

Abstract
Background
Duchenne muscular dystrophy (DMD) is the most common childhood form of muscular dystrophy. The incidence of cardiomyopathy in DMD increases with age, so its early detection is important because institution of cardioprotective medical therapies may slow adverse remodeling and attenuate heart failure symptoms in these patients.
Objective
To assess the cardiac functions in children clinically suspected to have DMD.
Methods
Over a one-year period, 28 male children aged from 3 to 18 years old, who met the criteria for diagnosis of DMD compared to 47 healthy controls children, were approached to participate in the study. The included children were subjected to full clinical examination, and blood samples were collected to determine creatinine phosphokinase (CPK), troponin I enzyme, myoglobin and lactate dehydrogenase (LDH) enzyme level. Echocardiography and 12-leads electrocardiogram (ECG) were also done for children in both groups. Data were analyzed using Independent-samples t-test, Mann-Whitney U, Chi square, and Fisher’s exact test.
Results
The mean age of the cases group was 7.29±3.24 years versus 8.06±2.86 years for controls. In DMD group, 25% had positive family history of DMD while 35.7% of them had positive consanguinity. All cases had elevated CPK level while CPK level in controls was normal (p<0.0001). LDH level was elevated in 19 cases (67.86%) of DMD while all controls children had normal LDH level (p<0.0001). Furthermore, the mean serum myoglobin level of DMD patients was higher relative to that of healthy controls (39.39±7.25 versus 33.68 ±12.38 ng/ml respectively) (p=0.01). Echocardiography of our patients revealed that seven cases (25%) had low ejection fraction (EF) and fraction shortening (FS). In addition, all controls children had normal EF (p<0.0001) and normal FS (p<0.0001). Interestingly, ECG showed that 28.57% of cases had sinus tachycardia vs. 6.88% for controls (p=0.0001). Prolonged QTc interval was present in 39.29% of cases (mean 431.39±43.60) while all controls had normal QTc duration for age (mean of 415.17±25.2) (p<0.0001).
Conclusion
ECG manifestations in children with DMD in the form of sinus tachycardia and prolonged QTc interval are an early alarm for developing cardiomyopathy before overt echocardiographic findings appear.
Keywords: Duchenne muscular dystrophy (DMD), Echocardiography, Electrocardiography (ECG), Cardiac evaluations
1. Introduction
Duchenne muscular dystrophy (DMD) is a genetic muscle disorder affecting one per 3,500–5,000 live-born males with a prevalence of 21.2/100,000 school aged boys. It is the most common type of muscular dystrophy in childhood (1, 2). DMD pathogenesis is attributed to mutations of the DMD gene which is located on chromosome Xp21, and encodes for dystrophin, a 427 kDa protein expressed at the sarcolemma of cardiac, skeletal, and smooth muscles. Dystrophin, which is located at the muscle cell membrane, develops a mechanical link from the actin cytoskeleton of the cell to the extracellular matrix through the dystrophin-associated glycoprotein complex (DGC) (3). Loss of dystrophin endangers the consistency of the muscle cell membrane and leads to muscle fibers that are extremely vulnerable to contraction-induced injury. This can result in progressive rounds of degeneration and regeneration of the muscle, which can lead to muscle fiber replacement with non-contractile fibrotic tissue and fatty infiltrates. This process of disruption of muscle membrane, brings about clinical features of motor developmental delay, joint contractures, calf hypertrophy, and progressive muscle weakness in affected boys, together with noticeably elevated serum CK that reflects continuing muscle damage. Moreover, there can be a variable degree of speech delay, learning disability, and/or cognitive impairment in boys with DMD (4, 5). Progressive muscle degeneration will ultimately lead to loss of independent ambulation by early adolescence, cardiomyopathy, scoliosis, respiratory insufficiency, and reduced life expectancy, leading to death resulting from cardiorespiratory complications before the third or fourth decade of life, according to recent DMD natural history studies (6, 7). The clinical course of the disease manifests at about 3 years of age (typically, children with DMD display skeletal muscle weakness by age 2–6 years) (8), and with progressing muscle weakness, patients are often wheelchair bound by the early teen years, and suffer cardiac/respiratory failure in their mid- to late twenties (4). This condition segregates in families with an X-linked pattern of inheritance, though approximately 25% of cases are due to de novo gene mutations (9). Following the discovery of the DMD gene, there has been vastly extensive of research, but despite this, there is still no curative treatment for DMD. Nevertheless, many promising lines of research are currently being conducted, including cell-based therapies and pharmacological reagents to upregulate dystrophin via read-through of nonsense mutations or the up-regulation of the dystrophin homolog