

Abstract
Infected or transformed cells must present peptides derived from endogenous proteins on MHC class I molecules in order to be recognized and targeted for elimination by antigen-specific cytotoxic T cells. In the first step of peptide generation, proteins are degraded by the proteasome. Here, we investigated the role of the ubiquitin-specific protease 14 (Usp14), a proteasome-associated deubiquitinase, in direct antigen presentation using a ligand-stabilized model protein expressed as a self-antigen. Chemical inhibition of Usp14 diminished direct presentation of the model antigenic peptide, and the effect was especially pronounced when presentation was restricted to the Defective Ribosomal Product (or DRiP) form of the protein. Additionally, presentation specifically from DRiP antigens was diminished by expression of a catalytically inactive form of Usp14. Usp14 inhibition did not appreciably alter protein synthesis and only partially delayed protein degradation as measured by a slight increase in the half-life of the model protein when its degradation was induced. Taken together, these data indicate that functional Usp14 enhances direct antigen presentation, preferentially of DRiP-derived peptides, suggesting that the processing of DRiPs is in some ways different from other forms of antigen.
Introduction
Activated cytotoxic CD8+ T lymphocytes are able to recognize short antigenic peptides bound to MHC class I molecules expressed at the surface of infected or transformed cells. These peptides are generated as proteins synthesized within the target cell are degraded, and the peptides are subsequently transported into the endoplasmic reticulum where they bind to MHC class I molecules. The peptide-MHC complex then traffics to the cell surface where it can be surveyed by CD8+ T cells. The process of peptide generation, transport, loading, and migration of complexes to the cell surface is known as direct antigen presentation.
Protein degradation is the critical first step in antigen processing. Generation of antigenic peptides is the result of proteasome-mediated degradation of the precursor protein, though there are notable exceptions to this rule (1). Proteins are usually targeted to the proteasome through the addition of ubiquitin moieties to the substrate. Before the protein can efficiently be degraded by the proteolytic activities present in the 20S barrel of the proteasome, the ubiquitin chains are further processed and removed by deubiquitinating enzymes (DUBs). One DUB, Rpn11, is an integral part of the 19S lid, while two other DUBs, Uch37 and Usp14, reversibly interact with the 19S lid (2, 3). Covalent modification of antigenic substrates with ubiquitin molecules often enhances presentation of peptides derived from the ubiquitin-tagged protein (4, 5), and it is therefore likely that deubiquitination of the antigenic substrate by proteasomal-associated DUBs is a necessary step for antigen presentation.
Peptides for direct antigen presentation can be derived from two potential forms of a given protein: retirees and Defective Ribosomal Products (DRiPs) (6, 7). Retired proteins are degraded based upon the observed metabolic half-life specific to the protein whereas DRiPs are rapidly degraded immediately after the protein is synthesized by the ribosome (8, 9). While both forms of a protein can give rise to peptides that are presented at the cell surface, advanced mass spectrometry experiments have demonstrated that the bulk of peptides at the cell surface are derived from DRiPs (10, 11). How DRiPs gain preferential access to the antigen presentation machinery of a cell is unknown.
Here we investigated the role of Usp14 in direct antigen presentation. Using a cell line expressing a model antigen, we demonstrate that inhibition of Usp14 with small molecules negatively impacts direct antigen presentation, and inhibition of Usp14 disproportionally impacts presentation of peptides derived from DRiPs. Expression of dominant-negative forms of Usp14 also inhibited DRiP presentation. These data support the hypothesis that DRiPs are a distinct subset of substrates necessary for antigen presentation and the activity of Usp14 is required for their presentation.
Materials and Methods
Antibodies and reagents
The anti-Kb-SIINFEKL monoclonal antibody 25D-1.16 mAb (12) was a kind gift of Drs. Jack Bennink and Jonathan Yewdell (NIAID) and was coupled to the fluorescent dye Alexa 647 using protein labeling kits from Molecular probes (Life technologies) following manufacture’s protocol. Rabbit anti-cytoskeletal actin and rabbit anti-Usp14 Abs were from Bethyl laboratories, while goat mAb anti-GFP was from Novus Biologicals. Mouse mAb FK2 for polyubiquitin was from Enzo. IRDye 680LT goat anti- mouse, IRDye 800CW goat anti-rabbit, and IRDye 680LT donkey anti-goat secondary Abs were from LI-COR. MG-132 and emetine were from Calbiochem and Brefeldin A (BFA) was from Millipore. 1-[1-(4-Fluorophenyl)-2,5-dimethyl-1H-pyrrol-3-yl]-2-(1-pyrrolidinyl)ethanone (IU1) was from Cayman chemical. Compounds N-(2-(1-(4-fluorophenyl)-2,5-dimethyl-1H-pyrrol-3-yl)-2-oxoethyl)-N-methyl-2,3-dihydrobenzo[b][1,4]dioxine-6-sulfonamide (hereafter, 1D18) and 1-(1-(3-chloro-4-fluorophenyl)-2,5-dimethyl-1H-pyrrol-3-yl)-2-(piperidin-1-yl)ethan-1-one (1B10) were from Enamine. Genetest pre-coated parallel artificial membrane permeability assays (PAMPA) plate system was obtained through Corning. BSA was from Amresco and Shield-1 was obtained through Clontech.
Plasmids
The Shield-Controlled Recombinant Antigenic Protein (SCRAP)-mCherry construct was created using overlapping PCR reactions. The FKB12-destabilization domain containing the SIINFEKL sequence was PCR amplified from the original SCRAP plasmid (13) with Primer 1 5′-TCTAGAGAGCTCCCACCATGGGAGTGCAGGTGGAAACCA-3′ and Primer 2 5′-CTTTTCGAAGTTGATGATCGATTCCGGTTTTA-3′ while the SIINFEKL-mCherry portion of the fusion protein was PCR amplified from the plasmid pSC11-mCherry-Ub-SIINFEKL (14) using Primer 3 5′-TCGATCATCAACTTCGAAAAGCTAGTGAGCAAGGGCGAGGAGGATAAC-3′ and Primer 4 5′-AGATCTCTCGAGCTACTTGTACAGCTCGTCCATGCCGCCGGTGGA-3′. Individual PCR reactions were purified using a Qiagen PCR purification kit and 1 μl of each reaction was used as a template for the final PCR reaction where Primers 1 and 4 were used to amplify the entire cassette. The final PCR product was digested with SacI and XhoI restriction enzymes and cloned into the pCAGGS expression vector (15). GFP-Usp14 wild-type (wt) and dominant negative (DN) constructs were PCR amplified from previously published peGFP-C1 plasmids (16) using the following primers: 5′-TCATCGAGAGCTCCCACCATG-3′ and 5′-TCGATGAGCTAGCCTATTACT-3′ to introduce a SacI and NheI restriction sites. This PCR product was digested with both enzymes and ligated with pCAGGS plasmid. Plasmid DNA was purified using a HiSpeed Midi kit (Qiagen) and used for stable cell line generation.
