

Abstract
Cushing’s syndrome is characterized by the overproduction of adrenocortical steroids. Steroidogenesis inhibitors are mainstays of medical therapy for Cushing’s syndrome; unfortunately, adverse side effects and treatment failures are common with currently available drugs. The general anesthetic induction agent etomidate is among the most potent inhibitors of adrenocortical steroidogenesis. However, its use as a treatment of Cushing’s syndrome is complicated by its sedative-hypnotic activity and ability to produce myoclonus, central nervous system actions thought to be mediated by the GABAA receptor. Here, we describe the pharmacology of the novel etomidate analog (R)-ethyl 1-(1-(3,5-dimethoxyphenyl)ethyl)-1H-imidazole-5-carboxylate (dimethoxy-etomidate). In contrast to etomidate, dimethoxy-etomidate minimally enhanced GABA-evoked GABAA receptor–mediated currents even at a near-saturating aqueous concentration. In Sprague-Dawley rats, dimethoxy-etomidate’s potency for producing loss of righting reflexes—an animal model of sedation/hypnosis—was 2 orders of magnitude lower than that of etomidate, and it did not produce myoclonus. However, similar to etomidate, dimethoxy-etomidate potently suppressed adrenocortical steroid synthesis primarily by inhibiting 11β-hydroxylase. [3H]etomidate binding to rat adrenocortical membranes was inhibited by dimethoxy-etomidate in a biphasic manner with IC50 values of 8.2 and 3970 nM, whereas that by etomidate was monophasic with an IC50 of 22 nM. Our results demonstrate that, similar to etomidate, dimethoxy-etomidate potently and dose-dependently suppresses adrenocortical steroid synthesis by inhibiting 11β-hydroxylase. However, it is essentially devoid of etomidate’s GABAA receptor positive modulatory and sedative-hypnotic activities and produces no myoclonus, providing proof of concept for the design of etomidate analogs without important central nervous system actions for the pharmacologic treatment of Cushing’s syndrome.
Introduction
Cushing’s syndrome is characterized by the overproduction of adrenocortical steroids and manifests by a variety of signs and symptoms of hypercortisolemia, including hypertension, diabetes, immunosuppression, water retention, depression, poor wound healing, and cognitive impairment (Tritos and Biller, 2014a; Lacroix et al., 2015; Pivonello et al., 2016, 2017). When poorly controlled, it is associated with significant morbidity (e.g., coronary artery disease and stroke) and increased mortality (Mancini et al., 2004; Dekkers et al., 2013). It is most commonly caused by an adrenocorticotropic hormone (ACTH)–secreting pituitary tumor that stimulates steroid production by the adrenal gland (i.e., Cushing’s disease), but can also be the result of ACTH secretion by neuroendocrine tumors, adrenocortical adenomas, or hyperplasia (Lacroix et al., 2015). Although surgery is typically the first line of therapy for patients with Cushing’s syndrome, medical treatment is often necessary due to postsurgical local recurrence or metastatic disease (Atkinson et al., 2005; Fleseriu et al., 2007; Patil et al., 2008; Alexandraki et al., 2013; Dimopoulou et al., 2013; Pivonello et al., 2015). Medical treatment may also be instituted while awaiting the effects of radiation therapy or preoperatively to reduce the risk of surgical complications (Cuevas-Ramos and Fleseriu, 2014; Fleseriu and Castinetti, 2016).
Steroidogenesis inhibitors are mainstays of medical therapy in Cushing’s syndrome (Daniel and Newell-Price, 2015). They reduce steroid synthesis by reversibly inhibiting the function of one or more enzymes in the adrenocortical steroid biosynthetic pathway. Although there are currently no U.S. Food and Drug Administration–approved steroidogenesis inhibitors, ketoconazole, metyrapone, mitotane, and etomidate are clinically available for other uses and are used off-label to treat patients with Cushing’s syndrome (Daniel and Newell-Price, 2015; Ambrogio and Cavagnini, 2016). Unfortunately, adverse effects and treatment failures with these drugs are common, and only etomidate—which is the most potent of the inhibitors—is completely efficacious (Lambert et al., 1986; Lamberts et al., 1987; Preda et al., 2012; Cuevas-Ramos and Fleseriu, 2014; Gadelha and Vieira Neto, 2014; Ambrogio and Cavagnini, 2016). However, such efficacy is achieved with etomidate only by using doses that risk producing sedation or hypnosis (Engelhardt and Weber, 1994; Preda et al., 2012; Cuevas-Ramos and Fleseriu, 2014; Tritos and Biller, 2014b; Daniel and Newell-Price, 2015).
Etomidate produces sedation/hypnosis and inhibits cortisol synthesis by binding to distinct protein targets (Fig. 1A). The former activity results from etomidate binding to the GABAA receptor, producing enhanced receptor function (Belelli et al., 2003; Jurd et al., 2003; Franks, 2006; Forman, 2011). The latter results primarily from etomidate binding to the active site of the adrenocortical enzyme 11β-hydroxylase, producing inhibition of enzymatic activity (de Jong et al., 1984; Fry and Griffiths, 1984; Allolio et al., 1985). In our previous experimental work aimed at designing safer sedative-hypnotic agents (Cotten et al., 2010; Shanmugasundararaj et al., 2013; Pejo et al., 2016), we demonstrated that by changing a single atom in etomidate’s molecular structure (forming carboetomidate), we could reduce binding affinity to 11β-hydroxylase and adrenocortical inhibitory potency by 3 orders of magnitude while retaining potent GABAA receptor positive modulatory activity and sedative-hypnotic action (Fig. 1B). Based on this ability to uncouple etomidate’s steroidogenesis inhibiting and sedative-hypnotic activities, we hypothesized that other etomidate analogs could be designed that possess exactly the reverse pharmacology of carboetomidate on these two critical targets: they would retain etomidate’s exceptionally high potency as an inhibitor of steroid synthesis but lack its sedative-hypnotic activity. As such, these analogs might have utility as pharmacological therapies for Cushing’s syndrome. In this study, we describe the in vitro and in vivo pharmacology of (R)-ethyl 1-(1-(3,5-dimethoxyphenyl)ethyl)-1H-imidazole-5-carboxylate (dimethoxy-etomidate), a first-in-class phenyl ring–substituted etomidate analog that retains etomidate’s high potency as a steroidogenesis inhibitor but is essentially devoid of its sedative-hypnotic activity (Fig. 1C).
Fig. 1.

