

Abstract
Advances in next-generation sequencing technologies enable routine genome sequencing, generating millions of short reads. A crucial step for full genome analysis is the de novo assembly, and currently, performance of different assembly methods is measured by a metric called N50. However, the N50 value can produce skewed, inaccurate results when complex data are analyzed, especially for viral and microbial datasets. To provide a better assessment of assembly output, we developed a new metric called U50. The U50 identifies unique, target-specific contigs by using a reference genome as baseline, aiming at circumventing some limitations that are inherent to the N50 metric. Specifically, the U50 program removes overlapping sequence of multiple contigs by utilizing a mask array, so the performance of the assembly is only measured by unique contigs. We compared simulated and real datasets by using U50 and N50, and our results demonstrated that U50 has the following advantages over N50: (1) reducing erroneously large N50 values due to a poor assembly, (2) eliminating overinflated N50 values caused by large measurements from overlapping contigs, (3) eliminating diminished N50 values caused by an abundance of small contigs, and (4) allowing comparisons across different platforms or samples based on the new percentage-based metric UG50%. The use of the U50 metric allows for a more accurate measure of assembly performance by analyzing only the unique, non-overlapping contigs. In addition, most viral and microbial sequencing have high background noise (i.e., host and other non-targets), which contributes to having a skewed, misrepresented N50 value—this is corrected by U50. Also, the UG50% can be used to compare assembly results from different samples or studies, the cross-comparisons of which cannot be performed with N50.
Keywords: genome assembly, N50, next-generation sequencing, U50
1. INTRODUCTION
Next-Generation Sequencing (NGS) is becoming the laboratory standard for genome sequencing, pathogen discovery, and advanced molecular detection. NGS generates a tremendous amount of short sequence reads, and one of the most common ways to analyze these data is by de novo assembly. Unfortunately, genome assembly remains a very difficult problem, which is made more challenging by shorter reads, non-uniform coverage of the target, and unreliable long-range linking information (Miller et al. 2010). Assemblies are measured by the size and accuracy of their contigs and scaffolds (Miller et al. 2010). Currently, the performance of a de novo assembly is measured by a metric called N50.
The N50 value is a measurement of the assembly quality of NGS data by determining how well an assembler performs in forming contigs and scaffolds. N50 is defined as a weighted median statistic such that 50% of the entire assembly is contained in contigs that are equal to or larger than this value. Though assembly accuracy is extremely hard to measure, the N50 value has thus far been the most common metric to use for genomic assembly completeness. Other metrics can be used in determining overall assembly performance, but they are all based on the N50 statistic. Generally, it is assumed that the higher the N50 value, the more accurate the assembly.
Although calculation of the N50 is a common practice among studies analyzing NGS datasets, the N50 value has several major disadvantages that can sometimes produce inaccurate results (Scott 2014). First, a poor assembly can force unrelated reads and contigs into supercontigs, resulting in an erroneously large N50. Second, using N50 and NG50 (see Table 1 for definitions) for metagenomics, microbial or viral datasets are problematic because a small fraction of the reads are of the targeted genome (Naccache et al. 2014) and because of the background reads (mostly of cellular origin) skewing the results. Third, N50 does not account for resulting contigs that are non-unique or overlapping; these overlapping contigs can sometimes greatly inflate the N50 value and, hence, miscalculate true performance of the NGS assembly. Finally, comparing all assemblies based solely on the N50 is impossible, because N50 is a number-based metric that is calculated from total contig length and it does not allow a fair comparison across different platforms or samples (Miller et al. 2010). Therefore, an alternative formula is needed.
Table 1.
N50 and U50 Assembly Metric Definitions
| N50 Metrics | U50 Metrics | ||
|---|---|---|---|
| N50 | The length of the smallest contig such that 50% of the sum of all contigs is contained in contigs of size N50 or larger. | U50 | The length of the smallest contig such that 50% of the sum of all unique, target-specific contigs is contained in contigs of size U50 or larger. |
| L50 [LG50] | The number of contigs whose length sum produces N50 [NG50]. | UL50 [ULG50] | The number of contigs whose length sum produces U50 [UG50]. |
| NG50 | The length of the smallest contig such that 50% of the reference genome is contained in contigs of size NG50 or larger. NG50 estimates the genome size based on the input contig lengths, not a reference genome as input. | UG50 | The length of the smallest contig such that 50% of the reference genome is contained in unique, target-specific contigs of size UG50 or larger. |
| UG50% | The estimated coverage length of the UG50 in direct relation to the length of the reference genome. |
This article describes a new metric called U50, as well as a computer algorithm that can calculate U50 automatically for any NGS data, for applications mainly targeting viral and microbial datasets. Using contig sequences that are generated by an assembly program as input, the U50 algorithm identifies unique regions from those contigs, then applies a brute force, cumulative sum method to calculate the U50 metric. Our U50 metric aims at circumventing the limitations of N50 by identifying unique, target-specific contigs by using a reference sequence as baseline.
