

Abstract
Fluorescent protein biosensors are typically implemented via genetic encoding which makes the examination of scarce cell samples impractical. By directly delivering the protein form of the biosensor into cells using electroporation, we detected intracellular molecular activity with the sample size down to ~100 cells with high spatiotemporal resolution.
Fluorescence resonance energy transfer (FRET) based fluorescent protein biosensors have been widely used for visualization of molecular activities in live cells in real time with high spatiotemporal resolution.1–3 FRET occurs when two fluorophores in close proximity undergo non-radiative transfer of energy from an excited donor fluorophore (more blue shifted) to an acceptor fluorophore (more red shifted). FRET-based biosensors exploit this physical phenomenon and translate a specific biochemical event, such as protein phosphorylation,4,5 GTPase activities,6,7 and ion concentration,8 into a conformational change in the biosensor (i.e. alteration in the distance and/or orientation between two fluorescent proteins) and subsequently a change in the optical signal. However, the use of fluorescent protein biosensors (i.e. biosensors constructed by fusing fluorescent proteins to biopolymers) has been largely limited to genetically modified cells created by delivering and expressing a plasmid form of the biosensor. Genetic encoding typically requires a sizable cell population to start in order to achieve successful transfection and high cell viability and functionality after the procedure. This limitation renders the application of fluorescent protein biosensor technology to scarce cell samples (e.g. primary cells from animals and patients) impractical. To further complicate the situation, primary cells are in general harder to transfect than cell lines because they divide slowly or do not divide. This requirement of genetic encoding needs to be removed in order to extend the use of fluorescent protein biosensors to clinical diagnosis and prognosis.
In this study, we deliver a FRET biosensor in its protein form into cells using electroporation. Electroporation refers to the use of an external electric field for permeabilizing the cell membrane and permitting cellular uptake of macromolecules or nanoparticles.9 Electroporation is most commonly used for gene transfer and drug delivery in vitro and in vivo9–11 and several studies have also demonstrated its use in protein delivery.12–15 Electroporation does not involve endocytosis (i.e. encapsulation of delivered molecules in vesicles) so that the delivered biosensor can be directly exposed to intracellular molecules for detection. Furthermore, electroporation is a physical method that has little dependence on cell types and is suitable for processing cell populations of various sizes. By delivering the protein form of the biosensor, our procedure does not require transfection and expression of a plasmid vector and thus it is particularly suitable for samples containing a low number of cells. As a proof-of-principle, we deliver the ECFP/YPet (enhanced cyan fluorescent protein/a variant of yellow fluorescent protein) paired Src biosensor in its protein form into mouse embryonic fibroblast (MEF) cells by electroporation for detecting intracellular Src activity. Src is a protein tyrosine kinase that plays critical roles in a variety of cellular activities, including cell adhesion, migration, cancer invasion and metastasis.16 We show that the electroporation-delivered Src biosensor reports Src activity in the entire cell with very similar sensitivity and spatial resolution to those obtained by genetically encoded biosensors. Our technology will greatly expand the application of fluorescent protein biosensors to cell samples that are scarce and hard to transfect.
We used a sensitive Src FRET biosensor that contained a Src SH2 domain, a flexible linker, and a Src substrate peptide, concatenated between enhanced cyan fluorescent protein (ECFP) and a variant of yellow fluorescent protein (YPet).17 As shown in Fig. 1A, the design of the Src reporter allows the juxtaposition of ECFP and YPet to yield a high FRET emitting yellow fluorescence. Upon reaction with Src, the Src substrate peptide is phosphorylated and then binds to the phosphopeptide-binding pocket of the SH2 domain.4,18 The associated conformational change separates YPet from ECFP and decreases the FRET efficiency, hence increasing cyan fluorescence at the expense of yellow fluorescence emission. The reverse process occurs under phosphatase activity (Fig. 1A). In this way, the activity of Src within cells can be quantitatively detected and mapped by monitoring the fluorescence emission. The use of this fluorescent protein biosensor (under genetic encoding) to visualize spatiotemporal dynamics of Src activity at the subcellular level has been demonstrated experimentally19,20 and computationally.21,22
Fig. 1.