Cell culture and stable cell line generation
EL4 cells were cultured as previously described (17). Plasmids were digested with ScaI to linearize the vector, purified by ethanol precipitation, and resuspended in sterile water. Approximately 6 μg of linearized DNA was used to transfect 8 × 105 EL4 cells using the Amaxa 96-well shuttle electroporation system (Lonza). Cells were resuspended in transfection solution SF and electroporated using program DS-113. Following transfection, cells were cultured for 1 week before an initial round of fluorescent sorting at the Oregon State University Flow Cytometry Core Facility using a Beckman coulter MoFlo XDP cell sorter. Fluorescent protein positive cells were returned to culture and re-sorted within two weeks. The process was repeated until cells were more than 90% positive for fluorescent protein expression.
Chemical permeability assay
PAMPA were utilized to determine the capability of the USP14 inhibitors to passively diffuse across a cellular membrane. Following the manufacturer protocol with slight modifications, the plate was allowed to warm up to room temperature for at least 30 min. Stock solutions were diluted in PBS. A standard curve was made form 15.6 (limit of detection), 31.25, 62.5, 125, and 250 μM for each compound. Replicates of three wells were used for each compound including a PBS negative control. The donor plate contained 250 μM in 300 μL PBS per well of each compound. Then 200 μL of PBS was added to each well in the acceptor plate and placed carefully into the donor plate and incubated at room temperature for 6 h in the dark to prevent chemical deterioration. After incubation was complete the acceptor plate was removed from the donor plate and the concentration of inhibitors was determined from absorbance at 310 nm with UV/VIS spectroscopy and calculating permeability using the formula derived by the manufacturer.
Activity-based competition assay using ubiquitin-based DUB probe
EL4 cells were lysed by sonication (five cycles of 30 seconds sonication/30 seconds no sonication) in lysis buffer (50 mM Tris, 250 mM sucrose, 5 mM MgCl2, 1 mM DTT) at 4 °C, followed by spinning (16,000 g, 10 min, 4 °C). Cell lysates (1 mg/mL) were incubated with indicated concentrations of inhibitor in DMSO (60 minutes at 37 °C) followed by incubation with 0.5 μM Rhodamine-ubiquitin-propargylamide (Rho-Ub-PA) (18) for 60 minutes at 37 °C. Labeling reactions were terminated by addition of reducing sample buffer and heating (95 °C, 10 min). Proteins were resolved by SDS-PAGE, followed by in-gel fluorescence scanning with a GE healthcare life sciences Typhoon FLA 9500 imager and analyzed with ImageQuant software (ex/em λ=496/520).
Antigen presentation and flow cytometry
To study presentation of antigenic peptides, cells were chilled on ice for 10 minutes and resuspended in ice-cold citric acid buffer (0.13M citric acid and 0.056M dibasic sodium phosphate, pH=3) at 1 × 107 cells/ml for 2 minutes. Ice cold RPMI 1640 supplemented with HEPES buffer was added to neutralize the acid and cells were washed in PBS and resuspended in warm, complete RPMI 1640 media at 1 × 106 cells/ml. Cells were cultured and at indicated time points in the presence, absence, or removal of 1 μM shield-1 and processed for flow cytometry analysis. Usp14 chemical inhibitors IU1, 1D18, and 1B10 were used at indicated concentrations. MG-132, a proteasome inhibitor, BFA, an inhibitor of the secretory pathway, and emetine, a protein synthesis inhibitor were also used in some experiments at 10 μM and added to cells after acid washing had occurred to prevent antigen presentation. At indicated times, aliquots of treated cells (generally 105) were removed and stained with Alexa 647-labeled 25D-1.16 mAb and analyzed by flow cytometry. Briefly, cells were harvested and washed in HBSS (life technologies) supplemented with 0.1% BSA. Kb-SIINFEKL expression was measured by staining cells with Alexa 647 coupled 25D-1.16 mAb for 30 min at 4°C, washing cells and resuspending cells in HBSS/BSA. Cells were analyzed by flow cytometry using an Accuri C6 flow cytometer (BD biosciences) except for experiments measuring SCRAP-mCherry degradation after Shield-1 removal, in which case a BD cytoflex flow cytometer equipped with a yellow laser was used to measure mCherry fluorescence. Flow cytometry data were analyzed using the Accuri C6 software.
Calculations for Antigen Presentation
Since the SCRAP-mCherry transgene is constitutively expressed as a stable gene in the EL4 cell line, all antigen presentation experiments reported here relied on acid washing cells prior to the start of an experiment to remove existing Kb-SIINFEKL complexes. Cells were analyzed immediately after acid wash as described above by staining with the 25D-1.16 mAb as described above and the mean fluorescent intensity (MFI) levels of both mCherry and Kb-SIINFEKL at this time point were treated as background levels and subtracted from the MFI levels at later indicated time points. SIINFEKL peptides can be derived from different sources in this system, including nascent proteins sensitive to Shield-1 (referred to as non-DRiP substrates), nascent proteins insensitive to Shield-1 treatment (referred to as DRiPs), and previously synthesized protein, stabilized by Shield-1 treatment and subsequently “retired” by removing Shield-1 and inducing degradation (referred to as retirees). The relative contribution of each source of peptide can be calculated. To determine antigen presentation of non-DRiP substrates, the MFI of the population of cells treated with Shield-1 was subtracted from the MFI of the cell population treated with ethanol alone. To determine antigen presentation from DRiP substrates, the MFI of the BFA treated cells (representing the background levels of antibody staining) was subtracted from the MFI of Shield-1 treated cells. To determine peptide presentation from retirees following Shield-1 removal, the MFI of the population previously treated with ethanol (and therefore lacking a pool of stable substrate to be degraded) was subtracted from the MFI of the population previously treated with Shield-1. To determine the percent inhibition of presentation we used the following formula:
where MFI is the MFI for Kb-SIINFEKL staining for the indicated treatment (treated with Usp14 inhibitor, DMSO, or BFA treatment) and the denominator of the equation defines the range of antigen presentation. When measuring percent inhibition for DRiP substrates, the recorded MFI measurements were used. When measuring non-DRiP presentation, the MFI of cells treated with Shield-1 was subtracted from the MFI of cells treated with ethanol prior to calculating percent inhibition. For antigen presentation experiments utilizing EL4/SCRAP-mCherry cells lines stably expressing either WT or DN Usp14, the MFI of Kb-SIINFEKL staining was normalized to the MFI of mCherry signal to account for differences in SCRAP-mCherry protein in the two cell lines.