2. IMPLEMENTATION
2.1. Definitions
See Table 1.
2.2. Algorithm
The U50 program requires Python to be installed. Two input files are needed from the user: a multi-FASTA file containing all the contigs and a FASTA file including only the reference genome. First, the user can run the provided bash shell script to convert the two input files into a sorted BED file, using Bowtie2, SAMtools, and BEDtools (Table 2). The U50 script will then compare the sorted BED file listing all the contig coordinates to the reference genome, using the U50 calculation and mask array as described later. The Quality Assessment Tool for Genome Assemblies (QUAST) program is not necessary for the U50 program to execute, but the added output information from QUAST helps make informed decisions about the quality of the assembly when combined with the U50 output data.
Table 2.
Software Used in the U50 Package
| Program/language | Version | Application | Execution order |
|---|---|---|---|
| Bowtie2 | 2.2.4 | Maps contigs to reference, creating an SAM file (Langmead and Salzberg 2012) |
1 |
| SAMtools | 1.2 | Converts SAM to BAM (Li 2011; Li et al. 2009) | 2 |
| BEDTools | 2.17.0 | Converts BAM to BED (Quinlan and Hall 2010) | 3 |
|
Python (BioPython) |
2.7.3 1.66 |
Executes the U50 program (Cock et al. 2009; Lutz 2013) | 4 |
| QUAST | 2.3 | Calculates assembly metrics (N50, N75, L50, GC%, etc.) based on the input contig file (Gurevich et al. 2013) | 5 |
During execution, the U50 program first sorts contigs by their lengths, starting from the longest to the shortest. Starting with the longest contig, the start and stop coordinates are used to compare it with the reference sequence at those specific coordinate positions. The coordinates are used to align the contig to the reference and find overlapping regions. If an overlap is identified, then those coordinates are removed and only the coordinates that reflect a unique contig are retained. To maintain order and position of all contigs, a mask array is used.
2.3. Using a mask array to determine unique regions
We constructed a mask array (Fig. 1) to be used as a switch to find overlapping regions. The length of the mask array was assigned to be the length of the input reference sequence. The mask array acts as a running tally that keeps track of the unique coordinates, where “1” denotes coordinates that already have a mapped contig, and “0” denotes coordinates that have yet to be covered. The initial mask array was assigned all zeros in memory, because no contigs have been mapped yet.
FIG. 1.

The use of mask array in U50. A schematic diagram demonstrating the use of a mask array to identify the unique contig length for two contigs.
Starting from the first contig in the sorted list, the contig coordinate positions directly align with the index positions of the mask array. This is because both the BED file and the array have a zero-based counting system.
When the mask array is compared with each of the contigs in order, the contig coordinates are first aligned with the mask array index. When a contig coordinate occupies a mask array index containing “0” in its current stage, the contig coordinate is denoted as a unique region; then, the mask array is updated as “1” in this coordinate. On the other hand, when a mask array index contains a “1,” the contig coordinate is discarded as an overlapping region. After performing all contig alignments, if a “0” is still present in the mask array, this denotes a gap in coverage. All unique contigs are then retained for the U50 calculation.
The same calculation used for the N50 is performed on the unique contigs, thus producing the U50 value. A brute force, cumulative sum method is used to find the N50, NG50, L50, U50, UG50, and UL50 values.
2.4. N50 calculation
Step 1: The first step for calculating N50 involves ordering contigs by their lengths from the longest (c1) to the shortest (cn).
Step 2: Calculate the cutoff value by summing all contigs and multiplying by the threshold percentage (x); examples include: N25, N50, N75, N90, etc. This example is for N50, so the threshold percentage is 50%.
that is, for N50
Step 3: Starting from the longest contig, the lengths of each contig are summed, until this running sum is greater than or equal to the Nx cutoff (e.g., for N50, it would be 50% of the total length of all contigs in the assembly).