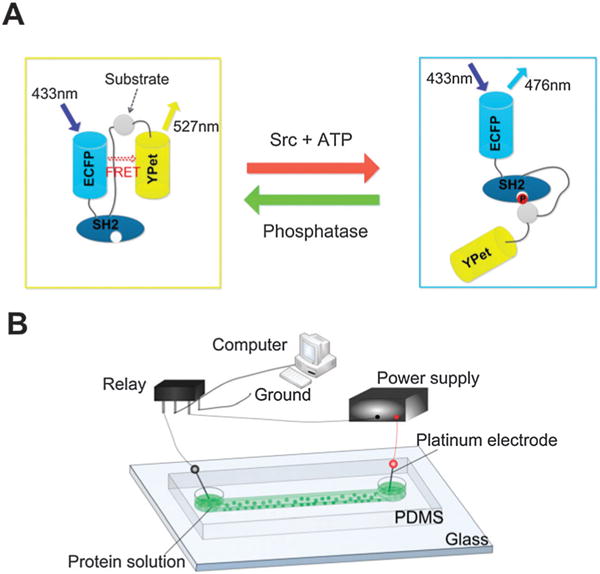
Schematic of the ECFP/YPet Src biosensor and its delivery by electroporation. (A) The mechanism of the Src FRET biosensor. The FRET signal varies with the Src activity and phosphatase treatment. (B) The setup for electroporation-based biosensor delivery in a microfluidic channel. A microfluidic channel facilitates applications of electric pulses of milliseconds and the observation of cellular dynamics. The dimensions of the channel were 150 μm (W) × 40 μm (D) × 3.8 mm (L).
In order to integrate electroporation delivery, cell culture, and fluorescence imaging, we conducted the experiment in a polydimethylsiloxane (PDMS) microfluidic channel (fabricated using soft lithography23) (Fig. 1B). Fibronectin was used to pretreat the channel to facilitate cell adhesion and culture.24–26 Wild type mouse embryonic fibroblasts (MEFs), or the Src/Yes/Fyn triple-knockout (SYF−/−) version of MEF cells, were suspended in the electroporation buffer that contained the Src reporter (in its protein form). The cell suspension was flowed into the microfluidic channel and millisecond pulses were applied to the cells immediately (before the cell adhesion fully occurred). After electroporation, cells were starved in fresh 0.5% FBS cell culture media for 36 h before stimulation and recording of the FRET signal. We used 100–200 cells in each experiment.
We show that electroporation is capable of delivering a significant amount of the fluorescent protein biosensor into cells and the delivered biosensor is highly functional for detecting the molecular activity of Src. Fig. 2A shows that Src reporters were delivered into both MEF and SYF−/− cells by electroporation. In our experiments, we loaded the cell sample into the microfluidic channel in a buffer containing the Src biosensor and then applied electroporation for the biosensor delivery. We then cultured the cells in the microfluidic channel for 36 h in a culture medium supplemented with only 0.5% FBS (serum starving to prepare the cells for stimulation treatment). We applied pervanadate (PVD), a tyrosine phosphatase inhibitor, for stimulation to promote Src activity.17,20 The level of phosphorylation on cellular proteins is regulated by the relative catalytic activities of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). PVD treatment increases the activities of intracelluar PTKs and consequently promotes Src Tyr416 phosphorylation by inhibiting the activities of PTPs. The phosphorylation in Tyr416 of Src promotes increased access by the Src substrate and facilitates substrate phosphorylation.27,28 In Fig. 2A, we observed a significant FRET signal change before and after the treatment by PVD in MEF cells, while no change in SYF−/− cells (with Src knockout). We used ratiometric FRET (i.e. ECFP/YPet emission ratio) to measure the FRET efficiency in order to minimize the influence on the result from the biosensor concentration, the cell size, the energy density of the excitation light, and the instability of optical devices.29 Fig. 2A shows that the FRET response was roughly uniform inside the MEF cells and this indicates that the delivered biosensor was taken up in both the cytosol and the nucleus. There was no FRET signal change related to PVD treatment when SYF−/− cells were used and this suggests that the biosensor was specific. In Fig. 2B, the ECFP/YPet ratio in MEF cells increased by 91% within 20 min after the PVD stimulation (compared to less than 13% in SYF−/− cells). Such temporal dynamics match well with previous work using the same biosensor implemented via genetic encoding.17 We found that with a single pulse of 15 ms and 1000 V cm−1 we were able to achieve 45–48% viability for both MEF and SYF−/− cells. It is worth noting that our method offered a rapid turnaround time for the biosensor assay. The electroporation-based delivery of the Src biosensor in its protein form was done within 1 h, while it usually takes several days to prepare a genetically encoded Src biosensor by transfecting cells with the plasmid form of the biosensor. In addition, because a significant percentage of cells survive the electroporation and become experimental subjects, our method may work with samples of tiny amounts.
