

Abstract
This Perspective describes the development of a family of copper(II)-catalyzed alkene difunctionalization reactions that enable stereoselective addition of amine derivatives and alcohols onto pendant unactivated alkenes to provide a range of valuable saturated nitrogen and oxygen heterocycles. 2-Vinylanilines and related substrates undergo alternative oxidative amination or allylic amination pathways, and these reactions will also be discussed. The involvement of both polar and radical steps in the reaction mechanisms have been implicated. Major product formation is a function of the lowest energy pathway, which in turn is a function of structural aspects of the various reaction components.
Graphical Abstract
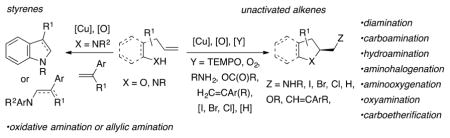
INTRODUCTION
Chiral saturated heterocycles are ubiquitous in nature and frequently found in small molecule therapeutics.1–3 Saturated heterocycles can enhance water solubility, provide conformational restriction, and provide points of H-bonding interaction with target biomolecules without increasing the number of aromatic rings, which some studies have shown should be minimized in lead compounds.4 Numerous strategies have been developed for de novo saturated nitrogen and oxygen heterocycle synthesis. One such strategy involves cyclization of amine derivatives and alcohols onto pendant alkenes. While such nucleophiles and electron-deficient α,β-unsaturated alkenes are natural coupling partners that have historically enjoyed facile and even spontaneous cyclization,5 cyclization of nucleophiles onto unactivated alkenes is not a spontaneous process. The latter process is attractive as it brings together simple, stable, easily installed, and ubiquitous functional groups. The bringing together of these electron-rich moieties, however, often requires application of π-activating electrophiles: halogens,6,7 protons,8,9 or transition metals.10,11 Significant progress has been made in each these areas. This Perspective will focus on copper-catalyzed cyclizations in particular.12 Transition-metal catalysis in general enables a broader range of transformations. and copper(II) catalysis in particular enables versatile reactivity that employs both polar and radical mechanisms, providing a powerful range of bond formations. The Earth abundance of copper, together with its versatile reactivity, facile access to oxidations states I–III, and excellent ligand coordination for tuning chirality and reactivity make it an excellent choice as catalyst.13
CONCEPTION
One of the key concerns in transition-metal-catalyzed alkene difunctionalization involves avoidance of competing β-hydride elimination, a process that tends to provide the Wacker and aza-Wacker products (Scheme 1a).14–16 When metals react primarily via two-electron mechanisms (e.g., often for [Pd], [Ni], and [Au]), alkene difunctionalization involves using ligands that prohibit β-hydride elimination,17 oxidants that oxidize the metal center,18–20 substrate chelation for conformational restriction21 or a comparatively higher reductive elimination rate.22 An alternative strategy involves application of paramagnetic transition metals. Alkene additions catalyzed by these metals result in transient alkyl radicals that can then engage in bond formations with radical acceptors or become oxidized to carbocations that then undergo reaction with nucleophiles (Scheme 1b). Cobalt(III),23–26 iron(III),27 and copper(II) (vide infra) have all demonstrated this propensity.28 A stoichiometric oxidant to turn over the catalyst is required to perform these net oxidative reactions.
Scheme 1.
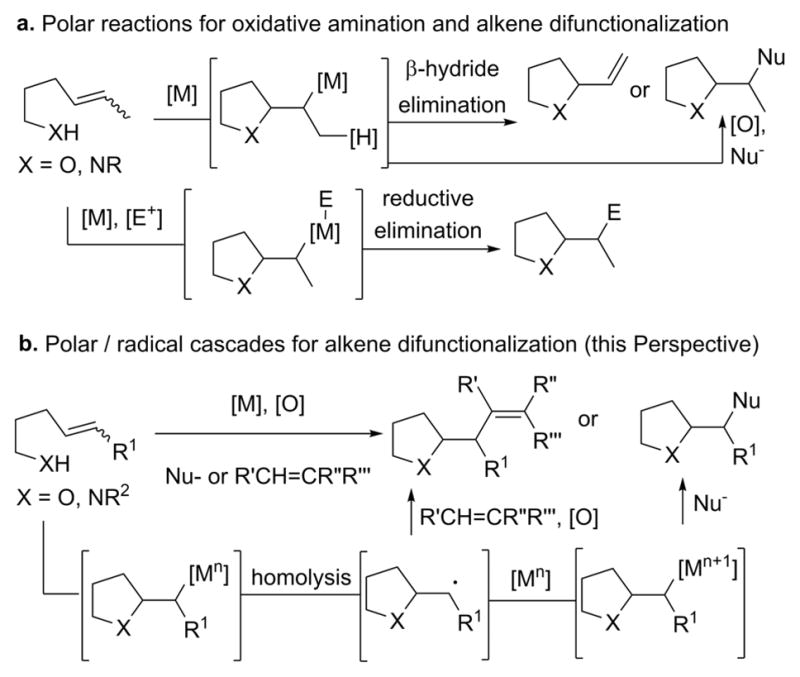
For copper, hints of these mechanistic possibilities can be found in the early organometallic literature, in particular, reactions of copper(I) and copper(II) salts with alkyl radicals and the respective decompositions of the corresponding organocopper intermediates. While elimination to give the alkene is always a possibility,29 alternative reactivity such as C–C and C–O bond formation was also shown to be possible.30–32
An important concern in transition-metal-catalyzed alkene difunctionalization involves control of absolute stereochemistry. Thus, in our work, coordination with chiral ligands to direct the initial aminocupration or oxycupration step was seen as essential. Fortuitously, ligands developed for copper’s application in asymmetric catalysis with polar alkenes and carbonyls33 were readily translated to the intramolecular difunctionalization of unactivated alkenes (vide infra). Herein is an account of the discovery and development of a family of reactions where polar copper-catalyzed additions of amine derivatives and alcohols to unactivated alkenes is followed by C–[Cu(II)] homolysis and additions of the resulting alkyl radical to various radical acceptors.
RESULTS
Discovery of the Copper(II)-Promoted Carboamination and Aminooxygenation of Alkenes
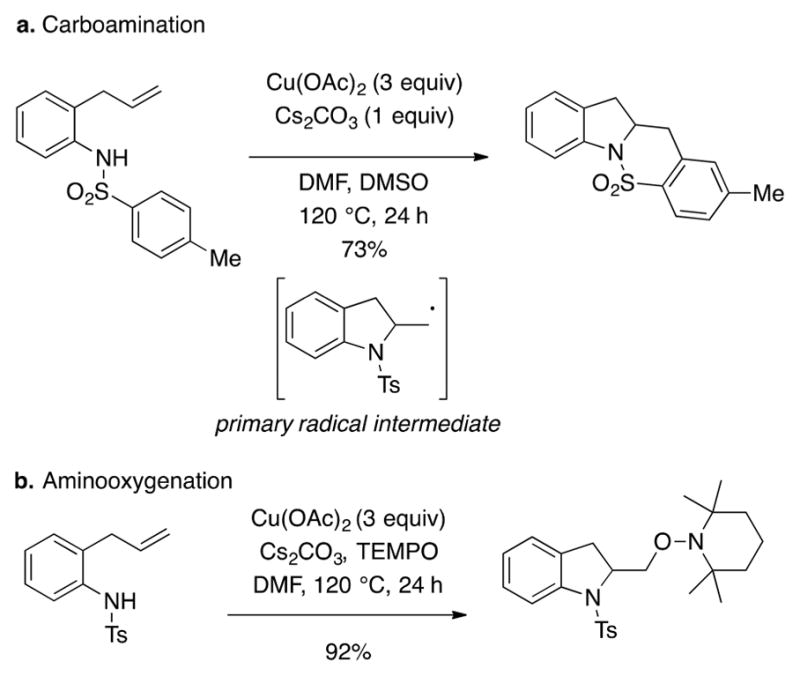
Prior to our work in the area, reactions involving aminocupration and oxycupration of unactivated alkenes with copper(II) complexes had not been reported. The first reaction discovered was the Cu(OAc)2-promoted doubly intramolecular carboamination of N-tosyl-2-allylanilines, providing sultams (Scheme 2a).34 At the time of its publication, we were uncertain whether the reaction initiated with oxidation of the sulfonamide to an amidyl radical or if it initiated with an aminocupration across the double bond (further mechanistic analysis will be discussed, vide infra). The copper-promoted aminooxygenation reaction was the second reaction in this class to be discovered and developed. The (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) derived aminooxygenation reaction was initially put forth as a probe to detect the presence of a primary carbon radical intermediate hypothesized to be involved in the related carboamination reaction (Scheme 2b).35
Scheme 2.
We recognized the potential utility of the aminooxygenation and expanded to it to include pyrrolidine synthesis. The cyclization of 4-pentenylsulfonamides worked best with the organic soluble copper(2-ethylhexanoate)2 (1.5 equiv) in xylenes, which could facilitate higher reaction temperatures when required (Scheme 3).36 2,5-cis-Pyrrolidines were generally favored with α-substituted 4-pentenylsulfonamides (Scheme 3a) except when the α-substituent and N-substituent were directly bonded to one another (Scheme 3b). In the latter case, selectivity for the 2,5-trans-pyrrolidine was observed. By applying a rationale based on empirical results, we proposed the selectivity pattern could emerge from cyclic transition states involving cis-aminocupration across the alkene (vide infra). To minimize steric interactions, the α- and N-substituents adopt a transdiaxial relationship when not directly connected. This reaction was applied to the formal synthesis of the ant pheromone (+)-monomorine (Scheme 3a). Oxidation of the TEMPO-adduct with m-CPBA provided the corresponding aldehyde that had previously been converted to (+)-monomorine by Backvall.37,38
Scheme 3.