Chemical structures of etomidate (A), carboetomidate (B), and dimethoxy-etomidate (C). Etomidate has two major targets: the GABAA receptor and 11β-hydroxylase. These two targets are responsible for etomidate’s sedative-hypnotic and steroidogenesis inhibitory activities, respectively. Carboetomidate and dimethoxy-etomidate are etomidate analogs whose respective potent 11β-hydroxylase and GABAA receptor actions have been “designed out.”
Materials and Methods
Sources of Drugs and Chemicals
Dexamethasone was obtained from American Regent (Shirley, NY), and ACTH1-24 and GABA were from Sigma-Aldrich Chemical Company (St. Louis, MO). Etomidate was purchased from Bachem (Torrance, CA). [3H]etomidate was prepared from unlabeled etomidate by PerkinElmer Life Sciences (Boston, MA) using a catalytic exchange reaction. The mass fragmentation pattern of [3H]etomidate showed that all of the tritium was located on the imidazole ring. The specific activity of [3H]etomidate was 8.6 Ci/mM. Enzyme-linked immunosorbent assay kits for corticosterone were purchased from IDS (Gaithersburg, MD), that for pregnenolone was purchased from Elabscience (Bethesda, MD), and those for progesterone and deoxycorticosterone were purchased from MyBioSource (Cambridge, MA). Dimethoxy-etomidate was synthesized by Aberjona Laboratories (Beverly, MA).
Animals
All studies were conducted with the approval of and in accordance with rules and regulations of the Institutional Animal Care and Use Committee at the Massachusetts General Hospital, Boston, MA. Adult male Sprague-Dawley rats (250–350 g) were purchased from Charles River Laboratories (Wilmington, MA). Xenopus laevis adult female frogs were purchased from Xenopus One (Ann Arbor, MI). All rat studies were conducted between 11 AM and 3 PM.
GABAA Receptor Electrophysiology Studies
Oocytes were harvested from Xenopus frogs and injected with messenger RNA encoding the α1, β3, and γ2L subunits of the human GABAA receptor (5 ng of messenger RNA total at a subunit ratio of 1:1:3). Oocytes were then incubated in ND96 buffer (96 mΜ NaCl, 2 mΜ KCl, 1.8 mΜ CaCl2, 1 mΜ MgCl2, 5 mM HEPES, pH = 7.4) containing 0.05 mg/ml of gentamicin for 18–48 hours at 18°C before electrophysiological study. Electrophysiological recordings were performed at room temperature using the whole-cell two-electrode voltage-clamp technique as previously described (Pejo et al., 2012). For each oocyte, a GABA concentration–peak current response curve was generated to define the GABA concentration that elicits 5% of the current evoked by 1 mM GABA (i.e., EC5 GABA). The effect of drug (etomidate or dimethoxy-etomidate) was then defined in that oocyte by perfusing it with EC5 GABA alone for 15–20 seconds followed immediately by EC5 GABA plus drug at the desired concentration for 20–60 seconds. The resulting current response was then recorded. The peak current response recorded in the presence of EC5 GABA plus drug was then normalized to the peak current response evoked by 1 mM GABA. Between electrophysiological recordings, oocytes were perfused with buffer for at least 3 minutes to remove GABA (and drug, if present) and to allow receptors to recover from desensitization.
The concentration-response relationships for EC5 GABA potentiation by etomidate and dimethoxy-etomidate were fit to a Hill equation using Prism 6 for Mac OS X (GraphPad, La Jolla, CA) to define an EC50 and maximal current response in the presence of high drug concentrations (and their respective S.E.M.):
where max and min are the maximum and minimum electrophysiological responses, and EC50 is the drug concentration that produces a response that is halfway between the max and min. In these fits, the minimum was constrained to 5% (by definition, for EC5-evoked currents).
Sedative-Hypnotic Activity Studies in Rats
The sedative-hypnotic potencies of etomidate and dimethyl-etomidate were assessed in Sprague-Dawley rats using a loss of righting reflexes (LoRR) assay (Cotten et al., 2009). In brief, the desired dose of drug in dimethylsulfoxide vehicle (0.1–0.3 ml) was rapidly (<5 seconds) injected through a 24-gauge intravenous catheter placed in a tail vein. This was followed by a 0.5-ml normal saline flush. Immediately after injection, rats were turned supine. A rat was judged to have LoRR if it failed to right (i.e., turn itself back onto all four paws) after drug administration. The duration of LoRR was defined as the time from drug injection until the animal spontaneously righted itself onto all four legs. For each drug, the median effective dose (ED50) for LoRR was determined by fitting the dose-response relationship to the following equation using the Waud approach for quantal (i.e., all or none) responses (Waud, 1972) with Igor Pro 6.37 (Wavemetrics, Lake Oswego, OR):
In this approach, a rat that has LoRR after receiving a given dose is assigned a value of 1, whereas one that does not is assigned a value of 0.
Steroidogenesis Inhibition by Etomidate and Dimethoxy-Etomidate
The in vivo adrenocortical inhibitory potencies of drugs were assessed in dexamethasone-suppressed rats (four per group) immediately after administering vehicle alone, a low etomidate or dimethoxy-etomidate dose (0.3 mg/kg i.v.), an intermediate etomidate or dimethoxy-etomidate dose (3 mg/kg i.v.), or a high dimethoxy-etomidate dose (50 mg/kg) using an ACTH-stimulation test as previously described (Cotten et al., 2010). In brief, rats were pretreated with dexamethasone (0.2 mg/kg i.v.) to reduce endogenous ACTH production and minimize baseline adrenocortical steroid concentrations. Two hours later and after receiving a second dexamethasone dose, rats received ACTH1–24 i.v. and the desired dose of either etomidate or dimethoxy-etomidate solubilized in water containing a 1:1 molar ratio of Captisol (Sulfobutylether-β-Cyclodextrin; Ligand Pharmaceuticals, San Diego, CA) or water containing Captisol vehicle alone as a control. Fifteen minutes later, a blood sample was drawn and ACTH-stimulated adrenocortical steroid concentrations in the plasma were determined using enzyme-linked immunosorbent assays and a 96-well plate reader (Molecular Devices, Sunnyvale, CA).
