

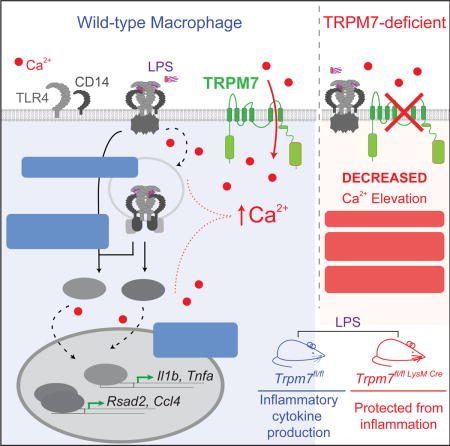
SUMMARY
Toll like receptors (TLRs) sense pathogen-associated molecular patterns to activate the production of inflammatory mediators. TLR4 recognizes lipopolysaccharide (LPS) and drives the secretion of inflammatory cytokines, often contributing to sepsis. We report that Transient receptor potential melastatin-like 7 (TRPM7), a non-selective but Ca2+-conducting ion channel, mediates the cytosolic Ca2+ elevations essential for LPS-induced macrophage activation. LPS triggered TRPM7-dependent Ca2+ elevations essential for TLR4 endocytosis and the subsequent activation of the transcription factor IRF3. In a parallel pathway, the Ca2+-signaling initiated by TRPM7 was also essential for the nuclear translocation of NFκB. Consequently, TRPM7-deficient macrophages exhibited major deficits in the LPS-induced transcriptional programs, failing to produce IL-1β and other key pro-inflammatory cytokines. In accord with these defects, mice with myeloid-specific deletion of Trpm7 are protected from LPS-induced peritonitis. Our study highlights the importance of Ca2+-signaling in macrophage activation and identifies the ion channel TRPM7 as a central component of TLR4 signaling.
Graphical abstract
INTRODUCTION
Cells of the innate immune system trigger finely-tuned cellular responses for host defense, while minimizing collateral damage to the host (Iwasaki and Medzhitov, 2015). An array of pattern-recognition receptors (PRRs) sense pathogen-associated molecular patterns (PAMPs) to activate the downstream intracellular signaling machinery necessary for mediating host defense (Brubaker et al., 2015). A family of membrane-resident PRRs called the Toll-like receptors (TLRs) are of central importance because their ectodomains sense the presence of PAMPs, eliciting cellular responses that fit the pattern of infection (Gay et al., 2014). According to an emerging framework, the subcellular location of activated TLRs plays a pivotal role in the choice of specific adaptors protein, linked signaling pathways, and elaboration of suitable transcriptional programs (Barton and Kagan, 2009; O’Neill and Bowie, 2007). This paradigm is clearly evident in the case of TLR4, a major sensor of bacterial lipopolysaccharide (LPS) (Poltorak et al., 1998). Using different adaptors, TLR4 signals distinctly from the cell membrane and early endosomes, expanding the repertoire of transcribed genes and refining the cellular processes for a potent immunological response.
LPS triggers dimerization of TLR4 at the cell membrane and promotes the assembly of “Myddosome”, the myeloid-differentiation factor 88 (MyD88)-containing adaptor complex, to initiate the activation of NFκB. The interaction of TLR4 with co-receptors, such as CD14 and MD2 (Tan et al., 2015; Zanoni et al., 2011), also promotes the endocytosis of LPS-bound TLR4 receptor complex. The formation of TLR4-containing signaling endosomes triggers the association of TLR4 to TRIF (and its coadaptor TRAM). Although both MyD88 and TRIF drive the activation and nuclear translocation of NFκB, TRIF uniquely promotes the activation of transcription factor IRF3, resulting in the expression of Type I interferon response genes (Kagan et al., 2008). Thus, TLR4 endocytosis serves as a pivotal regulatory event to amplify NFκB activation and modulate the cellular response through the additional activation of IRF3. The mechanisms that regulate the initiation and rate of TLR4 endocytosis during LPS stimulation are therefore of paramount importance to understanding the full spectrum of TLR4 signaling in inflammatory conditions. In addition to illuminating the underlying role of membrane traffic in inflammatory signaling, defining the mechanisms of TLR4 endocytosis may reveal novel pharmacological targets for the treatment of sepsis.
The LPS-induced endocytosis of TLR4 and the subsequent TRIF-mediated IRF3 activation are highly dependent on CD14, a GPI-linked cell surface protein. CD14 has been proposed to promote the activation of Syk and phospholipase C (PLC)-γ2 (PLC-γ2), which are necessary for the Ca2+-dependent endocytosis of TLR4 (Chiang et al., 2012; Zanoni et al., 2011). This model suggested a role for store-operated Ca2+ entry (SOCE) in the regulation of TLR4 signaling. Similarly, in addition to the essential role in TLR4 endocytosis, intracellular Ca2+ elevations regulate NFκB activity (Dolmetsch et al., 1997; Nelson et al., 2004) and NLRP3 inflammasome activation (Afonina et al., 2015; Brough et al., 2003). Interestingly, although the requirement for Ca2+ signaling in inflammatory signaling is abundantly clear, complete loss of SOCE in macrophages deficient in Stim1 and Stim2 does not affect their response to LPS (Vaeth et al., 2015). This indicates that Orai channels, the molecular conduits of SOCE, are dispensable for LPS-induced cytokine production and NLRP3 inflammasome activation in macrophages. The real identity of the ion channel(s) responsible for LPS-triggered Ca2+-signaling and TLR4 endocytosis, therefore, emerges as a mystery of biological and pharmacological significance.
Transient Receptor Potential (TRP) channels constitute a large family of cation-selective ion channels (Nilius and Szallasi, 2014). Despite the expression of many TRP channels in hematopoietic cells, their functions in the immune system remain largely undefined. The chanzyme TRPM7, a TRP channel that also contains an enzymatic kinase domain, is highly expressed in macrophages, but whether it has a role in processes pertinent to innate immunity is not known. TRPM7 is permeable to Na+, Ca2+, Mg2+ and Zn2+, and previously, it has been implicated in the endocytosis of Fas receptor (Desai et al., 2012) – a death receptor that, similar to TLR4, requires the generation of signaling endosomes for full activation during apoptosis. TRPM7 channel is regulated by various signals pertinent to inflammation, including caspase-dependent cleavage (Desai et al., 2012) and extracellular pH (Jiang et al., 2005). Furthermore, TRPM7 channel activity is highly sensitive to membrane PIP2 levels (Langeslag et al., 2007), which implies regulatory inputs through PLC activation. Based on these observations, we hypothesized that TRPM7 may have an important function in macrophage activation by PAMPs. Using a novel mouse line Trpm7fl/fl (LysM Cre) mice, wherein TRPM7 is selectively deleted in myeloid cells including macrophages, we have discovered that LPS-mediated macrophage activation is highly dependent on TRPM7. Mice with Trpm7 deleted selectively in macrophages are resistant to LPS-induced peritonitis. We show that TRPM7 mediates the LPS-induced Ca2+-elevations that are essential for TLR4 endocytosis (and therefore, IRF3 activation) and for NFκB nuclear translocation. Since TRP channels are highly attractive drug targets (Moran et al., 2011), these discoveries may stimulate novel pharmacological strategies for the treatment of chronic infections, septic shock, and autoinflammatory diseases.
RESULTS
Trpm7-deficient macrophages from Trpm7fl/fl (LysM Cre) mice develop normally
To study TRPM7 function in macrophages, we deleted Trpm7 selectively in myeloid cells by generating Trpm7fl/fl(LysM Cre) mice in a mixed background of sv129 and C57BL/6J (schematized in Fig. 1A). Through PCR-based genotyping, we confirmed that the LoxP-flanked exon 17 of Trpm7 was deleted in the bone marrow-derived macrophages (BMDMs) from Trpm7fl/fl (LysM Cre) (herein, Trpm7−/− or KO) mice, but not in the BMDMs derived from Trpm7fl/fl (herein, Trpm7+/+ or WT) or Trpm7fl/fl(Lck Cre) mice, which deletes Trpm7 in thymocytes (Fig. S1A). The qRT-PCR-based analysis of TRPM7 mRNA expression levels, using exon 17-directed primers, showed substantially decreased levels of TRPM7 mRNA in Trpm7−/− BMDMs, relative to WT BMDMs (Fig. S1B). We also confirmed that the deletion was not detectable in lymphoid cells derived from Trpm7fl/fl(LysM Cre) mice (data not shown). The deletion of exon 17 generates a frameshift in protein translation, resulting in a null mutant (Jin et al., 2008). To confirm the loss of TRPM7 channel activity in Trpm7−/− macrophages, we used whole-cell patch clamp electrophysiology to record Mg2+-inhibitable TRPM7 currents (ITRPM7) in Trpm7+/+ and Trpm7−/− peritoneal macrophages. The configuration and parameters used for these recordings are shown in Figure S1C. As shown in the current-voltage relationship (Fig. 1B, left), the Trpm7+/+ macrophages elicit robust outwardly rectifying (blue trace) and Mg2+-inhibitable (orange trace) ITRPM7, but this current is absent in Trpm7−/− macrophages (red trace). In Trpm7−/− macrophages, the mean current density for ITRPM7 was severely abrogated (WT, 38.9 pA/pF; KO, 9.8 pA/pF; n=5), and the residual current was not inhibitable by Mg2+ (Fig. 1B, right). Deletion of Trpm7 did not affect the cellularity of bone marrow; both Trpm7fl/fl and Trpm7fl/fl (LysM Cre) mice yielded equal numbers of isolated bone marrow cells (Fig S1D). The ability of bone marrow cells to differentiate ex vivo into macrophages, in the absence of TRPM7, was also normal based on flow cytometric analysis of differentiated cells. Expression of the characteristic macrophage surface markers CD11b and F4/80 (Geissman et al., 2010) was comparable between Trpm7+/+ and Trpm7−/− BMDMs (Fig. S1E) when immunophenotyped at day 4 (d4) and d7 in culture (Fig. S1F). The cultured Trpm7−/− BMDMs did not show any significant deficits in their proliferation (Fig. S1G). Collectively, these results demonstrate that Trpm7 is efficiently deleted in macrophages obtained from Trpm7fl/fl(LysM Cre) mice without causing any deficits in macrophage development and proliferation. These results, therefore, enable a systematic ‘loss of function’ analysis of TRPM7 in macrophage-mediated innate immunity.
Figure 1. Characterization of Trpm7fl/fl (LysM Cre) mice and Trpm7-deficient macrophages.
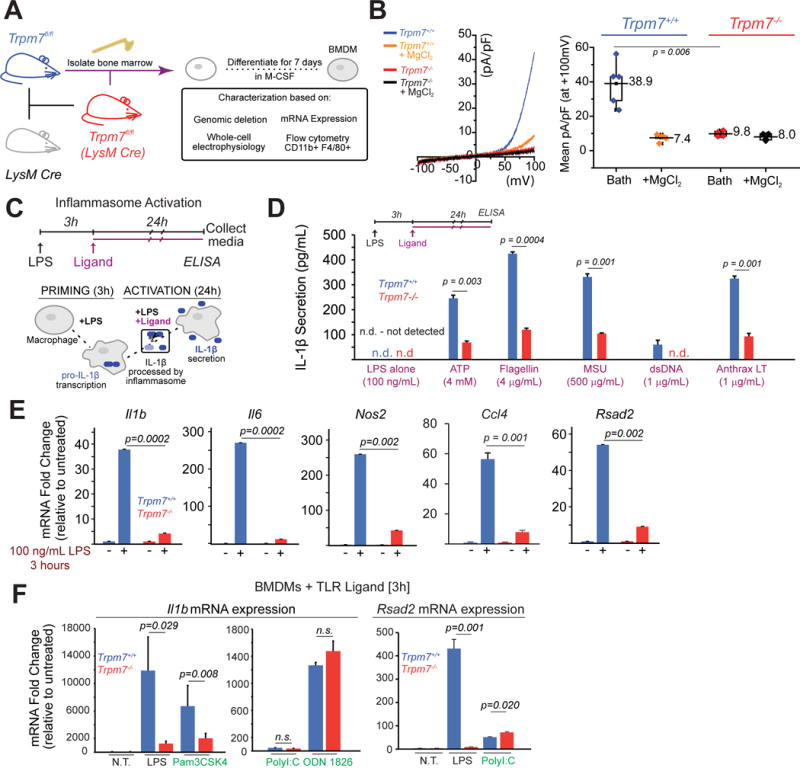
(A) Schematic showing generation of Trpm7fl/fl (LysM Cre) mice and bone-marrow-derived macrophage (BMDM) culture.
(B) A representative I-V relationship of whole-cell Mg2+-inhibitable ITRPM7 in freshly isolated Trpm7fl/fl [Trpm7+/+] and Trpm7fl/fl (LysM Cre) [Trpm7−/−] peritoneal macrophages (left panel). The statistics of TRPM7 current densities (n=5) are shown as box charts (right panel).
(C) Schematic of inflammasome activation assay used for results shown in panel D. The inflammasome activating ligands were added after 3h of LPS priming (100 ng/mL). The secreted IL-1β in the culture supernatants (collected at 24h) was quantified by ELISA.
(D) Quantification of secreted IL-1β in the supernatants of Trpm7+/+ and Trpm7−/− BMDMs, stimulated with indicated inflammasome activating ligands, as depicted in panel C. Error bars represent SEM, n=4.