Copper(II) Carboxylate Promoted Mechanism Analysis
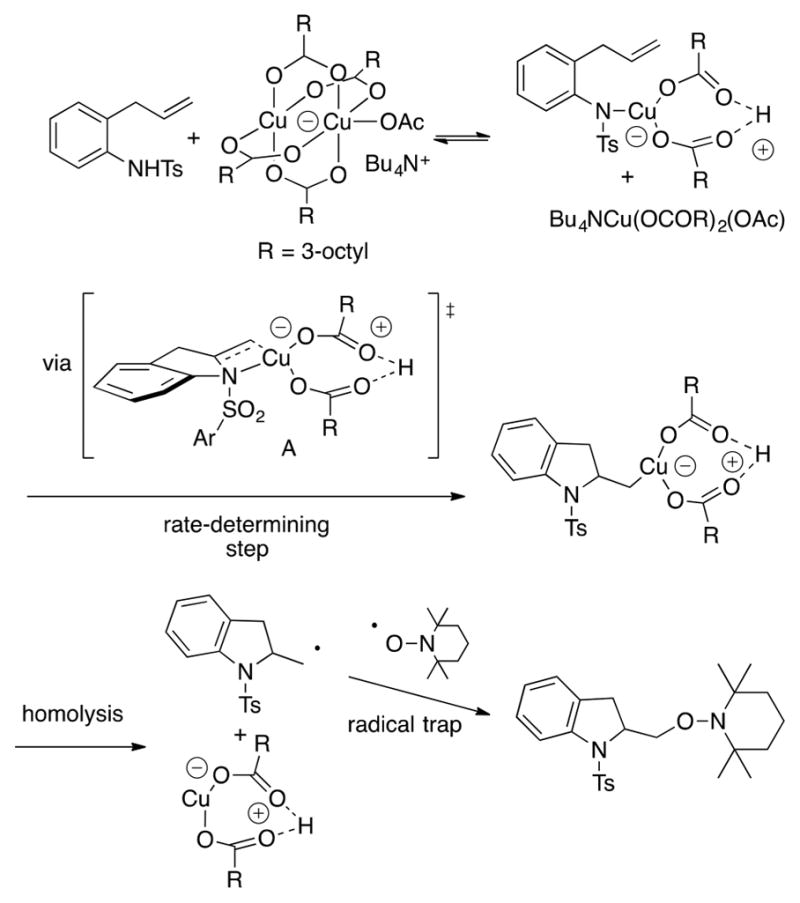
Although the stereoselectivity and the empirical reactivity trends we observed were helpful in forming a mechanistic framework for these reactions (involvement of radicals, cyclic, chairlike transition states), given the relative novelty of this reaction class, we pursued a more detailed mechanistic analysis. We explored the mechanism of the copper(II) carboxylate promoted alkene aminooxygenation using kinetic isotope effects, isotopic labeling, reaction kinetics, electron paramagnetic resonance (EPR) spectroscopy, and molecular modeling.39 Reaction kinetics were performed on the N-tosyl-2-allylaniline substrate for the copper(2-ethylhexanoate)2-promoted aminooxygenation reaction. In order for all reagents to be soluble for greater certainty of reaction concentrations, Cs2CO3, the base heretofore most frequently used in these reactions, was replaced with Bu4NOAc (Scheme 4). The reaction kinetics with respect to Bu4NOAc, copper(2-ethyl-hexanoate)2, N-tosyl-2-allylaniline (abbreviated R2NH), and TEMPO were examined. Early on in these studies, we observed that the rate of the reaction was affected by the concentration of base such that a peak in rate was observed around a 2:1 [Cu]/Bu4NOAc ratio, followed by a decrease in rate at higher base concentration. The effect of base (AcO–) on the copper species was further examined by UV–vis spectroscopy. We observed a change in the UV–vis spectra that could be interpreted to be a change in the copper species from dimeric to monomeric. We thus interpreted that the base serves as a ligand to break up the copper(II) carboxylate dimer to its more active monomeric species. All subsequent kinetic experiments for this reaction were performed with this 2:1 [Cu]/Bu4NOAc ratio.
Scheme 4.

We found the reaction to be 1/2 order in [Cu], 1/2 order in [R2NH], and zero order with respect to TEMPO (Scheme 4a). A secondary inverse kinetic isotope effect was observed with a labeled alkene substrate (Scheme 4b). This result indicates addition to the alkene is the rate-determining step. Although [R2N–Cu] could not be detected by EPR under the reaction conditions with Bu4NOAc, evidence for a productive [R2N–Cu] intermediate was obtained by independently synthesizing it [treatment with n-BuLi then copper(2-ethylhexanoate)2] and observing the N–[Cu(II)] bond by EPR spectroscopy (Scheme 4c). Upon heating this intermediate in the presence of TEMPO, the aminooxygenation product was produced. The reaction was first order with respect to [R2N–Cu]. These data are consistent with addition of both amine and copper across the alkene via an inner-sphere cis-aminocupration mechanism.
The mechanism most consistent with these data is illustrated in Scheme 5. The copper(II) carboxylate dimer, activated by –OAc, breaks into a monomeric species when it reacts with the N-tosyl-2-allylaniline. This intermediate undergoes rate-determining cis-aminocupration across the alkene. Homolysis of the C–Cu(II) bond forms the primary carbon radical that is rapidly trapped with TEMPO.
Scheme 5.
Fortuitously, the diastereoselective aminooxygenation reaction was easily rendered catalytic by employing an atmosphere of O2 (1 atm, balloon) (Scheme 6).40,41 High 2,5-diastereoselectivity was observed when Cu(2-ethylhexanoate)2 (20 mol %) (conditions A) was applied as catalyst (Scheme 6a). Substitution at the β-position of the 4-pentenylsulfonamide favored formation of the 2,4-cis-pyrrolidine, while substitution at the γ-position favored 2,3-trans diastereoselectivity (Scheme 6b). In some cases, the use of [Cu(bisoxazoline)](OTf)2 improved the reaction’s diastereoselectivity (conditions B, Scheme 6a).
Scheme 6.

A catalytic enantioselecitve aminooxygenation was also readily developed.42–44 Sulfonamides demonstrated the most promising properties with respect to reactivity and enantioselectivity. On small scale (ca. 50 mg of substrate), N-tosyl-2-allylanilines can undergo the catalytic aminooxygenation reaction with only TEMPO (300 mol %) used as both oxygen source and stoichiometric oxidant (Scheme 7).42 [Cu(I)] + TEMPO• → [Cu(II)] + TEMPO–, can account for the copper turnover under anaerobic conditions. Enantioselective amino-oxygenation reactions performed on scales larger than ca. 50 mg do require O2 (1 atm, balloon) as do aminooxygenations of 4-pentenylsulfonamides (Scheme 7a). N-Tosyl-2-allylanilines and 2,2-disubstituted 4-pentenylsulfonamides) reacted most efficiently in this reaction. While the (R,R)-Ph-box is an effective ligand for this reaction, the (4R,5S)-di-Ph-box ligand proved more effective with a broader substrate range (higher selectivity, higher reactivity), possibly due to enhanced catalyst solubility. Importantly, limiting the amount of TEMPO to a near stoichiometric amount (1.2 equiv), reducing the reaction temperature to no more than 110 °C, and application of O2 atmosphere (1 atm, balloon) were all critical to obtaining the maximum yield of the aminooxygenation product when the reaction was run on larger scales (Scheme 7b). After extensive experimentation, it became apparent to us that at elevated temperature and higher TEMPO concentrations side reactivity such as TEMPO disproportionation could manifest, resulting in low percent conversion.
Scheme 7.

We found the mechanism of the enantioselective [Cu(R,R-Ph-box)](OTf)2-catalyzed aminooxygenation of N-tosyl-2-allylaniline in the presence of excess TEMPO and no O2 to be relatively straightforward.43 Since K2CO3 is largely insoluble in PhCH3, however, we sought a replacement. Unfortunately, when Bu4NOAc was applied no enantioselectivity was observed (Scheme 8). When no base was used, however, a good yield and enantioselectivity was observed. In retrospect, the reaction with TEMPO as sole oxidant should make an equivalent of TEMPO anion, which can serve as the base. (In reactions that involve both TEMPO and O2 we believe O2 oxidizes the TEMPO anion to TEMPO radical since O2 alone is a poor oxidant for these [Cu(R,R)-Ph-box](OTf)2 catalyzed reactions.) This reaction was found to be first order in substrate, first order in catalyst, and zero order in TEMPO (Scheme 8a). An inverse kinetic isotope effect was observed with a deuteroalkene substrate. Taken together, these data are consistent with a rate-determining and enantiodetermining cis-aminocupration transition state. The cis-aminocupration transition state is put forth in analogy to the copper(II) carboxylate process described above. A density functional theory calculation from the R2N–[Cu] intermediate to the lowest energy cis-aminocupration transition state A (chemdraw rendition) indicated the activation energy was 18.2 kcal/mol (Scheme 8a).45 The experimentally determined ΔG of the reaction was determined to be 32 kcal/mol, where ΔH = 28 kcal/mol and ΔS = –11 cal/mol·K (reaction performed at 110 °C).46 This indicates that steps before the cis-aminocupration also contribute to the overall ΔG of the reaction. The transition state leading to the minor enantiomer (chemdraw depiction TS-B, Scheme 8b) was calculated to be 1.5 kcal/mol higher for the indoline series.45 A chairlike transition state (chemdraw depiction C) is more apparent in the formation of the pyrrolidine aminooxygenation product (Scheme 8c). Major transition state C (chemdraw rendition) was calculated to be 2.7 kcal/mol lower in energy than minor transition state D (Scheme 8c). In all of these transition states the copper(II) center adopts a distorted square planar geometry and the bulky sulfonamide avoids steric interactions with the ligand substituents.
Scheme 8.

