth patterns were observed in other cell types including endothelial (Fig. 4E) and stromal cells seeded on the COL membranes (Fig. 4F, 4G). Direct observation and visualization of the cultured cells were greatly facilitated by optical transparency of the ECM membranes (Fig. 4A, 4C). At typical flow rates used in these experiments (70–100 µl/h), media perfusion through the microchannels did not have any measurable adverse effects on membrane integrity. Under these perfusion culture conditions, the COL substrates maintained their original membrane architecture for extended periods (over one week) without a loss of structural integrity, demonstrating the long-term stability of our COL membranes and their resistance to cell-mediated proteolytic degradation (Fig. S5).
Effect of ECM membranes on cell adhesion and growth
After confirming the capacity of our membranes to support microfluidic cell culture, we then questioned whether the ECM membranes offer significant advantages over traditional membrane inserts for promoting physiological cell-matrix interactions. To address this question, we quantitatively analyzed the phosphorylation of focal adhesion kinase (FAK) in monolayers of HUVECs cultured on different types of membrane substrates in our microfluidic device (Fig. 5A). FAK is an important component of the focal adhesion complex that undergoes phosphorylation in response to integrin engagement, and serves as a key regulator of signaling pathways that mediate cell adhesion, proliferation, and a host of other critical cellular functions (31–33). When the cells were grown on bare Transwell PE membranes sandwiched between two microchannels, they showed uniformly low levels of phosphorylated FAK (pFAK) throughout the monolayer. Incubation of the PE membranes with fibronectin solutions before cell seeding led to a slight increase in phosphorylation, presumably due to the absorption of fibronectin onto the membrane surface that facilitated cell adhesion and integrin-mediated signaling. When the COL membranes were used in our device, however, pFAK staining became significantly more pronounced as evidenced by a more than 2.5-fold increase in fluorescence intensity on a per cell basis (P<0.05) (Fig. 5B). This promotive effect was further amplified by the incorporation of the COL-MAT membranes, in which case the levels of pFAK were more than 9 times higher than those measured in the bare PE membranes (P<0.01) (Fig. 5B). The increased FAK signaling on the composite membranes was also accompanied by the assembly of actin stress fibers into thick bundles at the cell periphery, resulting in intense actin staining along cell-cell junctions. As demonstrated by these data, our membranes engineered from native ECM proteins allow cells to engage the surface of their underlying substrates in a much more robust manner than is possible with commonly used synthetic membrane supports. Moreover, incorporation of basement membrane components into our membranes provides an effective means to further increase integrin engagement and to induce cytoskeletal rearrangement that contributes to the formation of barrier tissue with enhanced structural integrity.
Figure 5. Tunable cell adhesion and monolayer formation on engineered ECM membranes.

A. Impact of membrane composition on cytoskeletal organization (actin staining, top row) and focal adhesion-associated signaling (pFAK staining, bottom row) in cultured HUVECs. HUVECs were seeded at high density and cultured for 6 hours on the surface of polyester (PE), fibronectin-coated polyester (PE + FN), COL, or COL-MAT membranes. Localized actin staining (green) at cell borders indicates rapid barrier organization following seeding on COL-MAT membranes. Staining of the phosphorylated form of focal adhesion kinase (pFAK, red) localizes adhesion-associated signaling complexes activated by adhesion to the respective membranes. Cells cultured on COL-MAT membranes show significantly increased pFAK activities. Scale bar = 50 µm. B. Quantification of FAK phosphorylation on a per cell basis presented as relative fluorescence intensity normalized to values obtained for uncoated PE membranes. * and ** show P < 0.05 and P < 0.01, respectively. C. Phase contrast images of A549 cells growing as domed aggregates on COL membranes (left) or confluent monolayers on COL-MAT membranes (right). The cells were cultured for 72 hours under continuous perfusion of culture media at 100 µl/hour. Scale bar = 200 µm. D. Pericyte spreading on COL (left) and COL-MAT(right) membranes in microfluidic channels. Pericytes express a tomato red reporter of the Gli1 transcription factor. Scale bar = 100 µm. E. Alpha-6 integrin staining (green) of pericytes (red, Gli1 reporter) following 16 hours of culture on COL (left) and COL-MAT (right) membranes. Scale bar = 200 µm.