Fig. 2.
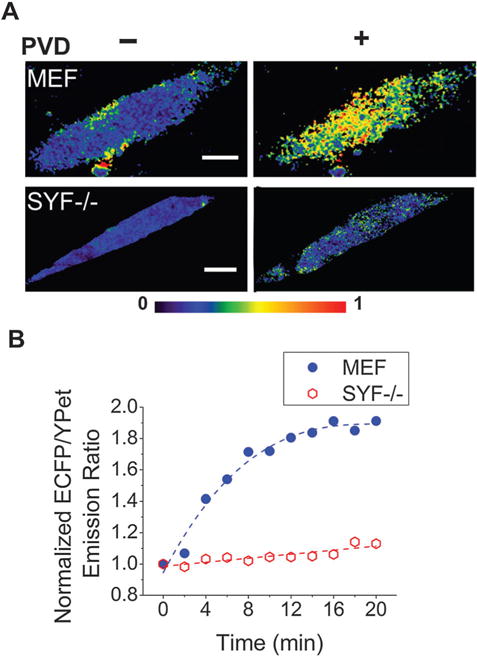
Intracellular Src activity in response to PVD stimulation was detected by the electroporation-delivered Src biosensor. (A) The color-coded ECFP/YPet emission ratio mapped over an entire cell before and 20 min after PVD treatment with MEF and SYF−/− cells. The images were generated using the pseudocolor mode in ImageJ by calculating the ECFP/YPet emission intensity ratio at each pixel. (B) Temporal dynamics of the normalized ECFP/YPet emission ratio upon PVD stimulation (at t = 0) with MEF and SYF−/− cells. The average emission ratio of a cell at t = 0 was taken as 1 and the other emission ratios were normalized against this value. Trend lines (broken lines) are added to guide the eye (scale bars, 10 μm).
Finally, we investigated the effects of the electroporation conditions on the biosensor performance. We used various field intensities (600–1200 V cm−1) with a fixed pulse duration (15 ms) for the electroporation delivery and recorded the FRET signals. As shown in Fig. 3A and B, the delivery and FRET change were weak when 600 V cm−1 was used, whereas significant Src activity detection was obtained when the field intensity was higher than 800 V cm−1, indicating successful delivery of the biosensor with the retention of cell functions. The cell viability after electroporation dropped when the field intensity increased (from 60% at 600 V cm−1 to 18% at 1200 V cm−1). To conclude, the electroporation conditions affect the biosensor performance mostly by the amount of the delivery. Sufficient amount of biosensor delivery is important for the biosensor detection. On the other hand, intensive electroporation conditions lead to increased cell death. Thus the electroporation conditions ought to be optimized to strike a balance between high cell viability and sufficient biosensor delivery.
Fig. 3.