When Less Is More
More recently, we explored the use of O2 as sole oxidant and oxygen source for the copper-catalyzed aminooxygenation of 4-pentenylsulfonamides (Scheme 9).47 In this case, molecular oxygen traps the carbon radical directly and subsequent peroxyradical decomposition provides the aldehyde. Use of CuCl as catalyst is critical to the success of this reaction; other copper salts such as Cu(OTf)2 or Cu(2-ethylhexanoate)2 provided much lower conversion to the desired aldehyde. The diastereoselectivity observed in these reactions mirrors that observed in the reactions involving TEMPO trapping of the carbon radical (vide supra). While promising enantioselectivity (45% ee) was observed in one example (not shown), the enantioselective variant requires further optimization.47 It should be noted that small amounts of aminochlorination and aminohydroxylation as well as lactams from over-oxidation are also formed.47
Scheme 9.
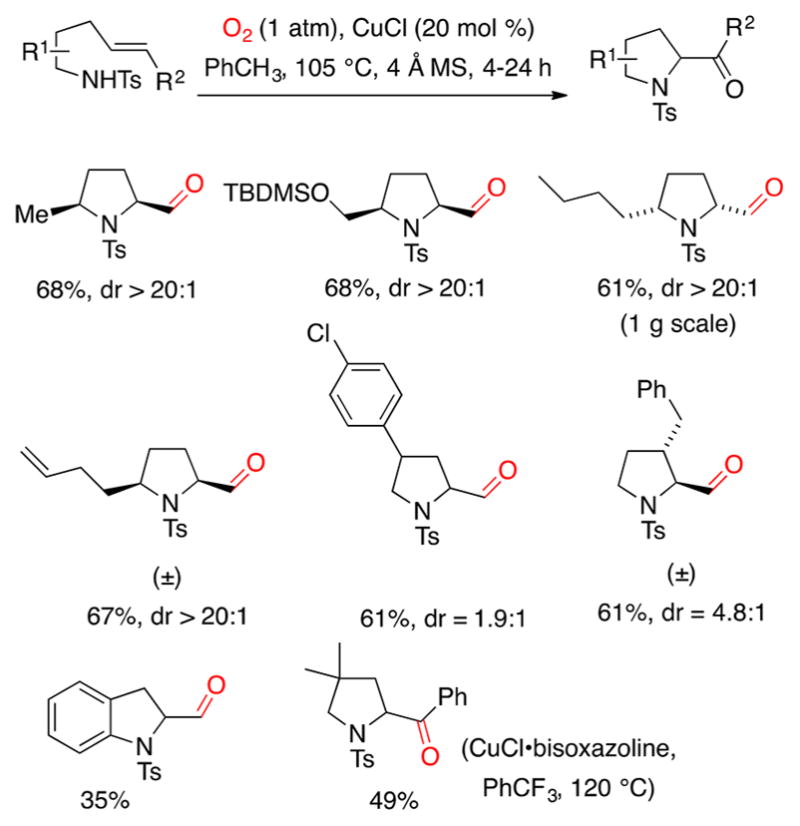
These copper-catalyzed aminooxygenation reactions are currently limited to the synthesis of five-membered rings. Substrates that could lead to six-membered ring products fail or give alternative products.
Carboaminations: Doubly Intramolecular or Inter-/ Intramolecular
Net addition of nitrogen-based and carbon-based moieties across an alkene can be defined as alkene carboamination. In the context of copper-catalyzed heterocycle synthesis, we have explored both doubly intramolecular48–51 and intra-/intermolecular52 carboamination approaches.
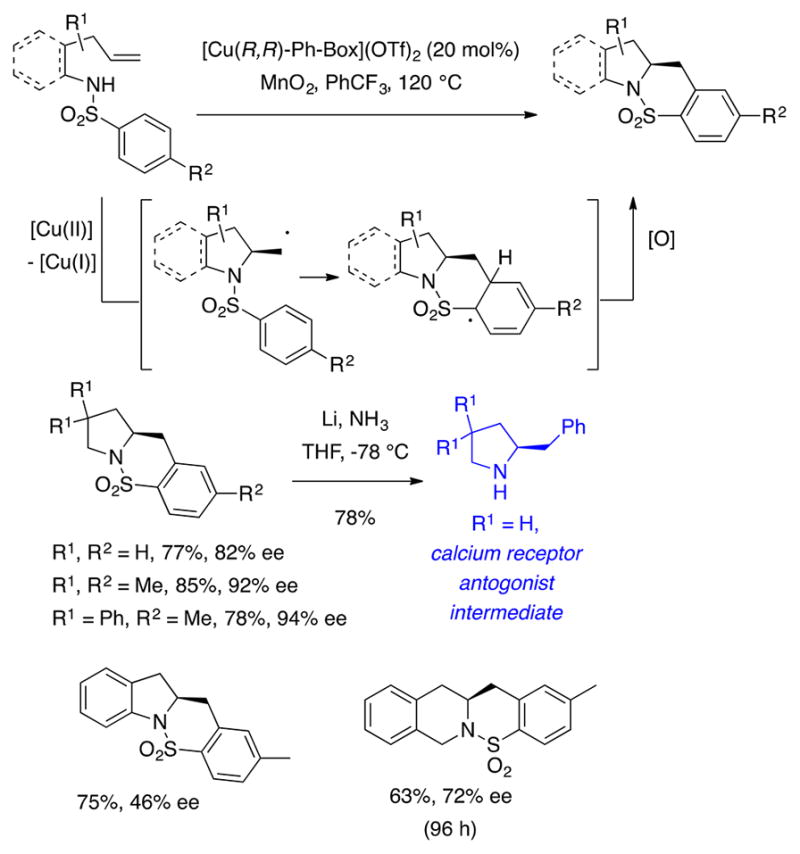
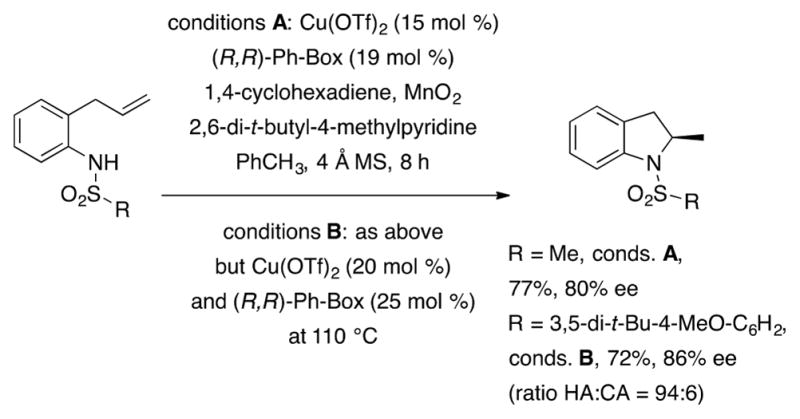
The enantioselective copper-catalyzed alkene difunctionalization was first developed in the synthesis of enantiomerically enriched sultams via doubly intramolecular alkene carboamination (Scheme 10).48 A ligand and oxidant screen led to the identification of (R,R)-Ph-Box and MnO2 as superior ligand and oxidant for this reaction. The oxidant is required to cycle [Cu(I)] back to [Cu(II)] and may also be involved in oxidation of the intermediate aryl radical. Use of Cu(OTf)2 resulted in enantiomerically enriched product, while Cu(OAc)2 did not. Oxygen was not an effective oxidant for the [Cu(R,R)-Box](OTf)2-catalyzed carboamination. While pyrrolidines were formed in 80–94% enantiomeric excess, an indoline was produced in 46% ee. The six-membered tetrahydroisoquinoline was produced in 72% ee. This reaction was applied to the synthesis of (S)-(+)-2-benzylpyrrolidine, an intermediate in the synthesis of a calcium-sensing receptor antagonist53 (Scheme 10), and to (S)-(+)-tylophorine, an anticancer alkaloid (not shown).49,54
Scheme 10.
Benz[f ]indoles appear to be the easiest bicyclic heterocycles to form from doubly intramolecular carboamination.50 The cis-benz[f ]indole is formed from 3,3-dibenzyl-4-pentenylsulfonamides irrespective of the presence of an N-arylsulfonyl group (Scheme 11a). 3-Benzyl substrates substituted on the arene with either electron-donating or electron-withdrawing groups were excellent substrates. While many of the reactions give straightforward arene addition products, 2-substituted arenes and the 4-CF3 arene gave regioisomeric mixtures that appear to be formed by ipso arene addition followed by differential C–C bond migration. Diastereoselective cyclization of a monobenzylated pentenylamine followed by N-desulfonylation and subsequent propylation provided a 5-HT1A receptor antagonist (Scheme 11b).55
Scheme 11.

