

Abstract
Currently, we have a poor understanding of the pathogenesis of neurodevelopmental disorders, owing to the fact that post-mortem and imaging studies are only capable of measuring the postnatal status quo and offer little insight into the processes that give rise to the observed outcomes. Human induced pluripotent stem cells (hiPSCs) in principle should prove powerful in elucidating the pathways that give rise to neurodevelopmental disorders. HiPSCs are embryonic stem cell-like cells that can be derived from somatic cells. They retain the unique genetic signature of the individual from whom they were derived from, and thus allow researchers to recapitulate that individual’s idiosyncratic neural development in a dish. In the case of diseased individuals, we can reenact the disease-altered trajectory of brain development and examine how and why phenotypic and molecular abnormalities arise in these diseased brains. In this paper, we review various aspects of hiPSC biology and experimental design as well as discuss existing hiPSC models of neurodevelopmental disorders. As already shown by some studies discussed in this review, our hope is that iPSCs will illuminate the pathophysiology of developmental disorders of the CNS and lead to therapeutic avenues for the millions that today suffer from neurodevelopmental disorders.
Keywords: neural stem cells, progenitor, human, development, genome, epigenome, mosaicism, brain, cerebral cortex, transcriptome, iPSC, organoid, monolayer, glutamate, GABA, synapse
1. Introduction
Normal development of neuronal networks requires a delicate balance of proliferation and differentiation of specific neuronal lineages. Furthermore, it requires the proper migration and integration of these specific neuronal subtypes into the appropriate neuronal circuits. The development of this neuronal circuitry and of the human brain as a whole has intrigued scientists for generations, yet we have not been able to explore the development of human-specific neural networks because of the limitations of current methodologies for investigating the human brain and disorders that affect it. A substantial portion of our understanding of the processes underlying normal brain development comes from our study of neurodevelopmental disorders, in which such critical developmental pathways are disrupted. However, the emerging use of human induced pluripotent stem cells (hiPSCs) can reproduce the disease-altered trajectory of early brain development while retaining a patient’s unique genetic signature, enabling us to disentangle the contribution of genetic and non-genetic factors to the construction or normal and diseased neuronal circuitry.
Here, we discuss biological and experimental challenges associated with hiPSC derivation and review applications of hiPSC derived neuronal cells to model neurodevelopmental diseases. We begin by discussing the current state of our understanding of neurodevelopmental disorders as derived from neuroimaging, genetic, and pathological studies. We then discuss the shortcomings of such studies and the power hiPSCs possess to overcome them and advance our understanding. Following this we discuss the specifics and mechanics behind building hiPSC model systems and end with a comprehensive overview of existing hiPSC models of neurodevelopmental disorders.
2. Neurodevelopmental disorders — the potential of hiPSCs
Neurodevelopmental disorders are a broad group of disorders in which development of the CNS is altered so that the way sensory, motor and cognitive information is acquired and processed in postnatal development is disturbed. In turn, these disturbances affect how neural networks are modified by ongoing neuronal activity, resulting in a wide spectrum of emotional, cognitive, and motor deficits – including, for example, impaired language and/or non-verbal communication, impaired memory and learning, and motor dysfunction.
Neurodevelopmental disorders, and more generally CNS development, are extraordinarily difficult to study for several reasons. Rodent models of brain development have proved insightful, but are limited by their lack of complexity and sophistication compared to the human brain, especially the human cortex. Mice and rats are estimated to have 3–4 orders of magnitude fewer neurons than humans1, and, owing to non-linear scaling of neuronal density and connections with brain size, the development and structure of the human brain differs considerably from that of a rodent’s. The development of rodent brains is also much faster than that of humans. As a result, the prefrontal and temporal cortices, along with other interconnected association areas, are highly-developed in humans compared with rodents2; these areas are often associated with higher cognitive functions such as language, planning, logical thought, expression of personality and emotions and are regions that are most relevant to neurodevelopmental disorders. The lack of efficacious CNS drugs discovered via rodent models supports the idea that preclinical animal models of neurodevelopmental disorders have limited validity 3–6.
In studying human brain development, post-mortem tissue offers only a single endpoint reading that provides little insight into the altered trajectory of early brain development in neurodevelopment disorders. Indeed, over two thirds of human genes are expressed in the developing brain itself 7. Therefore, even if symptoms develop postnatally, all genetic developmental disorders are likely to involve prenatal brain structural and/or functional alterations.
Genetic studies have helped to confirm the high heritability and within family recurrence risk of some neurodevelopmental disorders, such as autism and schizophrenia8–11. Improvements in genome sequencing have enabled numerous genome-wide association studies, candidate gene discoveries and exome-sequencing studies12–21. However, these genomic studies can identify loci that contribute to disease risk, but do not assess the functional consequences of genetic variants. Transcriptome analyses are able to assess these functional consequences to some degree, but are subject to environmental influence and other confounding variables. Furthermore, brain transcriptome studies cannot distinguish between primary causes and secondary consequences of disease. Ideally, genetic studies should, therefore, involve simultaneous transcriptome and genome analyses in a disease-relevant tissue, and should not be limited by the availability of disease-relevant tissue in the form of autopsy specimens from patients with neurodevelopmental disorders 22.
The ideal experimental model for studying human early brain development is represented by embryonic stem cell (ESC), but several ethical issues prevent their use in this way.
Furthermore, genetically affected embryos are accessible to only small number of laboratories, and the derivation of affected ESC lines requires connection to a PGD performing center. The discovery of human induced pluripotent stem cells (hiPSCs) has provided an alternative model, by allowing the derivation of ESC-like cells in vitro from virtually any type of somatic cell. Additionally, hiPSCs, as opposed to ESCs, can be generated from patients with defined clinical phenotypes, thus allowing to link in vitro phenotypes to the clinical presentation in vivo. The hiPSC model system shows great promise in overcoming many of the problems with the approaches discussed above and elucidating the pathogenesis of neurodevelopmental disorders.
In contrast to postmortem human brains, hiPSC-derived model systems are actively developing and express dynamic genetic programs that regulate the process of cell proliferation, differentiation into neural precursors and subsequently into mature neurons and glial cells. These systems consequently enable the study of genetic programs that are active in the prenatal brain, as gene expression changes dramatically at the time of birth23. As noted above, postmortem brain tissue is also often distorted by other disease processes, making it hard to distinguish causes from consequences and experimental artifacts. In principle, hiPSCs can recapitulate the progression of brain development from embryonic day zero to various stages of maturity. One drawback is that hiPSC-derived brain cells are not as complex as those in the brain, and technical reasons currently limit our ability to grow these cells long enough in vitro to recapitulate the perinatal and adult brain. Nevertheless, hiPSC-derived models can allow us to examine and understand how the aberrations in brain structure, composition and connectivity we observe in postmortem and imaging studies develop, and to derive quantifiable measures of neuronal morphology, function, electrophysiology, connectivity, and gene expression from multiple timepoints during embryonic brain development (Figure 1).
Figure 1.
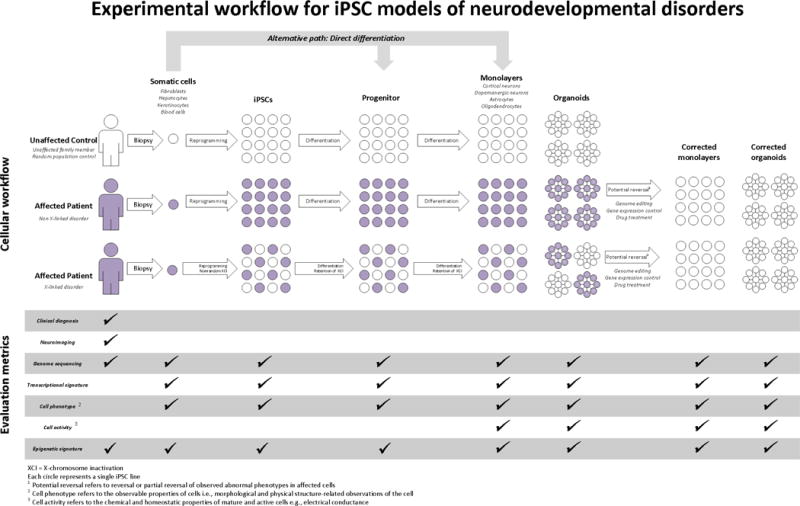
Experimental workflow for hiPSC models of neurodevelopmental disorders. Different experimental options are shown with regards to type of controls (cross-sectional, matched pair or family contrpol), choice of reprogrammed cell type, type of differentiation protocol, and outcome metrics. For patients with X-linked disorders, different colored cells represent cells with either the wild type or the mutated X allele. Corrected cells represent the same patient-derived cells after genome editing or drug treatment.
Similarly, implementation of genome-scale deep sequencing technologies with hiPSC model systems has increased the potential of these systems. These techniques can reveal the consequences of gene mutations on the entire cellular transcriptome, and, in turn, how changes in transcriptomics translate into cellular phenotypes. Genome engineering technologies should also help to determine which of the myriad developmental alterations are crucial for a given cellular and molecular phenotype. Cellular and molecular consequences of mutations can be explored in animal models and cultured human cell lines, but hiPSC-derived modeling provides information that is immediately applicable to humans because hiPSCs have a specific human genetic background and, given sufficient sample size, can reveal how inter-individual genetic variations influence phenotypes.
In summary, hiPSCs allow us to replicate the disease-altered trajectory of early brain development and examine when phenotypic and molecular abnormalities arise in these diseased brains. Furthermore, hiPSCs retain the patient’s unique genetic signature and thus can recapitulate the patient’s idiosyncratic neural development. In future, hiPSC-based studies, imaging studies, and perhaps other patient-based observational studies could be integrated in such a way that various technologies can inform each other22,24–26.
3. Generation of hiPSC models
HiPSC model generation is a two-step process. The process begins by taking a somatic cell (any cell that isn’t a sex cell) and reversing it (known as reverse differentiating and/or reprogramming) back to its embryonic stem cell-like state, known as the hiPSC state. The hiPSC then allows the experimenter to generate, through the differentiation process, the somatic cells required to model the disorder of interest (in the context of neurodevelopmental disorders this would likely be some brain region, neuronal network, or neuronal subtype).
a. Reprogramming
The reprogramming process involves the re-activation of key genes in the somatic cell, that are important in normal embryonic stem cells to maintain their characteristic pluripotent state. This is a highly specific, inefficient, and intricate processes triggered by introduction of a set of key transcription factors required to dedifferentiate a somatic cell into an hiPSC, a process which leads to a unique “open” chromatin profile (Box 1) 27–31. The efficiency of this process is influenced by many other factors including bioenergetic factors, cell cycle regulators and microRNAs 32 as well as dosage of reprogramming factors 33. Far less is understood about establishing and maintaining a pluripotency circuitry than is understood about the initial reprogramming step. What is known is that several new genes are activated and consequently several developmental transcription factor cascades turned on. We refer the reader to an excellent review by Buganim et al. for a more in-depth discussion of the subject 34. This process is far from perfect and hiPSCs may still retain some of their originator somatic cell’s gene expression patterns and chromatin modification patterns, biasing their differentiation potential towards their originator cell lineage. However, when human hiPSCs and ESCs were compared controlling for genetic background, clonality and sex, no consistent differential gene expression or epigenetic signature could be found, suggesting that hESCs and hiPSCs are molecularly and functionally equivalent 35.
Box 1. Epigenetics and hiPSCs.
The potential of hiPSCs to mitotically divide indefinitely in vitro and to differentiate into any cell type (including placental, see 182), derives from their unique epigenetic state characterized mainly by open chromatin structure, similar to that of embryonic stem cells 31,35,183,184. Open chromatin structure, or euchromatin, refers to a loose packing of the DNA around histone proteins in nucleosomes and easier access of transcription factors and other regulatory factors to the DNA template. The result is usually active transcription.