Myoclonic Movements Produced by Etomidate and Dimethoxy-Etomidate
The abilities of the two drugs to produce myoclonus were assessed in Sprague-Dawley rats using a two-way crossover protocol. Each rat was randomized to receive either etomidate or dimethoxy-etomidate (3 mg/kg i.v.) solubilized in water containing a 1:1 molar ratio of Captisol. The desired drug was rapidly injected through a 24-gauge intravenous catheter placed in a tail vein followed by a 0.5-ml normal saline flush. Rats were immediately tested for LoRR, and the number of myoclonic events was recorded during each 5-minute epoch for 30 minutes. A myoclonic event was defined as an unproductive (i.e., not associated with LoRR, feeding, or grooming behaviors) movement. Each distinct jerk or tremor constituted a single event. To avoid potentially confounding movements associated with bruxing, boggling, sniffing, and grooming, myoclonus was only considered to occur in the head and jaw in the presence of abnormal tongue lolling and jaw movement, or a sharp head movement out of sync with the rat’s breathing. At the end of the 30-minute observation period, rats were returned to their cages. After a 24-hour recovery period, each rat was crossed over to the other drug group, and the experiment was repeated.
Displacement of [3H]Etomidate from Adrenal Membranes by Etomidate and Dimethoxy-Etomidate
Adrenal glands from Sprague-Dawley rats were purchased from BioreclamationIVT (Baltimore, MD), shipped frozen to our laboratory, and prepared as previously described (Pejo et al., 2016). In brief, four adrenal glands were thawed for each preparation, placed on a glass stand, cut into pieces, and then added to ice-cold preparation buffer (10 mM HEPES, 1 mM EDTA, 10 μg/ml leupeptin, 10 μg/ml chymostatin, 10 μg/ml pepstatin A, 2 μg/ml aprotinin, 1 mM polymethanesulfonyl fluoride, and 10 μl/ml ethanol). After homogenization, carboxylesterases were inactivated by incubating with 1 mM diisopropyl fluorophosphate (Sigma-Aldrich) for 60 minutes. The mixture was then centrifuged and the pellet washed twice. The final pellet was then resuspended in buffer by manual homogenization, passed through a 23-gauge needle three times, aliquoted into 1-ml eppendorf tubes, and stored at −80°C until used. The protein concentration in these samples was quantified using a Pierce BCA Protein Assay Kit (ThermoFisher, Rockford, IL). All binding experiments were performed in glass vials at room temperature with the final concentration of protein adjusted to 0.07 mg/ml. The reported data were obtained using three separate preparations.
After thawing, adrenal membranes were equilibrated for 30 minutes at room temperature with 2 nM [3H]etomidate along with ranging concentrations of etomidate or dimethoxy-etomidate (total volume 0.5 ml) in a phosphate assay buffer (11.9 mM phosphates, 137 mM NaCl, 200 mM KCl, and 1 mM EDTA at pH 7.4). After equilibration, the mixture was passed through a presoaked (with 0.5% polyethylenimine in water for 2 hours) 25-mm GF/B glass fiber filter under suction, and the filter was immediately washed twice with 5 ml of assay buffer. After drying under a heat lamp for 2 hours, each filter was transferred to a scintillation vial. Liquiscint scintillation cocktail (National Diagnostics, Atlanta, GA) was added to the vial, and the radioactivity in the vial was quantified using a Tri-Carb liquid scintillation counter (Packard, Meriden, CT). At each concentration, the measured radioactivity was normalized to that measured in the absence of competing etomidate or dimethoxy-etomidate. The concentration-normalized response relationships for inhibition of [3H]etomidate by etomidate and dimethoxy-etomidate were fit to one- and two-site models, respectively, using Prism 6 for Mac OS ×.
One-Site Model Equation.
where min is the radioactivity counts in the presence of high competing ligand concentrations, and IC50 is the half-inhibitory concentration.
Two-Site Model Equation.
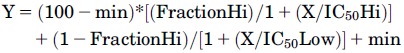 |
where min is the radioactivity counts in the presence of high competing ligand concentrations; IC50Hi and IC50Low are the half-inhibitory concentrations at the high and low affinity sites, respectively; and FractionHi is the fraction of sites that are high affinity.
Statistical Analysis
All individual data points are expressed as the mean ± S.E.M. of three to eight independent measurements. The results of nonlinear least-squares fitting of concentration-response curves are reported as the fitted value ± S.E.M., whereas those of quantal rat LoRR dose-response curves are reported as the fitted value ± S.D. as described by Waud (1972). Statistical analyses to define a preferred binding model (one site vs. two site) for competitive binding studies were carried out using an extra sum-of-squares F test, whereas those to assess differences in plasma steroid concentrations were carried out using either a Mann-Whitney U test (for a single comparison) or a Kruskal-Wallis test followed by a Dunn’s multiple comparisons test using Prism 6 for Mac OS ×. A two-way analysis of variance was used to compare the number of myoclonic movements produced by etomidate versus dimethoxy-etomidate during each 5-minute epoch after drug administration. Statistical significance was assumed for P < 0.05.
Results
Enhancement of EC5 GABA–Evoked Currents by Etomidate and Dimethoxy-Etomidate.
To assess and compare the abilities of etomidate and dimethoxy-etomidate to enhance GABAA receptor function, we perfused oocytes expressing α1β3γ2L GABAA receptors with EC5 GABA alone followed immediately by EC5 GABA plus either etomidate (Fig. 2A) or dimethoxy-etomidate (Fig. 2B) and recorded the resulting electrophysiological current responses. Although peak currents evoked by this low GABA concentration were enhanced by both drugs, the magnitude of enhancement produced by dimethoxy-etomidate was relatively small, less than or equal to one-eighth that produced by the same concentration of etomidate. Figure 2C plots the etomidate and dimethoxy-etomidate concentration-response relationships for this peak current enhancement. Etomidate enhanced peak currents in a manner that was concentration-dependent, potent, and highly efficacious. A fit of the etomidate data set to a Hill equation yielded an EC50 of 1.1 ± 0.2 µM and a maximum peak current that was 103% ± 3% of that produced by 1 mM GABA, a maximally activating agonist concentration. Although dimethoxy-etomidate also enhanced peak currents in a concentration-dependent manner, this action was neither potent nor efficacious, as fit of the dimethoxy-etomidate data set to a Hill equation yielded an EC50 of 210 ± 140 µM and a maximum peak current that was only 11% ± 1% of that produced by 1 mM GABA.
Fig. 2.