(E) Gene expression analysis (qRT-PCR) of Trpm7+/+ and Trpm7−/− BMDMs after stimulation with 100 ng/mL LPS (3h). Changes in mRNA levels, relative to untreated BMDMs are shown. Error bars represent SD (Means representative of n=3 independent experiments).
(F) Gene expression analysis (qRT-PCR) of Trpm7+/+ and Trpm7−/− BMDMs after stimulation with indicated ligand (3h). BMDMs treated with LPS (100 ng/ml), Pam3CSK4 (100 ng/mL), PolyI:C (25 μg/mL), or ODN 1826 (1 μM). Changes in mRNA levels, relative to untreated BMDMs are shown. Error bars represent SD (Means representative of n=3 independent experiments).
TRPM7 is required for LPS-induced inflammatory gene expression in macrophages
Activation of macrophages by PAMPs induces a program of inflammatory gene expression and the eventual secretion of inflammatory cytokines (Medzhitov and Horng, 2009). The secretion of potent inflammatory cytokines IL-1β and IL-18 however, also requires additional regulatory inputs from the inflammasome machinery (Garlanda et al., 2013). The “activating ligands” of various inflammasome complexes trigger the caspase-mediated maturation and unconventional secretion of IL-1β and IL-18 (Broz and Dixit, 2016). Interestingly, both the LPS-induced priming of Il1b gene and the inflammasome-mediated secretion of IL-1β are dependent on Ca2+-signaling (Murakami et al., 2012; Rada et al., 2014), but the identity of the pertinent ion channels is not known. To test the hypothesis that TRPM7 controls inflammasome-dependent IL-1β secretion, we primed Trpm7+/+ and Trpm7−/− BMDMs with LPS (100ng/mL, 3h) and then stimulated the BMDMs with various inflammasome activating ligands (Broz and Dixit, 2016; Perregaux and Gabel, 1994) (schematized in Fig. 1C). Trpm7−/− BMDMs displayed a drastic reduction (~75% decrease) in IL-1β secretion, relative to Trpm7+/+ BMDMs, across all activation conditions (Fig. 1D). Concomitantly, we also tested the hypothesis that TRPM7 regulates LPS-induced inflammatory gene expression in macrophages. After LPS treatment (100 ng/mL, 3h), we observed substantially reduced upregulation of genes involved in the inflammatory response, with striking reductions in Il1b, Nlrp3, Il6, Tnfa, Nos2, Ccl4, and Rsad2 (Fig. 1E and Fig. S1J). Treatment of Trpm7+/+ or Trpm7−/− macrophages with LPS did not alter Trpm7 mRNA expression (Fig. S1H), nor did expression of LysM Cre in mixed-background mice with normal Trpm7 alleles (Trpm7+/+) decrease LPS responsiveness in BMDMs (Fig. S1I).
Next, we reasoned that if TRPM7 was directly regulating TLR4 signaling, acute siRNA-mediated knockdown of Trpm7 mRNA should also result in defective LPS response. We successfully knocked down Trpm7 mRNA with siRNA (30 nM) in RAW 264.7 cells, a murine macrophage line (Fig. S2A). RAW 264.7 cells transfected with anti-TRPM7 siRNA, but not control siRNA, exhibited significant reductions in Nos2, Tnfa, and Il6 mRNA expression after LPS treatment (Fig. S2A). Similarly, treatment of BMDMs with a potent TRPM7 channel inhibitor FTY720 (Qin et al., 2013) reduced LPS-induced expression of Il6 and Il1b mRNA by over 80% relative to LPS alone (Fig. S2B). Interestingly, in RAW 264.7 cells, FTY720 reduced LPS-induced TNFα transcription only modestly (Fig. S2C), suggesting that TRPM7 may exert additional effects through its kinase domain, independent of ITRPM7. Use of FTY720 at concentrations sufficient to activate sphingosine-1-phosphate receptors (Brinkmann et al., 2002) did not recapitulate these effects (Fig. S2D), indicating that FTY720 suppresses the LPS response predominantly through the inhibition of TRPM7 channel.
To examine whether TRPM7 was required for signaling through other TLRs, we stimulated Trpm7+/+ and Trpm7−/− BMDMs with LPS (a TLR4 ligand), Pam3CSK4 (TLR 1/2 ligand), PolyI:C (TLR3 ligand), and ODN 1826 (TLR9 ligand, also “CpG DNA”) (Fig. 1F). We observed a robust decrease in ligand-induced transcription Il1b mRNA by LPS and Pam3CSK4 in Trpm7−/− BMDMs, but not in response to ODN 1826. Moreover, PolyI:C was able to induce robust Rsad2 expression, a readout of TRIF-dependent signaling, in Trpm7−/−. As expected, PolyI:C did not induce robust Il1b gene expression, nor did Pam3CSK4 and ODN 1826 strongly induce Rsad2 mRNA (Fig 1F and Fig. S1K) These results indicate that TRPM7 is essential for LPS-induced macrophage activation, but Trpm7−/− macrophages do not have a general defect in inflammatory signaling. Thus, the drastically reduced gene expression of Il1b, amongst many other inflammatory genes, underlies the defect in the secretion of IL-1β in Trpm7−/− macrophages.
TRPM7 controls TLR4 receptor endocytosis
Internalization of the TLR4-CD14 receptor complex is a pivotal signaling checkpoint during LPS-signaling (Rosadini and Kagan, 2016). Endosomal TLR4-TRIF complexes are required to initiate the transcription of IRF3-regulated genes in response to LPS (Kagan et al., 2008). Moreover, TLR4 association with TRAM-TRIF-dependent signaling amplifies the activation of NFκB upon LPS stimulation (Yamamoto et al., 2003a; Yamamoto et al., 2003b). Expression of TRIF-dependent gene transcripts (i.e. Ccl4 and Rsad2) was decreased in Trpm7−/− macrophages (Fig. 1E). Since TRPM7 has been previously shown to regulate Fas receptor endocytosis (Desai et al., 2012), we hypothesized that TRPM7 regulates LPS signal transduction through the control of TLR4 endocytosis. A monoclonal anti-TLR4 antibody (clone: MTS510) binds to TLR4-MD2 dimers only in the absence of LPS-induced assembly of TLR4-signaling complex (which includes dimerization of TLR4/MD2 dimers) (Akashi et al., 2000). The earliest events in LPS-induced TLR4 signaling can, therefore, be inferred and quantified from the loss of MTS510 signal in flow cytometry. Both Trpm7+/+ and Trpm7−/− BMDMs rapidly initiated the assembly of TLR4 signaling complex after LPS treatment (Fig. 2A), indicating that the initial assembly of LPS-induced TLR4-signaling complex occurs independently of TRPM7. The monoclonal antibody clone SA15-21 binds to TLR4 regardless of LPS-induced allosteric changes and can be used to track the loss of TLR4 at the cell surface (Akashi et al., 2003) and thus, infer and quantify TLR4 endocytosis through flow cytometry. Using this method, we measured the LPS-induced internalization of TLR4 over time. As shown (Fig. 2B), Trpm7−/− macrophages showed significantly reduced internalization of TLR4 after LPS stimulation, displaying nearly 2-fold higher surface TLR4 levels compared to Trpm7+/+ BMDMs (Trpm7+/+, 37%; Trpm7−/−, 66%, n=4). We then measured the internalization of CD14, which controls LPS-TLR4 endocytosis and formation of LPS-TLR4 signaling endosomes (Zanoni et al., 2011). After 30m of LPS treatment, Trpm7+/+ BMDMs displayed rapid endocytosis of CD14 (37% of CD14 remaining at the cell surface) and then, CD14 levels recovered over time, presumably due to receptor recycling. In striking contrast, CD14 internalization was severely abated in Trpm7−/− macrophages (81% of surface CD14 remaining, 30m post-LPS) and there was no observable recovery of surface CD14 levels (Fig. 2C). Prior to LPS treatment, there were no differences in the total levels of CD14 or TLR4 at the cell surface in Trpm7+/+ and Trpm7−/− BMDMs (Fig. S3A and S3B). These results indicate that TRPM7 is not required for the initial assembly of LPS-TLR4-signaling complex at the plasma membrane but it is essential for the endocytosis of the LPS-TLR4-CD14 signaling complex.
Figure 2. TRPM7 regulates TLR4 endocytosis and downstream NFκB and IRF3 phosphorylation.
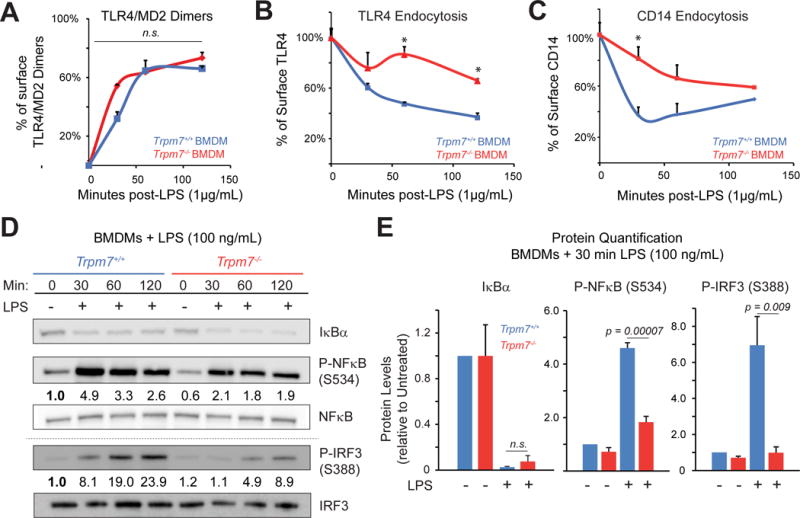
(A) Flow cytometry-based quantification of TLR4/MD2 dimers on the BMDM cell surface at indicated times after LPS (1 μg/mL) treatment. The change in percentage of cell surface TLR4/MD2 dimers was derived by staining the cells with an antibody specific for TLR4/MD2 dimers and calculations were based on the Mean fluorescence intensities (MFI). Error bars represent SEM (n=3).
(B) Flow cytometry-based quantification of TLR4 on the BMDM cell surface at indicated times after LPS (1 μg/mL) treatment. An anti-TLR4 antibody was used for staining. The error bars represent SEM (n=4).
(C) Flow cytometry-based quantification of CD14 on the BMDM cell surface at indicated times after LPS (1 μg/mL) treatment using an anti-CD14 antibody. Analysis was similar to panel B. Error bars represent SEM (n=5).
(D) Immunoblot analysis of NFκB p65 phosphorylation at S276 and S534 (in humans S536), P-IRF3 at S388, and IκBα protein levels from whole cells lysates of indicated BMDMs. Cells were stimulated with LPS (100 ng/mL) as indicated. Blots are representative of at least three independent experiments (n>3). Densitometry values indicate phospho-protein levels relative to total protein (ratios) which were then normalized to Trpm7+/+ at 0 min. Grey-dotted line denotes that the bottom gels were obtained from independent samples obtained using identical conditions.
(E) Quantification of immunoblots shown in Panel D. The densitometric values were calculated by taking the ratios of phospho-protein levels relative to total protein and then normalizing the ratios to Trpm7+/+ at 0 min. Bar charts represent means from at least three independent experiments (n= 4 for P-IRF3, n = 3 for others). Error bars are SEM. See also Fig. S3D.
LPS-induced phosphorylation of NFκB and IRF3 is regulated by TRPM7
LPS-initiated signaling triggers multiple post-translational modifications in the p65 subunit of NFκB (NFκB p65) prior to nuclear translocation (Vallabhapurapu and Karin, 2009). The phosphorylation of NFκB p65 at S276 and S536 (in mice, S534) is associated with nuclear translocation (Zhong et al., 1997) and transcriptional transactivation (Yang et al., 2003), respectively. Likewise, phosphorylation at IRF3 S396 (in mice, S388) is crucial for the induction of Type I IFNs (Lin et al., 1999). We examined if LPS-induced phosphorylation of either NFκB p65 or IRF3 was impaired in Trpm7−/− BMDMs. LPS treatment (100 ng/ml) induced the phosphorylation of NFκB p65 in Trpm7+/+ BMDMs, and this effect was significantly increased compared to Trpm7−/− BMDMs at both S276 (Trpm7+/+, 6.5-fold increase; Trpm7−/−, 3.4-fold increase) (Fig. S3C) and at S534 (Trpm7+/+, 4.5-fold; Trpm7−/− 1.8-fold increase). Similarly, LPS-induced phosphorylation of IRF3 (S388) increased (7.0-fold) after LPS treatment in Trpm7+/+ BMDMs, relative to 0 min, but Trpm7−/− BMDMs showed delayed kinetics of phosphorylation and lower levels of P-IRF3 after LPS stimulation in (Fig. 2D, 2E, and S3D). We also measured P-IRF3 levels by flow cytometry (using a different anti-P-IRF3 antibody clone validated for flow cytometry). This analysis revealed that LPS treatment for up to 3h did not increase P-IRF3 levels in Trpm7−/− BMDMs, while it increased P-IRF3 (S388) levels significantly in Trpm7+/+ BMDMs (Fig. S3E and S3F). As a proximal readout, degradation of IκBα may indicate the degree of signal transduction upstream of NFκB p65. Despite clear signaling defects at the level of NFκB and IRF3 in Trpm7−/− BMDMs, we did not observe defects in IκBα degradation, suggesting that TRPM7 regulates LPS signaling downstream of IκBα (Fig. 2D and 2E). Thus, in the absence of TRPM7, LPS-induced phosphorylation of the key transcription factors, NFκB and IRF3, are significantly compromised.
