

Abstract
The involvement of vascular endothelial injury with the pathophysiological process of heart failure has been identified. Baicalein (BAI), a flavonoid extracted from the root of Scutellaria baicalensis, is reported to exert antibacterial, antiviral, antithrombotic and antioxidant effects. The aim of the present study was to investigate the effects of BAI on lipopolysaccharide (LPS)-induced vascular endothelial injury. Human umbilical vein endothelial cells (HUVECs) were stimulated by LPS (10 µM) in the presence or absence of BAI. The expressions of the inflammatory cytokines interleukin (IL)-lβ, IL-6, tumor necrosis factor-α (TNF-α) and monocyte chemoattractant protein-1 (MCP-1) were analyzed by reverse transcription-quantitative polymerase chain reaction, western blotting and enzyme-linked immunosorbent assay. Cell apoptosis was assessed by flow cytometry and terminal deoxynucleotidyl transferase dUTP nick end labeling assay. The results showed that BAI significantly inhibited the LPS-induced inflammatory response and apoptosis in HUVECs. BAI suppressed the LPS-induced upregulation of IL-1β, IL-6, TNF-α and MCP-1. Furthermore, BAI decreased the expression of B-cell lymphoma 2 (Bcl-2)-associated X protein and cleaved caspase-3; however, it increased the protein level of Bcl-2. The inhibitory effect of BAI may occur through the suppression of the Toll-like receptor 4 (TLR4)/phosphorylated (p)-transforming growth factor β-activated kinase 1/tumor necrosis factor receptor-associated family member associated nuclear factor (NF)-κB activator-binding kinase 1 (p-TBK1)/NF-κB signaling pathway. An increase in the level of p-TBK1 by MRT67307 abolished the effect of BAI on p-p65. In conclusion, the results of the present research suggested that BAI ameliorated endothelial cell injury associated with TLR4/NF-κB signaling, and highlighted the potential clinical use of BAI in blocking endothelial dysfunction and preventing heart failure.
Keywords: baicalein, endothelial cells, lipopolysaccharide, inflammation, apoptosis, Toll-like receptor 4/nuclear factor-κB
Introduction
Vascular endothelial dysfunction is generally caused by pressure or volume overload, such as hypertension, myocardial infarction, valve stenosis or regurgitation (1), as well as the increased secretion and release of inflammatory cytokines, such as interleukin-lβ (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), and angiotensin II (Ang II). Vascular endothelial dysfunction induces or exacerbates cardiac disease, which will eventually result in heart failure (HF) (2–4). Although previous studies have implicated nuclear transcription factor-κB (NF-κB) (5), p38 mitogen-activated protein kinase (MAPK) (6) and phosphatidylinositol 3-hydroxy kinase (PI3K) (7) as potential targets in vascular endothelial dysfunction, the precise mechanisms are still incompletely understood. A human umbilical vein endothelial cells (HUVECs) have been widely used to establish experimental models for vascular inflammation in vitro, and the lipopolysaccharide (LPS)-induced HUVEC injury is commonly used as an in vitro model (4), we used LPS-stimulated HUVECs as the model for our study.
Baicalein (5,6,7-trihydroxy-2-phenyl-4H-1-benzop yran-4-one, BAI) is a flavonoid extracted from the root of Scutellaria baicalensis (3). Many evidences have suggested that BAI possesses many pharmacological activities. It is reported that BAI inhibits agonist-induced and tumor cell-induced platelet aggregation (8), and could suppress tumor growth and metastasis (9). BAI also combines synergistically with cefotaxime to prevent Klebsiella pneumoniae through the inhibition of CTX-M-1 gene expression (10). Tsai et al (11) proved that BAI protects against oxLDL-induced oxidative stress and inflammation via the modulation of AMPK-α. BAI reduced angiogenesis in the inflammatory microenvironment through the inhibition of the expression of AP-1 (12); and according to Zong et al (13), BAI exerts protection against cardiac hypertrophy and fibrosis through the suppression of mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase 1/2 (ERK1/2) signaling.