Potentiation of GABAA receptor currents by etomidate and dimethoxy-etomidate. Electrophysiological traces showing the potentiating effect of etomidate (A) or dimethoxy-etomidate (B) on currents evoked by a GABA concentration that elicits 5% of the current evoked by 1 mM GABA (EC5 GABA). For each set of traces, currents at all drug concentrations were obtained using the same oocyte. (C) Etomidate and dimethoxy-etomidate concentration-response curves for potentiation of EC5 GABA–evoked currents. Each symbol is the mean ± S.E.M. derived from four different oocytes. The curves are fits of the data sets to a Hill equation with the minimum constrained to 5%. For etomidate, the fit yielded a half-maximal potentiating concentration of 1.1 ± 0.2 µM and a maximum peak current amplitude at high etomidate concentrations of 103% ± 3% of that produced by 1 mM GABA. For dimethoxy-etomidate, the fit yielded a half-maximal potentiating concentration of 210 ± 140 µM and a maximum peak current value at high concentrations of 11% ± 1%.
Sedative-Hypnotic Activities of Etomidate and Dimethoxy-Etomidate.
To define the sedative-hypnotic potencies of etomidate and dimethoxy-etomidate, we administered these drugs to rats intravenously at various doses and assessed whether they caused LoRR (Fig. 3A). Etomidate produced LoRR in all rats receiving doses of 1 mg/kg or greater. A fit of the etomidate dose-response relationship for LoRR yielded an ED50 of 0.77 ± 0.17 mg/kg. In contrast, dimethoxy-etomidate produced LoRR in all rats only when administered at a dose of 100 mg/kg, the highest dimethoxy-etomidate dose studied. A fit of the dimethoxy-etomidate dose-response relationship for LoRR yielded an ED50 of 72 ± 13 mg/kg, a 2-order-of-magnitude reduction in sedative-hypnotic potency compared with etomidate. The time required for righting reflexes to return after administering etomidate increased with the dose, reaching 16.3 ± 0.5 minutes at 3 mg/kg (the highest etomidate dose studied). In contrast, even after administering a 100-mg/kg dose of dimethoxy-etomidate, righting reflexes returned in 2.2 ± 0.7 minutes (Fig. 3B).
Fig. 3.

(A) Dose-response curves for LoRR in rats. A fit of the dose-response relationships yielded ED50 values of 0.77 ± 0.17 mg/kg for etomidate and 72 ± 13 mg/kg for dimethoxy-etomidate. (B) Dose-response curves for LoRR duration in rats. In both panels, each symbol is the result obtained from a single rat experiment.
Suppression of Steroidogenesis by Etomidate and Dimethoxy-Etomidate.
We used an ACTH-stimulation test to evaluate and compare the abilities of etomidate and dimethoxy-etomidate to suppress adrenocortical steroidogenesis in rats. At an i.v. dose of 0.3 mg/kg, dimethoxy-etomidate reduced the ACTH-stimulated plasma corticosterone concentration by 63% from a control value of 152 ± 35 to 56 ± 13 ng/ml, whereas etomidate had no significant effect (Fig. 4A). At this dose, neither compound altered the ACTH-stimulated plasma concentrations of 11-deoxycorticosterone (Fig. 4B), pregnenolone (Fig. 4C), or progesterone (Fig. 4D). At an i.v. dose of 3 mg/kg, both etomidate and dimethoxy-etomidate reduced the ACTH-stimulated plasma corticosterone concentrations by 87% from 150 ± 48 ng/ml to values of 19 ± 11 and 19 ± 10 ng/ml, respectively (Fig. 4E), without significantly affecting the ACTH-stimulated plasma concentrations of 11-deoxycorticosterone (Fig. 4F), pregnenolone (Fig. 4G), or progesterone (Fig. 4H). At an i.v. dose of 50 mg/kg, dimethoxy-etomidate reduced the ACTH-stimulated plasma corticosterone concentration by 95% from a control value of 240 ± 69 to 12 ± 1 ng/ml (Fig. 5A) and the 11-deoxycorticosterone by 69% from 78 ± 5 to 24 ± 12 ng/ml (Fig. 5B). The ACTH-stimulated plasma concentrations of pregnenolone (Fig. 5C) and progesterone (Fig. 5D) were unaffected by this high dimethoxy-etomidate dose. We did not study etomidate at this high dose because it is lethal (Pejo et al., 2014).
Fig. 4.

Adrenocorticotropic hormone–stimulated plasma adrenocortical steroid concentrations following administration of vehicle (V), etomidate (E), or dimethoxy-etomidate (D-E) in rats. Intravenous doses of etomidate and dimethoxy-etomidate were 0.3 mg/kg (A–D) or 3 mg/kg (E–H). Each bar represents the mean ± S.E.M. obtained from four rat experiments. *P < 0.05.
Fig. 5.

Adrenocorticotropic hormone-stimulated plasma adrenocortical steroid concentrations following administration of vehicle (V) or 50 mg/kg IV dimethoxy-etomidate in rats. Corticosterone (A), 11-deoxycorticosterone (B), Pregnenolone (C), Progesterone D). Each bar represents the mean ± SEM from 4 rat experiments. *p < 0.05.
Myoclonic Movements Produced by Etomidate and Dimethoxy-Etomidate.
Etomidate (3 mg/kg i.v.) produced myoclonus (and LoRR) in all rats (n = 8). The mean (± S.E.M.) total number of myoclonic events in each rat during the 30-minute observation period after etomidate administration was 34 ± 2.4. The mean (± S.E.M.) number of myoclonic events reached a maximum value of 13.8 ± 2.2 per rat during the 5–10-minute epoch after administering the drug and then progressively decreased during the remaining 30-minute observation period (Fig. 6). In contrast, dimethoxy-etomidate (3 mg/kg i.v.) produced no myoclonic events (or LoRR) in any rat during the 30-minute observation period after drug administration.
Fig. 6.

Myoclonic activity produced by etomidate and dimethoxy-etomidate. Each rat was randomized to receive either etomidate or dimethoxy-etomidate (3 mg/kg i.v.). After drug administration, the number of myoclonic events observed during each 5-minute epoch was recorded for 30 minutes. After 24 hours, the rat was crossed over to the other drug and the experiment was repeated. Each symbol represents the mean ± S.E.M. number of myoclonic events recorded from eight rat experiments. **P < 0.01; ****P < 0.0001.
Displacement of [3H]Etomidate from Adrenal Homogenates by Etomidate and Dimethoxy-Etomidate.