utrophin (10). Cardiomyopathy is a leading cause of death in DMD patients although the age of onset varies considerably (8, 11, 12). The incidence of cardiomyopathy in DMD increases with age, affecting 30 % of patients by 14 years of age, 50 % of patients by 18 years of age, and approximately 95% of patients develop cardiomyopathy by 20 years of age (13, 14). Cardiomyopathy presents in the early stages of the disease as electrocardiogram abnormalities and sinus tachycardia (15). In adulthood, cardiovascular magnetic resonance (CMR) reveals fibrosis of the left ventricle and ventricular dilation (16, 17). This has been accompanied by abnormalities of rhythm including sinus arrhythmia, atrial flutter, and frequent premature beats atrial or ventricular (18). Ventricular arrhythmias are common in patients with impaired function of the ventricles and are indicative of progressive myocardial decline (19, 20). The clinical guidelines recommend that evaluations for cardiomyopathy, arrhythmia, and heart failure must be done upon diagnosis, every 2 years until age 10 years, and then yearly thereafter (11). Early detection of muscular dystrophy associated cardiomyopathy is important; because institution of cardioprotective medical therapies may slow adverse remodeling and attenuate heart failure symptoms in these patients. So, we conducted this study to evaluate cardiac functions in clinically suspected DMD children by clinical assessment, cardiac biomarkers, echocardiography, and electrocardiography (ECG).
2. Material and Methods
2.1. Study design
A prospective case control study was conducted in the Neuropediatric Outpatient Clinic and Pediatric Cardiology Clinic at Sohag University Hospital over a one-year period from January 2015 through December 2015. The Hospital is a tertiary health institution, located in the south zone of Egypt. A total of 28 male children aged from 3 to 18 years who met criteria for diagnosis of Duchenne muscular dystrophy were compared to 47 healthy controls male children were included in the study.
2.2. Selection criteria
2.2.1. Inclusion criteria
The following were set as the inclusion criteria of the study:
Male children
Fulfilling the criteria for diagnosis of Duchenne muscular dystrophy
Aged from 3 to 18 years
Parents acceptance of participation in the study
Elevated serum creatine kinase (CK) level
Myopathic EMG pattern
2.2.2. Exclusion criteria
We excluded from the study children with other types of neuromuscular disorders e.g. spinal muscle atrophy, neuropathy, other types of myopathy and myasthenia, children less than 3 years and more than 18 years and female children with muscular dystrophy.
2.3. Diagnosis and data collection
The diagnosis of Duchenne muscular dystrophy was made on the basis of the clinical characteristics (Male child with lordotic posture in standing, Gowers sign, Trendelenburg gait or hip waddle, delayed walking, falling, toe walking, trouble running or walking upstairs and developmental delay) ascertained by two experts in child neurology, together with serum creatine kinase (CK) determinations and electromyography (EMG) confirmation. For all the patients enrolled in the study, a checklist that included the needed data was filled by the investigator. The checklist included data of the patients concerning age, sex, duration of weakness, with special attention to family history of neuromuscular disorder and motor development, examination of vital signs, full cardiac and neurologic examination. Blood samples were collected from the patients. They were analyzed for serum CPK (creatinine phosphokinase), and myoglobin level using Cobas c311 (Cobas c311; Roche-Hitachi, Tokyo, Japan). Also assay of troponin I enzyme level (using Architect i1000SR; Abbott Diagnostic, USA), and LDH enzyme level were done and the results were documented in the checklist. All patients had 2-D and M-mode echocardiographic examinations which included evaluation of left ventricular systolic function. Assessment of ejection fraction (Ef) and fractional shortening (FS) and left ventricular dilatation. Results were analyzed in accordance with the American Society of Echocardiography standards. Furthermore, 12 leads ECG was performed to all children (Fukuda Denshi CardiMax ECG device model FCP-7101 with a 25 mm/s paper speed, gain 10 mm/mV). All electrocardiograms were evaluated through the construction of descriptive reports, and determination of variables such as heart rate, QRS duration and QRS amplitude. QT interval was calculated by the onset of the QRS complex to the end of the T wave, defined by the return of the terminal T wave to the isoelectric TP baseline. The end of the T wave was taken as the nadir between the T and U waves, when U waves were present. Next, using Bazett’s formula, QT interval was corrected for heart rate (21). Interpretation of all ECG papers was carried out using specific centile tables for normal values of ECG waves and intervals according to age (22). ECG was assessed for detection of abnormal rhythm (ectopics, supraventricular or ventricular tachycardia).