In the present study, we found that BAI protects HUVECs from LPS-induced injury associated with the Toll-like receptor 4 (TLR4), phosphorylation of TGF-β-activated kinase 1 (p-TAK1), phosphorylation of TANK-binding kinase 1 (p-TBK1) and phosphorylation of nuclear transcription factor-κB (NF-κB) p65 (p-p65) signaling, which inhibits inflammation and apoptosis in HUVECs. Further, using MRT67307, a IKKε and TBK1 inhibitor, we verified that BAI targets the expression of p-TBK1 to suppress TLR4/NF-κB signaling. These results suggested that BAI could protect against vascular endothelial injury and exert potential therapeutic effects for the prevention of HF.
Materials and methods
Cell culture
HUVECs (8000; ScienCell Research Laboratories, Carlsbad, CA, USA) were grown in Dulbecco's modified Eagle's medium (DMEM/F-12) supplemented with 10% fetal bovine serum (FBS) (both from Gibco, Grand Island, NY, USA) and endothelial cell growth supplement (ECGS, 1052; ScienCell Research Laboratories) under standard conditions (5% CO2; temperature, 37°C). The cells were passaged when 70–80% confluent and starved in serum-free DMEM for 12 h before LPS stimulation. LPS (L2630) and BAI (≥98%, CAS 491-67-8) were purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). MRT67307 (S7948) was bought from Selleck Chemicals (Houston, TX, USA).
Determination of cytotoxicity and efficacy
To determine the cytotoxicity, the cell viability of HUVECs was tested after treatment with different concentrations of BAI (0, 3.125, 6.25, 12.5, 25, 50, 100, 200, or 400 µM) for a specific period of time. The same process was repeated in HUVECs stimulated by LPS (10 µM) and simultaneously treated by different concentrations of BAI (0, 3.125, 6.25, 12.5, 25, or 50 µM) to determine the efficacy of BAI. Both the cytotoxicity and efficacy of BAI were determined by the cell counting kit assay (Enhanced Cell Counting Kit-8, C0042; Beyotime Institute of Biotechnology, Haimen, China). The absorbance at 450 nm was measured using the Bio-Tek Synergy HT Multi-Detection Microplate Reader (BioTek Instruments, Inc., Winooski, VT, USA).
Detection of apoptosis through TUNEL assay
The HUVECs were divided into three groups: i) Control: Cells treated with neither LPS nor BAI; ii) LPS: Cells treated with LPS (10 µM); iii) LPS + BAI: Cells treated with LPS (10 µM) and BAI (6.25 µM). Apoptosis was identified through the DNA fragmentation in the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (14). The TUNEL assay works through the incorporation of modified dUTPs at the 3′-OH ends of fragmented DNA by the enzyme terminal deoxynucleotidyl transferase (TdT), which subsequently tags cells with damaged DNA (15). After the TUNEL assay treatment, damaged DNA is stained green, whereas intact DNA is not stained. HUVECs were detected using the Apo-Direct TUNEL Assay kit (EMD Millipore, Billerica, MA, USA). All steps were conducted in strict accordance with the specifications and photographed using a fluorescent microscope.