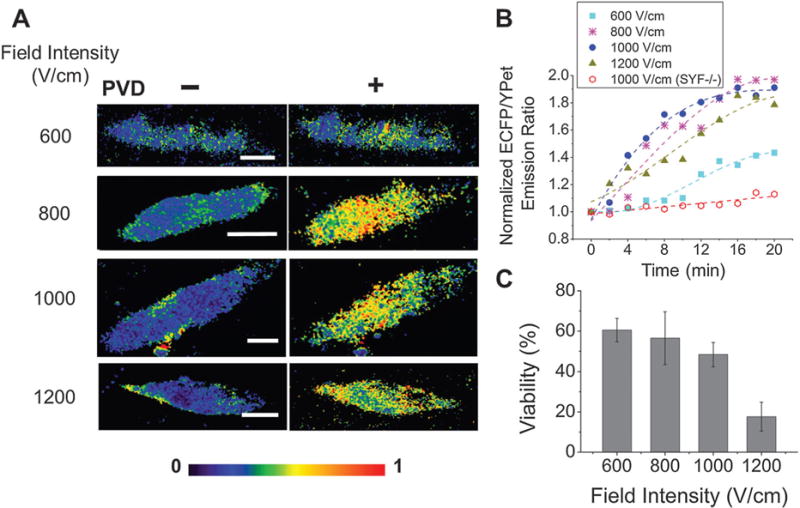
The signal from the Src biosensor under various pulse intensities. (A) Color-coded images of ECFP/YPet emission ratios before and 20 min after PVD stimulation. (B) Temporal dynamics of the normalized emission ratio of ECFP/YPet upon PVD stimulation at t = 0. Trend lines (broken lines) are added to guide the eye. (C) The dependence of the cell viability on the field intensities. The Src biosensor was delivered into MEF cells by electroporation under the field intensities of 600, 800, 1000 and 1200 V cm−1 in (A), (B) and (C), and into SYF−/− cells by 1000 V cm−1 in (B). The pulse duration was 15 ms (scale bars, 10 μm).
In conclusion, we demonstrated that a fluorescent protein biosensor in its protein form could be delivered into cells by electroporation with high efficiency and reproducibility. The electroporation-delivered Src reporter showed significant FRET signal variations in response to Src activity change. Such a FRET signal could be mapped spatially and temporally at the single cell level. The delivered amount of the biosensor depended on the electroporation conditions. Our electroporation-based approach circumvents issues associated with genetic encoding that is currently used in the majority of the protein biosensor studies and will enable application of the biosensor technology to cells from scarce sources such as animals and human patients.
Supplementary Material
Acknowledgments
This work was supported by ICTAS of Virginia Tech and NSF grants 1016547, 0967069 and 1344298.
Footnotes
Electronic supplementary information (ESI) available: Materials and methods.
Notes and references
- 1.Carlson HJ, Campbell RE. Curr Opin Biotechnol. 2009;20:19–27. doi: 10.1016/j.copbio.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Li IT, Pham E, Truong K. Biotechnol Lett. 2006;28:1971–1982. doi: 10.1007/s10529-006-9193-5. [DOI] [PubMed] [Google Scholar]
- 3.Piston DW, Kremers GJ. Trends Biochem Sci. 2007;32:407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Wang YX, Botvinick EL, Zhao YH, Berns MW, Usami S, Tsien RY, Chien S. Nature. 2005;434:1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 5.Ting AY, Kain KH, Klemke RL, Tsien RY. Proc Natl Acad Sci U S A. 2001;98:15003–15008. doi: 10.1073/pnas.211564598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirata E, Yukinaga H, Kamioka Y, Arakawa Y, Miyamoto S, Okada T, Sahai E, Matsuda M. J Cell Sci. 2012;125:858–868. doi: 10.1242/jcs.089995. [DOI] [PubMed] [Google Scholar]
- 7.Itoh RE, Kurokawa K, Ohba Y, Yoshizaki H, Mochizuki N, Matsuda M. Mol Cell Biol. 2002;22:6582–6591. doi: 10.1128/MCB.22.18.6582-6591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 9.Geng T, Lu C. Lab Chip. 2013;13:3803–3821. doi: 10.1039/c3lc50566a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neumann E, Schaeferridder M, Wang Y, Hofschneider PH. EMBO J. 1982;1:841–845. doi: 10.1002/j.1460-2075.1982.tb01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Hwang I, Britain D, Chung TD, Sun Y, Kim DH. Lab Chip. 2011;11:3941–3948. doi: 10.1039/c1lc20766k. [DOI] [PubMed] [Google Scholar]