Formation of 6-azabicyclo[3.2.1]octanes appears to be the most challenging of the doubly intramolecular carboaminations as this reaction (Scheme 11a) does not occur when cyclization onto the sulfonylarene is a possibility (Scheme 10).51 When N-alkylsulfonyl-2-aryl-4-pentenylamines are applied, enantioselective carboamination occurs smoothly to form variously substituted 6-azabicyclo[3.2.1]octanes (Scheme 11).51
We have primarily employed arene radical acceptors in these doubly intramolecular carboaminations, but alkenes and alkynes are also good radical acceptors in these reactions and some such examples will be illustrated in subsequent sections.
Intra-/Intermolecular Carboamination
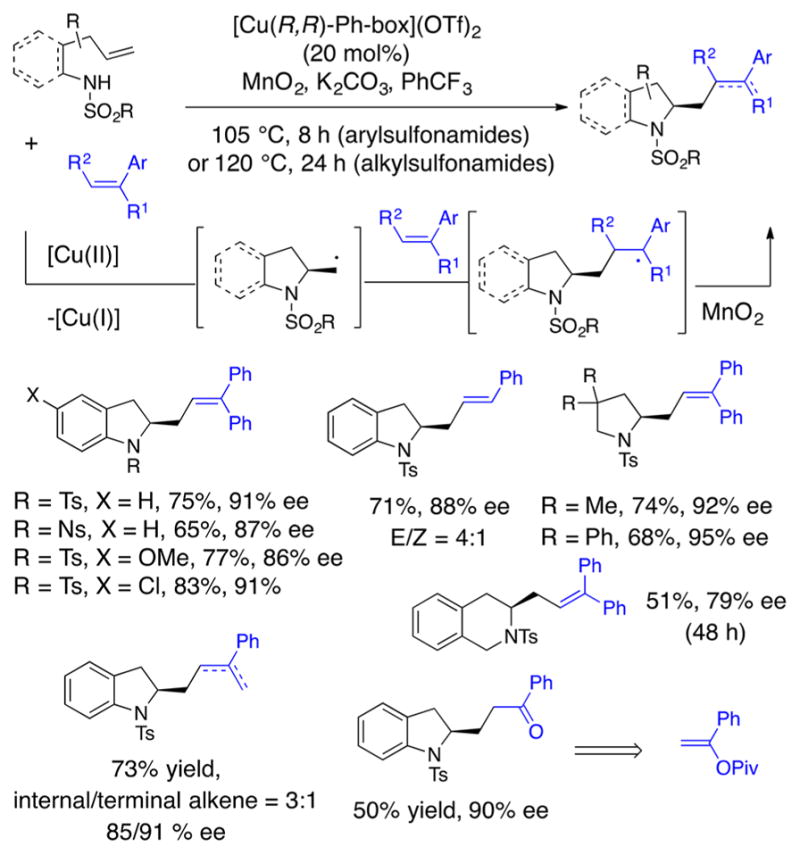
Catalytic enantioselective intra-/intermolecular alkene carboaminations were accomplished by cyclization of the sulfonamide onto the alkene via aminocupration followed by addition of the resulting primary carbon radical to exogenous alkene radical acceptors.56 1,1-Diphenylethylene and related styrenes proved to be excellent radical acceptors. The resulting stabilized benzylic radical intermediates undergo oxidation (either MnO2 or [Cu] facilitated) to provide higher substituted alkenes in a net alkyl-Heck type bond formation (Scheme 12). Several enantioenriched indolines, pyrrolidines, and a tetrahydroisoquinoline were formed using this method (Scheme 12). When a vinylpivalate radical acceptor was used, a ketone-functionalized pyrrolidine was obtained.
Scheme 12.
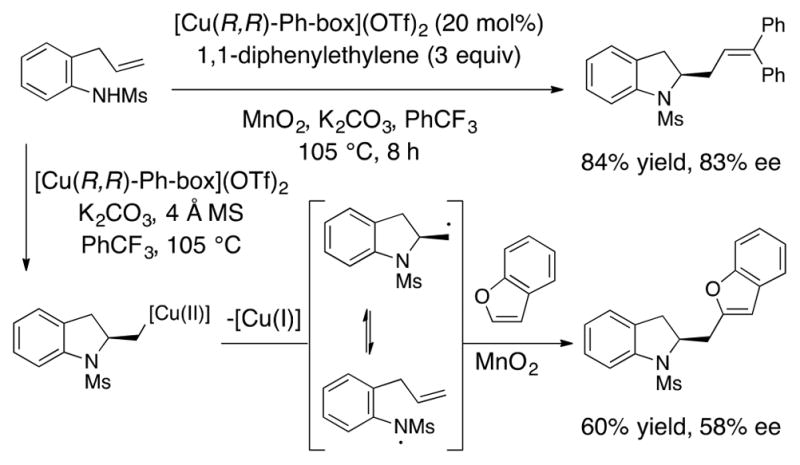
While the most reactive exogenous styrenes enabled the desired intermolecular radical addition over the competing intramolecular radical addition onto the pendant arylsulfonyl group, less reactive radical acceptors like benzofuran required use of the N-methanesulfonyl derivative to remove the possibility of intramolecular radical cyclization. A surprising effect on enantioselectivity was also observed: reaction of N-mesyl-2-allylaniline with 1,1-diphenylethylene gave the carboamination adduct in 83% ee while the benzofuran adduct was obtained in 58% ee (Scheme 13). We rationalized this by envisioning N–C bond cleavage of the alkyl carbon radical intermediate to give an aminyl radical intermediate. Any subsequent ring closure of the resulting aminyl radical onto its alkene would give a racemic carbon radical, and this side reactivity would be evident in the product enantiomeric excess. The comparatively lower reactivity of benzofuran and likely the stability of the arylsulfonyl aminyl radical (vide infra) renders the N–C bond-cleavage pathway competitive.
Scheme 13.
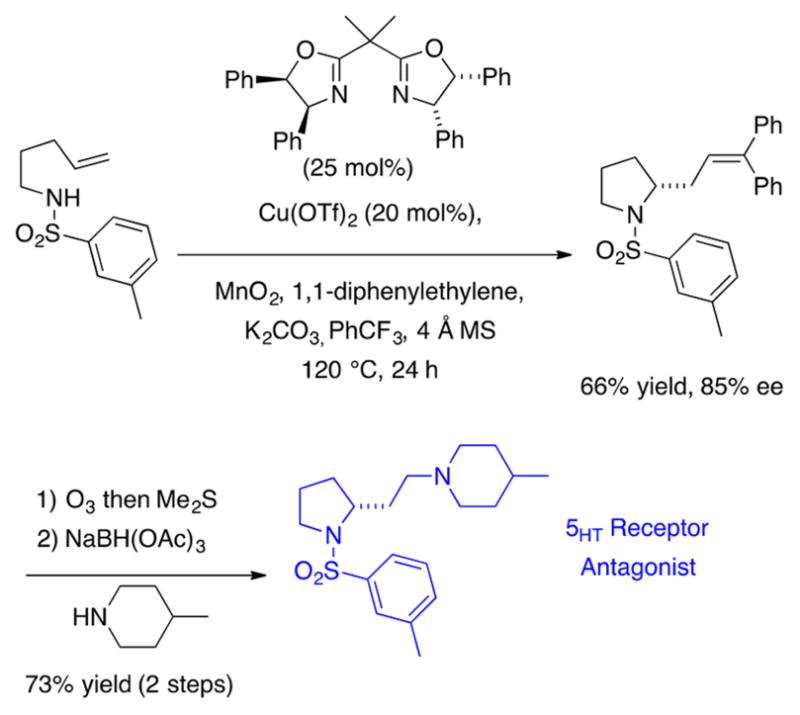
The catalytic enantioselective intermolecular carboamination was applied to the concise synthesis of an N-sulfonylpyrrolidine 5-HT7 receptor antagonist (Scheme 14).57
Scheme 14.
Alkene Diamination
The catalytic diamination of alkenes has been one of the more challenging alkene difunctionalization reactions to invent.58 This is due to a requirement to design a reaction that brings three nucleophiles together (two amines and one alkene) which involves advances in oxidation strategy and consideration for potential catalyst poisoning. Significant advances in alkene diamination technology have been developed over the last 15 years. Many of the strategies are based on palladium catalysis.59–61 Our contribution, based on copper-facilitated cyclization/oxidative C–N bond formation, stands out as being one of the more diastereoselective processes.62,63 We have developed both copper-promoted and copper-catalyzed alkene diamination protocols. Some examples of copper(II) promoted intra-/intra- and intra-/intermolecular alkene diaminations are shown in Scheme 15. A reaction mechanism for α-benzyl-4-pentenyltosamide that accounts for the observed 2,5-cis-pyrrolidine diastereoselectivity is shown in Scheme 15a. The best diamination conditions using this sulfonamide with aniline as exogenous amine involved use of 300 mol % of copper(2-ethylhexanoate)2 as both reaction promoter and oxidant.64 This reaction provided a differentially protected vicinal diamine. Under similar conditions 7-membered ring 1,4-benzodiazepinones can be formed (Scheme 15b).65 Although we have not yet developed a diamination that can employ electron-rich primary amines, a protocol employing sulfamide substrates can provide electron-rich vicinal diamines upon reduction (Scheme 15c).62,63 Less reactive substrates undergo diamination more efficiently when organic soluble copper(II) carboxylates are used in nonpolar solvents. For example, six-membered rings are formed more efficiently when copper(neodecanoate)2 (sourced as a toluene solution) is used as promoter in 1,2-dichloroethane (DCE) (Scheme 15d).63 Copper(2-ethylhexanoate)2 was subsequently identified as a superior copper(II) carboxylate as it is sold which is easier to handle.
Scheme 15.