2.4. Statistical analysis
Data was analyzed by STATA intercooled version 12.1 using descriptive statistics and Independent-samples t-test, Mann-Whitney U test, Chi square, and Fisher’s exact test. Graphs were produced by using Excel or STATA program. P value was considered significant if it was less than 0.05.
2.5. Ethics of research
Informed consent of the parents of children was taken to conduct this research in addition to the approval of the Faculty of Medicine, Sohag University Ethics committee (Declaration No 185 Date 16/2/2015, Faculty of Medicine, Sohag University Meeting). This work was carried out in accordance with The Code of Ethics of The World Medical Association (Declaration of Helsinki) for experiments in humans.
3. Results
Our study involved 28 clinically suspected Duchenne muscular dystrophy children and 47 age-matched healthy male children as controls. The mean age of the study group was 7.29±3.24 years, with range from 3–14 years and the mean age of the control group was 8.06±2.86 years, with age range from 3 to 14 years (p=0 .28). Twenty-five percent of cases had positive family history of DMD while 35.7% of cases had positive consanguinity. All reported cases had shown the onset of symptoms and muscle weakness between 4–6 years of age. Twenty-five patients had abnormal gait, three were already wheelchair- bound. Heart rate was above normal ranges in 7 cases (25%). In our study we, found that all the cases had elevated CPK level (creatine phosphokinase). The mean CPK level of cases was 2495.79±3457.02 and range was from 425 to 114 52. While CPK level in controls was normal, (p<0.0001). LDH level was elevated in 19 cases (67.86%). The mean serum LDH level in cases was 1232.07±617.72. All controls children had normal LDH level with mean 163.02±109.62 (p<0.0001).The mean serum myoglobin level of (DMD) patients was higher relative to that of healthy controls (but still within normal limits; 39.39±7.25 versus 33.68 ±12.38 ng/ml respectively) with statistically significant difference (p=0.01).There was no statistically significant difference between mean serum troponin I level of DMD patients and that of controls (0.03±0.05 vs. 0.01±0.02 U/L respectively) (p=0.37) (Table 1). Echocardiography of our patients revealed that one patient (3.57%) had mild dilation of the left ventricular cavity which was statistically not significant, (p value: 0.37). A further 7 cases (25%) of patients had low ejection fraction (EF) and fraction shortening (FS) with mean (EF) 59.86±10.20 and range from 45 to 80. All controls children had normal EF with mean 73.30±5.08 and range from 75 to 80, (p<0.0001). The mean FS was 20.29±5.59 with range from 22 to 41. All controls children had normal (FS) with mean ±SD of 36.17±2.58 and range from 28 to 40, (p<0.0001) (Table 2). Electrocardiogram showed that 28.57% of cases had sinus tachycardia with average heart rate 107±12.32 while 6.88% of controls had sinus tachycardia with average heart rate of control group being 96.02±12.35 (p=0.0001). Prolonged QTc interval was present in 39.29% of cases, the mean QTc interval was 431.39±43.60 and that of controls was 415.17±25.24 (p<0.0001). Correlating QTc interval to ejection fraction revealed that no significant correlation was present (p value= 0.8). All QTc duration values of controls were normal for age. Other parameters of ECG including (p-wave amplitude and duration, PR interval QRS duration, axis and voltage and QT interval) were normal for age; no type of arrhythmia could be detected apart from sinus tachycardia (Table 3, Figure 1).