Flow cytometric detection of HUVEC apoptosis
The cells were resuspended, rinsed and processed with Annexin V-FITC/PI Apoptosis Detection kit (KGA108; Nanjing KeyGen Biotech Co., Ltd., Nanjing, China). Annexin V was considered as a sensitive index of early stage apoptotic cells, while PI was the indicator of advanced stage apopotic cells. Briefly, the cells were re-suspended in 500 µl binding buffers and stained consecutively by 5 µl Annexin V-FITC and 5 µl PI. We then analyzed HUVECs by flow cytometry (FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA) to differentiate apoptotic cells from necrotic cells. Both HUVECs stained Annexin+/PI− and Annexin+/PI+ in the flow cytometric analyses were considered as apoptotic cells.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
The mRNA expression levels of IL-1β, IL-6, TNF-α, MCP-1 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were analyzed by RT-qPCR. Total RNA was extracted from HUVECs using TRIzol reagent in accordance with the manufacturer's instructions (Invitrogen Life Technologies, Carlsbad, CA, USA), and the purity of the RNA was evaluated based on the OD260/OD280 ratios detected using the SmartSpec Plus Spectrophotometer (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The mRNA was reverse transcribed into cDNA using oligo(dT) primers and the Transcriptor First Strand cDNA Synthesis kit (Roche, Basel, Switzerland). The following primer sequences were used: IL-1β forward, 5′-ATTTGAGTCTGCCCAGTTCCC−3′ and reverse, 5′-CCAGGAAGACGGGCATGTTT-3′; IL-6 forward, 5′-CAATGAGGAGACTTGCCTGG-3′ and reverse, 5′-GGCATTTGTGGTTGGGTCAG-3′; TNF-α forward, 5′-TCTGGGCAGGTCTACTTTGG-3′ and reverse, 5′-GGTTGAGGGTGTCTGAAGGA-3′; MCP-1 forward, 5′-AATCAATGCCCCAGTCACCT-3′ and reverse, 5′-CTTCTTTGGGACACTTGCTGC−3′. The transcripts were mixed with LightCycler 480 SYBR-Green I Master Mix and quantified in the LightCycler® 480 Real-Time Quantitative PCR System (both from Roche) using the double standard curve method. The results were normalized to the expression of GAPDH.
Inflammatory cytokines expression measured by enzyme-linked immunosorbent assay (ELISA)
We detected the expressions of IL-1β, IL-6, and TNF-α in HUVECs with Human Quantikine ELISA kits (R&D Systems, Inc., Minneapolis, MN, USA). HUVECs were divided into four groups: i) Control: Cells treated with neither LPS nor BAI; ii) LPS: Cells stimulated by LPS (10 µM); iii) LPS + BAI: Cells stimulated by LPS (10 µM) and treated with BAI (6.25 µM); and iv) BAI: Cells treated with BAI (6.25 µM). We collected the culture supernatant and measured the inflammatory cytokine expression in strict accordance with the manufacturer's specifications and read the absorbance of each well at 450 nm using a microplate reader; 550 nm was used as the reference wavelength. The values of the samples were calculated through a comparison of the absorbance values with prediluted inflammatory cytokines standards (R&D Systems, Inc.).
Immunofluorescent staining of p65
The HUVECs were fixed, contained, blocked, and incubated with a dilution of the p65 antibody, and then incubated with green fluorescent secondary antibody in the dark. The nuclei were stained with DAPI. The cells were photographed using a fluorescent microscope; green fluorescence represents p65, whereas the nuclei are shown in blue. Overlapping areas of green and blue indicate the occurrence of the nuclei translocation of NF-κB.
Western blotting
HUVECs were lysed with lysis buffer RIPA (G2002; Wuhan Goodbio Biotechnology Co., Ltd., Wuhan, China). Protein quantification was conducted using the Pierce BCA protein assay kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The following primary antibodies were used: Cleaved-caspase-3 (9664), caspase-3 (9665), B-cell lymphoma 2-associated X protein (Bax; 2722), and Bcl-2 (2870) (all from Cell Signaling Technology, Inc., Danvers, MA, USA), TLR4 (sc-10741; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), p-TAK1 (4508), p-TBK1 (5483), p-p65 (3033), TAK1 (5206), TBK1 (3013), p65 (8242), and GAPDH (2118) (all from Cell Signaling Technology, Inc.). The corresponding peroxidase-conjugated secondary antibodies used were IRdye 800CW-conjugated goat anti-mouse immunoglobulin G (926–32210) and IRDye 800CW-conjugated goat anti-rabbit immunoglobulin G (926–32211) (both from LI-COR Biosciences, Lincoln, NE, USA). The blots were scanned using a two-color infrared imaging system (Odyssey; LI-COR Biosciences) and the expression of target proteins were normalized to the expression of GAPDH.