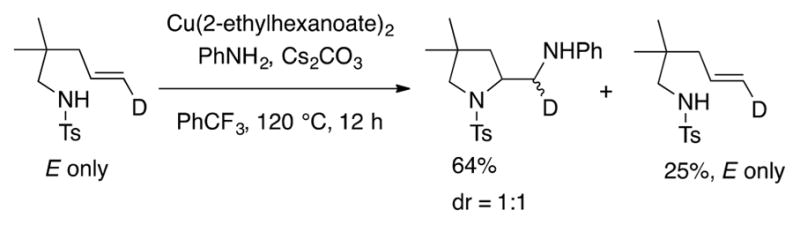
An isotopic labeling experiment indicated the second C–N bond formation occurs in stereorandom fashion, consistent with a transient carbon radical intermediate (Scheme 16). For the 4-pentenylsulfonamide the carbon radical does not appear to revert to an aminyl radical and regenerate the substrate since any substrate isolated from this partial conversion reaction still contained only E alkene geometry (Scheme 16).
Scheme 16.
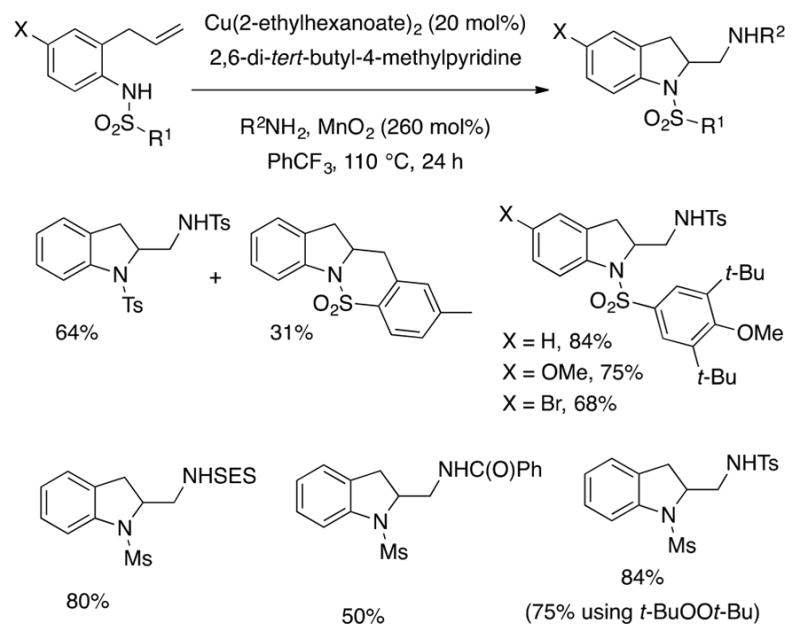
Catalytic diaminations were conducted with N-sulfonyl-2-allylanilines and unsaturated hydroxylamine derivatives using sulfonamides as exogenous amines and MnO2 as the stoichiometric oxidant (Schemes 17 and 18).66,67 For the catalytic diastereoselective or racemic process, copper(2-ethylhexanoate)2 is the most efficient copper salt, MnO2 is the most efficient oxidant and the hindered pyridine, 2,6-di-tert-butyl-4-methylpyridine, is the most efficient base (this more soluble base reduces a hydroamination side product likely formed from proton-promoted cyclization). For N-sulfonyl-2-allylanilines, use of (t-BuO)2 can be a lower yielding replacement for MnO2 and K2CO3 or Cs2CO3 can be replacements for the pyridine base in some instances (Scheme 17). Under the catalytic conditions, diamination of N-tosyl-2-allylaniline occurred with concomitant formation of 31% of the sultam side product. Substrates functionalized with the hindered 3,5-di-tert-butyl-4-methoxyarylsulfonyl group or alkyl-sulfonamides could be used instead to discourage the carboamination pathway.
Scheme 17.
Scheme 18.
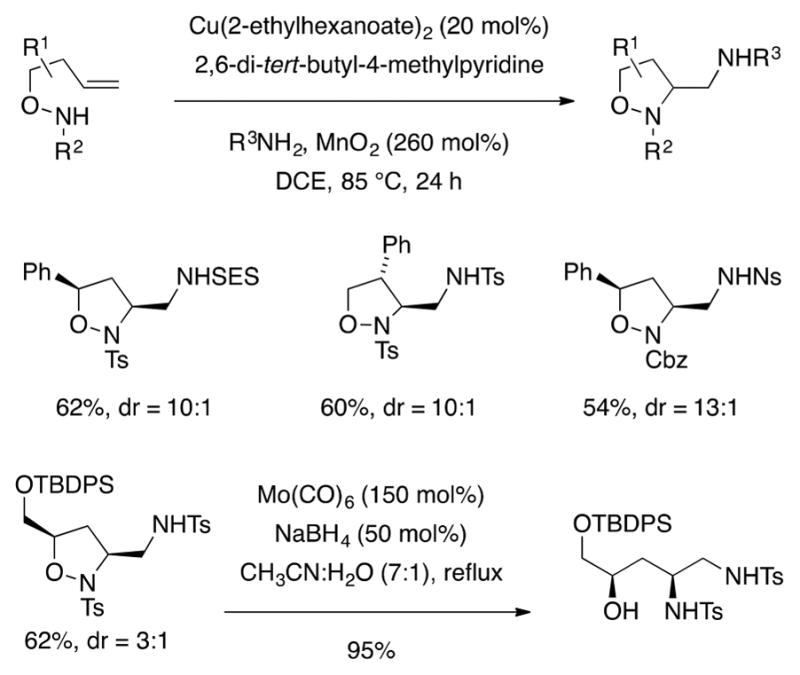
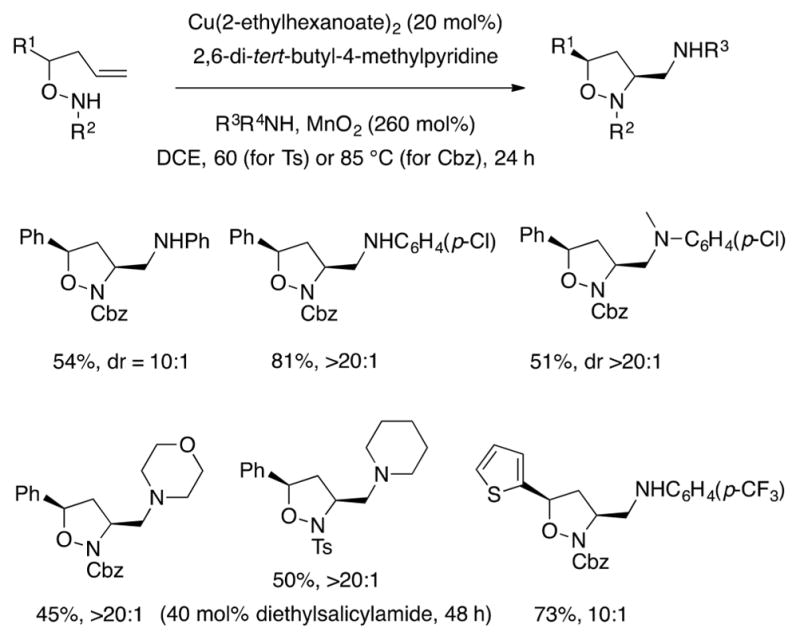
Sultam formation was not observed with hydroxylamine substrates, and Cbz-protected hydroxylamines also underwent the diamination reaction (Scheme 18). Carbamates are not commonly successful in these copper-promoted alkene difunctionalization reactions due to low reactivity and decomposition pathways at higher temperatures. The isoxazolidine N–O bond can be reduced to reveal the diamino alcohol product (Scheme 18).
Unsaturated hydroxylamine substrates react at lower temperatures (60–85 °C) than other unsaturated amine derivatives (typically 110–130 °C). Lower reaction temperature facilitated employment of a catalytic diamination with more electron-rich exogenous amines.67 Electron-rich amines are susceptible to oxidation (to give imines, for example) by copper(II) salts at elevated temperatures.68 They can also contribute to catalyst poisoning. Use of the diethylsalicylamide ligand appeared helpful to prevent catalyst poisioning when piperidine was used as the exogenous amine (Scheme 19).
Scheme 19.
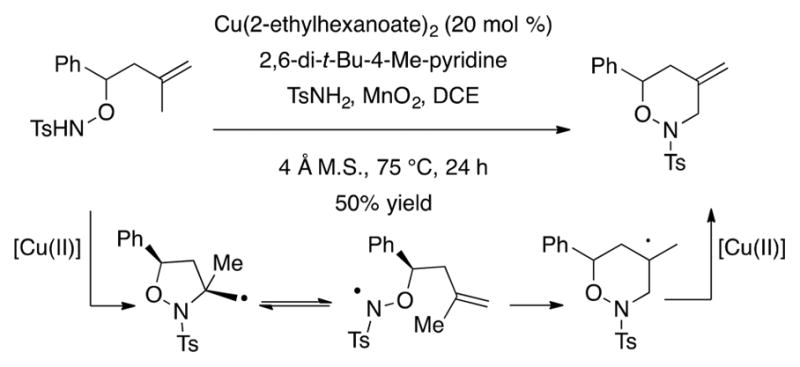
Interestingly, reaction of a 1,1-disubstituted alkenylsulfonamide resulted in oxidative amination involving endo cyclization (Scheme 20). Since the same substrate underwent amino-oxygenation uneventfully (vide supra, Scheme 6), it seems probable in the case of the diamination reaction that the second C–N bond formation requires a higher energy path for this substrate. This leads to an alternative path, conversion of the primary carbon radical intermediate to an aminyl radical intermediate that can undergo endo cyclization on the alkene (Scheme 20). Subsequent copper-assisted oxidation of the resulting radical then provides the observed terminal alkene. (Oxidation of the carbon radical by copper(II) via an associative mechanism proposed by Kochi29 is more likely to give the terminal alkene than oxidation to the carbocation by MnO2 followed by elimination). This path is facilitated by the fact that the hydroxylamine radical could have greater stability due to resonance with the oxygen atom.
Scheme 20.
Our catalytic enantioselective intra/intermolecular alkene diamination employs [Cu(R,R)-Ph-Box](OTf)2 as catalyst (Scheme 21).66 Although diamination catalysts based on Cu(OTf)2 gave poorer reactivity and are more prone to give sultam products from competing intramolecular carboamination than copper(2-ethylhexanoate)2 in the diastereoselective diaminations discussed above, when copper(2-ethylhexanoate)2 was complexed with (R,R)-Ph-Box and applied in an enantioselective attempt, the product was racemic. Replacement of MnO2 with (t-BuO)2 also significantly diminished the enantioselectivity of the reaction. Unsaturated alkyl- and arylamines with arylsulfonyl groups provide the highest level of enantioselectivity, but substantial competing intramolecular carboamination occurs. Substrates bearing the hindered 3,5-di-tert-butyl-4-methoxyarylsulfonyl group provided the best balance of diamine yield and enantiomeric excess in these reactions. In addition to stoichiometric amounts of MnO2 (260 mol %), substoichiometric KMnO4 (8 mol % for anilines, 15 mol % for 4-pentenylamines) was added to improve the diamination/carboamination ratio, presumably by increasing the rate of Cu(I) to Cu(II) oxidation as part of the catalyst turnover process, thereby increasing the rate of alkylcopper(III) formation required for the second C–N bond formation (for example, like in Scheme 15). Reactions run in 1,2-dichloroethane (DCE) provided a higher diamine to sultam ratio than reactions run in PhCF3, but the enantioselectivity for anilines was greater in PhCF3. The optimal solvent for reactions of N-sulfonyl-2-allylanilines was 1:1 DCE/PhCF3, while the optimal solvent for 4-pentenylsulfonamides was DCE. N-Tosyl-O-3-butenylhydroxylamine reacted with low enantioselectivity (11% ee).67 Under similar conditions, N-tosyl-4-pentenylureas were demonstrated to be good substrates for copper-catalyzed doubly intramolecular diamination by Zeng (an example is shown in Scheme 21).69
Scheme 21.

The arylsulfonyl group was readily removed from a diaminoindoline adduct by reduction with Mg in MeOH (Scheme 22).66
Scheme 22.