Table 1.
Serum biomarkers in studied populations
| Parameter | Cases (n=28) | Controls (n=47) | p-value | |
|---|---|---|---|---|
| CPK level; Mean±SD | 2495.79±3457.02 | 36.19±10.11 | <0.0001 | |
| CPK group; n (%) | High | 28 (100%) | 0 | <0.0001 |
| Normal | 0 | 47 (100%) | ||
| Troponin I; Mean±SD | 0.03±0.05 | 0.01±0.02 | 0.37 | |
| Troponin I group; n (%) | 28 (100%) | 47 (100%) | 1.00 | |
| Myoglobin level; Mean±SD | 39.39±7.25 | 33.68±12.38 | 0.01 | |
| Myoglobin group; n (%) | 28 (100%) | 47 (100%) | 1.00 | |
| LDH level; Mean±SD | 1232.07±617.72 | 163.02±109.62 | <0.0001 | |
| LDH group; n (%) | High | 19 (67.86%) | 0 | <0.0001 |
| Normal | 9 (32.14%) | 47 (100%) | ||
Table 2.
Echocardiographic parameters in studied populations
| Parameter | Cases | Controls | p-value | |
|---|---|---|---|---|
| EF; Mean ± SD | 59.86±10.20 | 73.30±5.08 | <0.0001 | |
| EF group; n (%) | Low | 7 (25.00%) | 0 | 0.0001 |
| Normal | 21 (75.00%) | 47 (100%) | ||
| FS; Mean ± SD | 20.29±5.59 | 36.17±2.58 | <0.0001 | |
| FS group; n (%) | Low | 7 (25.00%) | 0 | 0.001 |
| Normal | 21 (75.00%) | 47 (100%) | ||
| Left ventricular dilation; n (%) | Yes | 1 (3.57%) | 0 | 0.37 |
| No | 27 (96.43%) | 47 (100%) | ||
Table 3.
Electrocardiographic parameters in studied populations
| Parameter | Cases | Controls | p-value | |
|---|---|---|---|---|
| Heart rate (BPM) | Mean ± SD | 107.82±12.32 | 96.02±12.35 | 0.0001 |
| High | 8 (28.57%) | 3 (6.88%) | 0.009 | |
| Normal | 20 (71.43%) | 44 (93.62%) | ||
| Rhythm | Normal | 28 (100%) | 47 (100%) | 1.00 |
| p wave amplitude (mv) in Lead II | Mean ± SD | 0.12±0.01 | 0.12±0.01 | 0.17 |
| Normal | 28 (100%) | 47 (100%) | 1.00 | |
| p wave amplitude (mv) in Lead V1 | Mean ± SD | 0.11±0.005 | 0.11±0.004 | 0.66 |
| Normal | 28 (100%) | 47 (100%) | 1.00 | |
| p wave amplitude (mv) in Lead V2 | Mean ± SD | 0.11±0.006 | 0.11±0.005 | 0.77 |
| Normal | 28 (100%) | 47 (100%) | 1.00 | |
| P duration (ms) | Mean ± SD | 80.75±7.86 | 85.47±12.74 | 0.08 |
| Normal | 27 (96.43%) | 47 (100%) | 0.37 | |
| Low | 1 (3.57%) | 0 | ||
| PR interval | Mean ± SD | 123.96±17.03 | 130.32±17.99 | 0.14 |
| Normal | 28 (100%) | 47 (100%) | 1.00 | |
| QRS duration | Mean ± SD | 86.14±17.82 | 90.87±15.71 | 0.23 |
| Normal | 26 (92.86%) | 46 (97.87%) | 0.55 | |
| High | 2 (7.14%) | 1 (2.13%) | ||
| QT duration (ms) | Mean ± SD | 336.71±30.13 | 335.45±26.42 | 0.84 |
| Normal | 28 (100%) | 47 (100%) | 1.00 | |
| QTC duration (ms) | Mean ± SD | 431.39±43.60 | 415.17±25.24 | 0.04 |
| High | 11 (39.29%) | 2 (4.26%) | <0.0001 | |
| Normal | 17 (60.71%) | 45 (95.74%) | ||
| QRS axis | Mean ± SD | 56.61±22.08 | 56.32±22.62 | 0.81 |
| Normal | 28 (100%) | 47 (100%) | 1.00 | |
Figure 1.