Western blots
For Western blot analysis, cells were treated and collected at the indicated times. 106 cells were collected and lysed by boiling in 100 μl 4× Bolt LDS sample buffer (Thermo-Fisher) containing 10 nM protease inhibitor N-Ethylmaleimide (NEM) and Pierce EDTA-Free Protease Inhibitor for 20 minutes with periodic vortexing. After complete lysis, 100 μl of water containing 1.0 μM DTT was added to each sample and boiled for an additional 10 minutes. Samples were resolved on 4-12% Bolt® Bis-Tris polyacrylamide gels (Thermo Fisher) followed by blotting onto nitrocellulose membranes using the iBlot system and reagents according to the manufacturers recommendations. Membranes were blocked for 1h with 5% milk solution in TBS with 0.1% Tween 20 (TBS-T). After blocking, the membranes were incubated with primary antibodies in 0.5% milk solution in TBS-T overnight. Membranes were washed with TBS-T for 10 min and incubated with secondary antibodies also in 0.5% milk TBS-T for 45 min. The membranes were washed twice with TBS-T then once with water, and analyzed using an Odyssey infrared imager (LI-COR) and LI-COR software.
Statistics
Student’s t-test analysis, standard error, one-way ANOVA, linear regression, and one-phase decay were analyzed using GraphPad Prism software. All experiments were repeated a minimum of three times and representative results are depicted. Unless otherwise noted, standard errors on each graph depicted were determined from within experiment variation.
Results
The small molecule known as IU1 is a known Usp14 inhibitor (19). Two other compounds with structural similarity to IU1 (Figure 1A), which we term 1D18 and 1B10, were tested for their ability to inhibit Usp14 in an in vitro competition assay. EL4 cell lysates were incubated with the small molecule-inhibitors and then mixed with a fluorescent ubiquitin based probe, Rho-Ub-PA, which binds to the active site of DUBs in cell lysates and can be visualized by fluorescent scanning after resolving the proteins by SDS-PAGE (16, 18). Inhibitors of DUBs compete with the probe for binding to the DUB and loss of fluorescence signal indicates that a particular inhibitor targets a specific DUB or DUBs. All three molecules were able to inhibit the activity of a DUB approximately 60 kDa in size (Figure 1B) which corresponds to the size of Usp14 as determined by western blot (Figure 1C). Note the slight difference in apparent molecular weight of the band in 1B and 1C is due to the binding of the fluorescent probe, increasing the apparent size of Usp14 in Figure 1B relative to Figure 1C. Both 1B10 and 1D18 inhibited probe binding to Usp14 at concentrations similar to previously reported Usp14 inhibitors in similar assays (16) and statistically significant decreases in probe binding to Usp14 was noted at the highest concentration tested (Figure 1D, P < 0.05). While IU1 did decrease the level of fluorescent probe interacting with Usp14, the decrease was not statistically significant. We also tested the three small-molecule inhibitors in a membrane-permeability assay (Figure 1E). Both 1B10 and 1D18 traversed an artificial membrane to a greater extant then IU1.
Figure 1. Compounds 1D18 and 1B10 are comparable to IU1.
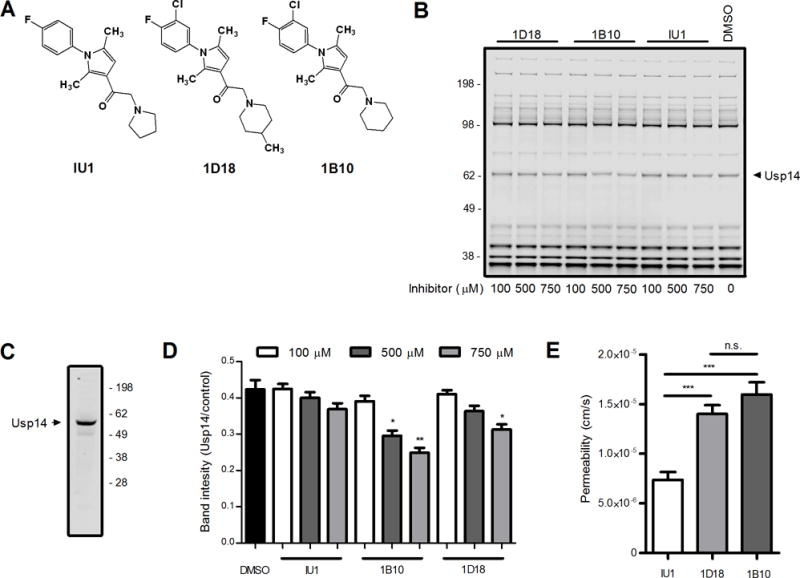
Compounds 1D18 and 1B10 were directly assessed against the selective Usp14 inhibitor IU1. A. Schematic depiction of the chemical structures of all three compounds. B. EL4 cell lysates were incubated with each compound of interest at various concentrations indicated for 60 min. Lysates were probed with Rho-Ub-PA and then resolved by SDS-PAGE to assess levels of deubiquitinating activity found throughout the cell. Usp14 is indicated in the figure based upon previous experiments (16) and Western blot of Usp14 (C). D. Analysis of DUB activity was determined by comparing Usp14 band intensity to the band intensity of a control band. E. Permeability (cm/s) was determined by passive diffusion of chemical compounds from one well to another through an artificial membrane after 6 h incubation at room temperature in the dark (* (* p < 0.01, ** p < 0.001, *** p < 0.0001, n.s. = not significant).
To test direct presentation of self-antigens from DRiP and non-DRiP sources of peptides, we generated a modified form of a model protein known as the Shield-controlled recombinant antigenic protein (SCRAP) (13) and replaced the original GFP with mCherry. The complete construct contained the FKBP12 destabilization domain (20), followed by the peptide SIINFEKL and mCherry fluorescent protein. EL4 cells were stably transfected with the SCRAP-mCherry construct and tested for both mCherry expression (Figure 2A) and Kb-SIINFEKL expression (Figure 2B). Fluorescent protein in EL4/SCRAP-mCherry cells increased following Shield-1 treatment (Figure 2A) compared to the parental cell line and did so in a dose-dependent manner (Figure 2C). Treatment with equivalent amounts of ethanol (the carrier for Shield-1) did not alter the cells in any appreciably manner. A 1.0 μM dose of Shield-1 is sufficient to saturate the cells, similar to other constructs (21). To measure antigen presentation, EL4/SCRAP-mCherry cells were washed in a mild citric acid buffer to remove existing Kb-SIINFEKL from the cell surface and cells were stained with the 25D-1.1.6 mAb at different times. Cells were treated with different compounds to inhibit antigen presentation, such as BFA, MG-132, and emetine, and compared to cells treated with Shield-1 or with ethanol alone. When SCRAP-mCherry degradation was prevented by treatment with Shield-1, there was a decrease in Kb-SIINFEKL levels compared to ethanol treated cells, though on-going antigen presentation was still observed and levels of peptide-MHC were higher than in cells treated with inhibitors which completely block antigen presentation (Figure 2D and 2E). This presentation can be observed in either kinetic (Figure 2D) or static experiments examining a single time point (Figure 2E). Because on-going antigen presentation was detected at a saturating dose of Shield-1 treatment, we conclude some portion of newly synthesized SCRAP-mCherry is inherently defective, degraded, and yields peptides for antigen presentation, and is thus likely a DRiP. This is consistent with data generated with similar constructs (13, 21, 22).
Figure 2. Shield-1 prevents the degradation of SCRAP-mCherry in EL4 cells.