Interestingly, differential cell adhesion responses observed in endothelial cells were also found in other types of adherent cells. For example, human lung cancer cells (A549) cultured on the COL membranes in our device exhibited elongated morphology and 3D growth into dome-shaped aggregates, whereas they formed confluent 2D monolayers on the surface of COL-MAT membranes (Fig. 5C). Pericytes, which serve as a key cellular component of the endothelial basement membrane niche (34), are another cell population tested in our study that showed distinct responses to different membrane compositions. When these cells were seeded on the COL membranes, they showed poor adhesion and remained rounded without spreading by 4 hours post-seeding. In contrast, the COL-MAT membranes allowed the cells to rapidly attach, spread, and extend cellular projections within the same time (Fig. 5D). We hypothesized that rapid pericyte adhesion and spreading observed on COL-MAT membranes was due to the engagement of laminin-specific adhesion receptors such as the α6 integrin subunit (35). Allowing sufficient time for pericytes to adhere and spread on pure COL by 16 hours post-seeding, we then compared α6 abundance and localization by immunofluorescence and observed markedly increased α6 staining on COL+MAT membranes (Fig. 5E). These simple examples suggest the possibility of using our ECM membranes to control and rationally manipulate cell adhesion and growth in microfluidic cell culture models.
Construction of microfluidic tissue-tissue interfaces
In microfluidic cell culture, semipermeable membrane inserts are used predominantly as physical barriers that separate two or more adjacent cell culture chambers, while their porosity allows for active and passive transport of fluids and various soluble factors between the chambers. This design is commonly implemented in constructing compartmentalized cell culture models in which two distinct cell types are cultured on either side of a porous membrane to replicate multicellular interfaces between two adjacent tissue compartments (3, 8). Motivated by this widespread use of semipermeable membrane inserts, we created 3-layer microfluidic devices containing COL-MAT membranes as a platform to engineer various types of tissue-tissue interfaces without requirement of any cell type-specific membrane coatings or other preprocessing.
As a preliminary study, we carried out microfluidic culture of human lung epithelial cells in our device to recreate the air-lung interface (Fig. 6A). To engineer this model, we first cultured A549 lung cells to confluence on the membrane surface of the upper chamber under continuous medium perfusion on both sides of the membrane. When the epithelial barrier was formed, medium was gently aspirated from the upper chamber to expose the apical side of the cells to air. Owing to the permeability of our membranes, this configuration permitted basolateral feeding of the epithelial tissue, as illustrated by nearly 100% cell viability after 3 days of air-liquid interface (ALI) culture (Fig. 6A). The lung epithelial barrier in our system remained viable for prolonged periods and effectively prevented leakage of culture medium from the lower chamber, allowing for stable maintenance of the microengineered air-lung interface.
Figure 6. On-chip engineering of tissue-tissue interfaces using ECM membranes.