Step 4: The N50 of the assembly is the length of the shortest contig at the first instance where the running sum becomes greater than or equal to the N50 cutoff.
whereas L50 = the length of contig number L
NG50 follows the same steps except that Step 2 is modified to the following:
2.5. U50 calculation
Our U50 metric aims at circumventing the limitations of N50 by identifying unique, target-specific contigs by using a reference sequence as baseline.
Step 1: The first step for calculating U50 involves ordering contigs by their lengths from the longest (c1) to the shortest (cn).
Step 2: All contigs are mapped to a reference genome, starting with c1. Using the mask array, only the unique portions of each contig are preserved (c′), and all other regions and non-mapping contigs are removed. Then, these modified contigs are sorted by length from the longest (c′1) to the shortest (c′n).
Step 3: From the modified list of contigs, the summation of all contigs is found and multiplied by the threshold percentage. This example is for U50, so the threshold percentage is 50%.
c′ = modified contigs with overlapping regions removed, and all non-target contigs removed.
Step 4: Starting from the longest contig, the lengths of each contig are summed, until this running sum is greater than or equal to the U50 cutoff (50% of the total length of all contigs in the assembly).
Step 5: The U50 of the assembly is the length of the shortest contig at the first instance where the running sum becomes greater than or equal to the U50 cutoff.
whereas UL50 = the length of contig number L
UG50 follows the same steps except:
Step 3 is modified to the following:
UG50% follows the same steps as UG50 with one additional step:
Step 6 involves calculating the UG50%.
The output contains four files in total: AssemblyStatistics.txt, contigs.txt, gaps.txt, and overlaps.txt. The AssemblyStatistics.txt file will show the standard output that prints to the terminal screen. The contigs.txt file shows the contig coordinates and a count of the unique base positions that a particular contig covers. The overlaps.txt file shows the index position of an overlap and which contig that overlap belongs to. The gaps.txt file shows the index position of any gaps in coverage.
3. RESULTS
3.1. U50 implementation with theoretical datasets
We constructed six different types of theoretical assembly results (A–F), increasing in complexity (Fig. 2), to compare the values for N50, NG50, U50, UG50, and UG50%. For the assemblies that generate long contigs with little overlap or gaps (e.g., A–C), N50 appears approximately equal to the U50 value. The U50 metric really excels in situations where assemblies generate short, fragmented contigs (e.g., E and F), skewing the N50 downward, or non-target-specific long contigs (e.g, D) that skew the N50 upward.
FIG. 2.

Simulated output of different assemblies, with the N50, NG50, U50, UG50, and UG50% values calculated. Blue denotes the reference sequence, black denotes a unique contig, red denotes an overlapping region of a contig, and green denotes a non-target contig.
Figure 2A is the simplest example. When a de novo assembly produces one contig that is the length of the genome, then the N50, NG50, U50, and UG50 all return the same values.
Figure 2B simulates an assembly producing two contigs totaling the length of the genome, with a single overlapping region. The N50, NG50, U50, and UG50 values are identical, even after removing the overlapping region to calculate U50 and UG50.
Figure 2C simulates an assembly producing multiple overlapping contigs totaling the length of the genome. This is often observed in earlier de Bruijn graph assemblers that produce hundreds or thousands of overlapping small contigs (Deng et al. 2015), skewing the result toward a lower N50. By removing the short overlaps, the U50 estimate corrects for the underestimation caused by the small contigs.
Figure 2D simulates a scenario where the final assembly contains long non-targeted sequences. This is exemplified by sequencing bacterial and/or viral genomes with a high background of host cellular nucleic acid. In this example, the N50 is overinflated, demonstrated by having an N50 that is much longer than the targeted genome. The U50 is a better reflection of the actual assembly performance.
Figure 2E simulates a scenario where the final assembly contains numerous small contigs. The over-abundance of these small contigs generally skews the N50 toward the smaller contig size. When removing the overlapping regions, the U50 is a better representation of the assembly performance.
Figure 2F simulates the most realistic example of a de novo assembly’s contig output. There are duplicated contigs, overlapping regions, and a gap region. There are varying sizes of contigs, and many contain overlapping regions, which skew the N50 downward. Once these duplicated contigs and overlapping regions are removed, the U50 provides a slightly better representation of the assembly performance.
Generally, it is assumed that the higher the N50, the better the assembly. However, it is important to keep in mind that a poor assembly that has forced unrelated reads and contigs into scaffolds can have an erroneously large N50. Using the U50 metric to discount any reads that do not belong to the given reference genome may minimize the effects of overabundant small, fragmented contigs.