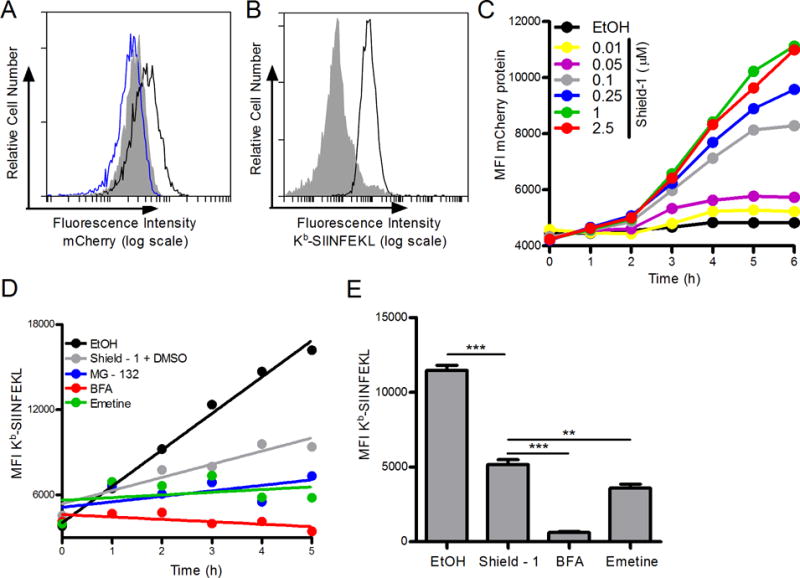
A. Fluorescent mCherry protein accumulation after 18 h treatment of EL4/SCRAP-mCherry cells with either ethanol (blue trace) or Shield-1 (black trace) compared to parental EL4 cells (shaded). B. Kb-SIINFEKL expressed in SCRAP-mCherry cells first washed in mild acidic buffer (shaded histogram) then treated with ethanol for 5 h (black trace). C. Kinetic accumulation of mCherry protein over 6 h with the addition of various concentrations of Shield-1. D. SCRAP-mCherry cells were treated with vehicle, 1.0 μM Shield-1, MG-132 (10 μM), Brefeldin A (10 μM), or emetine (10 μM) and aliquots of cell suspensions were collected every hour for 5 hours. Cells were stained as in B. A linear regression line of best fit is shown. E. Similar to D above except only one time point (5 hours) was measured in triplicate after cells were acid washed and treated with ethanol, Shield-1, Brefeldin A, or emetine. Analysis was performed by flow cytometry. (** p˂ 0.01, *** p ˂ 0.001)
To determine what impact Usp14 inhibition would have on direct antigen presentation, we incubated acid-washed EL4/SCRAP-mCherry cells with varying concentrations of each inhibitor in the presence or absence of Shield-1 and measured Kb-SIINFEKL after 5 hours of treatment. As shown in Figure 3A, treatment with Usp14 inhibitors diminished, but did not abolish, antigen presentation of peptides from DRiP substrates (ie 25D-1.16 staining in the presence of Shield-1 relative to BFA controls). Presentation of peptides from non-DRiP substrates was determined by subtracting the MFI of Shield-1 treated cells from the MFI of ethanol treated cells (Figure 3A). While compounds 1D18 and 1B10 slightly diminished presentation of peptides from non-DRiP substrates at the higher concentrations of inhibitor, inhibition of Usp14 by IU1 treatment had no impact on non-DRiP presentation. We also determined the percent of presentation inhibition for each class of substrate (Figure 3B) and found that DRiP presentation was more sensitive to Usp14 inhibition than presentation of peptides from non-DRiP substrates.
Figure 3. Chemical inhibition of Usp14 diminished DRiP Kb-SIINFEKL antigenic presentation.
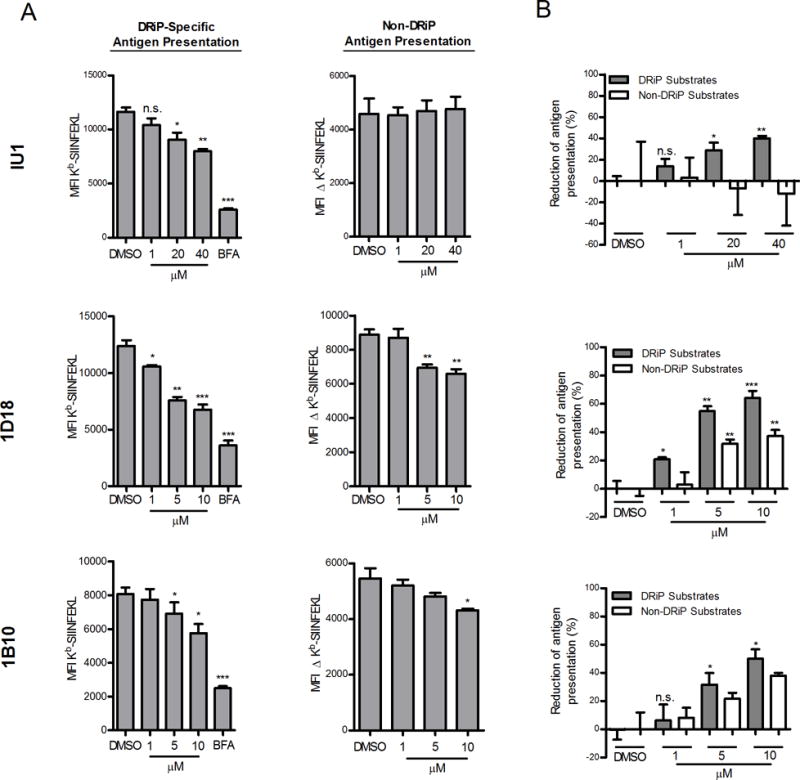
EL4/SCRAP-mCherry cells were utilized to monitor the effects of antigen presentation with Usp14 inhibition by IU1, 1D18, and 1B10. A. Cells were acid-washed to remove existing Kb-SIINFEKL complexes and cultured in the presence of Shield-1 or ethanol and various concentrations of IU1, 1D18 and 1B10. After 5 hours of culture, cells were stained in triplicate with 25D-1.16 mAb. Antigen presentation levels from non-DRiP substrates was determined by subtracting the MFI of 25D-1.16 staining of Shield-1 treated cells from the MFI of ethanol-treated cells. (B). Data from (A) is expressed as a percent inhibition of antigen presentation. The range of Kb-SIINFEKL for non-DRiP substrates was determined by subtracting the MFI of 25D-1.16 staining of Shield-1 treated cells from ethanol treated cells. For DRiP substrates (shaded bars) the range was determined by subtracting the MFI of 25D-1.16 staining of BFA treated cells from Shield-1 treated cells. (*p ˂ 0.05, **p ˂ 0.01, ***p ˂ 0.001, n.s. = not significant).