We have previously shown that [3H]etomidate binds specifically (i.e., etomidate-displacably), saturably, and reversibly to a single class of high-affinity sites in adrenal homogenates (Pejo et al., 2016). The dissociation constant for this binding is within the range previously reported for etomidate inhibition of adrenocortical function in vitro and in vivo, strongly suggesting that it reflects [3H]etomidate binding to 11β-hydroxylase (Lamberts et al., 1987; Crozier et al., 1988; Cotten et al., 2010; Campagna et al., 2014). To test whether etomidate and dimethoxy-etomidate bind to the same high-affinity sites in adrenal homogenates, we compared their abilities to displace [3H]etomidate (2 nM) from these homogenates. Both etomidate and dimethoxy-etomidate displaced [3H]etomidate from adrenal homogenates in a concentration-dependent manner, and at the highest concentration studied (100 µM), both drugs displaced essentially all (≥95%) [3H]etomidate binding (Fig. 7). However, the concentration-dependence of this displacement differed between the two drugs. Displacement of [3H]etomidate by etomidate increased (i.e., [3H]etomidate binding decreased) in a monophasic manner indicative of a single class of high-affinity sites, whereas that by dimethoxy-etomidate increased in a biphasic manner, indicating the existence of two classes of binding sites with affinities that differ by orders of magnitude. We used an F test to confirm this observation statistically and found that a one-site model was preferred for etomidate, whereas a two-site one was preferred for dimethoxy-etomidate [F(2,8) = 88.1] with P < 0.0001. The IC50 for the high-affinity etomidate binding site was 22 ± 5 nM. The IC50 values for the high- and low-affinity dimethoxy-etomidate binding sites were 8.2 ± 2.0 and 3970 ± 1530 nM, with the high-affinity site accounting for 54% ± 3% of all sites.
Fig. 7.

Displacement of [3H]etomidate by etomidate and dimethoxy-etomidate. [3H]etomidate (2 nM) and the desired concentration of etomidate and dimethoxy-etomidate were equilibrated with membranes. The mixture was then filtered, and radioactivity was measured in the washed filter. Each data point is the mean ± S.E.M. (n = 3) radioactivity measured in the washed filter normalized to that measured in the absence of competing etomidate or dimethoxy-etomidate. The S.E. are not visible in the graph because they are smaller than the data points. The curves are fits of each data set to a one-site (etomidate) or two-site (dimethoxy-etomidate) competitive binding equation. The IC50 of etomidate was 22 ± 5 nM, whereas those for dimethoxy-etomidate were 8.2 ± 2.0 and 3970 ± 1530 nM, with the high-affinity site accounting for 54% ± 3% of all sites.
Discussion
In this manuscript, we describe the pharmacology of dimethoxy-etomidate, a novel analog of etomidate that retains etomidate’s potent ability to suppress ACTH-stimulated adrenocortical steroid synthesis but minimally modulates GABAA receptor function, possesses negligible sedative-hypnotic activity, and does not produce myoclonus. Following the administration of low (0.3 mg/kg) and intermediate (3.0 mg/kg) dimethoxy-etomidate doses, our studies showed that ACTH-stimulated plasma concentrations of corticosterone were significantly reduced, whereas those of its precursors (i.e., 11-deoxycorticosterone, pregnenolone, and progesterone) were unaffected. This pattern of steroidogenesis suppression implicates 11β-hydroxylase as the most sensitive target of dimethoxy-etomidate in the adrenocortical steroid biosynthetic pathway (Fig. 8). This enzyme is also the most sensitive target of etomidate. Following the administration of a high (50 mg/kg) but nonhypnotic intravenous dimethoxy-etomidate dose, ACTH-stimulated plasma concentrations of 11-deoxycorticosterone were also significantly reduced. This is indicative of a lower-potency inhibitory action by dimethoxy-etomidate on the hydroxylation of progesterone by 21-hydroxylase.
Fig. 8.

Adrenocortical steroid biosynthetic pathway. Etomidate and dimethoxy-etomidate inhibit 11β-hydroxylase most potently. With high doses, dimethoxy-etomidate also inhibits 21-hydroxylase.
Our [3H]etomidate displacement studies resolved two distinct dimethoxy-etomidate binding sites (but only one etomidate binding site) in adrenal homogenates with vastly different apparent affinities for the etomidate analog. One site had a high apparent affinity for dimethoxy-etomidate with an IC50 that was similar to that of etomidate (8.2 and 22 nM, respectively). The other site had an apparent affinity for dimethoxy-etomidate that was 3 orders of magnitude lower, with an IC50 of 3970 nM. These two sites bound 2 nM [3H]etomidate in approximately equal proportions. Competitive binding studies using radiolabeled metyrapol and metomidate analogs have similarly identified two distinct binding sites in adrenal homogenates that bind etomidate with high apparent affinities (IC50 values: 3.02 ± 1.82 and 0.97 ± 0.39 nM, respectively, for displacing the two radioligands) but bind certain other inhibitors of 11β-hydroxylase with different apparent affinities (Berger et al., 2013). These nonidentical sites—which may correspond to our high- and low-affinity sites—are thought to be overlapping and located within the active site 11β-hydroxylase.
Currently available steroidogenesis inhibitors vary in the mechanisms by which they produce their therapeutic effects and commonly produce adverse effects that limit their dosing and efficacy (Daniel and Newell-Price, 2015). Consequently, long-term control of hypercortisolemia is not achieved in 30% of patients with Cushing’s disease, even when pharmacotherapy is combined with pituitary resection(s), radiation, and bilateral adrenalectomy (Geer et al., 2017). Ketoconazole acts primarily by blocking 17α-hydroxylase, but also blocks multiple other cytochrome p450 enzymes in the adrenocortical and gonadal steroid biosynthetic pathway, including cholesterol side chain cleavage enzyme, 11β-hydroxylase, and 17,20-lyase (Pont et al., 1982, 1984; Engelhardt and Weber, 1994). It also inhibits the hepatic enzymes CYP34A and CYP2C9, potentially reducing the metabolism of coadministered drugs, including some with low therapeutic indices such as phenytoin and warfarin (Zhang et al., 2002). Common side effects of systemic ketoconazole administration include gastrointestinal disturbances (Castinetti et al., 2014); male gynecomastia and impotence, which are likely the result of reduced testosterone production (Sonino et al., 1991); and hepatotoxicity, which led the U.S. Food and Drug Administration to issue a black-box warning. Mitotane similarly blocks multiple enzymes in the adrenocortical steroid biosynthetic pathway, primarily cholesterol side chain cleavage enzyme but also 11β-hydroxylase, 18-hydroxylase, and 3β-hydroxysteroid-dehydrogenase (Young et al., 1973). It has antiproliferative effects on adrenocortical cells, offering potential advantages for patients with adrenocortical carcinoma (Hahner and Fassnacht, 2005). Unfortunately, it commonly produces gastrointestinal and neurologic side effects (Luton et al., 1979). It is an extremely lipophilic drug that accumulates in adipose tissue. Consequently, therapeutic levels are achieved only after weeks or months of oral administration (Fleseriu and Petersenn, 2012). Because mitotane is also teratogenic, it is advised that pregnancy be avoided for up to 5 years after discontinuing its use (Leiba et al., 1989). By comparison, metyrapone and etomidate are relatively selective p450 enzyme inhibitors. Both drugs inhibit 11β-hydroxylase and the closely related enzyme aldosterone synthase, although the potency of etomidate is approximately an order of magnitude greater (Lamberts et al., 1987). At higher (i.e., sedative-hypnotic) concentrations, etomidate also inhibits cholesterol side chain cleavage enzyme and, similar to mitotane, has antiproliferative effects on adrenocortical cells (Fassnacht et al., 2000; Hahner et al., 2010).