- 12.Graziadei L, Burfeind P, Barsagi D. Anal Biochem. 1991;194:198–203. doi: 10.1016/0003-2697(91)90168-s. [DOI] [PubMed] [Google Scholar]
- 13.Eksioglu-Demiralp E, Kitada S, Carson D, Garland J, Andreef M, Reed JC. J Immunol Methods. 2003;275:41–56. doi: 10.1016/s0022-1759(02)00554-9. [DOI] [PubMed] [Google Scholar]
- 14.Puc M, Kotnik T, Mir LM, Miklavcic D. Bioelectrochemistry. 2003;60:1–10. doi: 10.1016/s1567-5394(03)00021-5. [DOI] [PubMed] [Google Scholar]
- 15.Banga AK, Prausnitz MR. Trends Biotechnol. 1998;16:408–412. doi: 10.1016/s0167-7799(98)01238-4. [DOI] [PubMed] [Google Scholar]
- 16.Martin GS. Nat Rev Mol Cell Biol. 2001;2:467–475. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 17.Ouyang MX, Sun J, Chien S, Wang YX. Proc Natl Acad Sci U S A. 2008;105:14353–14358. doi: 10.1073/pnas.0807537105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C. J Biol Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- 19.Seong J, Lu S, Wang Y. Cell Mol Bioeng. 2011;4:138–147. doi: 10.1007/s12195-011-0161-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seong J, Lu S, Ouyang M, Huang H, Zhang J, Frarne MC, Wang Y. Chem Biol. 2009;16:48–57. doi: 10.1016/j.chembiol.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu S, Wang Y. In: Fluorescence in Vivo Imaging Based on Genetically Engineered Probes: From Living Cells to Whole Body Imaging Iv. Savitsky AP, Wang Y, editors. Vol. 7191 2009. [Google Scholar]
- 22.Lu S, Ouyang M, Seong J, Zhang J, Chien S, Wang Y. PLoS Comput Biol. 2008;4:e1000127. doi: 10.1371/journal.pcbi.1000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geng T, Zhan Y, Wang J, Lu C. Nat Protoc. 2011;6:1192–1208. doi: 10.1038/nprot.2011.360. [DOI] [PubMed] [Google Scholar]
- 24.Wang F, Wang H, Wang J, Wang HY, Rummel PL, Garimella SV, Lu C. Biotechnol Bioeng. 2008;100:150–158. doi: 10.1002/bit.21737. [DOI] [PubMed] [Google Scholar]
- 25.Wang HY, Bao N, Lu C. Biosens Bioelectron. 2008;24:613–617. doi: 10.1016/j.bios.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 26.Zhan Y, Sun C, Cao Z, Bao N, Xing J, Lu C. Anal Chem. 2012;84:8102–8105. doi: 10.1021/ac302462s. [DOI] [PubMed] [Google Scholar]
- 27.Secrist JP, Burns LA, Karnitz L, Koretzky GA, Abraham RT. J Biol Chem. 1993;268:5886–5893. [PubMed] [Google Scholar]
- 28.Boulven I, Robin P, Desmyter C, Harbon S, Leiber D. Cell Signalling. 2002;14:341–349. doi: 10.1016/s0898-6568(01)00269-8. [DOI] [PubMed] [Google Scholar]
- 29.Kardash E, Bandemer J, Raz E. Nat Protoc. 2011;6:1835–1846. doi: 10.1038/nprot.2011.395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.