Catalytic Enantioselective Aminohalogenation
The enantiomerically enriched carbon radical intermediates formed in these reactions can also participate in atom-transfer reactions with halogen and hydrogen atom donors to enable net aminohalogenation70 and hydroamination71 reactions, respectively. For the enantioselective aminohalogenation, employment of a halogen-atom donor that would not participate as a halonium-ion donor was critical to obtaining enantioenriched products. Isopropyl iodide was the simplest and most general halogen atom donor for this purpose (Scheme 23). Substrates with the highest rates of cyclization under the aminocupration conditions, monosubstituted terminal alkene bearing N-tosyl-2-allylanilines and N-tosyl-4-pentenes, reacted with the highest levels of enantioselectivity (Scheme 23). An aniline-derived substrate with a substituent ortho to the amine reacted with poor enantioselectiivty (in the aminooxygenation reaction such substrates were unreactive),42 which could be attributed to steric hindrance at the major enantiomer transitions state. Additional evidence for a competing background amino-halogenation became apparent with more challenging substrates (e.g., formation of a tertiary amine or 6-membered ring) which were also formed with much lower levels of enantioselectivity (Scheme 23). A tandem cyclization was achieved with a 3,3-diallyl-4-pentenylsulfonamide, and a bicyclic alkyl iodide was obtained in good yield, diastereoselectivity, and enantioselectivity (Scheme 23).
Scheme 23.

Hydroamination
1,4-Cyclohexadiene is an effective hydrogen atom donor for the enantioenriched carbon radical intermediates under the copper-catalyzed net hydroamination reaction conditions as long as the reaction temperature is below 120 °C.71 At 120 °C and higher the reactions become less efficient, likely due to direct oxidation of 1,4-cyclohexadiene by MnO2. Because of this limitation, the reaction is best for the more reactive N-sulfonyl-2-allylaniline substrates. Competing carboamination (which, based on isolated yield, occurs at approximately the same rate when the N-tosyl-2-allylaniniline is used) is discouraged by employing N-3,5-di-tert-butylphenyl-sulfonyl-2-allylaniline derivatives, although it was not completely eliminated (Scheme 24, hydroamination (HA):carboamination (CA) = 94:6). Competing background hydro-amination caused by adventitious acid sources was discouraged by use of the organic soluble base 2,6-di-tert-butyl-4-methylpyridine in place of K2CO3.
Scheme 24.
C–H Amination and Allylic Amination via Aminyl Radical Reactivity
Hints of aminyl radical reactivity have been noted in some of the above reactions (e.g., Scheme 20).67 Under these copper-catalyzed reaction conditions, aminyl radicals can be generated via (reversible) homolysis of the R2N–[Cu(II)] bond, oxidation of the amine by MnO2 and/or by reversible ring opening of a 2-methyl radical indoline or isoxazoline.72 These are likely more favored to occur when the amine derivative’s oxidation potential is sufficiently low (e.g., below ca. 1.3 V). A summary of experimentally determined oxidation potentials of related amines determined by us73 and Dinnocenzo74 is shown in Scheme 25. Oxidation of the amines would form aminyl radical cations while homolysis of the R2N-[Cu(II)] bond, the proposed path for “direct” formation of aminyl radicals under our reaction conditions, would form a neutral aminyl radical, so the systems are not identical. A correlation is most absent for BuONHTs where we did not detect the oxidation potential under the conditions we used (1 mM amine derivative in CH3CN), although we did observe hints of its amidyl radical reactivity (vide supra, Scheme 20). Perhaps the cationic [BuONHTs]•+, which would be formed by electrochemical oxidation, is much higher in energy than the neutral [BuONTs]• (oxygen’s electronegativity could destabilize the positive charge). The most notable trend is that N-phenyltosamide and N,N-diphenylamine are more easily oxidized to their respective radical cations than N-butyltosamide, whose oxidation potential under these conditions could also not be detected. This trend appears to be reflected in the relative reactivity we observed in copper-catalyzed C–H amination of alkenes (vide infra, Schemes 26 and 27). Electron-donating substituents on the arene make the amine more easily oxidized while electron-withdrawing substituents on the arene make the amine less easily oxidized (Scheme 25).74
Scheme 25.

Scheme 26.

Scheme 27.

It appears that amine derivatives that form relatively stable neutral amidyl radicals such as ArNHSO2R → [ArNSO2R]• and perhaps RONHSO2R → [RONSO2R]• manifest in these reactions by either enabling reversible N–C bond formation (forward aminocupration, C–Cu homolysis then C–N bond cleavage to give the alkenyl aminyl radical, Scheme 25a), which can lead to either low observed enantioselectivity (e.g., Schemes 10 and 13, vide supra) or formation of the endo cyclization product (e.g., Scheme 20, vide supra), or by engaging in aminyl radical addition to alkenes that serve as excellent radical acceptors, resulting in net C–H amination (Scheme 26).72 These amidyl radicals appear to be electron-poor and to react faster with electron-rich styrenes. Related reactions of amidyl radicals with alkenes and alkanes have been reviewed.75,76 While sulfonamide-tethered terminal alkene substrates favor the exocyclization, primarily via cis-amino-cupration (supported additionally by the enantioselectivity, e.g., Schemes 12 and 21, vide supra), when an exceptionally good radical acceptor is added [e.g., (p-MeOC6H4)2CH═CH2] the amidyl radical reactivity can be observed (Scheme 26a). When the 2-allyl substituent is removed, N-sulfonylanilines engage efficiently and selectively in C–H amination and allylic amination reactions with 1,1-disubstituted vinyl arenes (Scheme 26b). The proposed mechanism involves formation of the amidyl radical, addition of the amidyl radical to the alkene, oxidation of the resulting benzylic radical to a carbocation by MnO2, and elimination to the new alkene. Evidence for the carbon radical intermediate is provided by a radical clock substrate (Scheme 26c). Trapping of the radical with TEMPO or 1,4-cyclohexadiene, however, was not possible.
Interestingly, in the intermolecular C–H aminations with ArNHTs, product yield did not correlate strongly with the electronic nature of the para substituent on the aniline (Scheme 26), indicating that factors other than oxidation potential are at play. These factors could involve rates of N–Cu(II) bond formation and number of N–Cu(II) bonds formed per [Cu(II)] (e.g., possible catalyst poisioning). Amines that did not work in this intermolecular reaction include alkylsulfonamides, Ph2NH and TsNH2.
Endoselective Intramolecular C–H Amination
Unsaturated amine derivatives containing 1,1-disubstituted or conjugated alkenes and an amine with a comparatively lower oxidation potential (ArNHSO2R, Ar2NH) or otherwise more stable neutral amidyl radical (e.g., RONHTs) can favor endoselective amidyl radical-mediated cyclization [Scheme 27 and 20 (vide supra)].72 1,1-Disubstituted styrenes provide higher yields than monosubstituted styrenes; this argues against activation of the alkene by [Cu] since a metal typically prefers lower steric hindrance. 1,1-Diarylalkenes in particular are excellent radical acceptors. The less reactive cyclization of terminal alkene 2-vinylaniline was best facilitated when the second amine substituent was the electron-rich p-methoxybenzene (Scheme 27). Larger rings could be formed via this methodology (Scheme 27a). An alkylsulfonamide did not undergo cyclization under these conditions. Evidence for a carbon radical intermediate was observed in the formation of a seven-membered ring where a hydroamination side product, presumed to emerge from H atom abstraction from toluene, was also obtained. When the solvent was changed to PhCF3, no hydroamination occurred (Scheme 27a). In all of these reactions, MnO2 (2.6 equiv) was used as terminal oxidant. We subsequently found that O2 (1 atm) could serve as terminal oxidant in the presence of catalytic amounts of TEMPO (Scheme 27b).77 This catalytic reaction worked for N-sulfonyl-and N-arylanilines but not for primary or N-alkylanilines. A peroxy-TEMPO adduct was isolated in this reaction (Scheme 27b). In a related report by Cheng, primary 2-vinylanilines and N-alkyl-2-vinylanilines underwent Cu(OAc)2-promoted (200 mol % [Cu]) oxidative endo cyclization under air atmosphere (Scheme 27c).78 A protocol for [Co]/[Cu] co-catalyzed aerobic oxidative cyclization of 2-vinyl anilines and phenols has also been reported.79
Unsaturated Alcohol Derivatives
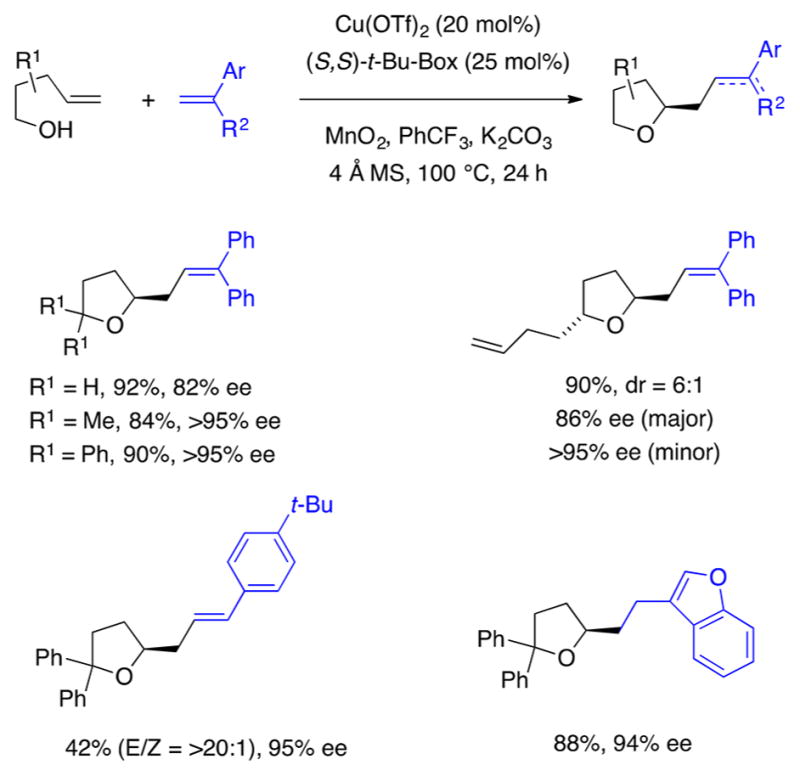
An analogous copper-catalyzed alkene carboetherification reaction was also developed. The reaction can occur via either a doubly intramolecular mode80 to create fused-ring and bridged-ring polycyclic ethers or an inter-/intramolecular mode6 to create predominantly (thus far) tetrahydrofuran derivatives (Scheme 28). While phenols are unreactive under our conditions, primary, secondary, and tertiary alkyl-substituted alcohols are all reactive in the copper-catalyzed cyclization. 2,2-Bis{[2-[4(S)-t-butyl-1,3-oxazolinyl]}propane [(S,S)-t-Bu-Box] and 2,2-bis{[2-(4(S)-isopropyl-1,3-oxazolinyl]}propane [(S,S)-i-Pr-Box] proved to be the best ligands for the enantioselective carboetherifications.6 Tertiary alcohol substrates tend to give the highest levels of enantioselectivity (ca. > 95% ee), while primary alcohols give somewhat lower levels (ca. 80% ee) (Scheme 28a). Terminal monosubstituted, 1,1-disubstituted, and 1,2-disubstituted alkenols are all good substrates for the carboetherification. 1,2-Disubstituted alkenols react with high enantioselectivity but give ca. 1:1 diastereomeric mixtures, indicative of the carbon radical intermediate involved in the reaction mechanism (Scheme 28c). Variously substituted arenes undergo C–C bond forming C–H functionalization in the doubly intramolecular manifold (Scheme 28b). The ortho-regioselectivity of the arene substitution provides evidence for a carbon radical addition mode (as opposed to aryl metalation which favors the less hindered para position). Reversible C–O bond formation appears to be possible if the C–C bond-forming step is comparatively higher in energy. This appears to be the case for the formation of an oxabicyclo[3.2.1]octane since the corresponding hydroetherification product, from reaction with 1,4-cyclohexadiene, is obtained with higher enantioselectivity than the carboetherification product (Scheme 28c).
Scheme 28.