Etomidate has central nervous system actions that are undesirable when the drug is used as a steroidogenesis inhibitor, leading to recommendations that it only be administered in a highly monitored environment such as an intensive care unit (Preda et al., 2012; Tritos and Biller, 2014b; Daniel and Newell-Price, 2015). Most prominently, it produces sedation and (at higher doses) hypnosis. Although these sedative-hypnotic actions typically occur with etomidate doses exceeding those necessary to suppress adrenocortical function in normal individuals or patients with ACTH-independent hypercortisolemia, the dosing window is sufficiently narrow to require sedating doses to achieve normal cortisol levels in some patients (Schulte et al., 1990). Such patients include those with ACTH-dependent hypercortisolemia (e.g., Cushing’s disease) who produce high levels of ACTH and are less sensitive to the actions of steroidogenesis inhibitors (Preda et al., 2012), and those who have low sensitivity to steroidogenesis inhibitors due to polymorphisms in genes coding for enzymes in the steroid biosynthetic pathway (Valassi et al., 2017). Where a “block and replace” strategy is planned, sedative-hypnotic etomidate doses may be needed to completely ablate the adrenocortical response to ACTH (Preda et al., 2012). Because other central nervous system depressants (e.g., opioids, benzodiazepines, alcohol) augment etomidate’s sedative-hypnotic activity, lower etomidate doses will produce sedation/hypnosis in patients receiving such drugs.
Myoclonus is another undesirable central nervous system action of etomidate and has been reported in patients receiving etomidate to produce anesthesia (Giese et al., 1985) or sedation (Kim et al., 2017) and to suppress steroidogenesis (Krakoff et al., 2001). The mechanism for this action is not well understood, but it has been speculated that it results from 1) disequilibrium of the drug, and/or 2) differential sensitivity to the drug among the various effect sites within the central nervous system (Modica et al., 1990; Doenicke et al., 1999). We failed to observe any myoclonic activity in rats that received dimethoxy-etomidate, supporting an important role for GABAA receptor positive modulation in mediating this side effect.
In summary, dimethoxy-etomidate is a novel phenyl ring–substituted etomidate analog that retains etomidate’s potent ability to suppress ACTH-stimulated adrenocortical steroid synthesis by inhibiting 11β-hydroxylase. It also weakly inhibits 21-hydroxylase, reducing the synthesis of 11-deoxycorticosterone. However, unlike etomidate, dimethoxy-etomidate produces little positive modulation of GABAA receptors, possesses negligible sedative-hypnotic activity, and does not produce myoclonus. It provides proof of concept that etomidate analogs without important central nervous system activities can be designed for the treatment of Cushing’s syndrome.
Abbreviations
- ACTH
adrenocorticotropic hormone
- dimethoxy-etomidate
(R)-ethyl 1-(1-(3,5-dimethoxyphenyl)ethyl)-1H-imidazole-5-carboxylate
- EC5 GABA
gamma-aminobutyric acid concentration that elicits 5% of the current evoked by 1 mM GABA
- etomidate
(R)-ethyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate
- LoRR
loss of righting reflexes
Authorship Contributions
Participated in research design: Raines, McGrath.
Conducted experiments: McGrath, Ma.
Performed data analyses: McGrath, Ma, Raines.
Wrote or contributed to the writing of the manuscript: Raines, McGrath.
Footnotes
This work was supported by the National Institutes of Health, Bethesda, MD [GM087316 and GM58448] and the Department of Anesthesia, Critical Care, and Pain Medicine, Massachusetts General Hospital, Boston, MA.
References
- Alexandraki KI, Kaltsas GA, Isidori AM, Storr HL, Afshar F, Sabin I, Akker SA, Chew SL, Drake WM, Monson JP, et al. (2013) Long-term remission and recurrence rates in Cushing’s disease: predictive factors in a single-centre study. Eur J Endocrinol 168:639–648. [DOI] [PubMed] [Google Scholar]
- Allolio B, Dörr H, Stuttmann R, Knorr D, Engelhardt D, Winkelmann W. (1985) Effect of a single bolus of etomidate upon eight major corticosteroid hormones and plasma ACTH. Clin Endocrinol (Oxf) 22:281–286. [DOI] [PubMed] [Google Scholar]
- Ambrogio AG, Cavagnini F. (2016) Role of “old” pharmacological agents in the treatment of Cushing’s syndrome. J Endocrinol Invest 39:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson AB, Kennedy A, Wiggam MI, McCance DR, Sheridan B. (2005) Long-term remission rates after pituitary surgery for Cushing’s disease: the need for long-term surveillance. Clin Endocrinol (Oxf) 63:549–559. [DOI] [PubMed] [Google Scholar]
- Belelli D, Muntoni AL, Merrywest SD, Gentet LJ, Casula A, Callachan H, Madau P, Gemmell DK, Hamilton NM, Lambert JJ, et al. (2003) The in vitro and in vivo enantioselectivity of etomidate implicates the GABAA receptor in general anaesthesia. Neuropharmacology 45:57–71. [DOI] [PubMed] [Google Scholar]
- Berger ML, Hammerschmidt F, Qian R, Hahner S, Schirbel A, Stichelberger M, Schibli R, Yu J, Arion VB, Woschek A, et al. (2013) [(3)H]metyrapol and 4-[(131)i]iodometomidate label overlapping, but not identical, binding sites on rat adrenal membranes. Mol Pharm 10:1119–1130. [DOI] [PubMed] [Google Scholar]