3.2. U50 implementation with published and in-house datasets
To demonstrate the U50 algorithm, various published and in-house datasets were compared, including four Illumina MiSeq bacteria samples that were analyzed in the GAGE-B paper (Magoc et al. 2013), four enterovirus D68 samples, three other types of picornaviruses, one Black Queen cell virus, one hepatitis E virus, one crAssphage, and one rhabdovirus Bas-Congo.
All 15 samples were assembled by using four different de novo assemblers: ABySS v.1.9 (Simpson et al. 2009); SOAPdenovo2 v.r240 (Luo et al. 2012); SPAdes v. 3.6.2 (Bankevich et al. 2012; Nurk et al. 2013); and Velvet v.1.2.10 (Zerbino and Birney 2008). Only the assembly output contig files were available from the four bacteria samples from the GAGE-B dataset, so the same approach was used for the other 11 samples. The UG50% was used to evaluate and compare samples across all assemblers. Our results show that the SPAdes assembler consistently generates a higher UG50% compared with the other three assemblers, suggesting that the SPAdes assembler generates the longest contigs and outperforms the others.
For most viral samples, the UG50% performance metric for SPAdes is consistently more than 90%, showing that it can produce full or near full genomes. In contrast, the UG50% performance metric for Velvet is always lower than 20%, indicating that the largest contig would not be more than one fifth of the genome, similar to previous findings (Magoc et al. 2013). For SOAPdenovo2 and ABySS, the performance varied from sample to sample, with the UG50% ranging from <1% to 100%. For the bacterial samples, a similar trend is apparent. The UG50% for SPAdes is higher than 12%, whereas the UG50% for the other three assemblers is lower than 5%.
For many samples, the U50 is within 10% of the N50. SOAPdenovo2 and Velvet have the largest differences when comparing the N50 and U50 values, because these assemblers produce many small contigs, as portrayed in Figure 2E. This typically results in the N50 value being skewed toward the smaller contig size. By removing the overlapping regions, the U50 value is much higher and would be a more accurate estimate (see picornaviruses SOAPdenovo2 and Velvet results for EV-C105 and EV-C117 in Figure 3). In these cases, the U50 is a better estimate compared with the N50 for SOAPdenovo2 and Velvet because of removal of the non-target specific contigs.
FIG. 3.
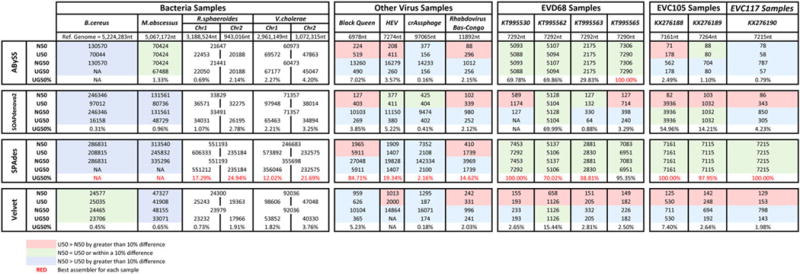
Comparison of the N50, U50, NG50, UG50, and UG50% for different bacterial and viral datasets using four different assemblers. Contigs for the bacteria dataset were retrievedfrom Magoc et al. (2013), whereas all other contigs were generated in-house. All datasets were sequenced by using the Illumina MiSeq.
When the assembler can generate long contigs covering almost the entire length of the reference genome, then the N50 and the U50 values are identical (see examples Figure 2A and B in the simulated data and the SPAdes results for EV-D68, EV-C105, and EV-C117). For the remaining of the SPAdes results, the N50 and U50 values are very close. The slight difference is most likely due to the removal of the duplicated and overlapping reads.
Noticeably, the NG50 value calculated for all Black Queen virus assemblies, hepatitis E virus assemblies, and the SPAdes assembly for crAssphage returned values that are longer than the reference genome. This is because NG50 estimates the reference genome length based on the input contigs themselves (Ghodsi et al. 2013), not on an input reference genome. This exemplifies that the overinflated NG50 results can be inaccurate and misleading.
3.3. Availability of data and material
The datasets generated during and/or analyzed during the current study and the U50 python script are available in the U50 Github repository, https://github.com/CDCgov/U50.