In the intra-/intermolecular manifold, styrenes have proven to be excellent radical acceptors (Scheme 29). The alkene reactivity and mechanism of their addition is similar to the inter-/intramolecular carboamination/alkyl-Heck type reactions discussed above (vide supra, Scheme 12).
Scheme 29.
Absolute Stereochemistry
The cis-oxycupration transition state differs from the cis-aminocupration transition state in that the products are pseudoenantiomeric. The (R,R)-Ph-Box ligand favors formation of the (S)-carboamination product, while the (R)-carboetherification product if favored with the same ligand.6 The (S,S)-t-Bu-Box ligand favors formation of the (S)-carboetherification product (this ligand does not work well in the corresponding carboamination reaction). In these two reactions, the “large” group of the substrate switches. For the sulfonamide, the NSO2R group is larger than the alkene; for the alcohol, the alkene is larger than the alkoxy group. On the basis of DFT calculations, the aminocupration has a more square planar [Cu(II)] at the transition state, while the oxycupration has a more tetrahedral [Cu(II)] at the transition state. ChemDraw representations of these transition states are shown in Scheme 30.
Scheme 30.

Oxyamination and Dioxygenation
Tetrahydrofurans and morpholines can be made with good diastereoselectivity via copper (2-ethylhexanoate)2 promoted oxyamination using alkenols and exogenous amine derivatives (Scheme 31a).81 In the absence of exogenous amines, alkene dioxygenation occurs where an ester group from the copper(II) salt transfers to the carbon (Scheme 31b). Similar copper(II)-promoted alkene dioxygenation can occur with allenyl alcohols to provide vinylcarboxytetrahydrofurans with good levels of diastereoselectivity (Scheme 31c).82 We hypothesized that the second C–N/C–O bond in these reactions is formed through reductive elimination of a copper(III) species.
Scheme 31.

SUMMARY
In summary, a range of heterocycle-forming reactions can be achieved when alkenols and alkenylamine derivatives are heated in the presence of copper(II) salts. Cis-oxycupration and cis-aminocupration are implicated in the addition/cyclizaton step with unactivated alkenes, and these tight transition states provide excellent diastereoselectivity, as well as enantioselectivity, when chiral [Cu(bisoxazoline)](OTf)2 complexes are applied. The copper-catalyzed oxidative cyclizations require stoichiometric oxidants to turnover the catalyst; MnO2, O2, and TEMPO have thus far demonstrated the most generality for this function. The organocopper(II) intermediates can undergo reversible C–Cu(II) homolysis, and the carbon radical can react with additional Cu(II) to form organocopper(III) intermediates. The subsequent bond formation depends upon the reaction components: TEMPO, alkenes, and pendant arenes as well as an H atom donor (1,4-cyclohexadiene) and halogen atom donors (e.g., isopropyl iodide) favor reaction with the carbon radical (where relative rates = TEMPO > alkenes, 1,4-cyclohexadiene, i-PrI > arenes) to form C–H, C–C, and C–I/C–X bonds while the presence of exogenous amine nucleophiles as well as the use of stoichiometric copper(II) salts can favor reaction via organocopper(III) intermediates to form C–N and C–O bonds. The copper salt can also be critical for reactivity where CuCl is unique in favoring catalytic aminooxygenation with O2 as both oxidant and oxygen source. Substitution of the alkene also critically affects the reactivity: terminal alkyl-substituted alkenes favor exocyclization via aminocupration while 1,1-disubstituted vinyl arenes favor endo cyclization or intermolecular oxidative amination via amidyl radical intermediates when a relatively stable amidyl radical can be formed.
CONCLUSION
The copper(II)-promoted and -catalyzed reactions developed in this program have good substrate scope and generally high levels of chemoselectivity and stereoselectivity. A strong driving force for reaction development has been mechanistic analysis, which has supported intentional reaction invention. An exciting aspect of this work, however, also involved the fortuitous discovery of new reactions found during mechanistic probes, scope extensions and process optimizations. Immediate future directions involve additional intra/intermolecular reaction development including aerobic processes and those designed for larger ring synthesis as well as development of more fully intermolecular reaction sequences83 using the reactivity concepts described in this Perspective and those yet to be developed.
Acknowledgments
Funding of this work was provided by the National Institutes of Health (GM078383) and the American Chemical Society (Petroleum Research Foundation). We thank the Egyptian Government and Helwan University for a graduate fellowship to Z.M.K. We thank Dr. David C. Lacy and Mr. Anthony F. Cannella for assistance with obtaining oxidation potentials. S.R.C. acknowledges Catherine A. Anderson for assistance with the cover.
Biography