- Campagna JA, Pojasek K, Grayzel D, Randle J, Raines DE. (2014) Advancing novel anesthetics: pharmacodynamic and pharmacokinetic studies of cyclopropyl-methoxycarbonyl metomidate in dogs. Anesthesiology 121:1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castinetti F, Guignat L, Giraud P, Muller M, Kamenicky P, Drui D, Caron P, Luca F, Donadille B, Vantyghem MC, et al. (2014) Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab 99:1623–1630. [DOI] [PubMed] [Google Scholar]
- Cotten JF, Forman SA, Laha JK, Cuny GD, Husain SS, Miller KW, Nguyen HH, Kelly EW, Stewart D, Liu A, et al. (2010) Carboetomidate: a pyrrole analog of etomidate designed not to suppress adrenocortical function. Anesthesiology 112:637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten JF, Husain SS, Forman SA, Miller KW, Kelly EW, Nguyen HH, Raines DE. (2009) Methoxycarbonyl-etomidate: a novel rapidly metabolized and ultra-short-acting etomidate analogue that does not produce prolonged adrenocortical suppression. Anesthesiology 111:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier TA, Beck D, Wuttke W, Kettler D. (1988) Relation of the inhibition of cortisol synthesis in vivo to plasma etomidate concentrations. Anaesthesist 37:337–339. [PubMed] [Google Scholar]
- Cuevas-Ramos D, Fleseriu M. (2014) Treatment of Cushing’s disease: a mechanistic update. J Endocrinol 223:R19–R39. [DOI] [PubMed] [Google Scholar]
- Daniel E, Newell-Price JD. (2015) Therapy of endocrine disease: steroidogenesis enzyme inhibitors in Cushing’s syndrome. Eur J Endocrinol 172:R263–R280. [DOI] [PubMed] [Google Scholar]
- de Jong FH, Mallios C, Jansen C, Scheck PA, Lamberts SW. (1984) Etomidate suppresses adrenocortical function by inhibition of 11 β-hydroxylation. J Clin Endocrinol Metab 59:1143–1147. [DOI] [PubMed] [Google Scholar]
- Dekkers OM, Horváth-Puhó E, Jørgensen JO, Cannegieter SC, Ehrenstein V, Vandenbroucke JP, Pereira AM, Sørensen HT. (2013) Multisystem morbidity and mortality in Cushing’s syndrome: a cohort study. J Clin Endocrinol Metab 98:2277–2284. [DOI] [PubMed] [Google Scholar]
- Dimopoulou C, Schopohl J, Rachinger W, Buchfelder M, Honegger J, Reincke M, Stalla GK. (2013) Long-term remission and recurrence rates after first and second transsphenoidal surgery for Cushing’s disease: care reality in the Munich metropolitan region. Eur J Endocrinol 170:283–292. [DOI] [PubMed] [Google Scholar]
- Doenicke AW, Roizen MF, Kugler J, Kroll H, Foss J, Ostwald P. (1999) Reducing myoclonus after etomidate. Anesthesiology 90:113–119. [DOI] [PubMed] [Google Scholar]
- Engelhardt D, Weber MM. (1994) Therapy of Cushing’s syndrome with steroid biosynthesis inhibitors. J Steroid Biochem Mol Biol 49:261–267. [DOI] [PubMed] [Google Scholar]
- Fassnacht M, Hahner S, Beuschlein F, Klink A, Reincke M, Allolio B. (2000) New mechanisms of adrenostatic compounds in a human adrenocortical cancer cell line. Eur J Clin Invest 30 (Suppl 3):76–82. [DOI] [PubMed] [Google Scholar]
- Fleseriu M, Castinetti F. (2016) Updates on the role of adrenal steroidogenesis inhibitors in Cushing’s syndrome: a focus on novel therapies. Pituitary 19:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleseriu M, Loriaux DL, Ludlam WH. (2007) Second-line treatment for Cushing’s disease when initial pituitary surgery is unsuccessful. Curr Opin Endocrinol Diabetes Obes 14:323–328. [DOI] [PubMed] [Google Scholar]
- Fleseriu M, Petersenn S. (2012) Medical management of Cushing’s disease: what is the future? Pituitary 15:330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman SA. (2011) Clinical and molecular pharmacology of etomidate. Anesthesiology 114:695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP. (2006) Molecular targets underlying general anaesthesia. Br J Pharmacol 147 (Suppl 1):S72–S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DE, Griffiths H. (1984) The inhibition by etomidate of the 11 β-hydroxylation of cortisol. Clin Endocrinol (Oxf) 20:625–629. [DOI] [PubMed] [Google Scholar]
- Gadelha MR, Vieira Neto L. (2014) Efficacy of medical treatment in Cushing’s disease: a systematic review. Clin Endocrinol (Oxf) 80:1–12. [DOI] [PubMed] [Google Scholar]
- Geer EB, Shafiq I, Gordon MB, Bonert V, Ayala A, Swerdloff RS, Katznelson L, Lalazar Y, Manuylova E, Pulaski-Liebert KJ, et al. (2017) Biochemical control during long-term follow-up of 230 adult patients with Cushing disease: a Multicenter Retrospective Study. Endocr Pract 23:962–970. [DOI] [PubMed] [Google Scholar]
- Giese JL, Stockham RJ, Stanley TH, Pace NL, Nelissen RH. (1985) Etomidate versus thiopental for induction of anesthesia. Anesth Analg 64:871–876. [PubMed] [Google Scholar]
- Hahner S, Fassnacht M. (2005) Mitotane for adrenocortical carcinoma treatment. Curr Opin Investig Drugs 6:386–394. [PubMed] [Google Scholar]
- Hahner S, Stürmer A, Fassnacht M, Hartmann RW, Schewe K, Cochran S, Zink M, Schirbel A, Allolio B. (2010) Etomidate unmasks intraadrenal regulation of steroidogenesis and proliferation in adrenal cortical cell lines. Horm Metab Res 42:528–534. [DOI] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, et al. (2003) General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor β3 subunit. FASEB J 17:250–252. [DOI] [PubMed] [Google Scholar]
- Kim MG, Park SW, Kim JH, Lee J, Kae SH, Jang HJ, Koh DH, Choi MH. (2017) Etomidate versus propofol sedation for complex upper endoscopic procedures: a prospective double-blinded randomized controlled trial. Gastrointest Endosc 86:452–461. [DOI] [PubMed] [Google Scholar]
- Krakoff J, Koch CA, Calis KA, Alexander RH, Nieman LK. (2001) Use of a parenteral propylene glycol-containing etomidate preparation for the long-term management of ectopic Cushing’s syndrome. J Clin Endocrinol Metab 86:4104–4108. [DOI] [PubMed] [Google Scholar]