We next determined the impact of Usp14 on SCRAP-mCherry protein degradation and presentation of peptides from these retired substrates. Cells were treated overnight with Shield-1 to build up a pool of SCRAP-mCherry and then acid washed to remove existing peptide-MHC complexes and cultured in the absence of Shield-1 with or without Usp14 inhibitors. There is a short lag time following Shield-1 removal before mCherry fluorescence rapidly decreases in cells (Figure 4A). We calculated the half-life of SCRAP-mCherry following Shield-1 removal to be 41 ± 5 minutes. Treatment with the Usp14 inhibitors led to a slight increase in the half-life of SCRAP-mCherry protein, though the results were not statistically significant (ANOVA, P >0.05). Conversely, inhibition of the proteasome with MG-132 not only prevented existing SCRAP-mCherry from being degraded but also rescued newly synthesized protein from degradation (Figure 4A), resulting in an increased fluorescence signal. As previously stabilized SCRAP-mCherry is degraded following Shield-1 removal, there is a statistically significant increase in Kb-SIINFEKL complexes detected at the cell surface as depicted in Figure 4B, where the number of precursors substrates degraded, as determined by mCherry fluorescence at time 0, yields an increase in peptide-MHC complexes at 2 hours post Shield-1 removal. The difference in 25D-1.16 staining between Shield-1 treated cells and those treated with ethanol alone can be used to infer the presentation of peptides from retired substrates. Usp14 inhibition by 1D18 and 1B10 resulted in a slight decrease in retiree presentation while IU1 treatment did not diminish retiree presentation (Figure 4C). Therefore, small molecule inhibition of Usp14 did not greatly alter the ability of the proteasome to degrade destabilized SCRAP-mCherry protein but did partially inhibit presentation of peptides from retired substrates.
Figure 4. Chemical inhibition of Usp14 with 1D18 and 1B10 reduces retiree protein degradation.
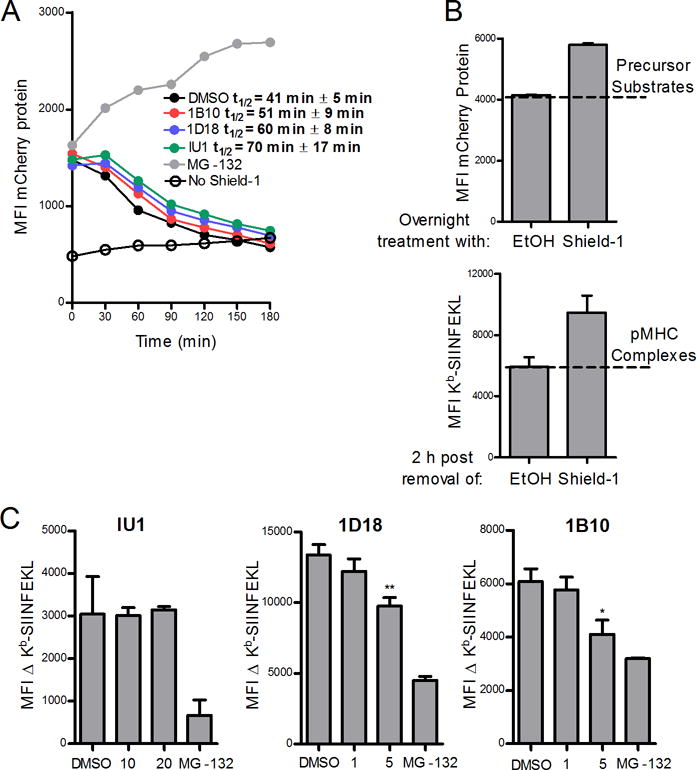
EL4/SCARP-mCherry cells were treated with Shield-1 for 18 h and then acid-washed to remove Kb-SIINFEKL complexes and cultured without Shield-1 and in the presence of MG-132 (10 μM) or the Usp14 inhibitors IU1 (20 μM), 1D18 (5 μM), or 1B10 (5 μM) for 3 hours (A) or 2 hours (B and C). A. SCRAP-mCherry protein degradation is shown at the indicated time points and the calculated half-life for the model protein in the presence of each inhibitor is listed. B. Representative graphs demonstrating (top) the amount of precursor substrates from mCherry that contribute to (bottom) the number of Kb-SIINFEKL complexes observed in the retiree antigen presentation model. The difference in MFI of 25D-1.16 staining between cells with retired SCRAP-mCherry (labeled Shield-1) and cells treated with ethanol alone is termed the ΔKb-SIINFEKL C. Usp14 inhibitors IU1, 1D18, and 1B10 at indicated concentrations were added to cells with retired SCRAP-mCherry and the ΔKb-SIINFEKL was determined 2 h post Shield-1 removal. (*p ˂ 0.05, **p ˂ 0.01)
To further examine the role of Usp14 in direct antigen presentation, we stably transfected DNA constructs encoding wild type (WT) and catalytically inactive human Usp14-GFP into EL4/SCRAP-mCherry cells. Catalytically inactive Usp14 contains a point mutation which changes the active site cysteine to an alanine residue and the gene product acts as a dominant negative (DN) form of Usp14 (16). We examined the effect of DN Usp14 in the presence of Shield-1 and compared it to transfected cells expressing the WT Usp14. We noted that cells expressed different levels of the SCRAP-mCherry protein (Figure 5A) and therefore normalized Kb-SIINFEKL expression to mCherry levels to account for differences in peptide-MHC complexes due to the abundance of the precursor substrate. Expression of DN Usp14 did not alter the degradation rate of SCRAP-mCherry as compared to control cells expressing the WT Usp14 (Figure 5A), and the half-life of SCRAP-mCherry in these cells is similar to the parental cell type (see Figure 4A). Presentation of SIINFEKL peptides from retired SCRAP-mCherry was similar between cells expressing either WT or DN Usp14 (Figure 5B). Similar to chemical inhibition of Usp14, expression of DN Usp14 resulted in a decrease in DRiP specific antigen presentation (Figure 5C). Because the levels of SCRAP-mCherry are different between cell types, it is necessary to normalize the levels of Kb-SIINFEKL to mCherry fluorescence to determine if lower peptide-MHC levels is due to loss of Usp14 function or simply due to lower substrate levels. As shown in Figure 5D, when Kb-SIINFEKL staining is normalized to mCherry signal, DRiP presentation is still reduced in cells expressing DN Usp14.
Figure 5. Catalytically inactive dominant negative Usp14 diminishes DRiP Kb-SIINFEKL presentation.