Statistical analysis
All statistical analyses were computed using the SPSS software (version 17.0; SPSS, Inc., Chicago, IL, USA) and the results are presented as the mean ± SD. For measurement data, a one-sample Kolmogorov-Smirnov test was utilized for normal distribution detection, all data detected being normal distribution (P>0.05). Then a homogeneity variance test followed. If the data with homoscedasticity (P>0.05), they were analyzed with one-way ANOVA and Tukey HSD test. If the data with heteroscedasticity (P<0.05), a Tamhane's T2 test was appropriate. Besides, enumeration data were compared with a Chi-square test. A value of P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of BAI on HUVEC cell survival
The characterization of the cytotoxicity of BAI (3.125–400 µM) in HUVECs revealed that high concentrations of BAI (400 µM) caused a reduction in cell viability by approximately 30%, but there was no significant difference of cell viability (P>0.05) occurred in HUVECs. Thus, BAI concentrations of 3.125–400 µM (Fig. 1A) were considered not to be cytotoxic to HUVECs. The cell viability of LPS-stimulated HUVECs (10 µM) was reduced by more than 30% compared with that of the control cells; this decrease was rescued by the addition of 3.125–50 µM BAI (Fig. 1B). We selected 6.25 µM BAI for further experiments, as it exerted the clearest rescue effect on cell viability. We also studied cells stimulated for 3, 6, 12, 24 and 48 h, finding out that HUVECs stimulated for 12 h started to have significant change of cell viability.
Figure 1.
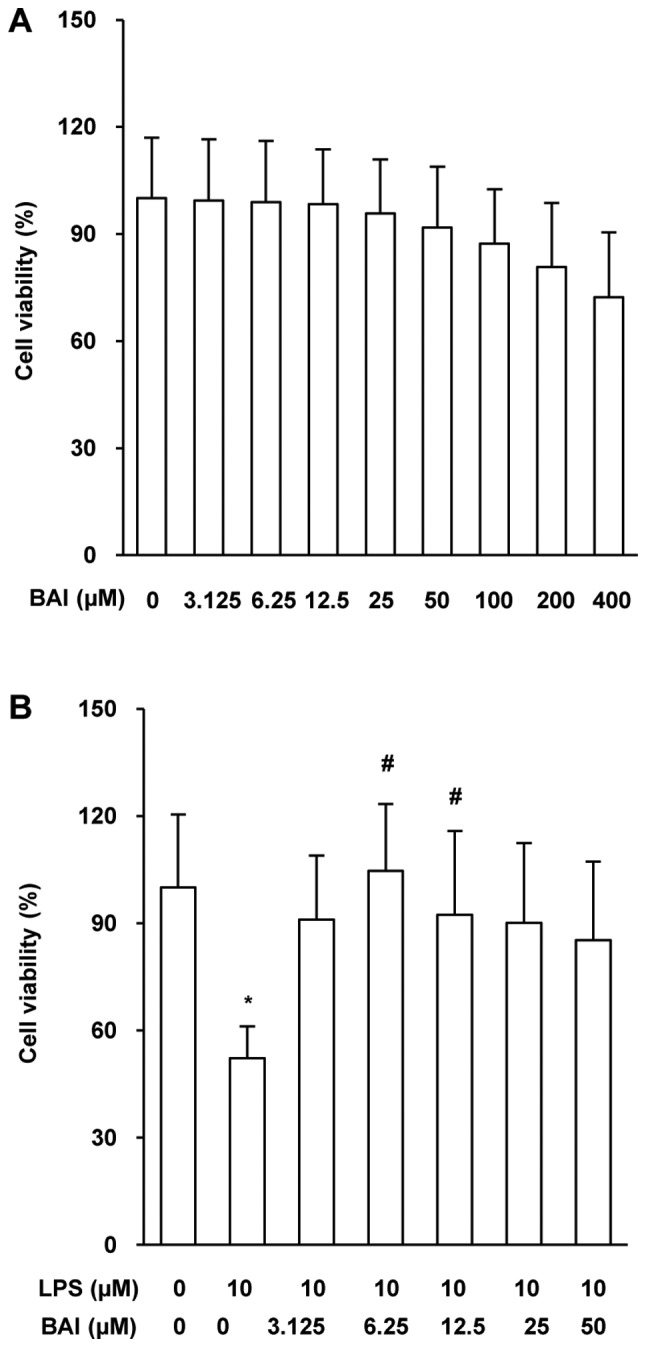
BAI protected against LPS-induced injury in HUVECs. HUVECs were stimulated by LPS or treated with BAI for 12 h. Cell viability was evaluated in HUVECs treated with (A) different concentrations of BAI (0, 3.125, 6.25, 12.5, 25, 50, 100, 200 and 400 µM) alone or (B) in combination (BAI, 0, 3.125, 6.25, 12.5, 25 or 50 µM) with 0 or 10 µM LPS. Cell viability was measured by Cell Counting Kit-8 assays and was calculated using the optical density values. The data are expressed as the mean ± standard deviation. *P<0.05 vs. control (0 µM BAI/LPS); #P<0.05 vs. LPS group (10 µM LPS only). BAI, baicalein; LPS, lipopolysaccharide; HUVECs, human umbilical vein endothelial cells.