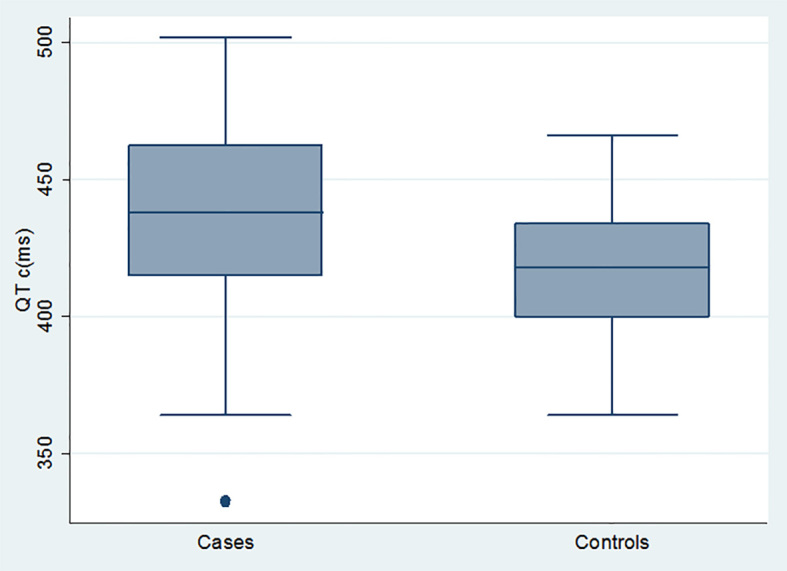
Comparison between cases and controls as regards QTc duration (ms).
4. Discussion
Basically, Duchenne muscular dystrophy is an inherited X-linked disease associated with absence of dystrophin (23) and characterized by progressive muscle weakness and inescapable cardiac involvement (24). Early detection of muscular dystrophy associated cardiomyopathy is paramount, as establishment of cardioprotective medical therapies may slow adverse remodeling and ameliorate heart failure symptoms in these patients (25–28). This study highlighted the cardiac affection in these children with DMD and focuses on early detection of subclinical cardiomyopathy as it has now become a major cause of morbidity and mortality with increased life expectancy of DMD patients. The age of our patients ranged from 3–14 years. In DMD group, 25% had positive family history of DMD while 35.7% of them had positive consanguinity. In a study by Gesmar et al., the age ranged from 6 to 14 years and 45% of their cases had a positive family history of DMD (29). Creatine kinase, troponin I, myoglobin, and lactate dehydrogenase are serum markers of muscle injury (30–32). All cases in our study had high CPK level, as it is a mainstay in diagnosis of DMD, there was also a statistically significant difference between cases and controls as regard LDH and myoglobin levels. Our results were relatively near to the results of Hathout et al. (33). All DMD patients’ serum levels of troponin I were normal, which disagrees with the Hathout study and can be explained by the lower age range of our patients. Cardiomyopathy that develops in DMD is characterized by normal or thinned left ventricular (LV) wall thickness and progressive decline in ejection fraction or fractional shortening. Variable degrees of LV dilation occur. Abnormal LV relaxation has also been identified as an early manifestation in DMD (34). Echocardiography of our patients revealed that 25% of patients had ejection fraction (EF) below 56% with mean 59.86±10.20 and range from 45 to 80. Our echocardiographic findings agreed with Jefferies et al., who documented that cardiac disease in a DMD population usually consists of dilated cardiomyopathy, which causes progressive decline in the ejection fraction and fractional shortening, and generally evolves to heart failure, even with concomitant arrhythmia (14). In the study of Thrush et al., 34% of DMD cases developed cardiomyopathy with follow up (35), and 30% of DMD cases in the study of Thomas et al. developed cardiomyopathy (36). Spurney et al. documented that the age of onset varies considerably, but abnormalities in systolic function are present in more than 80 % of boys older than 18 years of age (37). Electrocardiogram showed 28.57% of cases had sinus tachycardia with average heart rate (HR) of 107±12.32. These results were in agreement with that of the Thomas et al. study which hypothesized that an elevated heart rate would herald cardiomyopathy onset. Thomas’ study also established that heart rate elevation is a statistically significant risk factor for cardiomyopathy (38). In our study, we also found prolonged QTc interval in 39.29% of cases, the mean of QTc interval was 431.39 ± 43.60. All values QTc duration of controls were normal for age. In our study, QTc prolongation was present regardless of presence or absence of echocardiographic findings of cardiomyopathy, this agrees with findings of Thrush et al., who postulated that in their study, ECG changes are similar in patients with DMD regardless of presence of dilated cardiomyopathy (35). Martin et al. documented that a significant number of patients (24% to 32%) with dilated or hypertrophic cardiomyopathy may have a long QTc interval on the surface ECG, and ventricular hypertrophy/dilation may be additional rare causes of acquired prolongation of the QT interval. A lengthening of the QT interval predisposes to torsade de pointes ventricular tachycardia, which can degenerate into ventricular fibrillation (38).