Sherry Chemler received a B.A. in chemistry with Distinction from Boston University in 1994. She earned a Ph.D. from Indiana University in 1999, where she worked in the laboratory of William Roush. She undertook an NIH Postdoctoral Fellowship at the Memorial Sloan-Kettering Cancer Institute with Samuel Danishefsky from 1999–2002 and began as an Assistant Professor at the State University of New York at Buffalo in July 2001. Since 2013 she has been a Professor of Chemistry.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Vitaku E, Smith DT, Njardarson JT. J Med Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 2.Wolfe JP, Hay MB. Tetrahedron. 2007;63:261–290. doi: 10.1016/j.tet.2006.08.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Torre A, Cuyamendous C, Bultel-Ponce V, Durand T, Galano JM, Oger C. Tetrahedron. 2016;72:5003–5025. [Google Scholar]
- 4.Ritchie TJ, Macdonald SJF. Drug Discovery Today. 2009;14:1011–1020. doi: 10.1016/j.drudis.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez-Rosello M, Miro J, del Pozo C. Synthesis. 2017;49:2787–2802. [Google Scholar]
- 6.Bovino MT, Liwosz TW, Kendel NE, Miller Y, Tyminska N, Zurek E, Chemler SR. Angew Chem, Int Ed. 2014;53:6383–6387. doi: 10.1002/anie.201402462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denmark SE, Kuester WE, Burk MT. Angew Chem, Int Ed. 2012;51:10938–10953. doi: 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coric I, List B. Nature. 2012;483:315–319. doi: 10.1038/nature10932. [DOI] [PubMed] [Google Scholar]
- 9.Brown AR, Uyeda C, Brotherton CA, Jacobsen EN. J Am Chem Soc. 2013;135:6747–6749. doi: 10.1021/ja402893z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minatti A, Muniz K. Chem Soc Rev. 2007;36:1142–1152. doi: 10.1039/b607474j. [DOI] [PubMed] [Google Scholar]
- 11.McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chemler SR. J Organomet Chem. 2011;696:150–158. doi: 10.1016/j.jorganchem.2010.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chemler SR. Science. 2013;341:624–626. doi: 10.1126/science.1237175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dohanosova J, Gracza T. Molecules. 2013;18:6173–6192. doi: 10.3390/molecules18066173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotov V, Scarborough CC, Stahl SS. Inorg Chem. 2007;46:1910–1923. doi: 10.1021/ic061997v. [DOI] [PubMed] [Google Scholar]
- 16.Xiong P, Xu F, Qian X-Y, Yohannes Y, Song J, Lu X, Xu H-C. Chem - Eur J. 2016;22:4379–4383. doi: 10.1002/chem.201600329. [DOI] [PubMed] [Google Scholar]
- 17.Michael FE, Cochran BM. J Am Chem Soc. 2006;128:4246–4247. doi: 10.1021/ja060126h. [DOI] [PubMed] [Google Scholar]
- 18.Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690–7691. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]
- 19.Desai LV, Sanford MS. Angew Chem, Int Ed. 2007;46:5737–5740. doi: 10.1002/anie.200701454. [DOI] [PubMed] [Google Scholar]
- 20.Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w. [DOI] [PubMed] [Google Scholar]
- 21.Shrestha B, Basnet P, Dhungana RK, KCS, Thapa S, Sears JM, Giri R. J Am Chem Soc. 2017;139:10653–10656. doi: 10.1021/jacs.7b06340. [DOI] [PubMed] [Google Scholar]
- 22.Wolfe JP. Eur J Org Chem. 2007;2007:571–582. [PMC free article] [PubMed] [Google Scholar]
- 23.Schuch D, Fries P, Donges M, Perez BM, Hartung J. J Am Chem Soc. 2009;131:12918–12920. doi: 10.1021/ja904577c. [DOI] [PubMed] [Google Scholar]
- 24.Palmer C, Morra NA, Stevens AC, Bajtos B, Machin BP, Pagenkopf BL. Org Lett. 2009;11:5614–5617. doi: 10.1021/ol9023375. [DOI] [PubMed] [Google Scholar]
- 25.Shigehisa H, Koseki N, Shimizu N, Fujisawa M, Niitsu M, Hiroya K. J Am Chem Soc. 2014;136:13534–13537. doi: 10.1021/ja507295u. [DOI] [PubMed] [Google Scholar]
- 26.Shigehisa H, Hayashi M, Ohkawa H, Suzuki T, Okayasu H, Mukai M, Yamazaki A, Kawai R, Kikuchi H, Satoh Y, Fukuyama A, Hiroya K. J Am Chem Soc. 2016;138:10597–10604. doi: 10.1021/jacs.6b05720. [DOI] [PubMed] [Google Scholar]
- 27.Lu D-F, Zhu C-L, Sears JD, Xu H. J Am Chem Soc. 2016;138:11360–11367. doi: 10.1021/jacs.6b07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crossley SWM, Obradors C, Martinez RM, Shenvi RA. Chem Rev. 2016;116:8912–9000. doi: 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kochi JK. Acc Chem Res. 1974;7:351–360. [Google Scholar]
- 30.Navon N, Golub G, Cohen H, Meyerstein D. Organometallics. 1995;14:5670–5676. [Google Scholar]
- 31.Mansano-Weiss C, Epstein DM, Cohen H, Masarwa A, Meyerstein D. Inorg Chim Acta. 2002;339:283–291. [Google Scholar]
- 32.Andrus MB, Lashley JC. Tetrahedron. 2002;58:845–866. [Google Scholar]
- 33.Johnson JS, Evans DA. Acc Chem Res. 2000;33:325–335. doi: 10.1021/ar960062n. [DOI] [PubMed] [Google Scholar]
- 34.Sherman ES, Chemler SR, Tan TB, Gerlits O. Org Lett. 2004;6:1573–1575. doi: 10.1021/ol049702+. [DOI] [PubMed] [Google Scholar]
- 35.Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paderes MC, Chemler SR. Org Lett. 2009;11:1915–1918. doi: 10.1021/ol9003492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riesinger SW, Lofstedt J, Pettersson-Fasth H, Backvall J. Eur J Org Chem. 1999;1999:3277–3280. [Google Scholar]
- 38.Pettersson-Fasth H, Riesinger SW, Backvall J. J Org Chem. 1995;60:6091–6096. [Google Scholar]
- 39.Paderes MC, Belding L, Fanovic B, Dudding T, Keister JB, Chemler SR. Chem - Eur J. 2012;18:1711–1726. doi: 10.1002/chem.201101703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paderes MC, Chemler SR. Eur J Org Chem. 2011;2011:3679–3684. doi: 10.1002/ejoc.201100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karyakarte SD, Smith TP, Chemler SR. J Org Chem. 2012;77:7755–7760. doi: 10.1021/jo3013226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paderes MC, Keister JB, Chemler SR. J Org Chem. 2013;78:506–515. doi: 10.1021/jo3023632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sequeira FC, Bovino MT, Chipre AJ, Chemler SR. Synthesis. 2012;44:1481–1484. doi: 10.1055/s-0031-1289762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belding L, Chemler SR, Dudding T. J Org Chem. 2013;78:10288–10297. doi: 10.1021/jo401665n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karyakarte SD. PhD Thesis. State University of New York; Buffalo: 2016. Synthesis of Oxygen and Nitrogen Heterocycles via Copper Catalyzed and Promoted Difunctionalization of Unactivated Alkenes. [Google Scholar]
- 47.Wdowik T, Chemler SR. J Am Chem Soc. 2017;139:9515–9518. doi: 10.1021/jacs.7b05680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng W, Chemler SR. J Org Chem. 2008;73:6045–6047. doi: 10.1021/jo801024h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miao L, Haque I, Manzoni MR, Tham WS, Chemler SR. Org Lett. 2010;12:4739–4741. doi: 10.1021/ol102233g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casavant BJ, Hosseini AS, Chemler SR. Adv Synth Catal. 2014;356:2697–2702. doi: 10.1002/adsc.201400317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liwosz TW, Chemler SR. J Am Chem Soc. 2012;134:2020–2023. doi: 10.1021/ja211272v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang W, Wang Y, Roberge JY, Ma Z, Liu Y, Lawrence RM, Rotella DP, Seethala R, Feyen JHM, Dickson JKJ. Bioorg Med Chem Lett. 2005;15:1225–1228. doi: 10.1016/j.bmcl.2004.11.071. [DOI] [PubMed] [Google Scholar]
- 54.Chemler SR. Curr Bioact Compd. 2009;5:2–19. doi: 10.2174/157340709787580928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin C-H, Haadsma-Svensson SR, Phillips G, Lahti RA, McCall RB, Piercey MF, Schreur PJKD, Von Voigtlander PF, Smith MW, Chidester CG. J Med Chem. 1993;36:1069–1083. doi: 10.1021/jm00060a015. [DOI] [PubMed] [Google Scholar]
- 56.Liwosz TW, Chemler SR. Org Lett. 2013;15:3034–3037. doi: 10.1021/ol401220b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lovell PJ, Bromidge SM, Dabbs S, Duckworth DM, Forbes IT, Jennings AJ, King FD, Middlemiss DN, Rahman SK, Saunders DV, Collin LL, Hagan JJ, Riley GJ, Thomas DR. J Med Chem. 2000;43:342–345. doi: 10.1021/jm991151j. [DOI] [PubMed] [Google Scholar]
- 58.de Jong S, Nosal DG, Wardrop DJ. Tetrahedron. 2012;68:4067–4105. doi: 10.1016/j.tet.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu Y, Cornwall RG, Du H, Zhao B, Shi Y. Acc Chem Res. 2014;47:3665–3678. doi: 10.1021/ar500344t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muniz K, Martinez C. J Org Chem. 2013;78:2168–2174. doi: 10.1021/jo302472w. [DOI] [PubMed] [Google Scholar]
- 61.Ingalls EL, Sibbald PA, Kaminsky W, Michael FE. J Am Chem Soc. 2013;135:8854–8856. doi: 10.1021/ja4043406. [DOI] [PubMed] [Google Scholar]
- 62.Zabawa TP, Kasi D, Chemler SR. J Am Chem Soc. 2005;127:11250–11251. doi: 10.1021/ja053335v. [DOI] [PubMed] [Google Scholar]
- 63.Zabawa TP, Chemler SR. Org Lett. 2007;9:2035–2038. doi: 10.1021/ol0706713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sequeira FC, Turnpenny BW, Chemler SR. Angew Chem, Int Ed. 2010;49:6365–6368. doi: 10.1002/anie.201003499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karyakarte SD, Sequeira FC, Zibreg GH, Huang G, Matthew JP, Ferreira MMM, Chemler SR. Tetrahedron Lett. 2015;56:3686–3689. doi: 10.1016/j.tetlet.2015.01.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turnpenny BW, Chemler SR. Chem Sci. 2014;5:1786–1793. doi: 10.1039/C4SC00237G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khoder ZM, Wong CE, Chemler SR. ACS Catal. 2017;7:4775–4779. doi: 10.1021/acscatal.7b01362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu B, Hartigan EM, Feula G, Huang Z, Lumb J-P, Arndtsen BA. Angew Chem, Int Ed. 2016;55:15802–15806. doi: 10.1002/anie.201609255. [DOI] [PubMed] [Google Scholar]
- 69.Fu S, Yang H, Li G, Deng Y, Jiang H, Zeng W. Org Lett. 2015;17:1018–1021. doi: 10.1021/acs.orglett.5b00131. [DOI] [PubMed] [Google Scholar]
- 70.Bovino MT, Chemler SR. Angew Chem, Int Ed. 2012;51:3923–3927. doi: 10.1002/anie.201109044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Turnpenny BW, Hyman KL, Chemler SR. Organometallics. 2012;31:7819–7822. doi: 10.1021/om300744m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liwosz TW, Chemler SR. Chem - Eur J. 2013;19:12771–12777. doi: 10.1002/chem.201301800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khoder ZM, Chemler SR. Unpublished results. 2017 [Google Scholar]
- 74.Luo P, Feinberg AM, Guirado G, Farid S, Dinnocenzo JP. J Org Chem. 2014;79:9297–9304. doi: 10.1021/jo501761c. [DOI] [PubMed] [Google Scholar]
- 75.Xiong T, Zhang Q. Chem Soc Rev. 2016;45:3069–3087. doi: 10.1039/c5cs00852b. [DOI] [PubMed] [Google Scholar]
- 76.Zard SZ. Chem Soc Rev. 2008;37:1603–1618. doi: 10.1039/b613443m. [DOI] [PubMed] [Google Scholar]
- 77.Liwosz TW, Chemler SR. Synlett. 2015;26:335–339. doi: 10.1055/s-0034-1379015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang R, Yu J-T, Sun S, Zheng Q, Cheng J. Tetrahedron Lett. 2017;58:445–448. [Google Scholar]
- 79.Ghorai J, Reddy ACS, Anbarasan P. Chem - Eur J. 2016;22:16042–16046. doi: 10.1002/chem.201604111. [DOI] [PubMed] [Google Scholar]
- 80.Miller Y, Miao L, Hosseini AS, Chemler SR. J Am Chem Soc. 2012;134:12149–12156. doi: 10.1021/ja3034075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sequeira FC, Chemler SR. Org Lett. 2012;14:4482–4485. doi: 10.1021/ol301984b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Casavant BJ, Khoder ZM, Berhane IA, Chemler SR. Org Lett. 2015;17:5958–5961. doi: 10.1021/acs.orglett.5b02833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Um C, Chemler SR. Org Lett. 2016;18:2515–2518. doi: 10.1021/acs.orglett.6b01259. [DOI] [PMC free article] [PubMed] [Google Scholar]