Reprogramming of a somatic cell into an hiPSC is a three-step process from an epigenetic perspective. In the first step epigenetic and other transcription factors must work together to erase the somatic cell’s current epigenetic state. Following this, they must establish a pluripotent epigenetic state in the cell. Finally, they must maintain this pluripotency as well as immortality, i.e. ability for indefinite in vitro mitotic division. Lineage-committed somatic cells often have a relatively “closed” epigenetic state with many highly condensed heterochromatin foci 31,185–187. The tight packing of the DNA around histone proteins in nucleosomes restricts access of transcription factors to the DNA template and reduces overall transcriptional activity. As mentioned earlier, the polar opposite holds for chromatin in hiPSCs, which maintain an “open” epigenetic state with more accessible chromatin domains and less heterochromatin foci, resulting in more active transcription.
b. Differentiation
Any neuronal differentiation process consists of an initial neural induction of hiPSCs, mediated by appropriate concentrations and gradients of several morphogenetic factors that are normally expressed in the developing brain. In the case of autism, schizophrenia, intellectual disabilities, and in many other disorders primarily affecting higher cognitive functions, several in vitro protocols have been developed with the ultimate goal of mimicking the development of the cerebral cortex. The mature cerebral cortex is a six-layered structure composed of two main classes of neurons: glutamatergic excitatory neurons and GABAergic inhibitory interneurons. Moreover, different subtypes of neurons are located in different cortical layers. The fate specification and differentiation of the different subtypes of cortical neurons is determined by the expression of specific combinations of transcription factors in each subpopulation, responsible for their functional diversity. The hiPSC neuronal differentiation strategy should be chosen to mimic the differentiation process with the aim of understanding its derailment in developmental disorders.
c. Monolayers versus organoids
The two main neuronal differentiation models of hiPSCs are monolayer cultures or 3D organoid cultures (Figure 1). For each of these models, several protocols have been developed and are continually being improved.
In general, monolayer neuronal cultures are derived from dividing neuronal precursor cells that can be dissociated from each other, expanded under growth factor stimulation, and frozen, providing a useful source for later analyses. The advantages of these types of cultures are that a large number of neuronal progenitor cells can be obtained and differentiated into mature neurons, and analyses of cellular morphology (such as neuronal branching and spine quantification) are easier to perform, owing to the 2D nature of these in vitro systems. One of the most commonly used monolayer protocols in the ES and hiPSC fields is the dual SMAD inhibition method 36; the original version of this resulted in conversion of ES and hiPSCs to neuronal cells with an efficiency >80%, and an adaptation of this protocol has successfully been used to generate dopaminergic neurons to study Parkinson disease 37,38 and to generate cortical interneurons 39. An alternative protocol, that combined dual SMAD inhibition with retinoid signaling led to a 95% efficiency of neuronal differentiation 40 and has been used to model Alzheimer disease and schizophrenia 41–43. In addition, many more variations of 2D protocols exist, which have been developed and successfully used to generate specific neuronal subtypes, such as GABAergic inhibitory neurons 44, glutamatergic neurons 44, and hippocampal granule neurons 45 (see table 1 for more information on different monolayer protocols).
Table 1.
hiPSC models of Rett Disorder
| Mutated Gene | Differentiated cell type | Phenotype (compared to controls) | Phenotypic reversal or rescue with drug? | Isogenic control? | Organoid or monolayer? | Reference |
|---|---|---|---|---|---|---|
| MECP2 | Astrocytes | Mutant RTT astrocytes and their conditioned media have detrimental effects on the morphology and function of WT neurons; Glial effect on neuronal morphology is independent of the intrinsic deficits in mutant neurons. | IGF-1 is able to partially rescue the neuronal deficits caused by mutant RTT astrocytes | Yes | Monolayer | 91 |
| MECP2 | Neurons | Smaller neuron nuclear size | NA | Yes | Monolayer | 80 |
| MECP2 | Neurons | RTT hiPSCs displayed defects in neuronal maturation | NA | No | Monolayer | 81 |
| CDKL5 | Neurons | NA | NA | No | Monolayer | 87 |
| MECP2 | Neurons | Reduced glutamatergic synapse number and neurite spine number, smaller soma size, altered calcium signaling and decreased frequency of spontaneous postsynaptic currents | Overexpression of MECP2, addition of IGF1 or addition of gentamicin partially reversed RTT neuron phenotype and increased the glutamatergic synapse number | No | Monolayer | 74 |
| MECP2 | Neurons | Reduced soma size | NA | Yes | Monolayer | 82 |
| MECP2 | Neuronal progenitor cells | Increased frequency of L1 retrotransposition | NA | No | Monolayer | 83 |
| CDKL5 | Neurons | Significantly reduced number of synaptic contacts; Aberrant dendritic spine structures | NA | No | Monolayer | 89 |
| MECP2 | Neurons | Significant deficit in KCC2 (potassium chloride cotransporter) expression and consequently a delayed GABA functional switch from excitation to inhibition (GABA prenatally is excitatory and switches to being inhibitory postnatally) | Overexpression of MECP2 or IGF1 treatment rescued GABA functional deficits | No | Monolayer | 84 |
| FOXG1 | Neurons | Increased inhibitory synaptic markers and GluD1 (preferentially induces GABAergic differentiation); reduced excitatory synaptic markers | NA | No | Monolayer | 88 |
| MECP2 and CDKL5 | Neurons | Both MECP2 and CDKL5 mutated neurons had Increased GRID1 (encodes for GluD1). GluD1controls synaptic differentiation and shifts the balance toward inhibitory synapses (over excitatory synapses). Mutant hiPSCs had reduced GRID1. | NA | No | Monolayer | 85 |
| MECP2 | Astrocytes | Microtubule -dependent vesicle transport is altered | Epothilone D, a brain-penetrant microtubule stabilizing natural product, restored proper microtubule dynamics | No | Monolayer | 90 |
| MECP2 | Neurons | Deficits in neuronal maturation – decreased soma size, dendritic complexity, and cell capacitance; Dysfunction in action potential generation, voltage-gated sodium currents, and miniature excitatory synaptic currents’ frequency and amplitude |
Overexpression of appropriate MECP2 isoform resulted in reversion of reduction in soma size | No | Monolayer | 86 |
| MECP2 | hiPSC | This was the first hiPSC model of Rett Syndrome with known mutation in MECP2 | NA | NA | NA | 71 |
The main disadvantage of monolayer methods is the lack of spatial organization that is found in the brain. Moreover, monolayer approaches confer highly unphysiological mechanical and adhesive properties to differentiating neurons, which alter neuronal morphology and gene expression. As a result, the quantity and types of neurons produced are highly dependent upon subtle variations in cell-to cell contacts and interactions, which are in turn influenced by the initial plating density of the neuronal progenitors, the adhesive substrate, and the exogenous factors added to the preparation.
Organoid culture protocols have been developed in the past few years to overcome the drawbacks of monolayer cultures and better mimic the 3D structure, organization, composition, and connectivity of the human brain. The main difference with the monolayer culture is that cells are in direct contact with each other for much of their membrane surface. Thus, organoid cultures allow the recapitulation of a greater range and complexity of cellular interactions and morphogen gradients under minimally perturbed conditions and the preservation of cell-to-cell contacts allows the self-organization of progenitors and neurons in layers that resemble the early stages of mammalian forebrain development.
The advent of 3D cultures started with the pioneering work of Y. Sasai and colleagues in 2008, in which they derived 3D-organized aggregates of neuronal cells from mouse and human ES cells 46. In this study the authors also showed that the regional identity of specific cortical neurons could be specified and manipulated by the addition of patterning factors into the medium. Similar organoid protocols were refined and optimized for the 3D differentiation of human iPS cells 47,48. More recently Lancaster and colleagues developed an organoid method designed to mimic the organization of the entire brain and used this method to model intrinsic neuronal differentiation defects in individuals with microcephaly 49. Organoids obtained with this method are heterogeneous in the sense that they contain several different brain regions (such as cerebral cortex, brainstem, retina and choroid plexus) within each organoid. We have used 3D organoid cultures to study of ASD50, and used a method that differed in the initial enrichment of neural progenitor cells through the manual selection of neural rosettes, which allows a more homogeneous differentiation of each organoid in dorsal and ventral neurons mainly representing the telencephalon.
4. Experimental design in disease modeling with hiPSCs
Here we will only mention some experimental design either accessible only via hiPSCs based models, or where hiPSCs based models offer a distinct advantage. Matched pair design is a widely used and powerful experimental design in which cases are matched to controls based on the value of one or more factors, allowing to control for confounding variables, e.g. sex, age and genetic background, in the design stage as opposed to the analysis stage. It has been successfully applied to post-mortem tissue samples 51–53, as well as to hiPSC studies 35,54. Matching is more efficient than controlling for confounding in the analysis when the sample size is small, if cost of matching is lower than cost of increasing the sample size. This is certainly true in the case of hiPSCs. On the downside, it is not always possible to find the required match, and this is especially true in the case of post-mortem tissue samples, due to the limited availability of covariates-matched tissue samples. The family design can be considered a subclass of the matched pair design, where the family (genetic) background is the controlled factor. It has been extensively used in genomic studies to investigate genetic transmission within families; for example, to discover de novo mutations. It is a powerful approach that was successfully implemented in a recent hiPSCs based autism study 50. This design is not really accessible using post-mortem tissue. The advantage of a family design in the context of hiPSC studies is that it is better suited to a low number of samples in that it allows better control of genetic background effects, which have been shown to drive a large portion of variability across hiPSC lines. A potential drawback may be the intrinsic age difference between probands and controls, when children used as probands are opposed to their parents. Various groups have shown that the “age signature” of the cell donor appears to be reset by the reprogramming process 55,56, while this reversal does not appear to occur when the somatic cell is directly converted into neurons 57. In particular, hiPSCs derived from older donors do not seem to present significant transcriptional differences with respect to younger donors. Thus, age difference between probands and controls is less of concern when hiPSC-derived samples are compared. Another potential problem of the family design is that it cannot be used to study the influence on the phenotype of genetic risk factors that are shared within each family. To mitigate this, the design should incorporate “diseased” and “non-diseased” families, as commonly done in genomic studies. Indeed, a distinct advantage of a family design is that since it is commonly used in genomics, it may simplify correlations between the genetic variation and aspects of the phenotype that are investigated via hiPSC modeling.
Finally, hiPSC studies are increasingly using the isogenic design, usually implemented to analyze the functional effect of some (epi-)genetic perturbation on the same cell line (discussed earlier on in this review). This is an extremely powerful approach as it allows, for instance, the functional characterization of a genetic mutation by correcting it in the cells from the same patient carrying the mutation (e.g. genome engineering by CRISPR technology).
5. Induced-pluripotent stem cell models of neurodevelopmental disease
Many studies have used hiPSCs for modeling neurodevelopmental disorders (Tables 1–5). In many cases, the models have confirmed or expanded upon our existing understanding of the pathology gained from neuroimaging, postmortem and rodent model studies. However, hiPSC models have allowed us to understand, for the first time, the cellular and molecular mechanisms that give rise to these disorders and drive its progression throughout human development.