4. DISCUSSION AND CONCLUSIONS
In this study, we describe the U50 metric as a tool to evaluate assembly performance, aiming at circumventing some limitations that are inherent to the N50 metric. The core spirit of U50 is in removing noise and finding those unique regions that align to a targeted reference genome. Major advantages include the following: (1) reducing erroneously large N50 values due to a poor assembly, (2) eliminating overinflated N50 values caused by large measurements from overlapping contigs, (3) eliminating diminished N50 values caused by an abundance of small contigs, and (4) allowing comparisons across different platforms or samples based on the percentage-based metric UG50%.
The U50 assembly metric will be particularly useful for viral and microbial sequencing of samples with high background noise. This “needle-in-a-haystack” problem proves cumbersome when trying to assemble such small viral contigs into complete genomes with exponentially larger non-target contigs. The reads often do not overlap sufficiently to allow the de novo assembler to properly form long contigs (Kostic et al. 2011), and teasing out the true viral contigs from the rest is challenging. The U50 metric allows for only the proper viral contigs to be used when calculating the assembly performance, as opposed to using all of the contigs.
Despite the advantages, there are some limitations that prevent usage of the U50 under all circumstances. First, U50 can only be calculated if a reference is available. Therefore, this is not a metric to use during the assembling of unique prototype genomes. Second, because the overlapping regions are removed for the calculation, U50 does not account for coverage of the genome.
Since only unique, non-overlapping contigs are used in the calculation of U50, the sum of the unique contig length is usually less than the sum of the original contigs. In the instance where all the unique contigs do not sum to the percentage threshold of 50%, the U50 program returns a “0.” This is indicative of the input contigs not covering 50% of the reference genome. In such cases, we recommend using a lower threshold such as U10 or U25. These will typically return a result, but the overall assembly when aligned to a reference is still poor.
The U50 assembly metric is a tool to be used in conjunction with the commonly used N50, L50, and NG50 metrics. When used in unison, it gives a clearer picture of the performance and accuracy of the overall assembly. The UG50%, as a percentage-based metric, can be used to compare assembly results from different samples or studies. The use of this new metric and its software could facilitate a better comparison of assemblies, especially with viral and microbial datasets.
Acknowledgments
The authors thank Steve Oberste, Paul Rota, Greg Doho, Roman Tatusov, and Edward Ramos for their constructive comments. This work was supported by Federal appropriations to the Centers for Disease Control and Prevention (CDC), through the Advanced Molecular Detection Initiative line item. This research was also supported in part by an appointment to the Research Participation Program at the CDC administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S Department of Energy and CDC.
Footnotes
AUTHORS’ CONTRIBUTION
Both authors designed and performed research, analyzed data, and wrote and approved the final article.
AUTHOR DISCLOSURE STATEMENT
No competing financial interests exist.
References
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT, et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Naccache SN, Ng T, et al. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucl Acids Res. 2015;43:e46–e46. doi: 10.1093/nar/gkv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodsi M, Hill CM, Astrovskaya I, et al. De novo likelihood-based measures for comparing genome assemblies. BMC Res Notes. 2013;6:1–18. doi: 10.1186/1756-0500-6-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, et al. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Ojesina AI, Pedamallu CS, et al. PathSeq: A comprehensive computational tool for the identification or discovery of microorganisms by deep sequencing of human tissue. Nat Biotechnol. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:1–6. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz M. Learning Python. 5th. O’Reilly Media; Sebastobol, CA: 2013. [Google Scholar]
- Magoc T, Pabinger S, Canzar S, et al. GAGE-B: an evaluation of genome assemblers for bacterial organisms. Bioinformatics (Oxford, England) 2013;29:1718–1725. doi: 10.1093/bioinformatics/btt273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95:315–327. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naccache SN, Federman S, Veeraraghavan N, et al. A cloud-compatible bioinformatics pipeline for ultra-rapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014;24:1180–1192. doi: 10.1101/gr.171934.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Bankevich A, Antipov D, et al. Assembling genomes and mini-metagenomes from highly chimeric reads, 158–170. In: Deng M, Jiang R, Sun F, Zhang X, editors. Research in Computational Molecular Biology: 17th Annual International Conference, RECOMB 2013, Beijing, China, April 7–10, 2013. Springer Berlin Heidelberg; Berlin, Heidelberg: 2013. [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DC. Utilizing Next Generation Sequencing to Generate Bacterial Genomic Sequences for Evolutionary Analysis. Doctoral dissertation. 2014 Retrieved from http://scholarcommons.sc.edu/etd/2887.
- Simpson JT, Wong K, Jackman SD, et al. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]