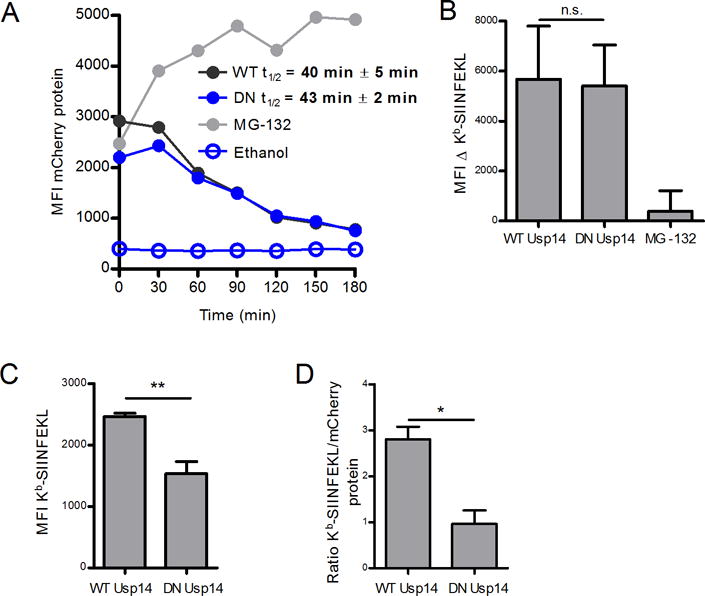
A catalytically deficient dominant negative (DN) Usp14 was utilized to measure effects of Usp14 on antigen presentation. A. Shield-1 treatment (1 μM) of EL4 SCRAP-mCherry cells expressing WT or DN Usp14 for 18 h and subsequent Shield-1 removal allowed for the determination of mCherry protein half-life. B. Kb-SIINFEKL differences between Shield-1 and ethanol treatment were measured in either Usp14 WT or DN cells 2 hours following the removal of Shield-1 and compared to MG-132 treated cells. C. Usp14 WT and Usp14 DN cells were first washed with mild acidic buffer then treated with Shield-1 or ethanol and monitored for Kb-SIINFEKL presentation after 5 h. Cells were stained in triplicate to determine Kb-SIINFEKL levels. D. To account for differences in mCherry levels between the cell types, the MFI of Kb-SIINFEKL signal was normalized to mCherry protein accumulation. (*p ˂ 0.05, **p ˂ 0.05, n.s. = not significant).
Despite its important role in trimming the poly-ubiquitin chains from substrates prior to proteasomal degradation, inhibition of Usp14 does not increase the levels of poly-ubiquitinated proteins in cells (19, 23). Our previous work has demonstrated that chemicals which specifically disrupt DRiP presentation can also increase levels of poly-ubiquitinated substrates in cells, suggesting a link between poly-ubiquitin chain disassembly and antigen presentation (13). To determine if Usp14 disruption increased poly-ubiquitinated proteins in our cells, we analyzed whole cell lysates by western blot for poly-ubiquitinated proteins. Cells were treated with IU1, 1D18, 1B10, or MG-132 for three hours and lysed by boiling SDS-PAGE buffer, which solubilizes nearly all proteins within the cell. Western blot analysis revealed that Usp14 inhibiting compounds did not increase levels of poly-ubiquitinated proteins within the cell when used at concentrations which inhibit direct antigen presentation (Figure 6A). Additionally, expression of DN Usp14 did not increase levels of poly-ubiquitinated substrates compared to cells expressing WT Usp14 (Figure 6B). These results confirm that Usp14 inhibition does not increase levels of poly-ubiquitinated proteins within cells in the same manner as proteasome inhibition.
Figure 6. Poly-ubiquitinated protein levels are not changed upon Usp14 inhibition.
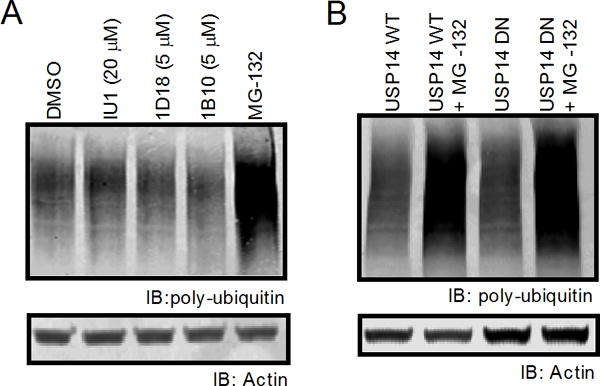
Poly-ubiquitinated protein levels of cells treated with either IU1, 1D18, 1B10, or cells containing the dominant negative phenotype for Usp14 were measured by western blot. A. Poly-ubiquitin staining with FK2 monoclonal mouse antibody after 2 h treatment with vehicle, IU1 (20 μM), 1D18 (5 μM), 1B10 (5 μM), or 10 μM MG-132. B. Cell lysates from WT Usp14 and DN Usp14 were untreated or incubated with MG-132 for 2 h then stained for poly-ubiquitin. Actin antibody was used as a loading control for all poly-ubiquitin western blots.
Finally, we tested the effect of Usp14 chemical-inhibitors on cells expressing DN Usp14. While compounds 1D18 and 1B10 inhibited DRiP antigen presentation in cells expressing WT Usp14, neither drug statistically reduced DRiP antigen presentation in cells expressing DN Usp14 (Figure 7). These data demonstrate that these agents do in fact target Usp14 in cells and that genetic inhibition of DRiP presentation cannot be further enhanced by treatment with the small molecule inhibitors.
Figure 7. Chemical inhibition of Usp14 in cells containing catalytically inactive Usp14 does not further diminish antigenic peptide presentation.
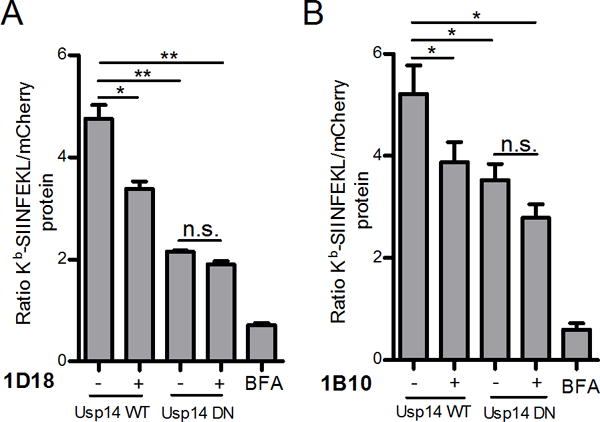
Effects of both Usp14 inhibition, with chemical compounds, and dominant negative form of Usp14 on DRiP antigen presentation was measured. WT Usp14 WT and DN Usp14 cells treated with 5 μM of 1B10 (A) or 1D18 (B) for 5 h and aliquots of cell suspension were analyzed by flow cytometry for the detection of Kb-SIINFEKL complexes and mCherry fluorescence. Kb-SIINFEKL was normalized to mCherry protein accumulation. BFA was added to a portion of cells to act as a positive control for complete reduction of antigen presentation. (p ˂ 0.05 =*, p ˂ 0.01 =**, n.s. = not significant)
Discussion
A functional proteasome is necessary for the degradation of many, though not all, proteins which contain antigenic peptides destined for presentation on MHC class I molecules. It is therefore necessary to understand how different subunits of the proteasome function in order to manipulate direct antigen presentation. In this manuscript we examine the role of a specific proteasome-associated DUB, Usp14, and find that chemical inhibition of Usp14 and expression of catalytically inactive forms of the protein negatively impact direct antigen presentation, which is particularly true for DRiP forms of the model protein when compared to retired SCRAP-mCherry. This corroborates the work of Fiebiger et al, which found that broadly inhibiting proteasomal-DUBs prevented direct antigen presentation (22), though it stands in contrast to Qian et al which found the presentation of vaccinia virus-derived DRiPs occurred independently of the DUB activity of the 19S subunit (24). While there are many differences between the experimental setups, both our data and the data of Fiebiger et al., were obtained from model antigens expressed as self-genes via transfection, whereas Qian et al measured direct presentation from viral genes, suggesting that perhaps the source of the antigenic peptide may dictate the necessity for proteasome deubiquitination. We have previously demonstrated that the efficiency of antigen presentation is different when a protein is expressed from a transfected plasmid compared to a viral expressed gene product (15), demonstrating that viral and self-antigens may be differentially recognized by the cell.