5. Conclusions
Cardiac affection, especially cardiomyopathy, is a common problem in children with DMD and could present in the early stages of the disease as electrocardiogram abnormalities and sinus tachycardia. This study reported ECG manifestations in children with DMD in the form of sinus tachycardia and prolonged QTc interval, are an early alarm for developing cardiomyopathy before overt echocardiographic findings appear. We recommend further exploration of this topic in the future, on a large series of patients with longer follow up periods.
Acknowledgments
Authors thank colleagues in the Pediatric Department, Faculty of Medicine, Sohag University (Egypt) for their assistance, and the parents and children who participated in this study.
Footnotes
iThenticate screening: August 26, 2017, English editing: October 13, 2017, Quality control: October 15, 2017
Conflict of Interest:
There is no conflict of interest to be declared.
Authors’ contributions:
All authors contributed to this project and article equally. All authors read and approved the final manuscript.
References
- 1.Emery AE. Population frequencies of inherited neuromuscular diseases – a world survey. NeuromusculDisord. 1991;1(1):19–29. doi: 10.1016/0960-8966(91)90039-U. [DOI] [PubMed] [Google Scholar]
- 2.Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. NeuromusculDisord. 2014;24(6):482–491. doi: 10.1016/j.nmd.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Matsumura K, Tomé FM, Collin H, Azibi K, Chaouch M, Kaplan JC, Fardeau M, Campbell KP. Deficiency of the 50K dystrophin-associated glycoprotein in severe childhood autosomal recessive muscular dystrophy. Nature. 1992;359(6393):320–2. doi: 10.1038/359320a0. [DOI] [PubMed] [Google Scholar]
- 4.Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 5.Pane M, Lombardo ME, Alfieri P, et al. Attention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: phenotype-genotype correlation. J Pediatr. 2012;161(4):705.e1–709.e1. doi: 10.1016/j.jpeds.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 6.McDonald CM, Henricson EK, Abresch RT, et al. The cooperative international neuromuscular research group Duchenne natural history study – a longitudinal investigation in the era of glucocorticoid therapy: design of protocol and the methods used. Muscle Nerve. 2013;48(1):32–54. doi: 10.1002/mus.23807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kieny P, Chollet S, Delalande P, et al. Evolution of life expectancy of patients with Duchenne muscular dystrophy at AFM Yolaine de Kepper Centre between 1981 and 2011. Ann PhysRehabil Med. 2013;56(6):443–454. doi: 10.1016/j.rehab.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Boland BJ, Silbert PL, Groover RV, Wollan PC, Silverstein MD. Skeletal, cardiac, and smooth muscle failure in Duchenne muscular dystrophy. Pediatr Neurol. 1996;14(1):7–12. doi: 10.1016/0887-8994(95)00251-0. [DOI] [PubMed] [Google Scholar]