LPS-induced nuclear translocation of NFκB p65 and IRF3 is dependent on TRPM7
Next, we tested whether Trpm7−/− macrophages are deficient in nuclear translocation of NFκB p65 and IRF3. Deletion of TLR4 signaling adaptors delays the kinetics of NFκB translocation resulting in reduced cytokine production (Kawai et al., 1999). Pre-treatment with the dynamin inhibitor Dynasore (80 μM) (Kirchhausen et al., 2008; Macia et al., 2006) inhibits TLR4 endocytosis, and we examined if this had effects on LPS signaling in Trpm7+/+ and Trpm7−/− BMDMs (Fig. S4A). With Dynasore treatment, we observed significantly decreased LPS-induced transcription of Il1b, Il6, and Rsad2 in Trpm7+/+ macrophages. However, there was no additional effect of Dynasore treatment on Trpm7−/− macrophages, further suggesting that TLR4 endocytosis and downstream signaling in response to LPS is dependent on TRPM7. It has been previously established that Dynasore treatment blocks TRIF-dependent signaling through IRF3 (Kagan et al., 2008). However, in Dynasore-treated WT BMDMs, the nuclear translocation of NFκB p65 and phosphorylation of NFkB p65 are both reduced by ~50% after LPS treatment (Fig. S4B and S4C).
We then examined the role of TRPM7 in NFκB translocation in response to LPS directly through ImageStream flow cytometric imaging, which permits highly quantitative measurements of nuclear localization on large cell populations (Maguire et al., 2011) (Fig. 3A). Indeed, Trpm7−/− BMDMs exhibit reduced (33% of Trpm7+/+ BMDMs) nuclear translocation of NFκB p65, 30m after LPS treatment (Fig. 3B and 3C). We also examined the subcellular distribution of NFκB p65 using confocal microscopy. Without stimulation, NFκB p65 was diffuse throughout the cytosol in Trpm7+/+ and Trpm7−/− BMDMs. Upon LPS treatment (100ng/mL), nearly 60% of cellular NFκB was localized to the nucleus within 30 min of stimulation in Trpm7+/+ macrophages. However, NFκB p65 remained diffusely distributed in Trpm7−/− BMDMs after 30m of LPS stimulation (Fig. 3D). The observed differences were not due to expression levels of NFκB p65, which was unaffected (mRNA) by the deletion of TRPM7 (Fig. S4D). We then examined the subcellular distribution of IRF3 using confocal microscopy. Similar to NFκB p65, IRF3 was diffuse throughout the cytosol in unstimulated BMDMs. Upon LPS treatment (100ng/mL), low levels of IRF3 were observed 30 min after LPS in Trpm7+/+ BMDMs, while nearly 50% of cellular IRF3 was localized to the nucleus within 60 min of stimulation in Trpm7+/+ macrophages (Fig. 3E). However, IRF3 remained excluded from the nucleus in Trpm7−/− BMDMs 60 min after LPS treatment, as evaluated by intensity analysis of IRF3 localization (Fig. 3G and Fig. S4G). To further confirm these findings, we examined total protein distribution using subcellular fractionation (Fig. S4E). NFκB p65 was observed in the nuclear fraction within 15 min of LPS stimulation in Trpm7+/+ BMDMs, with peak translocation at 30m post-LPS stimulation. However, NFκB p65 translocation was strikingly diminished in Trpm7−/− BMDMs, with very low levels of nuclear NFκB p65, up to 60 min after LPS treatment (Fig. 3F). Likewise, IRF3 nuclear translocation was significantly reduced in Trpm7−/− BMDMs (33% decrease from Trpm7+/+ by densitometry) after LPS treatment (Fig. S4F). Further, in response to ODN 1826 (TLR9 ligand), NFκB p65 translocation was not impaired in Trpm7−/− BMDMs, indicating that TRPM7 does not broadly regulate NFκB p65 nuclear translocation (Fig. S4H). Together, these complementary approaches demonstrate that TRPM7 regulates the nuclear translocation of LPS-induced transcription factors, downstream of TLR4 endocytosis. The transcriptional defects in Trpm7−/− macrophages are therefore attributable primarily to the role of TRPM7 in the early phases of TLR4 signaling and subsequently decreased nuclear translocation of LPS-inducible transcription factors.
Figure 3. LPS-induced nuclear translocation of NFκB p65 and IRF3 is defective in Trpm7-deficient BMDMs.
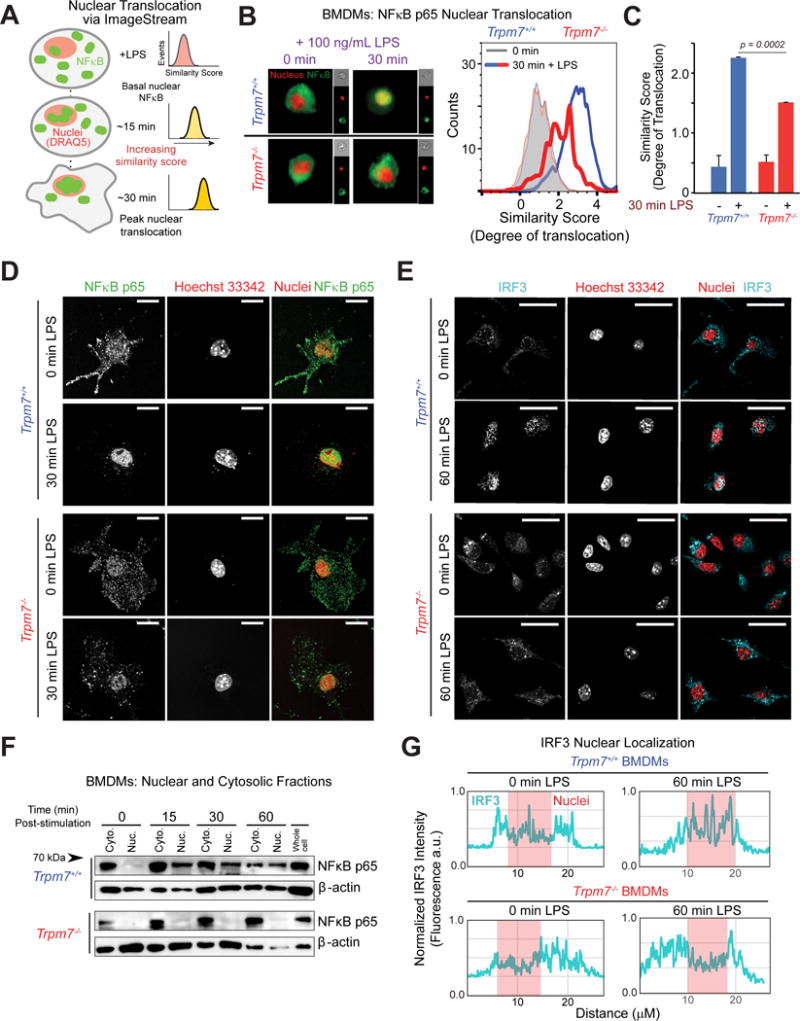
(A) Schematic of ImageStream analysis of LPS-induced NFκB translocation. Quantification of nuclear translocation is reflected by and directly proportional to the similarity scores of NFκB and nuclear staining. Similarity scores of individual cells are displayed together as a histogram.
(B) Representative images (n> 2000 cells) taken from ImageStream flow cytometry analysis of indicated BMDMs, stimulated with LPS (100 ng/mL, 30m) and stained with anti-NFκB p65 antibody and DRAQ5 (nuclear stain) (left panels). An overlaid image of NFκB p65 staining (green) and DRAQ5 (red) is shown and the single channel images of that cell are shown on the right in smaller sizes. The overlay of histograms depicts the representative similarity scores derived for each condition (right panel), quantified in Panel C (n=4).
(C) Quantification of ImageStream flow cytometry results depicted in Panel B. Error bars are SEM (n=4).
(D) Confocal immunofluorescence microscopy images of fixed BMDMs, treated or untreated with LPS (100 ng/ml, 30m) as indicated, and stained with anti-NFκB p65 and Hoechst nuclear stain. Images shown are a single optical section (0.25 μm). Scale bar = 15 μm. Typical results of n=3 experiments.
(E) Confocal immunofluorescence microscopy images of fixed BMDMs, treated or untreated with LPS (100 ng/ml, 60m) and stained with anti-IRF3 and Hoechst nuclear stain. Images shown are a single optical section (0.25 μm). Scale bar = 25 μm.
(F) Immunoblot analysis of NFκB p65 present in cytosolic and nuclear fractions obtained from indicated BMDMs at various time points after LPS (100 ng/ml) treatment. Immunoblots of β-actin in each fraction is also shown. These are representative results of independent experiments (n=3).
(G) Linear intensity analysis of IRF3 nuclear localization. Fluorescent intensity of IRF3 signal (cyan) was measured with a single x-y plane line trace; location of nuclei is overlayed (red). Line traces depict typical cells from panel E and are illustrated in Fig. S4H.
Clamping intracellular Ca2+ mimics the defects in LPS response seen in the absence of TRPM7
Stimulation of the T cell receptor (TCR), but not tumor necrosis factor receptor (TNFR), induces a Ca2+-dependent phosphorylation of NFκB p65 at S536 (Liu et al., 2016). It is, however, unknown if LPS-induced phosphorylation of NFκB p65 at S536 (S534 in mice) requires intracellular Ca2+ or [Ca2+]i elevations. We depleted and clamped [Ca2+]i by loading the macrophages with the high affinity Ca2+-chelator BAPTA-AM and then stimulated them with LPS. Preventing [Ca2+]i elevations during LPS stimulation decreased the phosphorylation of NFκB p65 at S534 (Fig. 4A and Fig. S5A). We then measured LPS-induced NFκB p65 nuclear translocation in BAPTA-AM loaded BMDMs using ImageStream flow cytometry. In Trpm7+/+ macrophages, NFκB translocation was significantly decreased (~60% of control) after LPS treatment (Fig. 4B and 4C). As shown previously (Fig. 3B), nuclear translocation of NFκB was greatly reduced in Trpm7−/− BMDMs, but notably, clamping [Ca2+]i did not further depress NFκB translocation. Gene expression analysis also demonstrated that BAPTA-AM severely inhibited LPS-induced transcription in Trpm7+/+ BMDMs, but had little additional effect in Trpm7−/− BMDMs (Fig. 4D). We then sought to “rescue” Ca2+-dependent transcriptional defects by mobilizing [Ca2+]i using the Ca2+ ionophore Ionomycin during LPS stimulation. Ionomycin provided no additional induction of gene expression in Trpm7+/+ BMDMs compared to LPS alone, suggesting that the LPS-induced [Ca2+]i elevations were functionally maximal. Ionomycin alone did not induce inflammatory gene expression either (data not shown). However, Ionomycin clearly rescued the LPS-induced gene expression in Trpm7−/− macrophages. This was the case for the upregulation of Il1b and Rsad2, downstream transcriptional targets of MyD88- and TRIF-signaling, respectively (Fig 4E). Interestingly, with the exception of Il18, not all LPS-induced genes regulated by TRPM7 were Ca2+ sensitive (Fig. S5B). However, Il1b mRNA expression induced by ODN 1826 was sensitive to Ca2+ depletion by BAPTA-AM (Fig. S5C), indicating that CpG DNA-induced gene transcription is intrinsically Ca2+ sensitive (although this Ca2+-influx is not mediated by TRPM7). Overall, these results argue that although the predominant function of TRPM7 during TLR4 signaling is to mediate [Ca2+]i elevation, it may also influence TLR4 signaling via other means (discussed later).
Figure 4. LPS signaling and NFκB translocation are abrogated by clamping intracellular Ca2+ but there is no further decrease in Trpm7-deficient macrophages.
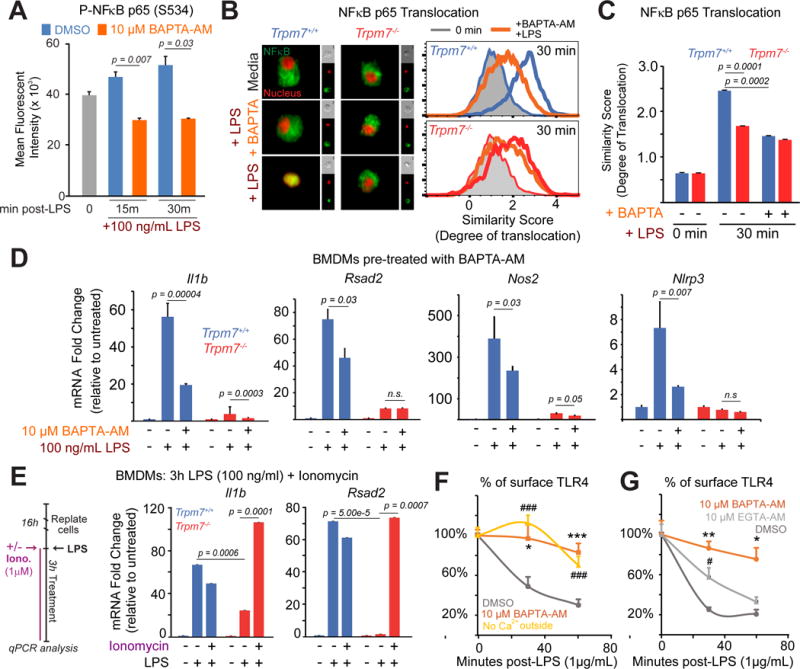
(A) Flow cytometry-based quantification of phospho-NFκB p65, as indicated by MFI, in DMSO (vehicle control) and BAPTA-AM loaded BMDMs in response to LPS (100 ng/mL, indicated time points). The error bars represent SEM (n=3).