A. Air-liquid interface culture of human adenocarcinoma cells (A549) in a 3-layer microfluidic device. The phase contrast images show confluent epithelial monolayers after 48 hours of submerged culture (“submerged”) and 72 hours of ALI culture (“ALI”). The fluorescence image of the cells stained with calcein-AM fluorescence (green) indicates virtually 100% viability. Scale bars = 200 µm. B. Human bronchial epithelial cells (BEAS-2b, red CellTracker) are cultured on the upper surface of ECM membrane with human lung fibroblasts (NHLFs, green CellTracker) seeded at low density on the lower membrane surface to recreate the airway epithelial-stromal interface in the lung. Scale bar = 200 µm. C. Epithelial-endothelial barrier comprised of BEAS-2b cells (red CellTracker) and HUVECs (GFP, green) cultured on the opposite sides of COL-MAT membrane. 3-D rendering was conducted to show angled view of the cell layers. Scale bar = 200 µm. D. Co-culture of HUVECs (green) and NHLFs (red). Scale bar = 200 µm. E. A preformed spheroid of human lung adenocarcinoma cells (A549, red CellTracker) is grown on the upper membrane surface in a static PDMS microwell. NHLFs (green) are cultured on the other side of the membrane in a microfluidic channel to mimic spatial arrangement of solid tumors and their associated stromal cells in the surrounding tissue. Scale bar = 200 µm. Inset: Confocal optical section of calcein-AM (green) staining in a representative spheroid cultured for 96 hours in our microdevice demonstrating viability of cells throughout the spheroid. Scale bar = 200 µm.
Building upon these results, we then sought to establish co-culture models and replicate the structural organization of biological interfaces between two different types of human tissue. Using the same 3-layer devices and perfusion culture techniques, for instance, we generated living bilayer tissues reminiscent of the epithelial-stromal interface that consisted of a monolayer of human bronchial epithelial cells (BEAS-2b) and a layer of primary human lung fibroblasts (NHLFs) separated by a COL-MAT membrane (Fig. 6B). In this model, NHLFs were seeded at low densities and maintained in low-serum media to minimize cell proliferation and to recapitulate the loose cellularity of the sub-epithelial connective tissue in many organs. Similar co-culture strategies were successfully applied to modeling the epithelial-endothelial (Fig. 6C) and vascular-stromal (Fig. 6D) interfaces in our microfluidic system.
Finally, we modified the design of our device to embed COL-MAT membranes between an open microwell and a bottom cell culture chamber to explore the possibility of combining 3D spheroids with 2D tissue layers (Fig. 6E). In this study, we pre-formed tumor spheroids by culturing A549 lung adenocarcinoma cells in agarose wells, and introduced them into the open well of our device. NHLFs were seeded into the bottom chamber and grown on the other side of the membrane to model the architecture of solid tumors and their associated fibroblasts. Although adhesion of the spheroids to the membrane surface induced outgrowth of cancer cells, the resultant spreading of the spheroids was not significant enough to cause tissue disintegration, making it possible to retain three-dimensionality and circularity of the spheroids as shown in Figure 6E. The vast majority of the cells in the spheroids remained viable throughout the culture period (Fig. 6E).
As demonstrated, our ECM membranes have the flexibility to accommodate co-culture of various cell types and provide stable structural scaffolds to reconstitute their relative spatial distributions in a physiologically relevant manner. These results highlight the potential of our membranes as an essential building block of microfluidic co-culture systems to model various kinds of tissue-tissue interfaces during health and disease.
Conclusions
In this paper, we have described a simple yet versatile hydrogel engineering technique to create semipermeable cell culture membranes that mimic the fibrous architecture and composition of native basement membranes. Structural integrity, optical transparency, and tunable biophysical properties of these biomimetic ECM analogs make them attractive for use in microfluidic cell culture systems. In comparison to traditional cell culture inserts made up of synthetic polymers, our membranes composed entirely of natural ECM proteins provide more physiological insoluble cellular microenvironments conducive to promoting cell adhesion and growth. We have also shown the utility of these membranes for constructing microphysiological models of multilayered tissue structures.
While further investigations are needed, the demonstrated effects of membrane composition on cell adhesion and tissue organization suggest the possibility of engineering the phenotype of cells using instructive cues contained in culture membranes. When combined with tissue-specific extracellular matrices (e.g., decellularized tissue) that have been increasingly recognized as critical determinants of cell fate and function, our membranes may enable new strategies to induce desired physiological or pathological responses in a programmable fashion. Such capabilities would allow cell culture membranes to serve as an externally controllable active regulator of biological processes for the development and optimization of more predictive microphysiological cell culture models. In addition, the simplicity, affordability, and controllability of our fabrication technique provide compelling advantages for widespread use and may offer a means to address the limited options of commercially available semipermeable membrane inserts.