- 9.Tuffery-Giraud S, Béroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009;30(6):934–45. doi: 10.1002/humu.20976. [DOI] [PubMed] [Google Scholar]
- 10.Goyenvalle A, Seto JT, Davies KE, Chamberlain J. Therapeutic approaches to muscular dystrophy. Hum Mol Genet. 2011;20(R1):R69–78. doi: 10.1093/hmg/ddr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bushby K, Muntoni F, Bourke JP. Neuromuscul Disord; 107th ENMC International Workshop: The Management of Cardiac Involvement in Muscular Dystrophy and Myotonic Dystrophy; 7th–9th June 2002; Naarden, the Netherlands. 2003. pp. 166–172. [DOI] [PubMed] [Google Scholar]
- 12.Wagner KR, Lechtzin N, Judge DP. Current treatment of adult Duchenne muscular dystrophy. BiochimBiophysActa. 2007;1772(2):229–37. doi: 10.1016/j.bbadis.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Kwon SW, Kang SW, Kim JY, Choi EY, Yoon YW, Park YM, et al. Outcomes of cardiac involvement in patients with late-stage Duchenne muscular dystrophy under management in the pulmonary rehabilitation center of a tertiary referral hospital. Cardiology. 2012;121(3):186–93. doi: 10.1159/000336810. [DOI] [PubMed] [Google Scholar]
- 14.Shirokova N, Niggli E. Cardiac phenotype of duchenne muscular dystrophy: Insights from cellular studies. J Mol Cell Cardiol. 2013;58:217–224. doi: 10.1016/j.yjmcc.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in duchenne muscular dystrophy. Int J Cardiol. 1990;26:271–277. doi: 10.1016/0167-5273(90)90082-G. [DOI] [PubMed] [Google Scholar]
- 16.Hor KN, Taylor MD, Al-Khalidi HR, Cripe LH, Raman SV, Jefferies JL, O’Donnell R, Benson DW, Mazur W. Prevalence and distribution of late gadolinium enhancement in a large population of patients with duchenne muscular dystrophy: Effect of age and left ventricular systolic function. J Cardiovasc Magn Reson. 2013;15:107. doi: 10.1186/1532-429X-15-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazur W, Hor KN, Germann JT, Fleck RJ, Al-Khalidi HR, Wansapura JP, Chung ES, Taylor MD, Jefferies JL, Benson DW, et al. Patterns of left ventricular remodeling in patients with duchenne muscular dystrophy: A cardiac mri study of ventricular geometry, global function, and strain. Int J Cardiovasc Imaging. 2012;28:99–107. doi: 10.1007/s10554-010-9781-2. [DOI] [PubMed] [Google Scholar]
- 18.Hermans MC, Pinto YM, Merkies IS, de Die-Smulders CE, Crijns HJ, Faber CG. Hereditary muscular dystrophies and the heart. Neuromusc Disord. 2010;20:479–492. doi: 10.1016/j.nmd.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Yanagisawa A, Miyagawa M, Yotsukura M, Tsuya T, Shirato C, Ishihara T, Aoyagi T, Ishikawa K. The prevalence and prognostic significance of arrhythmias in duchenne type muscular dystrophy. Am Heart J. 1992;124:1244–1250. doi: 10.1016/0002-8703(92)90407-M. [DOI] [PubMed] [Google Scholar]
- 20.Chenard AA, Becane HM, Tertrain F, de Kermadec JM, Weiss YA. Ventricular arrhythmia in duchenne muscular dystrophy: Prevalence, significance and prognosis. Neuromusc Disord. 1993;3:201–206. doi: 10.1016/0960-8966(93)90060-W. [DOI] [PubMed] [Google Scholar]
- 21.Hampton JR. The ECG Made Easy. 7th edition. London: Churchill Livingstone; 2008. [Google Scholar]
- 22.Rijnbeek PR, Witsenburg M, Schrama E, Hess J, Kors JA. New normal limits for the paediatric electrocardiogram. Eur Heart J. 2001;22(8):702–11. doi: 10.1053/euhj.2000.2399. [DOI] [PubMed] [Google Scholar]
- 23.Emery AEH. The Muscular Dystrophies. 2nd edition. Oxford: Oxford University Press; 1993. Duchenne muscular dystrophy or Meryon’s disease; pp. 55–71. [DOI] [Google Scholar]