Table 5.
hiPSC models of Schizophrenia
| Mutated Gene | Differentiat ed cell type | Phenotype (compared to controls) | Phenotypic reversal or rescue with drug? | Isogenic control? | Organoid or monolayer? | Reference |
|---|---|---|---|---|---|---|
| 22q11.2 Del | Neurons | Differential expression of several microRNAs | NA | No | Monolayer | 129 |
| 15q11.2 microdeletion | Neural progenitors | Defects in adherens junctions and apical polarity | NA | No | Monolayer | 43 |
| Idiopathic | Neurons | Increased catecholamines and elevated secretion of dopamine, norepinephrine and epinephrine; Tyrosine hydroylase, a catalyst for catecholamine biosynthesis, positive neurons was also increased. | NA | No | Monolayer | 133 |
| One had a 22q11.2del the other two idiopathic | Neurons | Delay in reduction of pluripotency marks during neuronal differentiation | NA | No | Monolayer | 194 |
| Idiopathic | Neurons | Decreased neuronal connectivity, neurite number, PSD-95 protein levels, and glutamate receptor expression; Altered expression of cyclic AMP and WNT signaling pathway genes |
Partial reversal following treatment with loxapine | No | Monolayer | 127 |
| Idiopathic | Neuronal progenitor cells | Abnormal expression of genes and proteins related to cytoskeletal remodeling and oxidative stress; Aberrant migration and increased oxidative stress |
NA | No | Monolayer | 128 |
| Idiopathic | Dopaminer gic and glutamater gic neurons | Affected dopaminergic cells showed severely impaired ability to differentiate; Affected glutamatergic cells were unable to mature; Affected cells showed perturbation in mitochondrial structure and function; |
NA | No | Monolayer | 131 |
| DISC1 | Cortical neurons | Synaptic vesicle release deficits and dysregulation of synapse-related genes | NA | No | Monolayer | 130 |
| 22q11 deletions | Neurons | Increased L1 retrotransposition | NA | No | Monolayer | 132 |
| Idiopathic | Hippocamp al dentate gyrus (DG) granule neurons | Reduced neuronal activity, spontaneous neurotransmitter release, hippocampal dentate gyrus granule neurons generation | NA | No | Monolayer | 45 |
| Idiopathic | NPCs | Perturbed responses (i.e., high variance in levels of heat shock factor 1 activation) to environmental stressors in SZ neurons | NA | No | Monolayer | 195 |
The engineering of specific disease-relevant penetrant mutations in hiPSCs is particularly powerful in studying disorders in which such a mutation exists (e.g., FMR1 in Fragile X syndrome or TSC1/2 in tuberous sclerosis). However, hiPSCs can be also useful for studying complex genetic disorders, such as schizophrenia and autism. In these disorders, given sufficient sample size, the derivation of hiPSCs from populations of diseased individuals with appropriate controls can be equally powerful.
Below, we will discuss existing hiPSC-based models of neurodevelopmental disorders, both those that involve a single disease-penetrant mutation and complex disorders in which hundreds of genes have been implicated. We define neurodevelopmental disorders as including any disorders that alter the course of normal human brain development and for which etiological factor(s) are active in prenatal or early postnatal life. Such disorders include Rett Syndrome, Timothy Syndrome, Fragile X, Autism, Tourette syndrome, and Schizophrenia. While there is some evidence that Bipolar disorder has developmental origins [see review 58 and a few prominent hiPSC studies 59–61 have been carried out] overall the evidence is not conclusively pointing to a definite developmental etiology. It is important to note that because of the high resource and time requirements of hiPSC studies, some of the published works on the subject are only able to conduct n=1 i.e., one control vs. one affected hiPSC without any genetic risk variant assessment. It is unlikely that these papers can add any conclusive evidence on disease pathology and so we purposely tried to exclude them from our analysis unless there is something especially novel about the experiment and/or its design.
a. Penetrant mutations
The most extensive work in hiPSC modeling of neurodevelopmental disorders has been done in the context of the monogenic disorder, Rett Syndrome (RTT). However, Fragile X syndrome (FXS) and Timothy syndrome (TS) are other monogenic neurodevelopmental disorders that have been studied with hiPSC models; the symptoms of both of these disorders include symptoms of autism spectrum disorder (ASD).
Rett syndrome
Rett Syndrome (RTT) is part of the family of Autism Spectrum Disorders. Classical RTT is a neurodevelopmental disorder clinically characterized by the initial appearance of normal early growth and development followed by a period of regression followed again by recovery or stabilization. Diagnosis is usually considered when deceleration of head growth is observed postnatally, though this may not be true in all cases. RTT patients often have partial or complete loss of acquired purposeful hand skills and/or acquired spoken language. It is also often accompanied by the advent of gait abnormalities and stereotypic hand movements 62. RTT almost exclusively affects females and is most often caused by X-linked mutations in the MECP2 gene, a transcription factor crucial to normal brain development. Rett syndrome is also associated with mutations in CDKL5 or FOXG1 though to a far lesser extent.
Pathological studies of RTT brains have found evidence of decreased neuronal and brain size, defects in synaptogenesis, and reduced dendritic arborizations and spines. Some studies have also identified altered (mostly reduced) neurotransmitter levels 63–65. All of these are indicators of less mature and less complex neurons. Defects in synaptogenesis may lead to alterations in neuronal processes 66 such as learning, memory, and information retrieval. Reduced dendritic arborizations can be considered as sign of a less complex neuron. Similarly, spine density on excitatory glutamatergic neurons is a strong indicator of these neuron’s maturity and ability to potentiate synaptic connectivity, as dendrites with reduced number of spines can receive less inputs. Furthermore, spine formation, plasticity, and maintenance depend on contact mediated and long range signals that are in part genetically encoded and in part regulated by synaptic activity and can be modulated by one’s sensory experiences 67–69. Since the synaptic activity and sensory experiences are likely abnormal for RTT patients, this implies at some level a vicious cycle if RTT patients do in fact have reduced spines as pathological studies so far suggest. Gene expression profiling of post mortem RTT brains also hinted at specific deficits in expression of presynaptic markers 70.
While the first derivation of Rett syndrome hiPSC lines with known mutations in MECP2 was performed by Hotta et al. in 2009 71, to our knowledge, the first of these models accompanied by extensive phenotypic characterization and later corroborated by other groups was created by Marchetto et al. who in 2010 derived hiPSCs from the fibroblasts of RTT patients with MECP2 mutations. They found that RTT patient-derived neurons had fewer synapses, reduced spine density, and smaller cell body compared to wild type (WT) controls. Furthermore cultures had a reduced density of the glutamate transporter VGLUT1 on glutamatergic neurons. Knockdown of MECP2, that is the reduction of the expression of the MECP2 gene so as to mimic RTT patient-derived neurons, in WT hiPSCs resulted in a similar glutamate transporter phenotype. Overexpression of MECP2 in WT and RTT patient-derived neurons resulted in increased density of VGLUT1, suggesting a dose-dependent relationship between the MECP2 gene and glutamate transport. Interestingly, overexpression of MECP2 is also pathogenic, and recently, a primate model of MECP2 overexpression showing autism-like social deficit has been generated72. A human hiPSC model for MECP duplication syndrome (MECP2dup), established by Nageshappa et al. showed that cortical neurons derived from affected hiPSC lines have increased synaptogenesis and dendritic complexity as well as altered neuronal network synchronization 73.
Similar to the RTT patient-derived neurons and RTT pathology previously described, WT neurons with MECP2 knockdown showed decreased spine density and soma size. RTT patient-derived neurons also exhibited functional alterations at the neural network level – a decrease in intracellular calcium oscillations and a decrease in frequency and amplitude of spontaneous post-synaptic excitatory currents compared to WT neurons 74,75. Calcium has wide-ranging effects in neurons ranging from microsecond level control of neurotransmitter secretion on the pre-synaptic side of the neurons to gene regulation in the neuron’s nucleus (which can last for hours or days). Calcium is also required for activity-dependent synaptic plasticity, a process crucial to learning and memory 76–79. The decrease of frequency and amplitude of spontaneous post-synaptic excitatory currents is consistent with the decrease in intracellular calcium transients and also indicates a decreased likelihood of action potential firing in the RTT patient-derived neurons. Note how these observations in RTT patient hiPSC-derived neurons would have been impossible to make in patients. From a translational perspective the Marchetto et al. study is also particularly promising as the group also showed rescue of certain neuron cell defects such as reduced VGLUT1 density with the administration of IGF-1 and/or gentamicin 74,75.
Several other studies 80–86 have also studied the morphology of hiPSC-derived neuronal progenitors and/or neurons from RTT patients with MECP2 mutations. Many of these of have largely corroborated the results of Marchetto et al., showing small cell body size, deficits in neuronal maturation, and decreased dendritic complexity. Djuric et al 86, went on to also show that RTT neurons had decreased cell capacitance, dysfunction in action potential generation, dysfunction in voltage-gated sodium currents, and dysfunction in miniature excitatory synaptic current frequency and amplitude. Two studies demonstrated particularly well the power of hiPSCs by showing that RTT neurons had a delayed functional switch in GABA from excitatory to inhibitory 84 and neuronal progenitors from RTT patients had an increased frequency of L1 retrotranspositions 83, conclusions almost impossible to derive from any other experimental medium. Tang et al. went on further to show that IGF1 treatment rescued GABA functional deficits 84. In addition, Rett gain of function by gene duplication (MECP2dup) has also been modeled in hiPSC lines, showing a phenotype in many respect opposite to that of Rett loss of function, namely increased synaptogenesis and dendritic complexity along with altered neuronal network synchrony 73.
HiPSC models from RTT patients with rarer mutations have also been created. Groups have derived hiPSCs and neurons from RTT patients with CDKL5 and FOXG1 mutations showing reduced number of synaptic contacts, aberrant spine structures, increased tendency to differentiate into inhibitory synapses, and reduced excitatory synaptic markers 85,87–89. Other studies modeling Rett Syndrome 90,91 have differentiated hiPSCs into astrocytes, supporting cells that form and maintain the environment and microstructure for neurons, and suggested that mutant RTT astrocytes have adverse effects on the morphology and function of WT neurons, which were partially rescuable with IGF1 treatment. Globally, the evidence suggests that Rett is a non-cell autonomous disorder (disorder in which genotypically mutant cells causes other non-genotypically mutant cells to exhibit a mutant phonetype) and that the glial effect on neuronal morphology is separate from the intrinsic neuronal deficit in RTT mutant neurons. Delepine et al. found that microtube-dependent vesicle transport in mutant astrocytes was altered and they were able to reverse this with Epothilone D 90.
Though Rett syndrome is a monogenic disorder and should theoretically be easier to model, being an X-linked disorder introduces unique challenges due to the phenomenon of somatic X chromosome inactivation (Box 2) Nevertheless, this same phenomenon creates the unique opportunity of an isogenic control (with the same genetic background) since hiPSC lines derived from one RTT patient could either express the mutant or WT allele. Cheung et al. exploited this opportunity and derived isogenic control hiPSC lines (from RTT patient fibroblasts) expressing only the WT or the mutant MECP2 allele 92. This pattern of X-chromosome inactivation was maintained through neuronal differentiation, and similar to Marchetto et al 74, Djuric et al 86, and Ananiev et al. 80 (who also had an isogenic control), these studies found that RTT patient derived neurons with the mutant MECP2 allele had a reduced soma size compared to the isogenic controls patient derived neurons expressing WT MECP2.
Box 2. X chromosome inactivation.