BAI attenuated LPS-induced inflammation in HUVECs
LPS-induced injury in HUVECs is related to increased inflammation. To determine the protective effect of BAI on inflammation, we measured the mRNA expression levels of IL-1β, IL-6, TNF-α and MCP-1 by quantitative PCR and the protein levels by ELISA. Our results showed that the expressions of the inflammatory markers was markedly suppressed in BAI-treated HUVECs (Fig. 2B-D). Besides, the changes of inflammatory cytokines mRNA level in HUVECs treated for 24 h, which were not shown in the article, were detected to be similar to cells treated for 12 h.
Figure 2.

BAI attenuated inflammation in HUVECs. HUVECs were cultured for 12 h and were then stimulated with LPS or treated with BAI for 12 h. (A) The mRNA expressions of IL-1β, IL-6, TNF-α and MCP-1 were detected by reverse transcription-quantitative polymerase chain reaction. The secretion levels of (B) IL-6, (C) IL-1β and (D) TNF-α were detected by ELISA. The data are expressed as the mean ± standard deviation. *P<0.05 vs. control (0 µM BAI/LPS); #P<0.05 vs. LPS group (10 µM LPS only). BAI, baicalein; LPS, lipopolysaccharide; HUVECs, human umbilical vein endothelial cells; IL, interleukin; TNF-α, tumor necrosis factor-α; MCP-1, monocyte chemoattractant protein-1.
BAI inhibited LPS-induced apoptosis in HUVECs
To examine the protective effect of BAI in the regulation of endothelial cell injury, flow cytometry was used to differentiate apoptotic cells and the TUNEL assay was performed to determine DNA fragmentation. Compared with the control group, treatment with BAI profoundly reduced apoptosis in LPS-stimulated cells (Fig. 3A-D).
Figure 3.

BAI inhibited HUVEC apoptosis. HUVECs were stimulated with LPS or treated with BAI for 12 h following culture for 12 h. (A) HUVECs were analyzed by flow cytometry and (B) the results revealed that BAI treatment lead to a reduction in apoptosis when compared with LPS stimulation only. The results are expressed as the cell ratio of propidium iodide staining and deoxyuridine triphosphate-fluorescein. (C) The representative images (magnification, ×200) as well as the (D) statistical results of TUNEL staining as verified these results; the results were calculated by determining the ratio of DNA damaged cells that were stained green. (E) Relative protein levels of c-caspase-3, Bax and Bcl-2 were determined by western blotting and (F) quantified by densitometry. BAI inhibited the increase of c-caspase-3 and Bax as well as the reduction of Bcl-2 induced by LPS stimulation. The data are expressed as the mean ± standard deviation. *P<0.05 vs. control (0 µM BAI/LPS); #P<0.05 vs. LPS group (10 µM LPS only). BAI, baicalein; LPS, lipopolysaccharide; HUVECs, human umbilical vein endothelial cells; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; c-caspase, cleaved-caspase.