Future work will focus on addressing the current limitations of our ECM membranes. More refined engineering control of membrane thickness is needed to model the full spectrum of basement membrane thickness observed across physiological and pathological conditions in vivo. Although our membranes composed of rodent-derived collagen type I and tumor-derived Matrigel demonstrated enhanced bioactivity for human cell culture, future studies should explore the possibility of utilizing human-derived ECM materials to unlock the full potential of our technology. A set of techniques described in this paper will provide a robust platform to test rationally-designed blends of different ECM and sacrificial materials and to precisely control critical membrane properties (e.g., ECM composition, permeability, and stiffness) that play a crucial role in regulating cellular phenotypes in vitro. Despite the limitations of our study, we believe that our work establishes a novel approach for application-specific and programmable engineering of cell culture membranes that will greatly enhance the physiological-relevance and predictive power of microfluidic in vitro models and organs-on-chips for a variety of biomedical, pharmaceutical, and environmental applications.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH) Director’s New Innovator Award to D.H. (1DP2HL127720-01). We thank Dr. Benjamin Humphreys for the Gli1-Tomato-expressing murine pericytes, Matthew Brukman for assistance with atomic force microscopy, and Raymond Meade for assistance with scanning electron microscopy. We also thank G. Alm, Brian Nam, David Conegliano, Jeongyn Seo, Cassidy Blundell, Eric Esch, and Megan Farrell for their valuable contributions.
References
- 1.Esch EW, Bahinski A, Huh D. Organs-on-chips at the frontiers of drug discovery. Nat Rev Drug Discov. 2015;14(4):248–260. doi: 10.1038/nrd4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32(8):760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- 3.Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011;21(12):745–754. doi: 10.1016/j.tcb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blundell C, et al. A microphysiological model of the human placental barrier. Lab Chip. 2016;16(16):3065–3073. doi: 10.1039/c6lc00259e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi Y, et al. A microengineered pathophysiological model of early-stage breast cancer. Lab Chip. 2015;15(16):3350–3357. doi: 10.1039/c5lc00514k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huh D, et al. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci Transl Med. 2012;4(159):159ra147. doi: 10.1126/scitranslmed.3004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huh D, et al. Reconstituting organ-level lung functions on a chip. Science. 2010;328(5986):1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huh D, et al. Microfabrication of human organs-on-chips. Nat Protoc. 2013;8(11):2135–2157. doi: 10.1038/nprot.2013.137. [DOI] [PubMed] [Google Scholar]
- 9.Huh D, Torisawa YS, Hamilton GA, Kim HJ, Ingber DE. Microengineered physiological biomimicry: organs-on-chips. Lab Chip. 2012;12(12):2156–2164. doi: 10.1039/c2lc40089h. [DOI] [PubMed] [Google Scholar]
- 10.Lo JH, Bassett EK, Penson EJ, Hoganson DM, Vacanti JP. Gas Transfer in Cellularized Collagen-Membrane Gas Exchange Devices. Tissue Eng Part A. 2015;21(15–16):2147–2155. doi: 10.1089/ten.tea.2014.0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richter L, et al. Monitoring cellular stress responses to nanoparticles using a lab-on-a-chip. Lab Chip. 2011;11(15):2551–2560. doi: 10.1039/c1lc20256a. [DOI] [PubMed] [Google Scholar]
- 12.Sun Y, Chen CS, Fu J. Forcing stem cells to behave: a biophysical perspective of the cellular microenvironment. Annu Rev Biophys. 2012;41:519–542. doi: 10.1146/annurev-biophys-042910-155306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dingal PC, Discher DE. Combining insoluble and soluble factors to steer stem cell fate. Nat Mater. 2014;13(6):532–537. doi: 10.1038/nmat3997. [DOI] [PubMed] [Google Scholar]