Females are the homogametic sex – that is, they have two of the same chromosome (XX). Males on the other hand are the heterogametic sex and are XY. In order for females not to receive double the dosage of genes on the X chromosome, reduction of gene expression via transcriptional silencing on one of the X chromosomes is required. This sex chromosome dosage compensation effect is commonly known as X chromosome inactivation (XCI) and is thought to be required for normal embryonic development. The process is mediated largely by the large noncoding RNA Xist, which coats the X chromosome in cis, mediating transcriptional silencing of X-linked genes and inducing repressive chromatin character along the entire chromosome 188. However, which X chromosome gets coated and consequently inactivated is random in the sense that the maternal X chromosome and paternal X chromosome each have equal 50% likelihood of being silenced 189. Interestingly, it was recently shown that X chromosome inactivation in female hiPSCs actually exhibits a non-random pattern 190. X-chromosome inactivation, which is retained in hiPSCs, presents a powerful addition to the potential of hiPSCs to model X-linked neurodevelopmental disorder such as Rett syndrome (Figure 1). For example, one can then derive hiPSCs from a heterozygous female patient with a mutation in an X-linked gene such that different hiPSC lines either express either the wild-type or the mutant form of that gene. In this way, both the mutant and wild-type form have the same genetic background, making the wild-type form an isogenic control within the experiment. To date the only neurodevelopmental disorder this has been done with is Lesch-Nyan 191 and Rett syndrome as discussed in the text. However, such an experimental design may not be as powerful as it initially seems. While matched genetic backgrounds give more statistical power, family studies are more informative, as they allow comparisons of functional effects of mutated alleles in different genetic backgrounds. Additionally, caution should be exerted though since female hiPSC undergo progressive “erosion” of the pattern of X chromosome inactivation over passage, with associated transcriptional derepression of genes on the inactive X 191.
Fragile X Syndrome
FXS is one of the most common causes of syndromic autism, and often also causes moderate to severe intellectual disability, speech delays, growth and motor abnormalities, hyperactivity, and anxiety 93,94. FXS is thought to be the result of decreased expression of the FMR1 gene, which encodes FMRP, an RNA-binding protein that inhibits mRNA translation. Regulation of mRNA translation is thought to be important for synaptic plasticity and neuronal maturation, thus making this gene crucial for normal brain development. More specifically, expansion of CGG repeats in the 5′ UTR of the FMR1 gene results in hyper-methylation and consequent silencing of the gene in an X-linked dominant manner. Healthy individuals have ~6–40 CGG repeats in their FMR1 gene whereas affected individuals have >200–230 CGG repeats 95. Elevated protein synthesis at baseline as a consequence of FMR1 reduced expression leads to an absence in synaptic activity-dependent protein synthesis, which in turns disrupts several higher cognitive processes 96–98. Without synaptic activation-dependent protein synthesis, processes that are involved in the physical encoding of information storage in the brain circuitry cannot function fully.
HiPSC modeling of Fragile-X syndrome is complicated in a similar way as is modeling of Rett syndrome. Eiges et al. showed that in ESCs derived from embryos carrying the FMR1 mutation (determined through preimplantation genetic diagnosis), the FMR1 gene is expressed in ESCs but undergoes transcriptional silencing following differentiation 99. The same group later showed that FXS patient-derived hiPSCs, following reprogramming of FXS-patient-derived somatic fibroblasts, had epigenetic inactivation of the FMR1 gene 100 (Box 1). The same group also tested in their FXS hiPSC several chromatin remodeling drugs to assess the reactivation of FMR1 gene expression. To our knowledge, only two groups 101,102 created hiPSC models of FXS and studied the phenotype of FXS neurons. Both reported aberrant neuronal differentiation of these hiPSCs and showed that FXS neurons had reduced neurite outgrowth and fewer and shorter processes, echoing reports on FMR1 knockout mouse models 103,104 and post-mortem brain tissue 105,101,102. Halevy et al. have also assessed and partially reversed the abnormal transcriptional signatures that may underlie the abnormalities 106. Interestingly these findings imply early neurodedevelopmental alterations, prior to synaptogenesis, although more studies are needed to understand in depth the pathophysiology of these defects. Four groups have used FXS 107–109,110 patient-derived hiPSCs to create proof of concept high-throughput drug discovery systems, though unfortunately the sensitivity and robustness of these screens is still poor and will require significant work. Promisingly though, Park et al. 111 used a genomic engineering technique known as CRISPR/Cas (Box 3) to ablate CGG repeats in FXS patient-derived hiPSCs and thus restore expression of FMR1 mRNA and consequently FMRP protein. Unfortunately, they did not assess for any phenotypic reversal in this study.
Box 3. CRISPR/Cas.
Genome engineering techniques allow researchers to edit sequences of DNA in their endogenous context, thus allowing for modulation of expression of the gene that that sequence of DNA encodes. The latest of genome engineering technologies, known as CRISPR/Cas, was discovered through the study of the prokaryotic immune system. Bacteria and archaea have a unique adaptive immune system in that they use RNA-guided enzymes to cleave and destroy foreign DNA. Prokaryotes are able to integrate pieces of foreign DNA (usually from invading viruses) into their own DNA into what are known as spacer regions. These spacer regions are regularly interspaced throughout the prokaryote’s DNA and serve as a memory of previous invaders’ DNA, thus allowing for acquired immunity. Spacer regions are preceded in the prokaryote’s DNA by short palindromic sequences. Segments of prokaryotic DNA containing these short palindromic sequences followed by spacer DNA are known as Clustered regularly interspaced short palindromic repeats, or CRISPR. CRISPR sequences serve as the RNA-guide for the DNA endonucleases known as CRISPR associated proteins, or Cas. These enzymes are the ones that actually destroy foreign pieces of DNA, which they recognize via the spacer regions of CRISPRs. Researchers today utilize known Cas proteins and engineer their own CRISPR sequences to act as RNA guides in order to target for endonuclease cleavage specific regions of cellular DNA. Such cleaved DNA can be repaired via endogenous non-homologous end joining which creates small insertions/deletion that typically lead to alteration of the DNA reading frame, generating gene knockouts. Alternatively, Cas-cleaved DNA can be repaired by homologous recombination, where the complementary strand is experimentally introduced in the cell, allowing for precise editing of the DNA sequence. CRISPR/Cas allows researchers the ability to knockdown/edit multiple genes simultaneously in a highly scalable fashion 192,193
Timothy Syndrome
Timothy Syndrome is much rarer than FXS but also often results in syndromic ASD. It is caused by a mutation in the CACNA1C gene, which encodes an L-type voltage gated calcium channel. The mutation leads to decreased calcium- and voltage-dependent inactivation of the channel. Calcium entry into excitable cells is a crucial cellular signal and calcium- and voltage-dependent inactivation, also known as CDI and VDI, are forms of negative feedback regulation following membrane depolarization. Calcium channels play an important role in neuronal development because they control dendritic growth and arborization and excessive calcium entry due to the loss of feed-back can lead to detrimental effects on brain development and growth as seen in Timothy Syndrome. Pasca et al. showed that hiPSC-derived Timothy Syndrome neurons showed impaired calcium signaling and electrophysiology, and had defects in activity-dependent transcription compared to WT 112. Interestingly, neurons also showed a high expression of tyrosine hydroxylase (an enzyme that converts the amino acid tyrosine to the dopamine precursor) and consequently had increased production of norepinephrine and dopamine. HiPSCs derived from these patients had abnormal differentiation tendencies and differentiated neurons had reduced expression of genes marking lower cortical layers and callosal projection neurons. The same group later used a bioinformatics approach, integrating co-expression network analysis and transcription factor binding analysis, to provide mechanisms by which altered calcium signaling generates altered level of calcium-dependent transcriptional regulators leading to the transcriptional network changes observed in TS patient-derived neurons 113. Another group reported that hiPSC-derived neurons from individuals with Timothy syndrome have activity-dependent dendrite retraction 114. Both groups promisingly were able to partially reverse some of the deficits in TS patient-derived neurons.
b. Complex genetic disorders
HiPSCs have proved quite helpful in further validating findings from previous pathological and rodent model studies of monogenic disorders, especially when coupled with a comparison between isogenic lines. Since most of the variation between hiPSC lines is due to genetic background effects 35, comparison between isogenic lines when investigating the phenotypic effect of single genes can be quite powerful. Yet, isogenic pairs do not completely resolve the issue of modeling disorders. One reason for this is that even in the so-called monogenic disorders, penetrance can vary substantially depending upon the genetic background. This requires the investigation of single genes’s mutations in multiple genetic backgrounds.
HiPSCs have proved to be useful in modeling of monogenic disorders, but the real power of these models lies in their ability to model complex genetic disorders, where multiple genes and multiple genetic backgrounds are typically at work in disease pathogenesis and for which it is not informative to engineer a single penetrant mutation into an ESC line or rodent. The approach for modeling complex genetic disorders with hiPSCs has been to compare phenotypes and gene expression in a sufficient number of patient-derived and control lines in order to get mechanistic insights into the neurobiological bases of the disorder (Table 1). In these studies, the choice of patients and controls, the overall design of the study and the clinical phenotyping are key.
Schizophrenia
Schizophrenia is a highly heterogeneous and complex disorder. The disorder has classically been thought of us as being caused by the interaction of predisposing genes and detrimental environmental factors. However, heritability has been estimated to be as high as 80% with a sibling recurrence risk ratio of 8.6 16. As a result, we primarily focus in this review on the genetic component and briefly discuss modeling environmental factors in a later section.
Schizophrenia comes in two phenomenological forms – Type 1 which is often accompanied by hallucinations, delusions and disorganized thinking and Type 2 which is often accompanied by cognitive deficits and disturbances in social and affective functions 115. The disorder is considered a developmental disorder because several perturbations during pregnancy are known to increase risk, and furthermore schizophrenia-associated gene and epigenetic factors point to early cortical development 116,117. In particular, schizophrenia-associated DNA methylation is closely related to cellular and transcriptome changes occurring during the transition between pre and postnatal life 118. Neuropathological studies of Schizophrenia have revealed reduced brain volume, cell size, and spine density as well as altered neuronal distribution in the prefrontal cortex and hippocampus compared to controls 119–123. Furthermore, pharmacological studies have shown that NMDA receptor antagonists, such as ketamine and phencyclidine (PCP), can cause psychotic and cognitive abnormalities reminiscent of schizophrenia, indicating some detrimental effect caused by reduced glutamate in SCZD 124–126.
In a pioneering study, Brennand et al. 127 constructed the first hiPSC model of Schizophrenia (SCZD). The Brennand et al. study interestingly echoed many of the neuropathological and pharmacological results showing diminished neuronal connectivity, decreased neurite number, decreased PSD95-protein levels, and decreased glutamate receptor expression. However they were also able to build on post-mortem studies in that they revealed further information about the cellular pathway underpinnings of SCZD – namely, their gene expression profiles of SCZD patient derived neurons had abnormal expression of components of the cyclic AMP and WNT signaling pathways 127. The same group later showed that SCZD patient-derived neuronal progenitor cells (NPCs) had aberrant migration, increased oxidative stress, and perturbed responses to environmental stresses 128. A separate study by this group suggests increased protein synthesis due to increased levels of ribosomal and translation initiation and elongation factor proteins in SCZD patient-derived cultures, hinting at yet another possible mechanism of disease 54.
Other hiPSC models of SCZD have gone even further in demonstrating cellular and molecular elements of the disorder’s pathogenesis previously undetectable through post-mortem, genetic, or neuroimaging studies. Some have focused on abnormal miRNA expression profiles 129, some on cellular level deficits in adherens junction and apical polarity 43, and synaptic vesicle release deficits have also been noted 130. Furthermore, Robicsek et al. studied patient-derived hiPSCs differentiated into dopaminergic and glutamatergic neurons, in contrast to the more frequently used general forebrain differentiation program, showing perturbations in neural differentiation and mitochondrial function 131.
Interestingly, some of the results from SCZD patient-derived hiPSCs and neurons are very similar to those of hiPSC model studies in other disorders. For example, SCZD hiPSC-derived neurons also show increased L1 retrotransposition (similar to Rett syndrome) 132, increased tyrosine hydroxylase, and consequent increased catecholamines, dopamine, norepinephrine and epinephrine (similar to Timothy syndrome) 133, potentially hinting at similar pathologies between these neurodevelopmental disorders.