(B) Representative images (n>5000 cells) from ImageStream analysis of DMSO and BAPTA-AM loaded cells are shown (left panel). Cells were stimulated with LPS (100 ng/ml, 30m) and then stained with anti-NFκB p65 antibody and DRAQ5 (a nuclear stain), prior to ImageStream analysis. To the right of the merged image of NFκB p65 (green) and DRAQ5 (red), single channel images of that cell are shown in smaller sizes. Overlaid histograms of similarity scores derived from each condition are shown (right panels); unstimulated (grey-filled), BAPTA-AM pre-treated (orange), and LPS treated (blue-wt; red-KO) are shown. The data are representative and typical of independent experiments (n=2).
(C) Quantification of similarity scores from panel B, reflecting the degree of NFκB p65 translocation in indicated conditions. Error bars represent SEM (n=3).
(D) qRT-PCR analysis of indicated inflammatory genes in Trpm7+/+ and Trpm7−/− BMDMs treated as depicted. Prior to LPS treatment (100 ng/mL, 3h), BMDMs were pre-treated with DMSO or BAPTA-AM for 30 min in serum-free media. Error bars represent SD (n=3).
(E) qRT-PCR analysis of indicated inflammatory genes in Trpm7+/+ and Trpm7−/− BMDMs treated as indicated in the schematic of the experimental design (left panel). Mean expression relative to untreated condition (right panel). Ionomycin is added with LPS to induce Ca2+ influx. Error bars represent SD (n=3).
(F) Flow cytometry-based quantification of cell surface TLR4 in RAW 264.7 cells at indicated times after LPS (1 μg/mL) treatment. Cells were treated with BAPTA-AM or DMSO as indicated prior to LPS treatment. Extracellular Ca2+-free conditions supplemented 10 mM EGTA. The error bars reflect SEM (n=3). * is BAPTA-AM treatment and # is low Ca2+ outside relative to wt. * indicates p<0.05, *** or ### indicates p<0.001.
(G) Flow cytometry-based quantification of cell surface TLR4 in RAW 264.7 cells at indicated times after LPS (1 μg/mL) treatment. Cells were treated with BAPTA-AM, EGTA-AM, or DMSO as indicated prior to LPS treatment. The error bars reflect SEM (n=3). * is BAPTA-AM treatment and # is EGTA-AM relative to wt. * or # indicates p<0.05, ** or ## indicates p<0.01.
The influx of extracellular Ca2+ is essential for TLR4 endocytosis
In macrophages, LPS initiates a rapid rise in [Ca2+]i (Letari et al., 1991). Based on the use of 100 M 1-Aminoethoxydiphenylborane (2-APB), it was proposed that the release of Ca2+ from intracellular stores is necessary for TLR4 endocytosis (Chiang et al., 2012). However, 2-APB is a highly non-specific ion channel inhibitor, and it blocks TRPM7 (Li et al., 2006) amongst many other ion channels (Lemonnier et al., 2004; Maruyama et al., 1997; Prakriya and Lewis, 2001). To better understand the nature of LPS-induced [Ca2+]i mobilization, we readdressed whether [Ca2+]i elevations are necessary for TLR4 endocytosis and also identified the source of that Ca2+ more definitively. To prevent cytosolic [Ca2+]i elevations, regardless of the source, RAW 264.7 cells were loaded with BAPTA-AM and then stimulated with LPS prior to the measurement of TLR4 endocytosis by flow cytometry. BAPTA-AM-loaded cells showed a drastic reduction in TLR4 endocytosis (96% and 86% of surface TLR4 remaining 30m and 60m, respectively), confirming the previous report that Ca2+-signaling is essential for TLR4 endocytosis (Fig. 4F). We also measured TLR4 endocytosis after acutely replacing the extracellular solution with a Ca2+-free solution. In this condition, Ca2+ in the intracellular stores would remain available for mobilization but there would be no influx of extracellular Ca2+. Strikingly, removal of extracellular Ca2+ also greatly diminished the endocytosis of TLR4, with over 70% of surface TLR4 remaining 60m after LPS treatment (compared to 30% in WT) (Fig. 4F). These results indicate that the influx of extracellular Ca2+ is essential for TLR4 endocytosis.
LPS-induced TLR4 endocytosis requires local elevations in cytosolic Ca2+
To further define the control of TLR4 endocytosis by Ca2+, we loaded the cells with either BAPTA-AM (fast-acting Ca2+ chelator) or the slow-acting EGTA-AM (both 10 M) prior to LPS treatment. This experimental design takes advantage of the kinetics of Ca2+-chelation to determine whether a local influx of Ca2+ controls TLR4 endocytosis. BAPTA-AM loaded RAW 264.7 cells were unable to internalize TLR4 after LPS treatment, displaying 86% (at 30 min) and 75% (at 60 min) of TLR4 at the plasma membrane after LPS treatment. Vehicle (DMSO) treated cells displayed 25% (at 30 min) and 20% (at 60 min) of surface TLR4 after LPS (Fig. 4G). This indicates that the rapid chelation of Ca2+ by BAPTA-AM, which clamps global as well as local Ca2+ elevations, abrogates TLR4 endocytosis almost entirely. In contrast, EGTA-AM loaded RAW 264.7 cells exhibited a moderately impaired rate of TLR4 endocytosis, but successfully internalized TLR4 60 min post-LPS (Fig. 4G). These results argue convincingly that the slow chelation of Ca2+ by EGTA-AM, which fails to prevent rapid, local Ca2+-elevations (likely Ca2+ in proximity to TLR4), permits the Ca2+-signaling necessary for TLR4 endocytosis. Overall, these results indicate that TLR4 endocytosis is controlled by a local rise in [Ca2+]i at the plasma membrane.
LPS-induced Ca2+ entry is mediated by TRPM7
The involvement of PLC-γ2 for the endocytosis of TLR4 (Chiang et al., 2012; Zanoni et al., 2011), had suggested a Ca2+ entry mechanism involving store-operated Ca2+ entry (SOCE). Surprisingly, the disruption of SOCE by the deletion of Stim1 and Stim2 does not affect TLR-ligand induced cytokine production in macrophages (Vaeth et al., 2015). These results indicate that Orai channels, the molecular conduits of SOCE, are not critical for LPS-induced macrophage activation, and the ion channels that mediate LPS-induced Ca2+ signaling for TLR endocytosis and downstream signaling remain elusive. Considering that TRPM7 is permeable to Ca2+ and is critical for TLR4 signaling, but not all TLRs, we hypothesized that TRPM7 directly mediates LPS-induced Ca2+ entry. Using BMDMs loaded with Ca2+-sensitive ratiometric fluorescent dye Fura2-AM, we measured [Ca2+] elevations in response to TLR ligands. A robust rise in cytosolic [Ca2+] was observed after stimulation with LPS (100 ng/ml) and Pam3CSK4 (100 ng/ml), with LPS yielding a 44.5% larger [Ca2+] elevation than Pam3CSK4 (Fig. 5A). Stimulation with ODN 1826 (1 μM), also induced [Ca2+] elevations, although to a significantly lower degree than LPS and Pam3CSK4 (down to 29% of LPS and 53% of Pam3CSK4).
Figure 5. LPS-induced Ca2+ elevations are highly compromised in Trpm7-deficient macrophages.
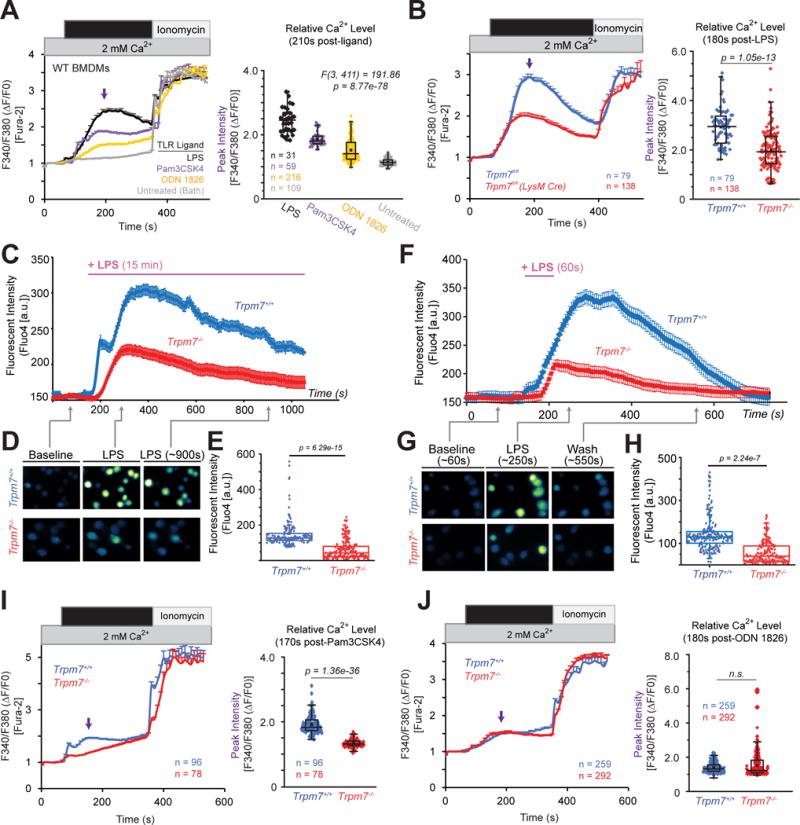
(A) Relative changes in [Ca2+]i over time in WT BMDMs treated with indicated TLR ligands for 5 min. Cells were treated with either LPS (100 ng/ml), Pam3CSK4 (100 ng/ml), ODN 1826 (1 μM), or bath solution (untreated). Left panel: trace represents mean ∆F340/F380 ratio from all samples and error bars are SEM; ionomycin (1 μM) was perfused as a positive control. The arrow indicates the time point of peak [Ca2+]i, these values were used for statistical analysis. Right panel: Quantification of peak [Ca2+]i after LPS stimulation; box-whisker plot is overlaid on individual measurements. One-way ANOVA statistical analysis and n values (number of cells) are indicated in the figure. Results are a compilation of three independent experiments.
(B) Relative changes in [Ca2+]i over time in Trpm7+/+ and Trpm7−/− BMDMs treated with LPS (100 ng/ml) for 5 min. Left panel: trace represents mean ∆F340/F380 ratio from all samples (n=79 and n=138 for Trpm7+/+ and Trpm7−/−, respectively) and error bars are SEM. Ionomycin (1 μM) was perfused as a positive control. Right panel: Quantification of peak [Ca2+]i after LPS stimulation; box-whisker plot is overlaid on individual measurements; n values (number of cells analyzed) indicated in figure. Results are a compilation of three independent experiments.
(C) Relative changes in the fluorescence of Fluo-4AM-loaded BMDMs, reflecting changes in intracellular Ca2+ concentration [Ca2+]i BMDMs after LPS (1 μg/mL) treatment for 15 min. Error bars reflect SEM (n=146 and n=179 for Trpm7+/+ and Trpm7−/−, respectively). These represent typical results from four independent experiments.
(D) Representative fluorescence images from indicated time points in the experiments shown in panel C.
(E) Statistical representation of mean peak intensities of Fluo-4 fluorescence after LPS treatment from panel C.
(F) Relative changes in [Ca2+]i over time in Trpm7+/+ and Trpm7−/− BMDMs treated with LPS (1 μg/mL) for 60s. Error bars are SEM (n=217 and n=190 for Trpm7+/+ and Trpm7−/−, respectively). These represent typical results from four independent experiments.
(G) Representative images from indicated time points in panel F.
(H) Statistical representation of mean peak intensities after LPS treatment from panel F.
(I) Relative changes in [Ca2+]i over time in Trpm7+/+ and Trpm7−/− BMDMs treated with Pam3CSK4 (100 ng/ml) for 5 min. Left and Right panels depicted as in Fig. 5B.
(J) Relative changes in [Ca2+]i over time in Trpm7+/+ and Trpm7−/− BMDMs treated with ODN 1826 (1 μM) for 5 min. Left and Right panels depicted as in Fig. 5B.
To examine the role of TRPM7 in LPS-induced Ca2+ entry, we performed live cell imagingTrpm7+/+ and Trpm7−/− BMDMs. Trpm7+/+ BMDMs display a rapid rise in [Ca2+]i, peaking less than 2 min after LPS stimulation (Fig. 5B). In contrast, Trpm7−/− BMDMs displayed a severely blunted rise in LPS-induced [Ca2+]i, which was decreased in both peak intensity and duration. However, both Trpm7+/+ and Trpm7−/− BMDMs responded normally to ionomycin. We made similar observations using both Fura-2-AM and Fluo-4-AM Ca2+ imaging dyes (data not shown). We also investigated the [Ca2+]i elevations in response to both sustained (15 min) (Fig. 5C–E) and a short pulse (60 sec) of LPS stimulation, after which, the extracellular bath solution was replaced by the LPS-free solution (Fig. F–H). In both stimulation conditions, Trpm7−/− BMDMs exhibited a significantly diminished [Ca2+]i elevations compared to Trpm7+/+ BMDMs (Fig. 5E and 5H) – the mean peak [Ca2+]i level was down to ~43% of that in WT or Trpm7+/+ BMDMs. With the caveat that LPS bound to TLR4 may not be easily washed out, it is interesting that a 60s stimulation with LPS results in a robust [Ca2+]i rise. We then measured whether Pam3CSK4 and ODN 1826 induced Ca2+ entry was dependent on TRPM7. Consistent with the observations from gene expression analysis (Fig. 1F), Trpm7−/− BMDMs displayed a significant reduction in Pam3CSK4-induced cytosolic [Ca2+] entry, relative to Trpm7+/+ BMDMs. However, ODN 1826-induced Ca2+ entry occurred independently of TRPM7. Overall, these results clearly demonstrate that TRPM7 controls the Ca2+-entry triggered by LPS and Pam3CSK3, but not the Ca2+-entry triggered by ODN 1826.