- 14.Li B, Chen J, Wang JH. RGD peptide-conjugated poly(dimethylsiloxane) promotes adhesion, proliferation, and collagen secretion of human fibroblasts. J Biomed Mater Res A. 2006;79(4):989–998. doi: 10.1002/jbm.a.30847. [DOI] [PubMed] [Google Scholar]
- 15.LeBleu VS, Macdonald B, Kalluri R. Structure and function of basement membranes. Exp Biol Med (Maywood) 2007;232(9):1121–1129. doi: 10.3181/0703-MR-72. [DOI] [PubMed] [Google Scholar]
- 16.Albuschies J, Vogel V. The role of filopodia in the recognition of nanotopographies. Sci Rep. 2013;3:1658. doi: 10.1038/srep01658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neeves KB, Diamond SL. A membrane-based microfluidic device for controlling the flux of platelet agonists into flowing blood. Lab Chip. 2008;8(5):701–709. doi: 10.1039/b717824g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Yang D, Yu T, Jiang X. Incorporation of electrospun nanofibrous PVDF membranes into a microfluidic chip assembled by PDMS and scotch tape for immunoassays. Electrophoresis. 2009;30(18):3269–3275. doi: 10.1002/elps.200900128. [DOI] [PubMed] [Google Scholar]
- 19.Iliescu C, Taylor H, Avram M, Miao J, Franssila S. A practical guide for the fabrication of microfluidic devices using glass and silicon. Biomicrofluidics. 2012;6(1):16505–1650516. doi: 10.1063/1.3689939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol. 2011;3(2) doi: 10.1101/cshperspect.a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon SR. Fibronectin antibody labels corneal stromal collagen fibrils in situ along their length and circumference and demonstrates distinct staining along the cell and stromal interfaces of Descemet's membrane. Curr Eye Res. 2014;39(3):312–316. doi: 10.3109/02713683.2013.841260. [DOI] [PubMed] [Google Scholar]
- 22.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3(6):422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 23.Randles MJ, Humphries MJ, Lennon R. Proteomic definitions of basement membrane composition in health and disease. Matrix Biol. 2016 doi: 10.1016/j.matbio.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3(12) doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10(7):445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 26.To M, et al. Diabetes-induced morphological, biomechanical, and compositional changes in ocular basement membranes. Exp Eye Res. 2013;116:298–307. doi: 10.1016/j.exer.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 27.Candiello J, et al. Biomechanical properties of native basement membranes. FEBS J. 2007;274(11):2897–2908. doi: 10.1111/j.1742-4658.2007.05823.x. [DOI] [PubMed] [Google Scholar]
- 28.Krag S, Olsen T, Andreassen TT. Biomechanical characteristics of the human anterior lens capsule in relation to age. Invest Ophthalmol Vis Sci. 1997;38(2):357–363. [PubMed] [Google Scholar]
- 29.Gueta R, Barlam D, Shneck RZ, Rousso I. Measurement of the mechanical properties of isolated tectorial membrane using atomic force microscopy. Proc Natl Acad Sci U S A. 2006;103(40):14790–14795. doi: 10.1073/pnas.0603429103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKee CT, Last JA, Russell P, Murphy CJ. Indentation versus tensile measurements of Young's modulus for soft biological tissues. Tissue Eng Part B Rev. 2011;17(3):155–164. doi: 10.1089/ten.teb.2010.0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6(1):56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 32.Panetti TS. Tyrosine phosphorylation of paxillin, FAK, and p130CAS: effects on cell spreading and migration. Front Biosci. 2002;7:d143–150. doi: 10.2741/A771. [DOI] [PubMed] [Google Scholar]
- 33.Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63(8):610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 35.Ramovs V, Te Molder L, Sonnenberg A. The opposing roles of laminin-binding integrins in cancer. Matrix Biol. 2016 doi: 10.1016/j.matbio.2016.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.