Autism
Our group recently reported on using hiPSC-derived organoids to model an idiopathic neurodevelopmental disorder – Autism Spectrum Disorder (ASD). This is, to our knowledge, the first published model of non-syndromic, idiopathic autism i.e., independent of any other disorder such as Rett Syndrome, Fragile X, that makes a phenotypic assessment.
Studies of rare exonic mutations have suggested that genes expressed during early fetal cortical development are etiologically implicated in ASD 134,135. In an effort to directly model early cortical development of patients with idiopathic autism, we produced hiPSC-derived telencephalic organoids from the fibroblasts of ASD patients with macrocephaly. The telencephalon is the embryonic structure that gives rise to the cerebral cortex, hippocampus, basal ganglia, and olfactory bulb. The ventral telencephalon gives rise to the basal ganglia, the principal source of cortical inhibitory interneurons, while the dorsal telencephalon gives rise to the excitatory projection neurons of the cerebral cortex. Telencephalic organoids contain both glutamatergic (excitatory) cortical neurons and GABAergic (inhibitory) cortical neurons. Macrocephaly refers to an increased head circumference due to increased brain size, and is one of the most consistently replicated phenotypes in ASD 136–138. Furthermore, ASD patients who present with macrocephaly usually have more severe symptoms and poorer outcomes. In this study, organoids were used to reflect early to mid-fetal telencephalic development in humans.
Gene network analyses of transcriptomes obtained from these organoids revealed upregulation of genes involved in cell proliferation, neuronal differentiation, and synaptic assembly in ASD. Interestingly, GABAergic genes were strongly upregulated. Immunocytochemical analyses confirmed that, indeed, ASD patient-derived organoids exhibited an accelerated cell cycle and an overproduction of GABAergic inhibitory neurons compared to control organoids derived from the patients’ unaffected parents. This study further showed, using RNA interference, that increased expression of the master regulatory transcription factor FOXG1 in ASD patient-derived organoids is the cause for the increased production of GABAergic neurons. The data are intriguing, because the model clearly mimics a very early stage of cortical development, suggesting that what is modeled though hiPSC is predisposition to disease. Yet, the altered expression of gene network modules positively correlated with symptom severity, albeit in a small patient sample. It is possible that increased production of GABA is responsible for aberrantly increased synaptogenesis, as GABA functions as an excitatory neurotransmitter promoting activity-dependent synapse formation in prenatal development 139. The increase in synapse-related transcripts detected in hiPSC-derived organoids echoes a neuropathological study where a morphological increase in synaptic connections was identified in unselected patients with idiopathic autism 140. Increased density of cortical neuron minicolumns 141 and areas of aberrant cortical neuron layering 142,143 have been shown to occur in autism, but there are as yet no neuropathological studies with sufficient number of cases to demonstrate an imbalance of inhibitory over excitatory neurons in the cortex. Remarkably, the increase in FOXG1 expression in idiopathic ASD with macrocephaly has been replicated in an independent set of cases using a non-organoid neuronal differentiation protocol, along with the increase in GABAergic neuronal progenitors 144, consolidating the idea of an involvement of FOXG1-mediated GABAergic disturbance in ASD and suggesting that hiPSC modeling is robust and can yield reproducible data across hiPSC lines, patients, and laboratories. The above mentioned study 144 has also hypothesized that the Wnt signalling pathway, which when elevated causes excessive proliferation of NPCs 145,146, is paradoxically reduced in ASD with macrocephaly, and somehow this is related to the aberrantly increased proliferative activity, as agents that increased the canonical b-catenin/BRN2 cascade normalized cell proliferation. Further support for an involvement of this signaling pathway in ASD is provided by the observation that ASD patients have mutation in CHD8, a negative regulator of canonical Wnt signalling pathway 17,147–149.
Other studies have engineered hiPSCs with specific mutations associated with rare cases of autism with variable penetrance. Griesi-Oliveira et al. 150 derived hiPSCs and consequently neurons from an individual with non-syndromic autism with a de novo balanced translocation disruption of TRPC6, a cation channel that plays a fundamental role in calcium homeostatsis. The group showed that these TRPC6 mutant hiPSC-derived neurons had shorter and less arborized neurites, reduced density of dendritic spines, significantly lower density of VGLUT1 puncta, and, predictably, impaired sodium and calcium currents. Promisingly, restoration of TRPC6 expression or treatment with IGF1 or hyperforin (a TRPC6-specific agonist) partially corrected these abnormal neuronal phenotypes. In contrast to the majority of hiPSC models, Wang et al. used CRISPR/Cas technology to engineer in hiPSCs a heterozygous loss of function mutation for CDH8, an ATP-dependent chromatin remodeler strongly associated with rare cases of autism. The mutation produced differential expression of thousands of genes enriched in GO functions such as β-catenin/Wnt signaling in mutated neurons, demonstrating the combined power of genome engineering and hiPSCs 151. This literature is still emerging, and it will be interesting to see whether a few common pathophysiological mechanisms underlying autism will emerge from these studies. In this respect, it will be essential to take into consideration technical and experimental variables that may influence the results.
Among these, one of the most important is the quality of the hiPSC lines and the potential presence of epigenetic abnormalities and somatic mutations 152–155. Poor quality hiPSC lines are more likely to fail or give variable outcomes using any differentiation method, complicating the interpretation of the results. A good practice is to differentiate between 2 and 3 different hiPSC lines for each individual in each experiment. The second most important variable is the method of differentiation. Variables that are often associated with different differentiation protocols include initial enrichment and selection of NPCs with different techniques such as manual isolation, FACS-sorting or bead-isolation, and whether NPCs are cultured in a monolayer or in a 3-D environment. For example, monolayer protocols typically involve a lower cell density, decreased cell-to-cell and increased cell-to-matrix signaling; these variables, along with matrix rigidity and stiffness, have all been shown to influence cell fate.
Regardless of the differentiation method used, it is imperative that the regional/cellular specificity of a neuronal differentiation protocol is properly analyzed. Few cellular markers are generally not sufficient for this purpose, and a better way to address this question is to perform global transcriptome analysis exploiting rich and well curated databases of developmental transcriptome of the human brain, like Brainspan 156, by classifying samples against it. A very simple classification algorithm, based on correlation analysis, showed that the organoid’s transcriptome resembled best human brain development at 8 to 10 post-conceptional weeks, with weaker correspondence to later stages of fetal development 50 suggesting that the in-vitro developmental timeline mimics early in-vivo brain development. A more sophisticated machine learning based algorithm, CoNTExT 157, was later developed, which identified strong conservation of transcriptomics network signatures between primary human neural progenitor cells and developing human fetal brain, but highlighted differences between these primary human neural progenitor cells and hiPSC-derived neural progenitors from multiple laboratories. However, CoNTExT had not yet been applied to transcriptome data derived from organoids, which may be different from neural progenitors dissociated and grown in 2D culture.
Several groups have identified CNVs in the context of ASD patients 158–162, although only a few have yet been modeled in hiPSCs. In particular it is interesting that de novo duplications of the 7q11.23 region is associated with ASD, whereas deletion of the same region causes Williams-Beuren syndrome, characterized increased sociability 163. Willliams syndrome has been modeled in hiPSC and it has been shown that Willliam syndrome progenitors have increased doubling time and apoptosis, while neurons show increased dendritic spines and synapses compared with typically developing neural cells 164 as well as prolonged repolarization times and a deficit in voltage-activated K+ currents 165.
c. Environmental factors
The models described above exploit the ability of hiPSCs to be differentiated into neurons while retaining the unique genetic signature of the individual from whom they were derived, thereby replicating the developmental trajectory of an individual’s brain development on the basis of their genetic code. So-called microenvironmental effects can also be studied in hiPSC-based organoid systems; these effects include cell-to-cell interactions, the effects of age and the effects of diffusible substances generated by developing cells; it is well known that the transcriptome and epigenome are dynamically altered by the cellular micro-environment. However, hiPSCs by themselves offer little to no insight into the macro-environmental component of the pathogenesis of neurodevelopmental disorders, such as organism-level effects and interactions (for example, hormones and sensory experiences). HiPSC organoid systems could, therefore, be exposed to toxic chemicals or immune challenges that mimic detrimental environmental effects to study the consequences on development.
One very recent example of modeling neurodevelopmental disorders primarily driven by environmental factors is the modeling of Zika infection with brain organoids. Modeling infection in early embryonic stages, as would be the case when modeling with hiPSCs, can be useful in predicting some problems later on in life. hiPSCs have allowed these researchers to investigate areas of the disease’ biology that were previously intractable – such as what the Zika virus actually targets in embryonic stages that results in the various physical birth anomalies (e.g., microecephaly) and later issues in brain development. In the small period of time since when Zika became a critical public health concern, researchers have been able to isolate exactly why babies born from Zika virus infected mothers had microcephaly. Tang et al. showed that the virus readily infects forebrain-specific cortical neural progenitors, the building blocks of the human cortex 166. The same study went further to show that at the molecular level the infection resulted in downregulation of cell-cycle genes and upregulation of apoptosis genes in NPCs. This was further evidenced by abnormal progression of cell cycle, increased cell death/apoptosis, and disrupted neurogenesis by the same study as well as many others 167–173. For a more in-depth overview of the subject see review by Ming et al. 174.
6. Conclusions
To date, hiPSC model systems have not only confirmed previous findings about the pathogenesis of neurodevelopmental disorders, but provided further detail and nuances, and shed light on the mechanisms underlying the associated manifestations. Altered synaptic connectivity and density, excitation inhibition imbalance, and altered neuronal activity had all been proposed as having a role as a result of postmortem, neuroimaging and genetic studies, and hiPSCs have been able to establish a level of causality for these mechanisms, because they provide the means for experimentally manipulating the system. Promisingly, these causal relationships have been used to successfully reverse observed abnormal phenotypes. Furthermore, hiPSCs have demonstrated when these phenotypic alterations first occur, the events leading up to their first occurrence, and how they progress, whereas postmortem studies were only able to examine a single time point, often remote from the genesis of the disorder. In some cases the phenotypic observations, such as neuron size and detailed spine morphology, made in patient-derived neurons are entirely novel and were previously unobservable. Still, we think there is much left to be discovered especially in using hiPSCs to generate non-neuronal brain cells, or glial cells, which in fact make up at least half of brain cells 175. Additionally, investigations must be made into reducing the resource and time requirements of conducting hiPSC studies so more labs can contribute to the field with high n studies. One potential avenue through which this can be achieved is direct conversion to neurons. A challenge for this approach is, however, that the factors that are required for the determination and maintenance of defined neuronal types have not been completely elucidated, and there is the risk of creating “hybrid’ or otherwise non-physiological neurons. There are still challenges in the iPSC field that will need to be addressed in the future years, to allow greater translational potential. An obvious difficulty is to reproduce the cellular and regional complexities of the human brain. Another is the fact that iPSC-derived neurons are often “timed” to a prenatal stage. It is important to recognize these limitations, and consider the complexity of these problems when trying to apply knowledge derived from iPSC studies to patients.