Blocking TRPM7 channel activity using FTY720 also diminishes LPS-induced cytosolic Ca2+-elevations, TLR4 endocytosis, and downstream signaling
While TRPM7 is required for LPS-induced Ca2+ elevations, TRPM7 could contribute to Ca2+ entry in an indirect manner, for instance by regulating another channel through its kinase domain. In Figure S2B, we used FTY720, a TRPM7 channel blocker (Qin et al., 2013), to demonstrate that TRPM7 channel activity played a critical role in LPS-induced gene expression. Here, we demonstrate that FTY720 can block LPS-induced Ca2+-influx. Treatment of BMDMs with FTY720 (5μM) decreased LPS-induced Ca2+-influx 6-fold, relative to untreated cells (Fig. 6A and 6B). In BMDMs, FTY720 (5μM) also decreased the rate of TLR4 endocytosis (Fig. 6C and S6E), with nearly 80% of surface TLR4 remaining 60 min after LPS treatment; BAPTA-AM pre-treatment completely ablated TLR4 endocytosis. FTY720 treatment also mimicked the effects of BAPTA-AM on the phosphorylation of NFκB p65 (Fig. S6A and S6B). Both FTY720 and BAPTA-AM decreased the phosphorylation of NFκB (S534) to ~50% of untreated cells. We also examined RAW 264.7 cells, a murine BALB/c macrophage cell line, in response to TRPM7 blockade by FTY720. In these cells, FTY720 treatment decreased LPS-induced Ca2+ influx to ~45% of untreated cells (Fig. S6C and S6D). FTY720 also drastically reduced the phosphorylation of NFκB p65 at S534 (Fig. S6F). Transcriptionally, the reduction in LPS-induced Il1b mRNA by FTY720 was comparable to clamping of [Ca2+]i with BAPTA- AM (Fig. S6G). Using cell fractionation, we determined that the nuclear translocation of NFκB p65 is greatly delayed in FTY720-treated RAW 264.7 cells, 15 min after LPS treatment (Fig. S6H). Some translocation was detectable at 30 min, but it was significantly less when compared to vehicle treated controls. The inhibition of NFκB p65 nuclear translocation by FTY720 was also evident using immunofluorescence confocal microscopy (Fig. S6I) and ImageStream flow cytometry (data not shown). Furthermore, we tested the effect of FTY720 on LPS-induced transcription in human THP-1 monocytes. In response to LPS, FTY720 (5μM) pre-treatment decreased the transcription of both Ccl4 (down to 51% of untreated), Il6 (to 14% of untreated), and Il1b (to 16% of untreated) (Fig. 6D), suggesting both MyD88 and TRIF-dependent signaling are compromised in human monocytes. Thus, blocking TRPM7 channel activity acutely inhibits LPS-induced Ca2+ entry, TLR4 endocytosis, and gene transcription, similar to Trpm7−/− macrophages. Together, these results indicate that TRPM7 channel directly conducts Ca2+ to mediate LPS-induced signaling.
Figure 6. Blocking TRPM7 channel activity abrogates LPS-induced Ca2+ entry and TLR4 endocytosis.
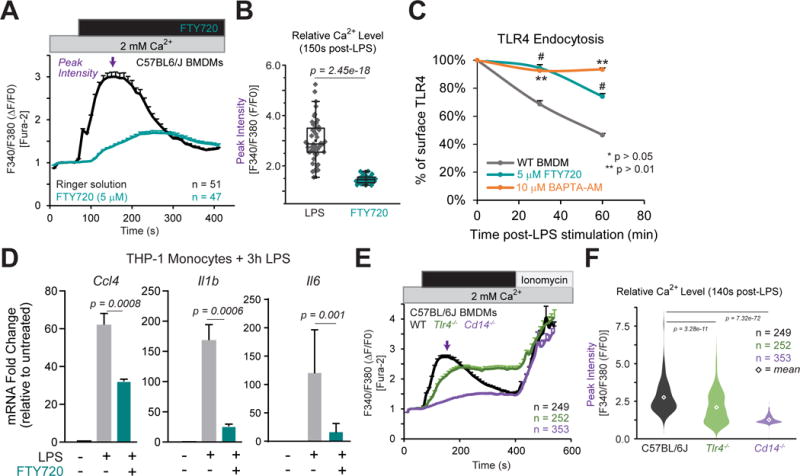
(A) Relative changes in [Ca2+]i over time in C57BL/6 BMDMs treated with LPS (100 ng/ml) or LPS+FTY720 (5μM) for 5 min. Trace represents mean ∆F340/F380 ratio from all samples and error bars are SEM. Ionomycin (1 μM) was perfused as a positive control. Results are from n=3 independent experiments; n value indicated in figure.
(B) Quantification of peak [Ca2+]i after LPS stimulation from Fig 6A. Box-whisker plot is overlaid on individual measurements.
(C) Flow cytometry based measurement of cell surface TLR4 levels in BMDMs, pre-treated with either vehicle (DMSO), FTY720, or BAPTA-AM for 15 min, and then stimulated with LPS as indicated. The relative change in the percentage of cell surface TLR4 levels was inferred based on mean MFI values. Error bars represent SEM (n=3; * indicates p < 0.05, ** p < 0.01). * indicates significance from WT relative to BAPTA-AM; #, FTY720 group. These results represent typical results obtained in n=2 independent experiments.
(D) Gene expression analysis (qRT-PCR) of human THP-1 monocytes stimulated with LPS (100 ng/ml; 3h) with FTY720 pre-treatment as indicated. Bar charts represent mean of n=3 independent experiments. Error bars are SEM (n=3).
(E) Relative changes in [Ca2+]i over time in BMDMs treated with LPS (100 ng/ml) for 5 min. Ionomycin (1 μM) was perfused as a positive control. Traces represent mean ∆F340/F380 ratio from all samples measured and error bars are SEM. Results are representative from n=5 independent experiments.
(F) Quantification of peak [Ca2+]i after LPS stimulation from Fig 6E. Violin plot is shown to illustrate range and distribution of data points; diamond indicates mean value for group. One-way ANOVA (F[2,851] = 266.23) indicates p = 1.6e–90. T-test used to compare groups with C57BL/6J BMDMs.
CD14, not TLR4, mediates LPS-induced Ca2+ entry in primary macrophages
Given that TRPM7 channel mediates LPS-induced Ca2+ entry, the molecular identity of the LPS receptor responsible for modulating TRPM7, and thereby Ca2+ entry, is unknown. To address this question, we performed live cell Ca2+ imaging on BMDMs from C57BL/6J (herein, “WT”), Tlr4−/−, and Cd14−/− mice. Upon stimulation with LPS (100 ng/ml), WT BMDMs responded with a rapid rise in [Ca2+]i over 5 min of stimulation (Fig 6E). Surprisingly, Tlr4−/− BMDMs exhibited only a modest decrease in LPS-induced [Ca2+]i elevation (down to ~76% of WT response at t = 140s). In striking contrast, Cd14−/− BMDMs failed to elevate [Ca2+]i in response to LPS (down to ~15% of WT) (Fig 6F). Thus, these results indicate that CD14 functions as the predominant trigger for LPS-induced Ca2+ influx.
Trpm7fl/fl (LysM Cre) mice are resistant to LPS-induced peritonitis
We evaluated the significance of macrophage-resident TRPM7 in vivo by challenging the mice with an intraperitoneal injection of LPS (0.2 mg/kg) (Fig. 7A). In vivo administration of LPS elicits a rapid cytokine response, and the increased serum levels of TNFα, IFN-γ, and IL-6 promote pathological inflammation in less than 6 hours after LPS administration (Rittirsch et al., 2008). After LPS challenge, mice were observed for pathological symptoms using a composite clinical score (Kadl et al., 2007) (Table S1), recorded in a double-blinded manner. Trpm7fl/fl (LysM Cre) mice displayed decreased sensitivity to LPS challenge over the course of 24h when compared to Trpm7fl/fl mice (Fig. 7B). Resistance to LPS challenge was also clearly evident at 4h after LPS administration, with a 4-fold increase in composite clinical score in Trpm7fl/fl mice compared to Trpm7fl/fl(LysM Cre) mice (Fig. 7C). Importantly, serum cytokine levels of TNFα, IFN-γ, and IL-6 were significantly decreased (to ~50%) in Trpm7fl/fl (LysM Cre) mice relative to LPS-treated Trpm7fl/fl mice (Fig. 7D). Likewise, we observed decreased transcription of Tnfa and Il1b mRNA levels in immune cells isolated via peritoneal lavage 4h after LPS administration (Fig. 7E). We then used flow cytometry to examine the differences, if any, in the infiltration of macrophages from LPS-treated Trpm7fl/fl and Trpm7fl/fl(LysM Cre) mice (Fig. S7A and S7B). There were no differences in the proportion of peritoneal macrophages (CD45+ CD11b+ F4/80+) between Trpm7fl/fl and Trpm7fl/fl (LysM Cre) mice, 24h after LPS challenge (Fig. 7F and 7G). However, the gross recruitment of immune cells was greatly reduced in LPS-treated Trpm7fl/fl (LysM Cre) mice. Trpm7fl/fl mice had a 2-fold increase in the number of CD45+ immune cells and macrophages into the peritoneum (24h post-LPS). In contrast, Trpm7fl/fl(LysM Cre) mice did not show a significant increase in CD45+ cells or macrophages (CD45+ CD11b+ F4/80+) (Fig. 7H). Overall, in comparison to Trpm7fl/fl mice, the Trpm7fl/fl(LysM Cre) mice are protected from LPS-induced pathology. We observed decreased cytokine production and reduced recruitment of immune cells in the peritoneum – rendering Trpm7fl/fl(LysM Cre) mice significantly resistant to LPS-induced peritonitis.
Figure 7. Trpm7fl/fl (LysM Cre) mice are resistant to LPS-induced peritonitis.
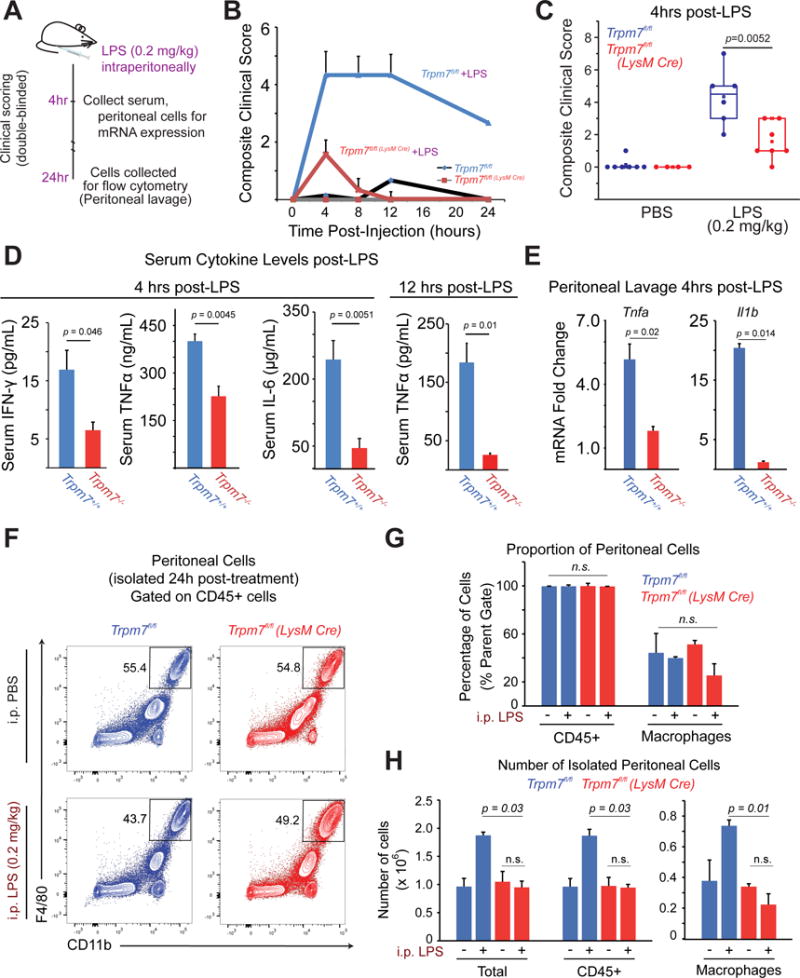
(A) Schematic of LPS-induced peritonitis mouse model and analysis.
(B) After a sub-lethal dose of LPS (0.2 mg/kg, i.p), the mice were observed for 24 hrs by a double-blinded experimenter to record clinical scores in accord with an index described in Table S1. Error bars represent SEM (n>5).