Inhibition of Usp14 is recognized to accelerate the degradation of substrates by the proteasome (19). Indeed, both chemical and genetic inhibition of Usp14 can help cells eliminate misfolded proteins, including clinically relevant proteins such as PrP, Tau, Htt, and TDP-43 which have been implicated in different neurological disorders (19, 25, 26). It would therefore seem contradictory that Usp14 inhibition would reduce presentation of peptides, especially from DRiPs which can be misfolded proteins (27) whose degradation should be accelerated when Usp14 is inhibited. Usp14 is also necessary for interacting with ubiquitin moieties and proteasome gate opening (28), allowing substrates to access the catalytic core of the proteasome. When compared to proteasome inhibition (Figure 4A), Usp14 inhibition with both chemical inhibitors and by expression of catalytically inactive Usp14, did not appreciably reduce the rate at which previously-stabilized SCRAP-mCherry was degraded upon Shield-1 removal, though a modest increase in substrate half-life was observed upon chemical inhibition. These data suggest that proteasome function was not compromised in the conditions of our experiments. However the effect of altered proteasome function may be more pronounced on peptide production than SCRAP-mCherry degradation. This may explain why direct antigen presentation was reduced when Usp14 was inhibited. Usp14 has also recently been shown to specifically recognize a substrate with multiple ubiquitin chains, suggesting that Usp14 has substrate specificity (29). We and others have suggested that DRiP antigen presentation may occur via different molecular mechanisms than presentation of peptides from retired proteins (13, 30, 31) and may in fact be compartmentalized (14). Perhaps DRiPs are marked in a particular manner to interact with Usp14 facilitating their degradation and efficient presentation.
Given that Usp14 can interact with and process ubiquitinated substrates, it is somewhat surprising that expression of dominant negative forms of ubiquitin within cells, in many instances, does not eliminate antigen presentation (32). Additionally, mutation of antigenic substrates to remove amino acids capable of being the target of ubiquitination (notably lysine, but also cysteine, seine, and threonine) does not eliminate a substrates ability to be degraded and for peptides to be presented (22). Furthermore, the effect of ubiquitin conjugation inhibitors on antigen presentation can vary based upon many factors including the source of the peptide and the particular MHC class I allele studied (33). Therefore, it may not be necessary for a substrate to be ubiquitinated to be degraded and presented. These findings complicate our interpretation of the role of Usp14 in direct antigen presentation. However, Usp14 is also known to inhibit the unfolded protein response (UPR) through its interactions with IRE1α (26, 34, 35). Inhibition or depletion of Usp14 is known to increase the UPR which can inhibit on-going protein synthesis, a process necessary for the creation of DRiPs. Indeed activating the UPR is known to decrease antigen presentation, especially of cytosolic antigens (36). It is therefore possible that the inhibition of Usp14 is due to induction of the UPR and may not involve the ubiquitin-proteasome system. In either case the mystery of substrate ubiquitination, proteasomal deubiquitination, and antigen presentation is far from solved and undoubtedly more complex than suspected.
Abbreviations
- DRiP
Defective Ribosomal Product
- DUB
Deubiquitinating enzyme
- BFA
Brefeldin A
- SCRAP
Shield-Controlled Recombinant Antigenic Protein
- PAMPA
Parallel artificial membrane permeability assays
- Usp14
Ubiquitin-specific protease 14
- DN Usp14
Dominant-Negative Usp14
- UPR
unfolded protein response
References
- 1.van Endert P. Post-proteasomal and proteasome-independent generation of MHC class I ligands. Cellular and Molecular Life Sciences. 2011;68:1553–1567. doi: 10.1007/s00018-011-0662-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee MJ, Lee BH, Hanna J, King RW, Finley D. Trimming of Ubiquitin Chains by Proteasome-associated Deubiquitinating Enzymes. Molecular & Cellular Proteomics. 2011;10 doi: 10.1074/mcp.R110.003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim HM, Yu Y, Cheng Y. Structure characterization of the 26S proteasome. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2011;1809:67–79. doi: 10.1016/j.bbagrm.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Setz C, Friedrich M, Hahn S, Dörrie J, Schaft N, Schuler G, Schubert U. Just One Position-Independent Lysine Residue Can Direct MelanA into Proteasomal Degradation following N-Terminal Fusion of Ubiquitin. PloS one. 2013;8:e55567. doi: 10.1371/journal.pone.0055567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez F, Zhang J, Whitton JL. DNA immunization: ubiquitination of a viral protein enhances cytotoxic T-lymphocyte induction and antiviral protection but abrogates antibody induction. Journal of virology. 1997;71:8497–8503. doi: 10.1128/jvi.71.11.8497-8503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antón LC, Yewdell JW. Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. Journal of leukocyte biology. 2014;95:551–562. doi: 10.1189/jlb.1113599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolan BP, Bennink JR, Yewdell JW. Translating DRiPs: progress in understanding viral and cellular sources of MHC class I peptide ligands. Cellular and molecular life sciences: CMLS. 2011;68:1481–1489. doi: 10.1007/s00018-011-0656-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, Bennink JR, Yewdell JW. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity. 2003;18:343–354. doi: 10.1016/s1074-7613(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 9.Qian SB, Reits E, Neefjes J, Deslich JM, Bennink JR, Yewdell JW. Tight linkage between translation and MHC class I peptide ligand generation implies specialized antigen processing for defective ribosomal products. J Immunol. 2006;177:227–233. doi: 10.4049/jimmunol.177.1.227. [DOI] [PubMed] [Google Scholar]