Our long-term goal should be to make hiPSC-based drug discovery systems a reality. The modern field of tissue engineering has advanced considerably and, although the human brain is far more difficult to model than mechanical tissue, keeping this as our long-term goal will likely help us develop more mature and sophisticated human neuron differentiation protocols. Such hiPSC-based drug discovery systems are also already well on their way. In some cases, abnormal neurobiological phenotypes have already been reversed with existing drugs 74,176,177. It is likely that the predictive validity of this system will only increase as the neurons used become more sophisticated and more accurately representative of human neurons. Groups have exploited neurobiological phenotypes they found in patient-derived neurons to conduct high-throughput genetic screens for compensatory mutations and potential drug targets 176–180. In contrast, the majority of current subtype characterization for clinical trials is done almost entirely through notoriously imprecise clinical diagnostic criteria. Gene network and neurobiological analyses on hiPSC-derived neurodevelopmental models could be used instead to identify subtypes based on specific genes/pathways that are altered. Finally, another therapeutic avenue may be through transplantation. While this is already in clinical trial for some adult neurological disorders such as Parkinson’s disorder, we believe it is challenging for disorders of childhood, especially for neurodevelopmental disorders where, as demonstrated by this review, there isn’t much evidence of neuronal loss or neural tissue damage that is in need of being regenerated. One recent clinical trial, not directly regarding neurodevelopmental disorders but in the area of CNS disorders, is the use of hiPSC-derived retinal pigment epithelial cells for the treatment of macular degeneration 181. Broadly, the idea of editing mutations in patient-derived hiPSCs and then transplanting them back into the patient is exciting. This is mainly because transplantation normally is accompanied by host rejection risk, however this would not the case with hiPSCs as the cells are derived from the patient themselves.
All of these areas of growth will require the collaboration of multiple scientific and engineering disciplines. As we move from studying single genes and single mutations to studying networks that integrate the two, we will require collaboration among neurobiologists, bioinformaticians, statisticians, and computer scientists. And as we move to push hiPSCs into the translational space, we will require not only the expertise but the support of clinicians.
Table 2.
hiPSC models of Fragile X Syndrome
| Mutated Gene | Differentiated cell type | Phenotype (compared to controls) | Phenotypic reversal or rescue with drug? | Isogenic control? | Organoid or monolayer? | Reference |
|---|---|---|---|---|---|---|
| FMR1 | Neurons and glia | Aberrant neuronal differentiation; Glial differentiation was variable (some looked like controls in morphology and number others didn’t); FXS neurons had fewer and shorter processes |
NA | No | Monolayer | 102 |
| FMR1 | Neurons | NA | NA | No | Monolayer | 100 |
| FMR1 | Neurons | FXS neurons had decreased expression of neuronal differentiation and axon guidance genes; REST, a transcription factor which acts to suppress neural genes in non-neuronal tissues (and thus its expression levels must be downregulated as neural differentiation progresses) fails to undergo downregulation and silencing as differentiation progresses in FXS-derived neurons; REST is regulated by microRNA has-mir-382 which has lower levels in FXS-derived neurons |
Introduction of a miR-382 mimic, repressed REST and upregulated its target axon guidance genes | No | Monolayer | 106 |
| FMR1 | Neurons | NA | Tested several chromatin remodeling drugs to assess re-activation of FMR1 gene expression; proof of concept for hiPSC-based drug screen model | No | Monolayer | 110 |
| FMR1 | Forebrain neurons | Reduced neurite outgrowth; Significantly fewer and shorter processes | NA | No | Monolayer | 101 |
| FMR1 | Neurons | NA | FXS hiPSCs CGG repeats were completely ablated using CRISPR/Cas9 resulting in expression of FMR1 mRNA. FMR1 promoter showed near complete demethylation and differentiated mature neurons showed reactivation of FMRP | No | Monolayer | 111 |
| FMR1 | Neural stem cells | NA | Proof of concept high throughput drug discovery screen | No | Monolayer | 108 |
| FMR1 | Neuronal progenitor | NA | Proof of concept high throughput drug discovery screen | No | Monolayer | 107 |
| FMR1 | Neural progenitor cells | NA | Proof of concept high throughput drug discovery screen | No | Monolayer | 109 |
Table 3.
hiPSC models of Timothy Syndrome
| Mutated Gene | Differentiated cell type | Phenotype (compared to controls) | Phenotypic reversal or rescue with drug? | Isogenic control? | Organoid or monolayer? | Reference |
|---|---|---|---|---|---|---|
| CACNA1 | Neurons | Defects in calcium signaling and activity-dependent gene expression, abnormalities in differentiation (decreased expression of lower cortical layer and callosal projection neurons), and abnormal expression of tyrosine hydroxylase and consequent increased in production of norephineprine and dopamine | Roscovitine, a cyclin-dependent kinase inhibitor and atypical L-type-channel blocker, reversed the phonotype | No | Monolayer | 112 |
| CACNA1 | Neurons | Altered calcium signaling in patient-derived neurons resulted in an alteration in calcium-dependent transcriptional regulators which led to molecular dysregulation and transcriptional changes in TS patient derived neurons | NA | No | Monolayer | 113 |
| CACNA1 | Neurons | Activity-dependent dendrite retraction | Dendrite retraction was inhibited by overexpression of the channel-associated GTPase Gem | No | Monolayer | 114 |
Table 4.
hiPSC models of Autism
| Mutated Gene | Differentiated cell type | Phenotype (compared to controls) | Phenotypic reversal or rescue with drug? | Isogenic control? | Organoid or monolayer? | Reference |
|---|---|---|---|---|---|---|
| TRPC6 | Neurons | Shorter and less arborized neurites, density of dendritic spines was reduced, significantly lower density of VGLUT1 puncta, impaired sodium and calcium currents | Restoration of TRPC6 expression or treatment with IGF1 or hyperforin (a TRPC6-specific agonist) corrected abnormal neuronal phenotypes | No | Monolayer | 150 |
| Idiopathic | Cortical neurons | Decreased cell cycle length of undifferentiated hiPSCs, overproduction of synapses, and imbalanced overproduction of GABAergic inhibitory neurons, increased expression of FOXG1 and other GABA neurons transcription factors | Dowregulation of FOXG1 overactivity by shRNA was able to revert the overproduction of GABAergic inhibitory neurons to physiological levels | Yes | Organoid | 50 |
| CHD8 | Neurons | Differential expression of genes enriched in GO functions: neural development, β-catenin/Wnt signaling, extracellular matrix, and skeletal system development | NA | No | Monolayer | 151 |
| Idiopathic | GABAergic neurons | First derivation of hiPSC (and subsequent differentiation into GABAergic neurons) from peripheral blood mononuclear cells derived from the whole blood of autistic children | NA | No | Monolayer | 44 |
| Idiopathic | Cortical neurons | Increased proliferation of progenitors, increased number of GABA neuron progenitors, decreased neurite arborization, decreased beta-catenin-Brn2 canonical pathway, increased expression of FOXG1 and other GABA neurons transcription factors | Partial correction of deficit in network activity with insulin growth factor 1 (IGF-1) | No | Monolayer | 144 |
Glossary
Glossary 1: Neurobiology
- Neurons
Core component cells of the brain. The function of these electrically excitable cells is to receive, conduct, and transmit signals. Neurons typically have three parts – the soma, dendrites, and an axon.
- Cortical neurons
Neurons of the cerebral cortex
- Glial cells
Non-neuronal brain cells that provide support and protection to neuronal cells
- Soma
Cell body – contains the cell nucleus
- Axon
Conducts signals from the cell body to other target neurons
- Dendrites
Extend and branch out from the cell body like antennae providing an enlarged surface area through which neurons receive signals from other neurons
- Synapse/Synaptic connection
Neuronal structure that allows a neuron to pass an electrical or chemical signal to another neuron. Neurons are connected to each other through what are known as synaptic connections.
- Neuronal network
A groups of neurons and their synaptic connections is referred to as a neuronal network. It is classically thought that neurons form these networks in order to execute some function or process or store information.
- Connectivity
The number of synaptic connections between a group of neurons or in a neuronal network
- Synaptogenesis
The generation of new synapses
- Dendritic arborization
Branching of dendrites, increased branching results in increased surface area through which neurons can receive signals
- Spines
Protrusions from dendrites that receive and help transmit to the cell body, signals from single synapses. Spines are hypothesized to serve as the storage site for memories and be crucial to learning processes. Spine plasticity is hypothesized to mediate synaptic plasticity.
- Spine plasticity
The process by which neuronal activity affects the size, shape, and density of dendritic spines. These changes in dendritic spines can be short or long term. This process is hypothesized to play a crucial role and learning and memory processes.
- Synaptic plasticity
The process by which neuronal activity affects the strength of synaptic connections. This change can arise through changes in the quantity or sensitivity of neurotransmitter receptors or through the quantity of neurotransmitter release.
- Neurite
Can refer to any projection from the soma i.e., axon or dendrite. This term is frequently used when describing neurons in development as it sometimes hard to distinguish axon vs. dendrites
- Glutamate/Glutamatergic
Excitatory neuron
- GABA/GABAergic
Inhibitory neuron
- PSD95
Post-synaptic density protein
- Monogenic disorders
Disorders that are the result of a single defective gene
- Complex disorders
Disorders that are the result of >1 defective genes (more commonly the result of 1000s of genes interacting with each other through complex and dynamic pathways and networks)
Glossary 2: Stem Cells
- Proliferation
Reproduction of cells, results in an increased number of cells
- Differentiation
The process by which a stem cell or less specialized cell becomes a more specialized cell. This occurs through regulation of gene expression as different genes are turned on and off at different levels in different cells.
- Embryonic stem cells
These pluripotent cells are derived from the inner cell mass of the blastocyst
- Epigenetic
Changes in gene expression or regulation that do not involve alterations in primary sequence or copy number of DNA
- Pluripotent
Able to differentiate into any adult cell type
- Induced Pluripotent stem cell
Pluripotent stem cells, resembles embryonic stem cell, and can be derived via reprogramming from virtually any somatic cell type
- Somatic cell
Any cell that is not a sex cell (i.e., not sperm or zygote)
- Neural/Neuronal progenitor cell
These cells are somewhere in-between a stem cell and neuron in terms of differentiation and specialization. They have less replication capacity than stem cells but are more primed to differentiate into neurons.
- Organoids
-
Tridimensional aggregates generated from pluripotent ES or iPS cells, which differentiate over time in a self-directed and organized fashion into a particular tissue or organ, under appropriate conditions in vitro.
In the case of the brain, these 3D cultures mimic the brain’s cell type composition and layer/tissue cytoarchitecture.
- Reprogramming
The process by which a somatic cell is dedifferentiated into a iPSC. The dedifferentiation step involves erasing a somatic cell’s epigenetic signatures, switching the cell’s epigenetic state from closed to open, and establishing pluripotency networks.
- Differentiation
Follows reprogramming and is the conversion of an iPSC into the desired type of somatic cell. The differentiation step involves establishing the epigenetic signatures and transcription factor networks associated with the desired somatic cell type.
- Direct differentiation/direct lineage reprogramming
The process of a converting a somatic cell into another desired somatic cell type, but skipping the pluripotent stage
- Embryoid bodies
Tridimensional aggregates that can be generated from pluripotent ES and iPS cells, with the potential to differentiate in the three embryonic germ layers under appropriate stimulations in vitro.
- Neural rosettes
Radially self-organized neuroepithelial structures composed of early neural progenitor cells, resembling the developing neural tube.