(C) Statistical box charts showing composite clinical scores from individual mice, at 4h of observation with indicated treatments (see panel A for details). Data were compiled from three independent cohorts of mice. Box chart parameters are described in methods.
(D) ELISA measurements of indicated cytokines in the serum collected after LPS (0.2 mg/kg) injections at indicated time points. Error bars represent SEM (n>6 compiled from three independent cohorts).
(E) Gene expression analysis (qRT-PCR) of cells collected via a peritoneal lavage, after LPS (0.2 mg/kg, 4h) injections. Quantification is relative to PBS-injected controls (not shown). Error bars represent SEM (n=4 mice).
(F) Flow cytometry-based immunophenotypic analysis of peritoneal macrophages for the indicated cell surface markers, after gating on CD45+ cells. The legacy of gates is shown in Figure S5. Cells were isolated from peritoneal lavage after LPS (0.2 mg/kg, 24h). The data represent typical results from independent experiments (n=3 mice).
(G) The mean percentage of peritoneal hematopoietic (CD45+) and macrophages (CD45+ CD11b+ F4/80+) determined by flow cytometry analysis shown in Figure S5. Error bars represent SEM (n=3 mice).
(H) Mean numbers of total cells, hematopoietic (CD45+) and macrophages (CD45+ CD11b+ F4/80+) in peritoneal lavages, as determined by flow cytometry analysis shown in Panel F. Cell counts were calculated by accounting for the number of cells analyzed, percentages of cells in legacy gates, and recovery volume of peritoneal lavage. Error bars represent SEM (n=3 mice). See also Figure S7 and Table S1.
DISCUSSION
We have uncovered a novel TRPM7-mediated Ca2+-signaling pathway that is essential for LPS-induced macrophage activation and is, therefore, central to innate immunity. TRPM7 channel mediates cytosolic Ca2+ elevations in response to LPS, and this Ca2+ influx is necessary for TLR4 receptor endocytosis and IRF3 activation. Similarly, in a presumably parallel arm of LPS-signaling, the Ca2+-influx also regulates the activation and nuclear translocation of NFκB. Thus, TRPM7 controls the transcriptional programs mediated by the two key transcription factors, NFκB and IRF3, during LPS-induced macrophage activation. In accord with these findings, the selective deletion of Trpm7 in myeloid cells renders the mice resistant to LPS-induced peritonitis, revealing TRPM7 as a major controller of pro-inflammatory cytokine production by myeloid cells in vivo. These discoveries are highly significant because TRPM7 now emerges as a pharmacologically tractable molecular target for developing novel therapeutic strategies directed against sepsis and auto-inflammatory diseases.
LPS has long been known to trigger a rapid elevation in cytosolic Ca2+ (Letari et al., 1991), but the identity of the ion channel responsible for initiating this Ca2+-signaling pathway has remained elusive. Likewise, the endocytosis of the TLR4 signaling complex is a crucial early event in cellular LPS response, and this process was known to be Ca2+-dependent (Chiang et al., 2012). However, due to the non-specific properties of pharmacological inhibitors, it was previously concluded that the release of Ca2+ from intracellular stores and through IP3 receptors, is the key process necessary for TLR4 endocytosis. In that model, the activation of PLCγ2 (and the generation of IP3) during LPS signaling leads to the release of ER Ca2+ through the IP3 receptors and subsequently results in store-operated Ca2+-entry (SOCE) through the Orai channels. Surprisingly, macrophages that are completely deficient in Stim1-Stim2 and SOCE show no defects in LPS-induced production of inflammatory cytokines, indicating that SOCE is not a major determinant of TLR4-signaling (Vaeth et al., 2015). The release of intracellular Ca2+ (and subsequent SOCE) may occur concomitantly with TRPM7 activity, but these processes are not the salient regulators of TLR4 signaling. This is also supported by the observation that although Ca2+-elevations are severely diminished in Trpm7−/− macrophages, they are not entirely absent. We emphasize that our findings do not negate a role for other ion channels in regulating various aspects of macrophage activation (Desai and Leitinger, 2014; Feske et al., 2015; Santoni et al., 2013; Tauseef et al., 2012; Yamamoto et al., 2008). While pharmacological approaches have suggested a vague role for TRPM7 in macrophages (Schilling et al., 2014), our study is the first to define the function of TRPM7 in a mechanistic manner using mice with a targeted genetic deletion of TRPM7 in macrophages.
Given our observations that CD14, not TLR4, is vital to LPS-induced Ca2+ entry, we propose that the activation of TRPM7 through a CD14-mediated event, likely located in close proximity to TLR4, is the essential Ca2+-signaling component of this pathway. The resulting local Ca2+-influx promotes the endocytosis of TLR4 at rates that are physiologically meaningful for downstream signaling, including IRF3 activation. Similarly, TRPM7-mediated Ca2+-signaling also regulates the parallel activation of NFκB, but this process is likely influenced by additional inputs from the TLR4 signaling endosomes. This idea is supported by our observation that the inhibition of TLR4 endocytosis by the dynamin inhibitor Dynasore substantially reduces LPS-induced NFκB translocation. However, given the multitude of dynamin-family GTPases in the cell, Dynasore will likely abrogate multiple cellular processes, and thus, we are cautious in our conclusions from Dynasore-treated cells. Interestingly, IκBα degradation appears to be intact in Trpm7−/− macrophages, but NFκB p65 phosphorylation is decreased. This observation suggests that as-yet unidentified Ca2+-triggered kinase likely modulates NFκB p65 activity during LPS signaling. Recent evidence suggests that phosphorylation at S534 NFκB p65 is crucial for the stability of p65 signaling. We observed a striking Ca2+ sensitivity of this phospho-site, suggesting that the spatiotemporal aspects of Ca2+-signaling can greatly modify NFκB signaling. Thus, Ca2+ likely plays a dual role – a rapid role in “jump starting” TLR4 endocytosis and possibly a slower role in tailoring an appropriate inflammatory response by modifying the activity of NFκB. This study defines the function of TRPM7 channel activity in macrophage activation, but the role of the TRPM7 kinase activity in inflammatory signaling remains unexplored. Our experiments with the TRPM7 channel blocker FTY720 reveal that TRPM7 channel activity does not account for the entire spectrum of defects seen in Trpm7−/− macrophages. In the embryonic stem cells, the cleaved TRPM7 kinase domain has been shown to translocate to the nucleus and modify the chromatin landscape through the phosphorylation of histones (Krapivinsky et al., 2014). Since TRPM7 is cleaved by multiple proteases (including caspases), a similar function of the cleaved kinase domain may be of salience to inflammatory gene expression in macrophages.
In addition to the role of the kinase domain, a number of other important questions emerge from this study and are the focus of our ongoing and future studies. The molecular mechanism by which TRPM7 is activated during LPS-signaling is not yet clear. Previously, we have shown that the TRPM7 channel is activated by a caspase-mediated proteolytic event that also dissociates the kinase domain from the membrane resident channel (Desai et al., 2012). The function of TRPM7 kinase activity in the regulation of inflammatory gene expression is thus a major topic of interest. Intracellular LPS has been shown to activate caspase-11 (Shi et al., 2014), but LPS-induced, TRPM7-mediated Ca2+-influx is very rapid, and it is highly unlikely that a brief pulse of LPS can activate caspase-11 in less than a minute. Other possibilities include direct activation of TRPM7 by LPS or activation by the local depletion of PIP2 (Langeslag et al., 2007) in close proximity to TLR4. Lastly, once the Ca2+-influx is triggered in close proximity to TLR4, the identity of the Ca2+-sensitive molecular switch in the endocytic machinery is not yet known.
STAR Methods
Key Resources Table
| REAGENT | SOURCE | IDENTIFIER |
|---|---|---|
| Inflammatory Ligands | ||
| LPS (LPS-EB Ultrapure; E. coli strain 0111:B4) | Invivogen | tlrl-3pelps |
| ATP | Invivogen | tlrl-atp |
| Flagellin from S. typhimurium | Invivogen | tlrl-epstfla-5 |
| MSU Crystals | Invivogen | tlrl-msu |
| dsDNA [Poly(dA:dT)] | Invivogen | tlrl-patn |
| Anthrax LT | Gift of Dr. Molly Hughes (UVA) | n/a |
| Pam3CSK4 | Invivogen | tlrl-pms |
| PolyI:C | Enzo | ALX-746-021 |
| ODN 1826 | Invivogen | tlrl-1826 |
| Antibodies for Western Blot | ||
| Rabbit polyclonal P-NFκB p65 (S276) (#3037) | Cell Signaling Technologies | 3037 |
| Rabbit monoclonal P-NFκB p65 (S536) (clone: 93H1) | Cell Signaling Technologies | 3033 |
| Rabbit monoclonal NFκB p65 (clone: C22B4) | Cell Signaling Technologies | 4764 |
| Rabbit monoclonal NFκB p65 XP (clone: D14E12) | Cell Signaling Technologies | 8242 |
| Mouse monoclonal β -actin (clone: BA3R) | Thermo Fisher | MA515739 |
| Rabbit monoclonal P-IRF3 (S396) (clone: 4D4G) | Cell Signaling Technologies | 4947 |
| Rabbit monoclonal IRF3 (clone: D83B9) | Cell Signaling Technologies | 4302 |
| Anti-mouse HRP secondary | JacksonImmuno | 111035144 (Ms) |
| Anti-rabbit HRP secondary | JacksonImmuno | 111035144 (Rb) |
| Mouse monoclonal IκBα (clone: L35A5) | Cell Signaling Technologies | 4814 |
| Rabbit monoclonal LSD1 (C69G12) | Cell Signaling Technologies | 2184 |
| Rabbit monoclonal GAPDH XP (clone: D16H11) | Cell Signaling Technologies | 5174 |
| Antibodies for Flow Cytometry | ||
| Rabbit monoclonal P-IRF3 (S396) (clone: D6O1M) | Cell Signaling Technologies | 29047 |
| F4/80 (PE-Cy7) (clone: BM8) | eBioscience | 25480182 |
| CD11b (Alexa Fluor 488) (clone: M1/70) | eBioscience | 53011282 |
| TLR4/MD-2 (PE-Cy7) (clone: MTS510) | eBioscience | 25992482 |
| TLR4 (PE) (clone: SA15-21) | Biolegend | 145404 |
| CD14 (APC) (clone: Sa2-8) | eBioscience | 17014181 |
| CD45 (APC-eFluor 780) (clone: 30-F11) | eBioscience | 47045182 |
| TruStain FcX (anti-CD16/32) | Biolegend | 101320 |
| Antibodies for Immunocytochemistry | ||
| NFκB p65 (clone: C22B4) | Cell Signaling Technologies | 4764 |
| Anti-rabbit Alexa Fluor 488 secondary | JacksonImmuno | 711545152 |
| Chemical Reagents | ||
| Hoechst 33342 | Invitrogen | H3570 |
| DRAQ-5 | Molecular Probes | 62254 |
| FTY720 | Cayman Chemical | 162359560 |
| Ionomycin | Thermo Fisher | I24222 |
| BAPTA-AM | Thermo Fisher | B6769 |
| Fura-2-AM | Thermo Fisher | F1221 |
| Fluo-4-AM | Thermo Fisher | F14201 |
| Dynasore | ApexBio | A1605 |
| EGTA-AM | Thermo Fisher | E1219 |
| ELISA Kits | ||
| Mouse IL-6 ELISA MAX Deluxe | Biolegend | 431306 |
| Mouse IL-1β ELISA MAX Deluxe | Biolegend | 432606 |
| Mouse TNFα ELISA MAX Deluxe | Biolegend | 430906 |
| Gene Expression Analysis Kits | ||
| SensiMix SYBR NO-ROX Kit | Bioline | BIO-98020 |
| ISOLATE II RNA Mini Kit | Bioline | BIO-52073 |
| GoScript Reverse Transcriptase System | Promega | A5001 |
Contact for Reagent and Resource Sharing
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Bimal Desai (bdesai@virginia.edu).
Mice
Male and female mice between 6 and 12 weeks of age were used for all experiments. Trpm7fl/fl and Trpm7fl/fl (Lck Cre) mice were described previously (Jin et al., 2008). To generate Trpm7fl/fl (LysM Cre) mice, Trpm7fl/fl mice were crossed with Lyz2tm1(cre)lfo (“LysM Cre”) mice (Clausen et al., 1999) (Jackson Laboratories, Bar Harbor, ME) expressing Cre under the Lyz2 gene promoter/enhancer elements, which selectively deletes Trpm7-exon 17 in myeloid cell-specific manner. Male C57BL/6J [#000664], Tlr4−/− [B6.B10ScN-Tlr4lps-del/JthJ; #007227], and CD14−/− [B6.129S4-Cd14tm1Frm/J; #003726] mice were purchased from Jackson Laboratories; all mice were 6 to 8 weeks of age. Mice were housed and bred in accordance with policies and procedures of the University of Virginia Institutional Animal Care and Use Committee (IACUC).