- Lacroix A, Feelders RA, Stratakis CA, Nieman LK. (2015) Cushing’s syndrome. Lancet 386:913–927. [DOI] [PubMed] [Google Scholar]
- Lambert A, Frost J, Mitchell R, Robertson WR. (1986) On the assessment of the in vitro biopotency and site(s) of action of drugs affecting adrenal steroidogenesis. Ann Clin Biochem 23:225–229. [DOI] [PubMed] [Google Scholar]
- Lamberts SW, Bons EG, Bruining HA, de Jong FH. (1987) Differential effects of the imidazole derivatives etomidate, ketoconazole and miconazole and of metyrapone on the secretion of cortisol and its precursors by human adrenocortical cells. J Pharmacol Exp Ther 240:259–264. [PubMed] [Google Scholar]
- Leiba S, Weinstein R, Shindel B, Lapidot M, Stern E, Levavi H, Rusecki Y, Abramovici A. (1989) The protracted effect of o,p′-DDD in Cushing’s disease and its impact on adrenal morphogenesis of young human embryo. Ann Endocrinol (Paris) 50:49–53. [PubMed] [Google Scholar]
- Luton JP, Mahoudeau JA, Bouchard P, Thieblot P, Hautecouverture M, Simon D, Laudat MH, Touitou Y, Bricaire H. (1979) Treatment of Cushing’s disease by O,p’DDD. Survey of 62 cases. N Engl J Med 300:459–464. [DOI] [PubMed] [Google Scholar]
- Mancini T, Kola B, Mantero F, Boscaro M, Arnaldi G. (2004) High cardiovascular risk in patients with Cushing’s syndrome according to 1999 WHO/ISH guidelines. Clin Endocrinol (Oxf) 61:768–777. [DOI] [PubMed] [Google Scholar]
- Modica PA, Tempelhoff R, White PF. (1990) Pro- and anticonvulsant effects of anesthetics (Part II). Anesth Analg 70:433–444. [DOI] [PubMed] [Google Scholar]
- Patil CG, Prevedello DM, Lad SP, Vance ML, Thorner MO, Katznelson L, Laws ER., Jr (2008) Late recurrences of Cushing’s disease after initial successful transsphenoidal surgery. J Clin Endocrinol Metab 93:358–362. [DOI] [PubMed] [Google Scholar]
- Pejo E, Cotten JF, Kelly EW, Le Ge R, Cuny GD, Laha JK, Liu J, Lin XJ, Raines DE. (2012) In vivo and in vitro pharmacological studies of methoxycarbonyl-carboetomidate. Anesth Analg 115:297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejo E, Santer P, Jeffrey S, Gallin H, Husain SS, Raines DE. (2014) Analogues of etomidate: modifications around etomidate’s chiral carbon and the impact on in vitro and in vivo pharmacology. Anesthesiology 121:290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejo E, Zhou X, Husain SS, Raines DE. (2016) Sedative-hypnotic binding to 11β-hydroxylase. Anesthesiology 125:943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivonello R, De Leo M, Cozzolino A, Colao A. (2015) The treatment of Cushing’s disease. Endocr Rev 36:385–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivonello R, De Martino MC, De Leo M, Simeoli C, Colao A. (2017) Cushing’s disease: the burden of illness. Endocrine 56:10–18. [DOI] [PubMed] [Google Scholar]
- Pivonello R, Isidori AM, De Martino MC, Newell-Price J, Biller BM, Colao A. (2016) Complications of Cushing’s syndrome: state of the art. Lancet Diabetes Endocrinol 4:611–629. [DOI] [PubMed] [Google Scholar]
- Pont A, Graybill JR, Craven PC, Galgiani JN, Dismukes WE, Reitz RE, Stevens DA. (1984) High-dose ketoconazole therapy and adrenal and testicular function in humans. Arch Intern Med 144:2150–2153. [PubMed] [Google Scholar]
- Pont A, Williams PL, Azhar S, Reitz RE, Bochra C, Smith ER, Stevens DA. (1982) Ketoconazole blocks testosterone synthesis. Arch Intern Med 142:2137–2140. [PubMed] [Google Scholar]
- Preda VA, Sen J, Karavitaki N, Grossman AB. (2012) Etomidate in the management of hypercortisolaemia in Cushing’s syndrome: a review. Eur J Endocrinol 167:137–143. [DOI] [PubMed] [Google Scholar]
- Schulte HM, Benker G, Reinwein D, Sippell WG, Allolio B. (1990) Infusion of low dose etomidate: correction of hypercortisolemia in patients with Cushing’s syndrome and dose-response relationship in normal subjects. J Clin Endocrinol Metab 70:1426–1430. [DOI] [PubMed] [Google Scholar]
- Shanmugasundararaj S, Zhou X, Neunzig J, Bernhardt R, Cotten JF, Ge R, Miller KW, Raines DE. (2013) Carboetomidate: an analog of etomidate that interacts weakly with 11β-hydroxylase. Anesth Analg 116:1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonino N, Boscaro M, Paoletta A, Mantero F, Ziliotto D. (1991) Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients. Clin Endocrinol (Oxf) 35:347–352. [DOI] [PubMed] [Google Scholar]
- Tritos NA, Biller BM. (2014a) Cushing’s disease. Handb Clin Neurol 124:221–234. [DOI] [PubMed] [Google Scholar]
- Tritos NA, Biller BM. (2014b) Medical management of Cushing’s disease. J Neurooncol 117:407–414. [DOI] [PubMed] [Google Scholar]
- Valassi E, Aulinas A, Glad CA, Johannsson G, Ragnarsson O, Webb SM. (2017) A polymorphism in the CYP17A1 gene influences the therapeutic response to steroidogenesis inhibitors in Cushing’s syndrome. Clin Endocrinol (Oxf) 87:433–439. [DOI] [PubMed] [Google Scholar]
- Waud DR. (1972) On biological assays involving quantal responses. J Pharmacol Exp Ther 183:577–607. [PubMed] [Google Scholar]
- Young RB, Bryson MJ, Sweat ML, Street JC. (1973) Complexing of DDT and o,p’DDD with adrenal cytochrome P-450 hydroxylating systems. J Steroid Biochem 4:585–591. [DOI] [PubMed] [Google Scholar]
- Zhang W, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, Sellers EM. (2002) Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab Dispos 30:314–318. [DOI] [PubMed] [Google Scholar]