To further clarify the effect of BAI on LPS-induced injury, we measured the relative expressions of Bcl-2, Bax and cleaved caspase-3 by using western blotting. The data revealed that BAI increased the level of Bcl-2, but reduced the level of Bax and cleaved caspase-3, which contrasted with the changes induced by LPS (Fig. 3E and F).
BAI reduced LPS-induced HUVECs injury via TLR4/NF-κB signaling
The above results suggested that BAI might attenuate LPS induced endothelial cell injury. However, the molecular mechanism by which BAI exerted these protective effects was unclear. As the TLR4/NF-κB cascade signaling pathway is proven to play an important part in the development of endothelial inflammation and apoptosis, we examined whether BAI affected the LPS-induced activation of the TLR4/NF-κB signaling pathway. In comparison with the LPS group, the expression of TLR4 and the LPS-induced phosphorylation of TAK1, TBK1, and NF-κB p65 were reduced by BAI (Fig. 4A and B). MRT67307 removed the protective effects of BAI on the phosphorylation of TBK1 (Fig. 4C and D). In addition, the immunofluorescent staining of p65 revealed that nuclear translocation in HUVECs was inhibited by BAI treatment (Fig. 4E and F).
Figure 4.

BAI reduced LPS-induced damage via TLR4/NF-κB signaling. The protein expressions of TLR4/NF-κB signaling were established by (A-D) western blotting. (A) Blots for LPS stimulation demonstrated that (B) TLR4, p-TAK1, p-TBK1 and p-p65 were upregulated by LPS, then downregulated by BAI treatment. (C) MRT67307 treatment and (D) inhibited the effect of BAI on p-TBK1. (E) The immunofluorescent staining of p65 (magnification, ×200) revealed that (F) there was more cell nuclear translocation with LPS stimulation and less with BAI treatment. The data are expressed as the mean ± standard deviation. *P<0.05 vs. control (0 µM BAI/LPS); #P<0.05 vs. LPS group (10 µM LPS only); **P<0.05 vs. LPS+BAI (10 µM LPS, 6.25 µM BAI). BAI, baicalein; LPS, lipopolysaccharide; HUVECs, human umbilical vein endothelial cells; TLR4, Toll-like receptor 4; p, phosphorylated; TAK1, transforming growth factor β-activated kinase 1; TBK1, tumor necrosis factor receptor-associated family member associated nuclear factor-κB activator-binding kinase 1.
Discussion
In this study, we found that BAI protected against the LPS-induced injury in HUVECs. BAI reduced the activation of TLR4/NF-κB signaling in blunt inflammation and apoptosis. This discovery points to an important role for BAI in the regulation of vascular endothelial dysfunction.
Endothelial cells are reported to have a critical role in the process of HF (16). Due to their advantages of plasticity, secretion ability and repair function, endothelial cells have been confirmed as an important cardiac moiety (1). Traditionally, inflammatory cytokines are considered to be derived from immune cells, such as monocytes, but recent studies have indicated that endothelial cells could also be the source of inflammatory cytokines during inflammatory responses (17). Endothelial cells are reported to be involved in the adhesion and migration of immunocytes (18).
To elucidate the mechanisms of BAI in HUVECs, we detected the expression of certain proteins in HUVECs. This helped in revealing a reduction in the expression of TLR4 and in the phosphorylation of NF-κB p65 (p-p65) in BAI-treated HUVECs compared with those of the control group. Previous studies have also indicated that BAI reduced p-p65, including the BAI-induced prevention of cisplatin-induced acute kidney injury through the downregulation of the NF-κB pathway (19) and the BAI-induced suppression of p-p65 expression in the IL-1β-induced proliferation of human rheumatoid arthritis fibroblast-like synoviocytes (20). Wang et al (21) also reported, decreased expressions of TLR4 and p-p65 in their experiment, which investigated the ameliorative effects of BAI in early brain injury in rats induced by experimental subarachnoid hemorrhage. Our data showed that BAI downregulated the expression of TLR4 and p-p65 in LPS-stimulated HUVECs. We also found the reduced activation of TBK1 and TAK1 after BAI treatment.