Genotyping and Characterization of Mice
Tail samples were dissolved in 75 μl of digestion buffer (25 mM NaOH, 0.2 mM EDTA) for 30 min at 85°C, and digestion was stopped with 75 μl of 40 mM Tris-HCl. 1 μl of the reaction was used as a template for PCR using MyTaq Hot Start Polymerase and 5X MyTaq Red Reaction Buffer (Bioline; #BIO-21112 and #BIO-37112, respectively). PCR products were electrophoretically separated on a 1% agarose gel and visualized with ethidium bromide. Genotype was determined using the following primers:
Expression of Cre recombinase in Trpm7fl/fl (LysM Cre) mice. Primer sequences and method for analysis were as previously described (Clausen et al., 1999):
LysM Cre Primer: 5′-CCCAGAAATGCCAGATTACG-3′
Common Primer: 5′-CTTGGGCTGCCAGAATTTCTC-3′
Wild-type Primer: 5′-TTACAGTCGGCCAGGCTGAC-3′
Expression of Cre recombinase in Trpm7fl/fl (Lck Cre) mice
Forward Primer: 5′-GATTTCGACCAGGTTCGTTC-3′
Reverse Primer: 5′-GCTAACCAGCGTTTCGTTC-3′
The presence of absence of LoxP sites flanking Trpm7 exon 17:
Forward Primer: 5′-CAGAGGTACTGGCAATTGTG-3′
Reverse Primer: 5′-ACGAGGACTCAGCATATAGC-3′
BMDM characterization via Trpm7 Exon 17
DNA was isolated from 5 × 106 BMDMs according to protocols for “Genotyping” described above. PCR products were amplified via primers flanking Trpm7 exon 17.
RNA Interference
RAW 264.7 cells incubated as described in text with either anti-TRPM7 (siR-M7-776) or scrambled siRNA (Integrated DNA Technologies). siRNA were designed with the following sequences:
Sense: CAACUAAUUCUGUUCGUCUGAUGUU
Antisense: AACAUCAGACGAACAGAAUUAGUUG
Cell Culture
Primary murine BMDMs were isolated from bone marrow and differentiated in RPMI 1640 (Gibco) with 10% fetal bovine serum (FBS) and 20% L929-conditioned media and used between 7 and 10 days after isolation. RAW 264.7 cells (ATCC) were cultured in DMEM (Gibco) with 10% FBS. BMDMs and RAW 264.7 cells were gently scraped for detachment as needed. THP-1 cells (ATCC) were cultured in RPMI 1640 supplemented with 0.05 mM 2-mercaptorethanol and 10% FBS. All cells were cultured at 37°C and 5% CO2.
Patch Clamp Electrophysiology
TRPM7 currents (ITRPM7) were measured in whole cell configuration as illustrated (Fig. S1C). The standard external solution contained (in mM): 135 Na-methanesulfonate, 5 Cs-gluconate, 2.5 CaCl2, 10 HEPES, pH 7.3 (adjusted with NaOH), and osmolality 280–290 mOsm/Kg. The standard pipette solution contained (in mM): 110 Cs-gluconate, 0.5 NaCl, 0.75 CaCl2, 10 HEPES, 10 HEDTA, 1.8 Cs4-BAPTA, 2 Na2ATP, pH 7.3 (adjusted with CsOH), and osmolality 273 mOsm/Kg. Free [Ca2+] = ~100nM and was calculated via the Maxchelator algorithm (http://maxchelator.stanford.edu/webmaxc/webmaxcS.htm). Peritoneal macrophages were freshly-isolated prior to analysis by peritoneal lavage. MgCl2 (10 mM) was added to the external solution to inhibit TRPM7 currents. The recording protocol used 400 ms ramps from −100 mV to +100 mV and a holding potential (HP) of 0 mV. Signals were low-pass filtered at 5 kHz and sampled at 10 kHz. All electrophysiology experiments were conducted at RT (~23°C) using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA).
Inflammasome Activation
BMDMs were cultured at a density of 0.75 × 106 cells/mL in 6 or 24-well plates. Cells were washed with PBS prior to addition of culture media containing LPS (100 ng/mL) for 3 hrs. Media was removed and replaced with XVIVO 15 media (Lonza; #04744Q) containing LPS (100 ng/mL) and indicated concentrations of ligands. Cell media was collected, centrifuged at 13000g to remove dead cells and debris, and analyzed for IL-1β secretion via sandwich ELISA (Biolegend).
Gene Expression Analysis
Cells were cultured on 24 well plates at a density of 0.5 × 106/ml in a culture volume of 500 μL/well and stimulated as indicated. For pharmacological studies, BMDMs were pre-treated with the following compounds prior to addition of LPS: FTY720 for 15 min in culture media; Dynasore for 30 min in RPMI 1640; BAPTA-AM for 30 min in Ca2+-free Hanks’ buffered saline solution with 0.02% pluronic acid. Quantitative real-time PCR analysis of gene expression is described in below.
Quantitative Real-time PCR
Total RNA was isolated using the ISOLATE II RNA Mini Kit (Bioline). RNA concentrations were measured using NanoDrop 2000c (Thermo Scientific) and normalized amongst the experimental sample set. cDNA was reverse transcribed using the GoScript Reverse Transcription System (Promega Corporation) according to the manufacturer’s instructions. qPCR measurements were setup in triplicate using the SensiMix SYBR NO-ROX Kit (Bioline) and amplified in CFX connect Real-Time system (Bio-Rad, USA) according to the manufacturer’s instructions. Data were analyzed using the CFX manager 3.1 software (Bio-Rad, USA) according to the ΔΔCt method (Schmittgen and Livak, 2008). Β-2-microgobulin was used to normalize for cDNA input error.
| Gene Name | Primer Sequence (forward) 5′ – 3′ | Primer Sequence (reverse) 5′ – 3′ |
|
| ||
| Il1b | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
|
| ||
| Nlrp3 | CTCTCCCGCATCTCCATTTGT | GCTTGGATGCTCCTTGACCA |
|
| ||
| Il6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
|
| ||
| Tnfa | CCCTCACACTCAGATCATCTTCT | GCTACGACGTGGGCTACAG |
|
| ||
| Nos2 | GTTCTCAGCCCAACAATACAAGA | GTGGACGGGTCGATGTCAC |
|
| ||
| Rsad2 | AACAGGCTGGTTTGGAGAAGA | AGCAAGAATGTCCAAATACTCCC |
|
| ||
| Trpm7 | AGCAGTATTCCAATGATTTTGGC | TCATAGCCATCGTTTCATCCTGT |
|
| ||
| Rela | AGGCTTCTGGGCCTTATGTG | TGCTTCTCTCGCCAGGAATAC |
|
| ||
| Il18 | AAGAAAGCCGCCTCAAACCT | AGTGAAGTCGGCCAAAGTTGT |
|
| ||
| Ccl4 | TCTGTGCAAACCTAACCCCG | GAGGGTCAGAGCCCATTGGT |
|
| ||
| B2M | GGCCTGTATGCTATCCAGAA | GAAAGACCAGTCCTTGCTGA |
Receptor Endocytosis via Flow Cytometry
BMDMs (0.5 × 106) were treated as indicated at 37°C. For experiments depicted in Figure 5, cells were pre-treated in HBSS and treated in Ca2+-free HBSS with 2% FBS and other reagents as indicated. Cells were washed with cold PBS and collected at 4°C in FACS Buffer (0.5% BSA, 2 mM EDTA in PBS). Cells were stained on ice with TruStain FcX (Biolegend) according to the manufacturer’s recommendations for 10 min and then for 20 to 30 min using the appropriate antibodies. Stained cells were washed in cold FACS Buffer, resuspended in 200 μl of FACS Buffer, and placed on ice for immediate analysis. Cell surface staining was measured on the BD FACSCanto II flow cytometer.
The efficiency of surface receptor endocytosis was determined as previously described (Tan et al., 2015). For measurement of CD14 and TLR4 endocytosis, cells were stained on ice with TruStain FcX (as recommended by the manufacturer) and then stained for 20 min on ice with fluorescently conjugated anti-TLR4 (0.2 μg/mL; clone: SA15-21) or anti-CD14 (0.4 μg/mL; clone: Sa2-8) antibodies in FACS buffer. Cells were washed in cold FACS buffer prior to analysis via flow cytometry. Mean fluorescence intensity (MFI) of CD14 and TLR4 was measured from unstimulated stained and unstained cells on the BD FACSCanto II. The percentage of surface receptor expression is the ratio of MFI from stimulated cells to unstimulated cells at indicated time points. Ratios are plotted to express receptor endocytosis over time. For measuring the formation of TLR4/MD-2 dimers, the percent of TLR4/MD-2 dimers were determined by calculating the fraction of 100% of the inverse of TLR4/MD-2 monomers, as measured by MFI values (via anti-TLR4 clone: MTS510 staining). TLR4/MD-2 monomers were determined by the ratio of MFI stimulated to unstimulated cells.
Protein Immunoblots
Biochemical fractions were prepared with modification as described by Rockland, Inc. (http://www.rockland-inc.com/NuclearExtract.aspx). In brief, macrophages (5 × 106 cells) were treated as indicated and collected at 4°C. Cells were washed with 1 mL of cold PBS and resuspended in 6X pellet volumes (PV) of cytoplasmic buffer ([in mM] 10 HEPES, 60 KCl, 1 EDTA, and 0.075% NP40) for 5 min on ice. The suspension was centrifuged for 5 min at 1500 rpm at 4°C. Supernatant (cytoplasmic extract) was removed to a new tube. Pellet was washed in 12X PV in cytoplasmic buffer without NP40. Nuclei were collected by centrifugation and supernatants were discarded. The pellet was lysed in 1X PV with nuclear buffer ([in mM] 20 Tris-Cl, 420 NaCl, 1.5 MgCl2, 0.2 EDTA) for 10 min on ice. Both cytoplasmic extracts and nuclei mixture were centrifuged at maximum speed for 10 min to remove debris. Supernatants were collected for immunoblot analysis.
Whole cell lysates were prepared by collecting cells in Lysis Buffer (150 NaCl, 50 Tris-HCl pH 7.4, 2 EDTA, 0.5% TritonX-100) and lysis cells for 30 min on ice. Lysates were cleared of debris by centrifugation at 14000rpm for 10 min. Supernatants were collected, mixed with 5X Laemmli Sample Buffer (0.3M Tris-HCl pH 6.8, 10% SDS, 50% glycerol, 25% beta-mercaptoethanol, 0.05% bromophenol blue), and boiled at 95°C prior to SDS-PAGE analysis. Using cleared lysates, protein content was determined via BCA Assay (ThermoFisher; #23225) according to the manufacturer’s instructions. Samples were loaded into 4–20% or 12% ExpressPlus PAGE gels (GenScript; #M42012 and #M01210, respectively) and electrophoretically separated according to the manufacturer’s instructions (150 V for 60 min in MES Running Buffer).
Immunofluorescence Microscopy
BMDMs were seeded on plates seeded with coverslips overnights. BMDMs were then treated as indicated at 37°C. Cells were washed in cold PBS and fixed in cold PFA (4% PFA in PBS) for 15 min at RT. Coverslips were washed 3 times with wash buffer (0.05% tween-20 in PBS) to removed residual PFA. Coverslips were permeabilized for 5 min in 0.1% Triton X-100 in PBS and blocked for 1h at RT in blocking buffer (1% BSA, 0.1% fish gelatin, 0.1% Triton-X-100, 0.05% Tween-20, and 5% donkey serum in PBS). Coverslips were stained with the anti-NFκB or anti-IRF3 antibody (1:250 dilution in blocking buffer) (Cell Signaling; #4764 and #4302), respectively) for 2h at RT. Cells were washed 3X with wash buffer and stained with anti-rabbit-Alexa Fluor 488 conjugated antibody (1 μg/mL) (JacksonImmuno) for 1h at RT. After washing 3X in wash buffer, coverslips were stained with Hoechst 33342 (1:2000) for 10 min at RT. Coverslips were washed 2X in wash buffer prior to mounting with VectaMount medium (Vector Laboratories, CA; #H5000). Mounted samples were cured overnight at RT and imaged within 24h. Images were collected using an Olympus FluoView 1000 under 40X or 60X objective lens. Images were analyzed with ImageJ software.
Nuclear Translocation via ImageStream
Cells were treated as described at 37°C. Cells were then transferred to 4°C for 5 min, washed 2X with PBS, and collected by gentle scraping. Cells were fixed with cold paraformaldehyde (4%) for 15 min at RT. Cells were permeablized with Perm Buffer (2 mM EDTA, 0.5% BSA, 0.1% triton X-100 in PBS) for 5 min and stained with primary antibody and anti-rabbit secondary antibody as recommended in Perm Buffer. Cells were resuspended in FACS Buffer (2 mM EDTA, 0.5% BSA in PBS) with DRAQ5 prior to analysis via ImageStream.
Ca2+ Imaging
Cells were allowed to adhere to #1 thickness coverslips overnight (~16h) prior to analysis. For ratiometric Ca2+ imaging, cell were incubated for 30 min with agitation at RT with 5 μM of Fura-2-AM in presence of 0.02% of pluronic acid (BIOTIUM) and 500 μM probenecid (Enzo; ALX-430113G005) in Ringer solution ([in mM] 155 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES, 10 glucose, pH 7.4). Fura-2-AM emissions were collected at 510 nm and with 340/380 nm excitation. Ratio of 340/380 was calculated by the F/F0 method (Helmchen, 2011). For Fluo-4-AM imaging, cells were incubated for 2h at 37°C with 5 μM of Fluo-4-AM in presence of 0.02% of pluronic acid and 2.5 mM probenecid in standard extracellular solution (SES; [in mM] 145 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 10 glucose, pH 7.4). For Fluo-4 imaging, cells were excited at 488 nm and emissions were measured at 530 nm. In all cases, fluorescent excitation was performed using a DG4 Illuminator (Sutter Instruments, Canada) and fluorescence was detected using an ORCA-Flash 4.0 V2 CMOS camera (Hamamatsu) using SlideBook 6 software. Measurements were performed with continuous perfusion of bath solution with a gravity feed system and ionomycin (1 μM) was perfused as a positive control for cellular responsiveness. Experiments were performed in Ringer solution or SES with addition of TLR ligands as indicated in figures and legends.