- High throughput screening
A process through which a large number of compounds/molecules can be assessed systematically for their biological activity (or more commonly for their ability to reverse or partially reversed a targeted abnormal phenotype)
References
- 1.Herculano-Houzel S. The human brain in numbers: a linearly scaled-up primate brain. Frontiers in human neuroscience. 2009;3:31. doi: 10.3389/neuro.09.031.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18:383–388. doi: 10.1016/0166-2236(95)93934-p. doi:016622369593934P [pii] [DOI] [PubMed] [Google Scholar]
- 3.Spooren W, Lindemann L, Ghosh A, Santarelli L. Synapse dysfunction in autism: a molecular medicine approach to drug discovery in neurodevelopmental disorders. Trends Pharmacol Sci. 2012;33:669–684. doi: 10.1016/j.tips.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh A, Michalon A, Lindemann L, Fontoura P, Santarelli L. Drug discovery for autism spectrum disorder: challenges and opportunities. Nature reviews Drug discovery. 2013;12:777–790. doi: 10.1038/nrd4102. [DOI] [PubMed] [Google Scholar]
- 5.Dragunow M. The adult human brain in preclinical drug development. Nature reviews Drug discovery. 2008;7:659–666. doi: 10.1038/nrd2617. [DOI] [PubMed] [Google Scholar]
- 6.Dolmetsch R, Geschwind DH. The human brain in a dish: the promise of iPSC-derived neurons. Cell. 2011;145:831–834. doi: 10.1016/j.cell.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson MB, et al. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. doi:S0896-6273(09)00286-4 [pii]10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:255–274. doi: 10.1002/ajmg.b.31159. [DOI] [PubMed] [Google Scholar]
- 9.Ozonoff S, et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics. 2011;128:e488–495. doi: 10.1542/peds.2010-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallmayer J, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenwood TA, et al. Initial heritability analyses of endophenotypic measures for schizophrenia: the consortium on the genetics of schizophrenia. Arch Gen Psychiatry. 2007;64:1242–1250. doi: 10.1001/archpsyc.64.11.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu B, et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet. 2011;43:864–868. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schizophrenia Psychiatric Genome-Wide Association Study, C. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ripke S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Roak BJ, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.International Schizophrenia, C et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bakkaloglu B, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ardhanareeswaran K, Coppola G, Vaccarino F. The use of stem cells to study autism spectrum disorder. The Yale journal of biology and medicine. 2015;88:5–16. [PMC free article] [PubMed] [Google Scholar]
- 23.Colantuoni C, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524nature10524. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevens HE, Mariani J, Coppola G, Vaccarino FM. Neurobiology meets genomic science: the promise of human-induced pluripotent stem cells. Dev Psychopathol. 2012;24:1443–1451. doi: 10.1017/s095457941200082x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaccarino FM, et al. Annual Research Review: The promise of stem cell research for neuropsychiatric disorders. J Child Psychol Psychiatry. 2011;52:504–516. doi: 10.1111/j.1469-7610.2010.02348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaccarino FM, et al. Induced pluripotent stem cells: A new tool to confront the challenge of neuropsychiatric disorders. Neuropharmacology. 2011;60:1355–1363. doi: 10.1016/j.neuropharm.2011.02.021. doi:S0028-3908(11)00101-8 [pii]10.1016/j.neuropharm.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fussner E, et al. Constitutive heterochromatin reorganization during somatic cell reprogramming. EMBO J. 2011;30:1778–1789. doi: 10.1038/emboj.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maherali N, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 29.Mattout A, Biran A, Meshorer E. Global epigenetic changes during somatic cell reprogramming to iPS cells. Journal of molecular cell biology. 2011;3:341–350. doi: 10.1093/jmcb/mjr028. [DOI] [PubMed] [Google Scholar]
- 30.Mikkelsen TS, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang G, Zhang Y. Embryonic stem cell and induced pluripotent stem cell: an epigenetic perspective. Cell research. 2013;23:49–69. doi: 10.1038/cr.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anokye-Danso F, et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carey BW, et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell. 2011;9:588–598. doi: 10.1016/j.stem.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 34.Buganim Y, Faddah DA, Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat Rev Genet. 2013;14:427–439. doi: 10.1038/nrg3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi J, et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol. 2015;33:1173–1181. doi: 10.1038/nbt.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chambers SM, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. doi:nbt.1529 [pii]10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kriks S, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller JD, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maroof AM, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 2012;15:477–486. doi: 10.1038/nn.3041nn.3041. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi Y, et al. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med. 2012;4:124ra129. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 2012;15:477–486. S471. doi: 10.1038/nn.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoon KJ, et al. Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell. 2014;15:79–91. doi: 10.1016/j.stem.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeRosa BA, et al. Derivation of autism spectrum disorder-specific induced pluripotent stem cells from peripheral blood mononuclear cells. Neurosci Lett. 2012;516:9–14. doi: 10.1016/j.neulet.2012.02.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu DX, et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem cell reports. 2014;2:295–310. doi: 10.1016/j.stemcr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eiraku M, et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell. 2008;3:519–532. doi: 10.1016/j.stem.2008.09.002. doi:S1934-5909(08)00455-4 [pii]10.1016/j.stem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 47.Mariani J, et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109:12770–12775. doi: 10.1073/pnas.1202944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kadoshima T, et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci U S A. 2013;110:20284–20289. doi: 10.1073/pnas.1315710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mariani J, et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015;162:375–390. doi: 10.1016/j.cell.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voineagu I, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110nature10110. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fillman SG, et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry. 2013;18:206–214. doi: 10.1038/mp.2012.110. [DOI] [PubMed] [Google Scholar]
- 53.Lennington JB, et al. Transcriptome Analysis of the Human Striatum in Tourette Syndrome. Biol Psychiatry. 2016;79:372–382. doi: 10.1016/j.biopsych.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Topol A, et al. Increased abundance of translation machinery in stem cell-derived neural progenitor cells from four schizophrenia patients. Translational psychiatry. 2015;5:e662. doi: 10.1038/tp.2015.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rohani L, Johnson AA, Arnold A, Stolzing A. The aging signature: a hallmark of induced pluripotent stem cells? Aging Cell. 2014;13:2–7. doi: 10.1111/acel.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lapasset L, et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011;25:2248–2253. doi: 10.1101/gad.173922.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mertens J, et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell. 2015;17:705–718. doi: 10.1016/j.stem.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Shea KS, McInnis MG. Neurodevelopmental origins of bipolar disorder: iPSC models. Mol Cell Neurosci. 2016;73:63–83. doi: 10.1016/j.mcn.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 59.Mertens J, et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 2015;527:95–99. doi: 10.1038/nature15526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Madison JM, et al. Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol Psychiatry. 2015;20:703–717. doi: 10.1038/mp.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen HM, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Translational psychiatry. 2014;4:e375. doi: 10.1038/tp.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neul JL, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Armstrong DD. Neuropathology of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:72–76. doi: 10.1002/mrdd.10027. [DOI] [PubMed] [Google Scholar]
- 64.Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- 65.Jellinger KA. Rett Syndrome – an update. J Neural Transm (Vienna) 2003;110:681–701. doi: 10.1007/s00702-003-0822-z. [DOI] [PubMed] [Google Scholar]
- 66.Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 67.Yogev S, Shen K. Cellular and molecular mechanisms of synaptic specificity. Annu Rev Cell Dev Biol. 2014;30:417–437. doi: 10.1146/annurev-cellbio-100913-012953. [DOI] [PubMed] [Google Scholar]
- 68.Caroni P, Donato F, Muller D. Structural plasticity upon learning: regulation and functions. Nat Rev Neurosci. 2012;13:478–490. doi: 10.1038/nrn3258. [DOI] [PubMed] [Google Scholar]
- 69.Shen K, Scheiffele P. Genetics and cell biology of building specific synaptic connectivity. Annu Rev Neurosci. 2010;33:473–507. doi: 10.1146/annurev.neuro.051508.135302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Colantuoni C, et al. Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. Neurobiol Dis. 2001;8:847–865. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- 71.Hotta A, et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat Methods. 2009;6:370–376. doi: 10.1038/nmeth.1325. [DOI] [PubMed] [Google Scholar]
- 72.Liu Z, et al. Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature. 2016;530:98–102. doi: 10.1038/nature16533. [DOI] [PubMed] [Google Scholar]
- 73.Nageshappa S, et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol Psychiatry. 2016;21:178–188. doi: 10.1038/mp.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marchetto MC, et al. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. doi:S0092-8674(10)01186-4 [pii]10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Han SS, Williams LA, Eggan KC. Constructing and deconstructing stem cell models of neurological disease. Neuron. 2011;70:626–644. doi: 10.1016/j.neuron.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 76.Ross WN. Understanding calcium waves and sparks in central neurons. Nat Rev Neurosci. 2012;13:157–168. doi: 10.1038/nrn3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Burgoyne RD. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat Rev Neurosci. 2007;8:182–193. doi: 10.1038/nrn2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Budde T, Meuth S, Pape HC. Calcium-dependent inactivation of neuronal calcium channels. Nat Rev Neurosci. 2002;3:873–883. doi: 10.1038/nrn959. [DOI] [PubMed] [Google Scholar]
- 79.Bading H. Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci. 2013;14:593–608. doi: 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- 80.Ananiev G, Williams EC, Li H, Chang Q. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS One. 2011;6:e25255. doi: 10.1371/journal.pone.0025255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim KY, Hysolli E, Park IH. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc Natl Acad Sci U S A. 2011;108:14169–14174. doi: 10.1073/pnas.1018979108. doi:1018979108 [pii]10.1073/pnas.1018979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cheung AY, et al. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011;20:2103–2115. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Muotri AR, et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010;468:443–446. doi: 10.1038/nature09544. doi:nature09544 [pii]10.1038/nature09544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang X, et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc Natl Acad Sci U S A. 2016;113:751–756. doi: 10.1073/pnas.1524013113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Livide G, et al. GluD1 is a common altered player in neuronal differentiation from both MECP2-mutated and CDKL5-mutated iPS cells. Eur J Hum Genet. 2015;23:195–201. doi: 10.1038/ejhg.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Djuric U, et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol Dis. 2015;76:37–45. doi: 10.1016/j.nbd.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Amenduni M, et al. iPS cells to model CDKL5-related disorders. Eur J Hum Genet. 2011;19:1246–1255. doi: 10.1038/ejhg.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patriarchi T, et al. Imbalance of excitatory/inhibitory synaptic protein expression in iPSC-derived neurons from FOXG1 patients and in foxg1 mice. Eur J Hum Genet. 2015 doi: 10.1038/ejhg.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ricciardi S, et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol. 2012;14:911–923. doi: 10.1038/ncb2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Delepine C, et al. Altered microtubule dynamics and vesicular transport in mouse and human MeCP2-deficient astrocytes. Hum Mol Genet. 2016;25:146–157. doi: 10.1093/hmg/ddv464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Williams EC, et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum Mol Genet. 2014;23:2968–2980. doi: 10.1093/hmg/ddu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheung AY, Horvath LM, Carrel L, Ellis J. X-chromosome inactivation in rett syndrome human induced pluripotent stem cells. Frontiers in psychiatry. 2012;3:24. doi: 10.3389/fpsyt.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J Clin Invest. 2012;122:4314–4322. doi: 10.1172/JCI63141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harbor perspectives in biology. 2012;4 doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crawford DC, Acuna JM, Sherman SL. FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med. 2001;3:359–371. doi: 10.1097/00125817-200109000-00006. doi:10.109700125817-200109000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Willemsen R, Oostra BA, Bassell GJ, Dictenberg J. The fragile X syndrome: from molecular genetics to neurobiology. Ment Retard Dev Disabil Res Rev. 2004;10:60–67. doi: 10.1002/mrdd.20010. [DOI] [PubMed] [Google Scholar]