- 10.Bourdetsky D, Schmelzer CEH, Admon A. The nature and extent of contributions by defective ribosome products to the HLA peptidome. Proceedings of the National Academy of Sciences. 2014;111:E1591–E1599. doi: 10.1073/pnas.1321902111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croft NP, Smith SA, Wong YC, Tan CT, Dudek NL, Flesch IE, Lin LC, Tscharke DC, Purcell AW. Kinetics of antigen expression and epitope presentation during virus infection. PLoS pathogens. 2013;9:e1003129. doi: 10.1371/journal.ppat.1003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 13.Dolan BP, Li L, Veltri CA, Ireland CM, Bennink JR, Yewdell JW. Distinct pathways generate peptides from defective ribosomal products for CD8+ T cell immunosurveillance. J Immunol. 2011;186:2065–2072. doi: 10.4049/jimmunol.1003096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lev A, Princiotta MF, Zanker D, Takeda K, Gibbs JS, Kumagai C, Waffarn E, Dolan BP, Burgevin A, Van Endert P, Chen W, Bennink JR, Yewdell JW. Compartmentalized MHC class I antigen processing enhances immunosurveillance by circumventing the law of mass action. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6964–6969. doi: 10.1073/pnas.0910997107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolan BP, Sharma AA, Gibbs JS, Cunningham TJ, Bennink JR, Yewdell JW. MHC class I antigen processing distinguishes endogenous antigens based on their translation from cellular vs. viral mRNA. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:7025–7030. doi: 10.1073/pnas.1112387109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Jong A, Merkx R, Berlin I, Rodenko B, Wijdeven RHM, El Atmioui D, Yalçin Z, Robson CN, Neefjes JJ, Ovaa H. Ubiquitin-Based Probes Prepared by Total Synthesis To Profile the Activity of Deubiquitinating Enzymes. ChemBioChem. 2012;13:2251–2258. doi: 10.1002/cbic.201200497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmer AL, Dolan BP. MHC Class I Antigen Presentation of DRiP-Derived Peptides from a Model Antigen Is Not Dependent on the AAA ATPase p97. PloS one. 2013;8:e67796. doi: 10.1371/journal.pone.0067796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ekkebus R, van Kasteren SI, Kulathu Y, Scholten A, Berlin I, Geurink PP, de Jong A, Goerdayal S, Neefjes J, Heck AJR, Komander D, Ovaa H. On Terminal Alkynes That Can React with Active-Site Cysteine Nucleophiles in Proteases. Journal of the American Chemical Society. 2013;135:2867–2870. doi: 10.1021/ja309802n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banaszynski LA, Chen L-c, Maynard-Smith LA, Ooi AGL, Wandless TJ. A Rapid, Reversible, and Tunable Method to Regulate Protein Function in Living Cells Using Synthetic Small Molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cram ED, Simmons RS, Palmer AL, Hildebrand WH, Rockey DD, Dolan BP. Enhanced Direct Major Histocompatibility Complex Class I Self-Antigen Presentation Induced by Chlamydia Infection. Infection and Immunity. 2016;84:480–490. doi: 10.1128/IAI.01254-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiebiger BM, Moosmann A, Behrends U, Mautner J. Mature proteins derived from Epstein-Barr virus fail to feed into the MHC class I antigenic pool. European journal of immunology. 2012;42:3167–3173. doi: 10.1002/eji.201242627. [DOI] [PubMed] [Google Scholar]
- 23.Chen PC, Qin LN, Li XM, Walters BJ, Wilson JA, Mei L, Wilson SM. The Proteasome-Associated Deubiquitinating Enzyme Usp14 Is Essential for the Maintenance of Synaptic Ubiquitin Levels and the Development of Neuromuscular Junctions. The Journal of Neuroscience. 2009;29:10909–10919. doi: 10.1523/JNEUROSCI.2635-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian SB, Princiotta MF, Bennink JR, Yewdell JW. Characterization of rapidly degraded polypeptides in mammalian cells reveals a novel layer of nascent protein quality control. The Journal of biological chemistry. 2006;281:392–400. doi: 10.1074/jbc.M509126200. [DOI] [PubMed] [Google Scholar]
- 25.Homma T, Ishibashi D, Nakagaki T, Fuse T, Mori T, Satoh K, Atarashi R, Nishida N. Ubiquitin-specific protease 14 modulates degradation of cellular prion protein. Scientific Reports. 2015;5:11028. doi: 10.1038/srep11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyrskyluoto A, Bruelle C, Lundh SH, Do HT, Kivinen J, Rappou E, Reijonen S, Waltimo T, Petersén Å, Lindholm D, Korhonen L. Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: involvement of the proteasome and ER stress-activated kinase IRE1α. Human Molecular Genetics. 2014;23:5928–5939. doi: 10.1093/hmg/ddu317. [DOI] [PubMed] [Google Scholar]
- 27.Anton LC, Yewdell JW, Bennink JR. MHC class I-associated peptides produced from endogenous gene products with vastly different efficiencies. J Immunol. 1997;158:2535–2542. [PubMed] [Google Scholar]
- 28.Peth A, Besche HC, Goldberg AL. Ubiquitinated Proteins Activate the Proteasome by Binding to Usp14/Ubp6, which Causes 20S Gate Opening. Molecular cell. 2009;36:794–804. doi: 10.1016/j.molcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee BH, Lu Y, Prado MA, Shi Y, Tian G, Sun S, Elsasser S, Gygi SP, King RW, Finley D. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature. 2016;532:398–401. doi: 10.1038/nature17433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Apcher S, Daskalogianni C, Lejeune F, Manoury B, Imhoos G, Heslop L, Fåhraeus R. Major source of antigenic peptides for the MHC class I pathway is produced during the pioneer round of mRNA translation. Proceedings of the National Academy of Sciences. 2011;108:11572–11577. doi: 10.1073/pnas.1104104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Apcher S, Millot G, Daskalogianni C, Scherl A, Manoury B, Fåhraeus R. Translation of pre-spliced RNAs in the nuclear compartment generates peptides for the MHC class I pathway. Proceedings of the National Academy of Sciences. 2013;110:17951–17956. doi: 10.1073/pnas.1309956110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang L, Marvin JM, Tatsis N, Eisenlohr LC. Cutting Edge: Selective Role of Ubiquitin in MHC Class I Antigen Presentation. The Journal of Immunology. 2011;186:1904–1908. doi: 10.4049/jimmunol.1003411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J, Zanker D, Di Carluccio AR, Smelkinson MG, Takeda K, Seedhom MO, Dersh D, Gibbs JS, Yang N, Jadhav A, Chen W, Yewdell JW. Varied Role of Ubiquitylation in Generating MHC Class I Peptide Ligands. The Journal of Immunology. 2017;198:3835–3845. doi: 10.4049/jimmunol.1602122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagai A, Kadowaki H, Maruyama T, Takeda K, Nishitoh H, Ichijo H. USP14 inhibits ER-associated degradation via interaction with IRE1α. Biochemical and Biophysical Research Communications. 2009;379:995–1000. doi: 10.1016/j.bbrc.2008.12.182. [DOI] [PubMed] [Google Scholar]
- 35.Perry JW, Ahmed M, Chang KO, Donato NJ, Showalter HD, Wobus CE. Antiviral Activity of a Small Molecule Deubiquitinase Inhibitor Occurs via Induction of the Unfolded Protein Response. PLoS pathogens. 2012;8:e1002783. doi: 10.1371/journal.ppat.1002783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Granados DP, Tanguay PL, Hardy MP, Caron É, de Verteuil D, Meloche S, Perreault C. ER stress affects processing of MHC class I-associated peptides. BMC Immunology. 2009;10:1–15. doi: 10.1186/1471-2172-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]