In vivo LPS Challenge
Trpm7fl/fl and Trpm7fl/fl (LysM Cre) mice were injected intraperitoneally with 0.2 mg/kg LPS (200 μl) or PBS. Mice were weighed less than 1 hr prior to injection to determine dosage. Mice were randomly assigned to treatment groups and all observations were performed by a double-blinded experimenter (blind to treatment and genotype). Mice were observed for up to 24h. Mice were euthanized at 4 hours to collect blood serum and peritoneal immune cells. Serum was collected after centrifugation of whole blood at 4°C for 10 min at 2500g. Serum concentrations of IL-6 and TNFα were measured via ELISA (Biolegend). IFNγ was measured via Luminex analysis conducted by the UVA Flow Cytometry Core Facility.
Peritoneal cells were isolated by injection of 8 mL of cold PBS into the peritoneal cavity. The peritoneal lavage fluid was collected, and cells were centrifuged at 4°C for 6 min at 350g. The pellet was resuspended in ACK lysis buffer for 5 min to lyse red blood cells. Cells were collected and subjected to staining for flow cytometry. Cells were stained on ice with TruStain FcX (Biolegend, #101320) for 10 min and then for 30 min with anti-F4/80 (clone: BM8), CD11b (clone: M1/70), and CD45 (clone: 30-F11) antibodies at the manufacturer recommended concentrations. Stained cells were washed with 1 mL of cold FACS Buffer, resuspended in 200 μl of FACS Buffer, and placed on ice for immediate analysis. Cell surface staining was measured on the BD FACSCanto II. Gating strategy is described in Fig. S7.
Statistics
All data were analyzed using Origin Pro 9.1.0 (Origin Lab) or Excel (Microsoft) software. Data are presented as means with error bars, which reflect standard deviation (SD) or standard error of the mean (SEM) as indicated in figure legends. Bar charts were plotted with Excel, dot plots and box charts were plotted using Origin Pro, and Violin plots were plotted in R studio (R package ‘ggplot2’). Statistical box charts are shown as a box (range of 25–75 percentile) and whisker bars (1–99 percentile) with data points overlaid; the median is represented by a horizontal line and mean is shown as an empty square. Violin plots represent distribution of individual sample values and the diamond represents the mean value for the group. Normality and variance for data sets were tested graphically. The sample size is indicated in figures and legends. Statistical significance was determined using paired t-test (two-tailed), unless indicated otherwise in legend. A p value less than 0.05 was considered statistically significant.
Supplementary Material
In Brief.
Schappe et al. show genetic deletion of Trpm7 in macrophages or pharmacological inhibition of TRPM7 channel prevents macrophage activation due to the loss of TRPM7-mediated Ca2+ influx in response to LPS. The study identifies TRPM7 as a Ca2+ entry pathway required for macrophage activation.
HIGHLIGHTS.
TRPM7 is essential for LPS-induced macrophage activation
TRPM7 mediates the Ca2+-influx necessary for TLR4 endocytosis
LPS-induced phosphorylation and translocation of NFκB p65 and IRF3 depend on TRPM7
Mice with a myeloid-specific Trpm7 deletion are resistant to LPS-induced peritonitis
Acknowledgments
We thank members of the Desai and Leitinger laboratories, Prof. J. Casanova (UVA) and Prof. S. Ewald (UVA) for their helpful insights and discussions. We thank the following core facilities for their technical resources and support: UVA Flow Cytometry Core, Carter Immunology Center Flow Cytometry Core, and UVA Cardiovascular Research Center Microscopy Facility. We are also grateful for the funding that supported this work: B.N.D. (GM108989) and M.S.S. (5T32GM007055-41).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
EXPERIMENTAL PROCEDURES
See STAR Methods Section
AUTHOR CONTRIBUTIONS Conception: MSS, BND; Research Design: MSS, NL, BND; Investigation: MSS, KS, SKM, MES, PVS, MAM, TKD, SD, SEA, JKK; Data analysis: MSS, KS, PVS, SKM, MAM, TKD, BND; Resource assistance: JKK, EJS, JSR, NL; Writing – Draft and Editing: MSS, BND; Project Administration: BND
The authors declare that they have no conflicts of interest to disclose.
References
- Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity. 2015;42:991–1004. doi: 10.1016/j.immuni.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Akashi S, Saitoh S, Wakabayashi Y, Kikuchi T, Takamura N, Nagai Y, Kusumoto Y, Fukase K, Kusumoto S, Adachi Y, et al. Lipopolysaccharide Interaction with Cell Surface Toll-like Receptor 4-MD-2: Higher Affinity than That with MD-2 or CD14. The Journal of experimental medicine. 2003;198:1035–1042. doi: 10.1084/jem.20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi S, Shimazu R, Ogata H, Nagai Y, Takeda K, Kimoto M, Miyake K. Cutting edge: cell surface expression and lipopolysaccharide signaling via the toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. Journal of immunology. 2000;164:3471–3475. doi: 10.4049/jimmunol.164.7.3471. [DOI] [PubMed] [Google Scholar]
- Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nature reviews Immunology. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. The Journal of biological chemistry. 2002;277:21453–21457. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- Brough D, Le Feuvre RA, Wheeler RD, Solovyova N, Hilfiker S, Rothwell NJ, Verkhratsky A. Ca2+ Stores and Ca2+ Entry Differentially Contribute to the Release of IL-1 and IL-1 from Murine Macrophages. The Journal of Immunology. 2003;170:3029–3036. doi: 10.4049/jimmunol.170.6.3029. [DOI] [PubMed] [Google Scholar]
- Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nature reviews Immunology. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CY, Veckman V, Limmer K, David M. Phospholipase Cgamma-2 and intracellular calcium are required for lipopolysaccharide-induced Toll-like receptor 4 (TLR4) endocytosis and interferon regulatory factor 3 (IRF3) activation. The Journal of biological chemistry. 2012;287:3704–3709. doi: 10.1074/jbc.C111.328559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic research. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Desai BN, Krapivinsky G, Navarro B, Krapivinsky L, Carter BC, Febvay S, Delling M, Penumaka A, Ramsey IS, Manasian Y, et al. Cleavage of TRPM7 releases the kinase domain from the ion channel and regulates its participation in Fas-induced apoptosis. Developmental cell. 2012;22:1149–1162. doi: 10.1016/j.devcel.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai BN, Leitinger N. Purinergic and calcium signaling in macrophage function and plasticity. Frontiers in immunology. 2014;5:580. doi: 10.3389/fimmu.2014.00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. 2015;33:291–353. doi: 10.1146/annurev-immunol-032414-112212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nature reviews Immunology. 2014;14:546–558. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- Geissman F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F. Calibration of fluorescent calcium indicators. Cold Spring Harbor protocols. 2011;2011:923–930. doi: 10.1101/pdb.top120. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nature immunology. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Li M, Yue L. Potentiation of TRPM7 Inward Currents by Protons. J Gen Physiol. 2005;126:137–150. doi: 10.1085/jgp.200409185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Desai BN, Navarro B, Donovan A, Andrews NC, Clapham DE. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science. 2008;322:756–760. doi: 10.1126/science.1163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadl A, Pontiller J, Exner M, Leitinger N. Single bolus injection of bilirubin improves the clinical outcome in a mouse model of endotoxemia. Shock (Augusta, Ga) 2007;28:582–588. doi: 10.1097/shk.0b013e31804d41dd. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nature immunology. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T, Macia E, Pelish HE. Use of Dynasore, the Small Molecule Inhibitor of Dynamin, in the Regulation of Endocytosis. 4. 2008;38:77–93. doi: 10.1016/S0076-6879(07)38006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Clapham DE. The TRPM7 chanzyme is cleaved to release a chromatin-modifying kinase. Cell. 2014;157:1061–1072. doi: 10.1016/j.cell.2014.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langeslag M, Clark K, Moolenaar WH, van Leeuwen FN, Jalink K. Activation of TRPM7 Channels by Phospholipase C-coupled Receptor Agonists. Journal of Biological Chemistry. 2007;282:232–239. doi: 10.1074/jbc.M605300200. [DOI] [PubMed] [Google Scholar]
- Lemonnier L, Prevarskaya N, Mazurier J, Shuba Y, Skryma R. 2-APB inhibits volume-regulated anion channels independently from intracellular calcium signaling modulation. FEBS letters. 2004;556:121–126. doi: 10.1016/s0014-5793(03)01387-5. [DOI] [PubMed] [Google Scholar]
- Letari O, Nicosia S, Chiavaroli C, Vacher P, Schlegel W. Activation by bacterial lipopolysaccharide causes changes in the cytosolic free calcium concentration in single peritoneal macrophages. Journal of immunology. 1991;147:980–983. [PubMed] [Google Scholar]
- Li M, Jiang J, Yue L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. The Journal of general physiology. 2006;127:525–537. doi: 10.1085/jgp.200609502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol. 1999;19:2465–2474. doi: 10.1128/mcb.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Berry CT, Ruthel G, Madara JJ, MacGillivray K, Gray CM, Madge LA, McCorkell KA, Beiting DP, Hershberg U, et al. T Cell Receptor-induced Nuclear Factor kappaB (NF- kappaB) Signaling and Transcriptional Activation Are Regulated by STIM1- and Orai1-mediated Calcium Entry. The Journal of biological chemistry. 2016;291:8440–8452. doi: 10.1074/jbc.M115.713008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Developmental cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Maguire O, Collins C, O’Loughlin K, Miecznikowski J, Minderman H. Quantifying nuclear p65 as a parameter for NF-kappaB activation: Correlation between ImageStream cytometry, microscopy, and Western blot. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2011;79:461–469. doi: 10.1002/cyto.a.21068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-Aminoethoxydiphenyl Borate, a Membrane-Penetrable Modulator of Ins(1,4,5)P3-Induced Ca2+ Release. The Journal of Biochemistry. 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nature reviews Immunology. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- Moran MM, McAlexander MA, Biro T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nature reviews Drug discovery. 2011;10:601–620. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11282–11287. doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- Nilius B, Szallasi A. Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacological reviews. 2014;66:676–814. doi: 10.1124/pr.113.008268. [DOI] [PubMed] [Google Scholar]
- O’Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature reviews Immunology. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. The Journal of biological chemistry. 1994;269:15195–15203. [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signling in C3H/HeJ and C57BL/10ScCr Mice; mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. The Journal of Physiology. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Yue Z, Sun B, Yang W, Xie J, Ni E, Feng Y, Mahmood R, Zhang Y, Yue L. Sphingosine and FTY720 are potent inhibitors of the transient receptor potential melastatin 7 (TRPM7) channels. British journal of pharmacology. 2013;168:1294–1312. doi: 10.1111/bph.12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada B, Park JJ, Sil P, Geiszt M, Leto TL. NLRP3 inflammasome activation and interleukin-1β release in macrophages require calcium but are independent of calcium-activated NADPH oxidases. Inflammation Research. 2014;63:821–830. doi: 10.1007/s00011-014-0756-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nature reviews Immunology. 2008;8:776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosadini CV, Kagan JC. Early innate immune responses to bacterial LPS. Current opinion in immunology. 2016;44:14–19. doi: 10.1016/j.coi.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoni G, Farfariello V, Liberati S, Morelli MB, Nabissi M, Santoni M, Amantini C. The role of transient receptor potential vanilloid type-2 ion channels in innate and adaptive immune responses. Frontiers in immunology. 2013;4:34. doi: 10.3389/fimmu.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling T, Miralles F, Eder C. TRPM7 regulates proliferation and polarisation of macrophages. Journal of cell science. 2014;127:4561–4566. doi: 10.1242/jcs.151068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protocols. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- Tan Y, Zanoni I, Cullen Thomas W, Goodman Andrew L, Kagan Jonathan C. Mechanisms of Toll-like Receptor 4 Endocytosis Reveal a Common Immune-Evasion Strategy Used by Pathogenic and Commensal Bacteria. Immunity. 2015;43:909–922. doi: 10.1016/j.immuni.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauseef M, Knezevic N, Chava KR, Smith M, Sukriti S, Gianaris N, Obukhov AG, Vogel SM, Schraufnagel DE, Dietrich A, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. The Journal of experimental medicine. 2012;209:1953–1968. doi: 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M, Zee I, Concepcion AR, Maus M, Shaw P, Portal-Celhay C, Zahra A, Kozhaya L, Weidinger C, Philips J, et al. Ca2+ Signaling but Not Store-Operated Ca2+ Entry Is Required for the Function of Macrophages and Dendritic Cells. Journal of immunology. 2015;195:1202–1217. doi: 10.4049/jimmunol.1403013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003a;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nature immunology. 2003b;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nature medicine. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Tang E, Guan K, Wang CY. IKK Plays an Essential Role in the Phosphorylation of RelA/p65 on Serine 536 Induced by Lipopolysaccharide. The Journal of Immunology. 2003;170:5630–5635. doi: 10.4049/jimmunol.170.11.5630. [DOI] [PubMed] [Google Scholar]
- Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, Kagan JC. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147:868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The Transcriptional Activity of NF-κB Is Regulated by the IκB-Associated PKAc Subunit through a Cyclic AMP– Independent Mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.