- 97.Wang T, Bray SM, Warren ST. New perspectives on the biology of fragile X syndrome. Curr Opin Genet Dev. 2012;22:256–263. doi: 10.1016/j.gde.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sidorov MS, Auerbach BD, Bear MF. Fragile X mental retardation protein and synaptic plasticity. Molecular brain. 2013;6:15. doi: 10.1186/1756-6606-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eiges R, et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1:568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 100.Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. doi:S1934-5909(10)00160-8 [pii]10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Doers ME, et al. iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014;23:1777–1787. doi: 10.1089/scd.2014.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sheridan SD, et al. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Uutela M, et al. Reduction of BDNF expression in Fmr1 knockout mice worsens cognitive deficits but improves hyperactivity and sensorimotor deficits. Genes Brain Behav. 2012;11:513–523. doi: 10.1111/j.1601-183X.2012.00784.x. [DOI] [PubMed] [Google Scholar]
- 104.Louhivuori V, et al. BDNF and TrkB in neuronal differentiation of Fmr1-knockout mouse. Neurobiol Dis. 2011;41:469–480. doi: 10.1016/j.nbd.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 105.Castren M, et al. Altered differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci U S A. 2005;102:17834–17839. doi: 10.1073/pnas.0508995102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Halevy T, Czech C, Benvenisty N. Molecular mechanisms regulating the defects in fragile X syndrome neurons derived from human pluripotent stem cells. Stem cell reports. 2015;4:37–46. doi: 10.1016/j.stemcr.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kaufmann M, et al. High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. Journal of biomolecular screening. 2015;20:1101–1111. doi: 10.1177/1087057115588287. [DOI] [PubMed] [Google Scholar]
- 108.Kumari D, et al. High-Throughput Screening to Identify Compounds That Increase Fragile X Mental Retardation Protein Expression in Neural Stem Cells Differentiated From Fragile X Syndrome Patient-Derived Induced Pluripotent Stem Cells. Stem cells translational medicine. 2015;4:800–808. doi: 10.5966/sctm.2014-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li M, et al. Establishment of Reporter Lines for Detecting Fragile X Mental Retardation (FMR1) Gene Reactivation in Human Neural Cells. Stem Cells. 2017;35:158–169. doi: 10.1002/stem.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bar-Nur O, Caspi I, Benvenisty N. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. Journal of molecular cell biology. 2012;4:180–183. doi: 10.1093/jmcb/mjs007. [DOI] [PubMed] [Google Scholar]
- 111.Park CY, et al. Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell reports. 2015;13:234–241. doi: 10.1016/j.celrep.2015.08.084. [DOI] [PubMed] [Google Scholar]
- 112.Pasca SP, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tian Y, et al. Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy syndrome. Genome medicine. 2014;6:75. doi: 10.1186/s13073-014-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Krey JF, et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16:201–209. doi: 10.1038/nn.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–276. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- 116.Malkki H. Neurodevelopmental disorders. Altered epigenetic regulation in early development associated with schizophrenia. Nature reviews Neurology. 2016;12:1. doi: 10.1038/nrneurol.2015.239. [DOI] [PubMed] [Google Scholar]
- 117.Gulsuner S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jaffe AE, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19:40–47. doi: 10.1038/nn.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goldman-Rakic PS, Selemon LD. Functional and anatomical aspects of prefrontal pathology in schizophrenia. Schizophr Bull. 1997;23:437–458. doi: 10.1093/schbul/23.3.437. [DOI] [PubMed] [Google Scholar]
- 120.Selemon LD, Goldman-Rakic PS. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry. 1999;45:17–25. doi: 10.1016/s0006-3223(98)00281-9. [DOI] [PubMed] [Google Scholar]
- 121.Rajkowska G, Selemon LD, Goldman-Rakic PS. Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry. 1998;55:215–224. doi: 10.1001/archpsyc.55.3.215. [DOI] [PubMed] [Google Scholar]
- 122.Brennand KJ, Gage FH. Beyond Phenotype: The Promise of hiPSC-Based Studies of Schizophrenia. Stem Cells. 2011 doi: 10.1002/stem.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wong AH, Van Tol HH. Schizophrenia: from phenomenology to neurobiology. Neurosci Biobehav Rev. 2003;27:269–306. doi: 10.1016/s0149-7634(03)00035-6. [DOI] [PubMed] [Google Scholar]
- 124.Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69–108. doi: 10.1016/S0074-7742(06)78003-5. [DOI] [PubMed] [Google Scholar]
- 125.Javitt DC. Glutamatergic theories of schizophrenia. The Israel journal of psychiatry and related sciences. 2010;47:4–16. [PubMed] [Google Scholar]
- 126.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brennand KJ, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. doi:nature09915 [pii]10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brennand K, et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry. 2015;20:361–368. doi: 10.1038/mp.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao D, et al. MicroRNA Profiling of Neurons Generated Using Induced Pluripotent Stem Cells Derived from Patients with Schizophrenia and Schizoaffective Disorder, and 22q11.2 Del. PLoS One. 2015;10:e0132387. doi: 10.1371/journal.pone.0132387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wen Z, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 2014;515:414–418. doi: 10.1038/nature13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Robicsek O, et al. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol Psychiatry. 2013;18:1067–1076. doi: 10.1038/mp.2013.67. [DOI] [PubMed] [Google Scholar]
- 132.Bundo M, et al. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron. 2014;81:306–313. doi: 10.1016/j.neuron.2013.10.053. [DOI] [PubMed] [Google Scholar]
- 133.Hook V, et al. Human iPSC neurons display activity-dependent neurotransmitter secretion: aberrant catecholamine levels in schizophrenia neurons. Stem cell reports. 2014;3:531–538. doi: 10.1016/j.stemcr.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Parikshak NN, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Willsey AJ, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chaste P, et al. Adjusting head circumference for covariates in autism: clinical correlates of a highly heritable continuous trait. Biol Psychiatry. 2013;74:576–584. doi: 10.1016/j.biopsych.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chawarska K, et al. Early generalized overgrowth in boys with autism. Arch Gen Psychiatry. 2011;68:1021–1031. doi: 10.1001/archgenpsychiatry.2011.106. doi:68/10/1021 [pii]10.1001/archgenpsychiatry.2011.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lainhart JE, et al. Head circumference and height in autism: a study by the Collaborative Program of Excellence in Autism. Am J Med Genet A. 2006;140:2257–2274. doi: 10.1002/ajmg.a.31465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 140.Tang G, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83:1131–1143. doi: 10.1016/j.neuron.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Casanova MF, et al. Minicolumnar abnormalities in autism. Acta Neuropathol (Berl) 2006;112:287–303. doi: 10.1007/s00401-006-0085-5. [DOI] [PubMed] [Google Scholar]
- 142.Stoner R, et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med. 2014;370:1209–1219. doi: 10.1056/NEJMoa1307491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Casanova MF, et al. Focal cortical dysplasias in autism spectrum disorders. Acta neuropathologica communications. 2013;1:67. doi: 10.1186/2051-5960-1-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Marchetto MC, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chenn A, Walsh CA. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex. 2003;13:599–606. doi: 10.1093/cercor/13.6.599. [DOI] [PubMed] [Google Scholar]
- 146.Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors.[comment] Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- 147.Bernier R, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158:263–276. doi: 10.1016/j.cell.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cotney J, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nature communications. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sugathan A, et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci U S A. 2014;111:E4468–4477. doi: 10.1073/pnas.1405266111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Griesi-Oliveira K, et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry. 2015;20:1350–1365. doi: 10.1038/mp.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang P, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Molecular autism. 2015;6:55. doi: 10.1186/s13229-015-0048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bhutani K, et al. Whole-genome mutational burden analysis of three pluripotency induction methods. Nature communications. 2016;7:10536. doi: 10.1038/ncomms10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cheng L, et al. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell. 2012;10:337–344. doi: 10.1016/j.stem.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gore A, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Abyzov A, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kang HJ, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523nature10523. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stein JL, et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron. 2014;83:69–86. doi: 10.1016/j.neuron.2014.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Marshall CR, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Levy D, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 162.Glessner JT, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chailangkarn T, et al. A human neurodevelopmental model for Williams syndrome. Nature. 2016;536:338–343. doi: 10.1038/nature19067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Khattak S, et al. Human induced pluripotent stem cell derived neurons as a model for Williams-Beuren syndrome. Molecular brain. 2015;8:77. doi: 10.1186/s13041-015-0168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tang H, et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell. 2016;18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhang F, et al. Molecular signatures associated with ZIKV exposure in human cortical neural progenitors. Nucleic Acids Res. 2016;44:8610–8620. doi: 10.1093/nar/gkw765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Simonin Y, et al. Zika Virus Strains Potentially Display Different Infectious Profiles in Human Neural Cells. EBioMedicine. 2016;12:161–169. doi: 10.1016/j.ebiom.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liang Q, et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell. 2016;19:663–671. doi: 10.1016/j.stem.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Garcez PP, et al. Zika virus impairs growth in human neurospheres and brain organoids. Science. 2016;352:816–818. doi: 10.1126/science.aaf6116. [DOI] [PubMed] [Google Scholar]
- 171.Dang J, et al. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell. 2016;19:258–265. doi: 10.1016/j.stem.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cugola FR, et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature. 2016;534:267–271. doi: 10.1038/nature18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Brault JB, et al. Comparative Analysis Between Flaviviruses Reveals Specific Neural Stem Cell Tropism for Zika Virus in the Mouse Developing Neocortex. EBioMedicine. 2016;10:71–76. doi: 10.1016/j.ebiom.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ming GL, Tang H, Song H. Advances in Zika Virus Research: Stem Cell Models, Challenges, and Opportunities. Cell Stem Cell. 2016;19:690–702. doi: 10.1016/j.stem.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.von Bartheld CS, Bahney J, Herculano-Houzel S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J Comp Neurol. 2016;524:3865–3895. doi: 10.1002/cne.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee G, et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol. 2012;30:1244–1248. doi: 10.1038/nbt.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fattahi F, et al. Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature. 2016;531:105–109. doi: 10.1038/nature16951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Egawa N, et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med. 2012;4:145ra104. doi: 10.1126/scitranslmed.3004052. [DOI] [PubMed] [Google Scholar]
- 179.Xu X, et al. Prevention of beta-amyloid induced toxicity in human iPS cell-derived neurons by inhibition of Cyclin-dependent kinases and associated cell cycle events. Stem cell research. 2013;10:213–227. doi: 10.1016/j.scr.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 180.Hoing S, et al. Discovery of inhibitors of microglial neurotoxicity acting through multiple mechanisms using a stem-cell-based phenotypic assay. Cell Stem Cell. 2012;11:620–632. doi: 10.1016/j.stem.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 181.Kimbrel EA, Lanza R. Current status of pluripotent stem cells: moving the first therapies to the clinic. Nature reviews Drug discovery. 2015;14:681–692. doi: 10.1038/nrd4738. [DOI] [PubMed] [Google Scholar]
- 182.Xu RH, et al. BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat Biotechnol. 2002;20:1261–1264. doi: 10.1038/nbt761. [DOI] [PubMed] [Google Scholar]
- 183.Chin MH, Pellegrini M, Plath K, Lowry WE. Molecular analyses of human induced pluripotent stem cells and embryonic stem cells. Cell Stem Cell. 2010;7:263–269. doi: 10.1016/j.stem.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Guenther MG, et al. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell. 2010;7:249–257. doi: 10.1016/j.stem.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Efroni S, et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008;2:437–447. doi: 10.1016/j.stem.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol. 2006;7:540–546. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- 187.Meshorer E, et al. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379:131–137. doi: 10.1038/379131a0. [DOI] [PubMed] [Google Scholar]
- 189.Papp B, Plath K. Epigenetics of reprogramming to induced pluripotency. Cell. 2013;152:1324–1343. doi: 10.1016/j.cell.2013.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tchieu J, et al. Female human iPSCs retain an inactive X chromosome. Cell Stem Cell. 2010;7:329–342. doi: 10.1016/j.stem.2010.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mekhoubad S, et al. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell. 2012;10:595–609. doi: 10.1016/j.stem.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Heidenreich M, Zhang F. Applications of CRISPR-Cas systems in neuroscience. Nat Rev Neurosci. 2016;17:36–44. doi: 10.1038/nrn.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pedrosa E, et al. Development of patient-specific neurons in schizophrenia using induced pluripotent stem cells. Journal of neurogenetics. 2011;25:88–103. doi: 10.3109/01677063.2011.597908. [DOI] [PubMed] [Google Scholar]
- 195.Hashimoto-Torii K, et al. Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron. 2014;82:560–572. doi: 10.1016/j.neuron.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]