MRT67307 (Formula C26H36N6O2·xHCl) is a potent and dual IKKε and TBK1 inhibitor. According to the MRT67307 specification from Selleck Chemicals, MRT67307 prevents the phosphorylation of IRF3 and the production of IFNβ in macrophages; enhances the IL-1-stimulated phosphorylation of p105 and RelA, as well as enhances IL-1-stimulated activation of NF-κB-dependent gene transcription in wild-type MEFs; increases IL-10 production and suppresses proinflammatory cytokine production via a cAMP response element-binding protein (CREB)-regulated transcriptional coactivator (CRTC) 3 dependent mechanism; inhibit ULK and block autophagy in MEF cells. Furthermore, MRT67307 was reported to transiently enhance the protein level of p-TBK1 in 1 h, such as enhancing the LPS-stimulated p-TBK1 (22). In this study, we found that the protective effects of BAI were negated by MRT67307.
The present study has revealed the protective effect of BAI on endothelial cells injury and provided insights into the mechanism underlying the protective effect of BAI. BAI decreased LPS-induced cell apoptosis through the inactivation of TLR4/TBK/NF-κB signaling. Our observations provided further insights into the pathogenesis of vascular endothelial injury and HF and demonstrated that BAI may contribute to the inhibition of endothelial dysfunction and in the prevention of HF. However, we focused on only one reported signaling pathway of BAI; the study is also limited by the absence of research into the in vivo effects and mechanisms of BAI. Additional studies are required to conduct further exploration into the impact of BAI on LPS-induced injury in HUVECs.
Acknowledgements
This study was supported by National Natural Science Foundation of China (grant nos. 81270303, 81470516, 81530012); and Fundamental Research Funds of the Central Universities (grant no. 2042017kf0145).
Glossary
Abbreviations
- BAI
baicalein
- LPS
lipopolysaccharide
- HUVECs
human umbilical vein endothelial cells
References
- 1.McCarron JG, Lee MD, Wilson C. The endothelium solves problems that endothelial cells do not know exist. Trends Pharmacol Sci. 2017;38:322–338. doi: 10.1016/j.tips.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fraisl P, Mazzone M, Schmidt T, Carmeliet P. Regulation of angiogenesis by oxygen and metabolism. Dev Cell. 2009;16:167–179. doi: 10.1016/j.devcel.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Wei F, Liu S, Luo L, Gu N, Zeng Y, Chen X, Xu S, Zhang D. Anti-inflammatory mechanism of ulinastatin: Inhibiting the hyperpermeability of vascular endothelial cells induced by TNF-α via the RhoA/ROCK signal pathway. Int Immunopharmacol. 2017;46:220–227. doi: 10.1016/j.intimp.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Jang J, Jung Y, Kim Y, Jho EH, Yoon Y. LPS-induced inflammatory response is suppressed by Wnt inhibitors, Dickkopf-1 and LGK974. Sci Rep. 2017;7:41612. doi: 10.1038/srep41612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu X, Lan P, Hou X, Han Q, Lu N, Li T, Jiao C, Zhang J, Zhang C, Tian Z. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. J Hepatol. 2017;66:693–702. doi: 10.1016/j.jhep.2016.12.018. [DOI] [PubMed] [Google Scholar]
- 6.Peng T, Lu X, Lei M, Moe GW, Feng Q. Inhibition of p38 MAPK decreases myocardial TNF-alpha expression and improves myocardial function and survival in endotoxemia. Cardiovasc Res. 2003;59:893–900. doi: 10.1016/S0008-6363(03)00509-1. [DOI] [PubMed] [Google Scholar]