- 24.Melacini P, Vianello A, Villanova C, Fanin M, Miorin M, Angelini C, et al. Cardiac and respiratory involvement in advanced stage Duchenne muscular dystrophy. NeuromusculDisord. 1996;6(5):367–76. doi: 10.1016/0960-8966(96)00357-4. [DOI] [PubMed] [Google Scholar]
- 25.Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain A, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J. 2007;154(3):596–602. doi: 10.1016/j.ahj.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 26.Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, et al. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112(18):2799–804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- 27.Kajimoto H, Ishigaki K, Okumura K, Tomimatsu H, Nakazawa M, Saito K, et al. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ J. 2006;70(8):991–4. doi: 10.1253/circj.70.991. [DOI] [PubMed] [Google Scholar]
- 28.Ramaciotti C, Heistein LC, Coursey M, Lemler MS, Eapen RS, Iannaccone ST, et al. Left ventricular function and response to enalapril in patients with duchenne muscular dystrophy during the second decade of life. Am J Cardiol. 2006;98(6):825–7. doi: 10.1016/j.amjcard.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 29.Herdy Gesmar Volga Haddad, Pinto Roberta Duarte Bezerra, de Almeida Costa Guilherme, Torbey Ana Flavia Malheiros, Ramos Vivianne Galante, Vasconcelo Marcio Moacyr. Clinical and molecular study on Duchenne muscular dystrophy. International Journal of Cardiovascular Sciences. 2015;28(3):173–180. 2015. [Google Scholar]
- 30.Ohlendieck K. Proteomic identification of biomarkers of skeletal muscle disorders. Biomark Med. 2013;7(1):169–86. doi: 10.2217/bmm.12.96. [DOI] [PubMed] [Google Scholar]
- 31.Carp SJ, Barr AE, Barbe MF. Serum biomarkers as signals for risk and severity of work-related musculoskeletal injury. Biomark Med. 2008 Feb;2(1):67–79. doi: 10.2217/17520363.2.1.67. [DOI] [PubMed] [Google Scholar]
- 32.Brancaccio P, Lippi G, Maffulli N. Biochemical markers of muscular damage. ClinChem Lab Med. 2010;48(6):757–67. doi: 10.1515/CCLM.2010.179. [DOI] [PubMed] [Google Scholar]
- 33.Hathout Y, Brody E, Clemens PR, Cripe L, DeLisle RK, Furlong P, et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. ProcNatlAcadSci U S A. 2015;112(23):7153–8. doi: 10.1073/pnas.1507719112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Markham LW, Michelfelder EC, Border WL, Khoury PR, Spicer RL, Wong BL, et al. Abnormalities of diastolic function precede dilated cardiomyopathy associated with Duchenne muscular dystrophy. J Am SocEchocardiogr. 2006;19(7):865–71. doi: 10.1016/j.echo.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 35.Thrush PT, Allen HD, Viollet L, Mendell JR. Re-examination of the electrocardiogram in boys with Duchenne muscular dystrophy and correlation with its dilated cardiomyopathy. Am J Cardiol. 2009;103(2):262–5. doi: 10.1016/j.amjcard.2008.08.064. [DOI] [PubMed] [Google Scholar]
- 36.Thomas TO, Morgan TM, Burnette WB, Markham LW. Correlation of heart rate and cardiac dysfunction in Duchenne muscular dystrophy. PediatrCardiol. 2012;33(7):1175–9. doi: 10.1007/s00246-012-0281-0. [DOI] [PubMed] [Google Scholar]
- 37.Spurney CF. Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve. 2011;44(1):8–19. doi: 10.1002/mus.22097. [DOI] [PubMed] [Google Scholar]
- 38.Martin AB, Garson A, Jr, Perry JC. Prolonged QT interval in hypertrophic and dilated cardiomyopathy in children. Am Heart J. 1994;127(1):64–70. doi: 10.1016/0002-8703(94)90510-X. [DOI] [PubMed] [Google Scholar]