- 7.Ives A, Nomura J, Martinon F, Roger T, LeRoy D, Miner JN, Simon G, Busso N, So A. Xanthine oxidoreductase regulates macrophage IL-1β secretion upon NLRP3 inflammasome activation. Nat Commun. 2015;6:6555. doi: 10.1038/ncomms7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim SD, Lee YJ, Baik JS, Han JY, Lee CG, Heo K, Park YS, Kim JS, Ji HD, Park SI, et al. Baicalein inhibits agonist- and tumor cell-induced platelet aggregation while suppressing pulmonary tumor metastasis via cAMP-mediated VASP phosphorylation along with impaired MAPKs and PI3K-Akt activation. Biochem Pharmacol. 2014;92:251–265. doi: 10.1016/j.bcp.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Han E, Xing Q, Yan J, Arrington A, Wang C, Tully D, Kowolik CM, Lu DM, Frankel PH, et al. Baicalein upregulates DDIT4 expression which mediates mTOR inhibition and growth inhibition in cancer cells. Cancer Lett. 2015;358:170–179. doi: 10.1016/j.canlet.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai W, Fu Y, Zhang W, Chen X, Zhao J, Song W, Li Y, Huang Y, Wu Z, Sun R, et al. Synergistic effects of baicalein with cefotaxime against Klebsiella pneumoniae through inhibiting CTX-M-1 gene expression. BMC Microbiol. 2016;16:181. doi: 10.1186/s12866-016-0797-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai KL, Hung CH, Chan SH, Shih JY, Cheng YH, Tsai YJ, Lin HC, Chu PM. Baicalein protects against oxLDL-caused oxidative stress and inflammation by modulation of AMPK-alpha. Oncotarget. 2016;7:72458–72468. doi: 10.18632/oncotarget.12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Y, Miao Z, Hu Y, Yuan Y, Zhou Y, Wei L, Zhao K, Guo Q, Lu N. Baicalein reduces angiogenesis in the inflammatory microenvironment via inhibiting the expression of AP-1. Oncotarget. 2017;8:883–899. doi: 10.18632/oncotarget.13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zong J, Zhang DP, Zhou H, Bian ZY, Deng W, Dai J, Yuan Y, Gan HW, Guo HP, Tang QZ. Baicalein protects against cardiac hypertrophy through blocking MEK-ERK1/2 signaling. J Cell Biochem. 2013;114:1058–1065. doi: 10.1002/jcb.24445. [DOI] [PubMed] [Google Scholar]
- 14.Zheng K, Setyawati MI, Lim TP, Leong DT, Xie J. Antimicrobial cluster bombs: Silver nanoclusters packed with daptomycin. ACS Nano. 2016;10:7934–7942. doi: 10.1021/acsnano.6b03862. [DOI] [PubMed] [Google Scholar]
- 15.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: A cautionary note. Hepatology. 1995;21:1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 16.Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heim A, Zeuke S, Weiss S, Ruschewski W, Grumbach IM. Transient induction of cytokine production in human myocardial fibroblasts by coxsackievirus B3. Circ Res. 2000;86:753–759. doi: 10.1161/01.RES.86.7.753. [DOI] [PubMed] [Google Scholar]
- 18.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 19.Sahu BD, Mahesh Kumar J, Sistla R. Baicalein, a bioflavonoid, prevents cisplatin-induced acute kidney injury by up-regulating antioxidant defenses and down-regulating the MAPKs and NF-κB pathways. PLoS One. 2015;10:e134139. doi: 10.1371/journal.pone.0134139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S, Yang Y, Feng H, Wang H, Zhao R, Liu H. Baicalein inhibits interleukin-1-induced proliferation of human rheumatoid arthritis fibroblast-like synoviocytes. Inflammation. 2014;37:163–169. doi: 10.1007/s10753-013-9725-9. [DOI] [PubMed] [Google Scholar]
- 21.Wang CX, Xie GB, Zhou CH, Zhang XS, Li T, Xu JG, Li N, Ding K, Hang CH, Shi JX, Zhou ML. Baincalein alleviates early brain injury after experimental subarachnoid hemorrhage in rats: Possible involvement of TLR4/NF-κB-mediated inflammatory pathway. Brain Res. 2015;1594:245–255. doi: 10.1016/j.brainres.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 22.Clark K, Peggie M, Plater L, Sorcek RJ, Young ER, Madwed JB, Hough J, McIver EG, Cohen P. Novel cross-talk within the IKK family controls innate immunity. Biochem J. 2011;434:93–104. doi: 10.1042/BJ20101701. [DOI] [PubMed] [Google Scholar]