

Abstract
Precise control of the amplitude of protein kinase C signaling is essential for maintaining cellular homeostasis, and disruption of this control leads to pathophysiological states such as cancer and neurodegeneration. This amplitude is precisely tuned by multiple inputs that regulate the amount of protein kinase C in the cell, its ability to sense its allosteric activator diacylglycerol, and protein scaffolds that coordinate access to substrates. Key to regulation of the signaling output of most protein kinase C isozymes is the ability of cytosolic enzyme to respond to the membrane-embedded lipid second messenger, diacylglycerol, in a dynamic range that prevents signaling in the absence of agonists but allows efficient signaling in response to small changes in diacylglycerol. This review discusses the regulatory inputs that control the spatiotemporal dynamics of protein kinase C signaling, focusing on conventional and novel PKC isozymes.
Keywords: Protein kinase C, regulation, priming phosphorylation, second messengers, conformational change, scaffold
Introduction
The protein kinase C (PKC) family transduces a multitude of signals that control diverse cellular processes such as proliferation, migration, invasion, differentiation, apoptosis, transcription, and translation. Therefore, aberrant PKC activity or localization has been linked to numerous diseases, most notable of which are cancer, neurodegeneration, and diabetes [1]. This serine/threonine kinase family belongs on the AGC kinase branch of the kinome and comprises 9 genes that share a similar architecture with an N-terminal regulatory moeity and a C-terminal kinase domain (Figure 1). PKCs are classified according to their regulatory domains, and thus the 2nd messengers that regulate their activity. Conventional PKCs (cPKCs: α, β and γ) contain tandem C1 domains that bind diacylglycerol (DAG) and phosphatidylserine (PS) and a C2 domain that binds anionic phospholipids, including phosphatidylinositol 4,5-bisphosphate (PIP2), in a Ca2+-dependent manner. Novel PKCs (nPKCs: δ, ε, η, θ) contain tandem C1 domains that bind diacylglycerol and a novel C2 domain that is Ca2+-unresponsive and does not assist in membrane binding. Atypical PKCs (aPKCs: ι and ζ) have an atypical C1 domain that does not bind diacylglycerol and lack a C2 domain all together, but instead contain a PB1 domain that mediates protein-protein interactions. Atypical PKCs and PKCα contain a C-terminal PDZ ligand.
Figure 1. Schematic showing domain composition of the three classes of protein kinase C.
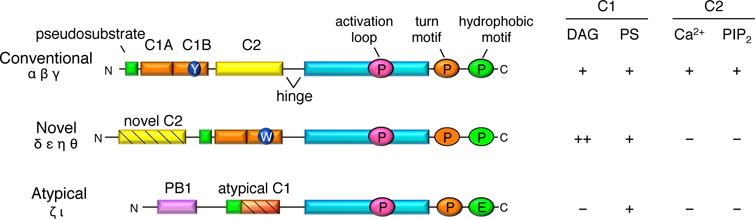
Conventional PKCs (α, β, and γ) contain an N-terminal pseudosubstrate (green), tandem C1 domains (orange) that bind diacylglycerol (DAG) and phosphatidylserine (PS), a C2 domain (yellow) that binds phosphatidylinositol 4,5-bisphosphate (PIP2) in a Ca2+-dependent manner, and a C-terminal kinase domain (cyan). Novel PKCs (δ, ε, η, and θ) have a similar domain composition except that their C2 domain cannot bind Ca2+ or PIP2 and their C1B domain has ~100-fold higher affinity DAG as it contains a Trp (W) at position 22 in the domain, as opposed to a Tyr (Y) as the C1B domain of conventional PKCs has. Atypical PKCs (ζ and ι) have a protein-binding module, PB1 domain, and an atypical C1 domain that cannot bind DAG. All PKCs are phosphorylated at 3 conserved sites: the activation loop within the kinase domain and the turn and hydrophobic motifs within the C-terminal tail (except for atypical PKCs which have a phosphomimetic at the hydrophobic motif).
Regulation by priming phosphorylation
Phosphorylation is absolutely critical 1] to render PKC in a catalytically competent conformation and 2] to protect PKC from degradation [2]. Because, unlike many other kinases, the phosphorylation of PKC is constiututive, its activity is not acutely regulated by phosphorylation. Rather, cellular levels of PKC are directly regulated by its phosphorylation. Conventional and novel PKCs are constitutively phosphorylated at 3 conserved residues: the activation loop, the turn motif, and the hydrophobic motif [3] (Figure 1). Atypical PKCs are also phosphorylated at the activation loop and turn motif, but contain a phosphomimetic glutamic acid at the hydrophobic motif. The first priming phosphorylation occurs at the activation loop within the kinase domain and is catalyzed by the phosphoinositide-dependent kinase, PDK-1 [4, 5]. Phosphorylation at this site properly aligns residues within the active site for catalysis, an event that induces two tightly-coupled and ordered phosphorylations on the C-terminal tail: phosphorylation at the turn motif and intramolecular autophosphorylation at the hydrophobic motif. For conventional and novel PKCs such as PKCe, but not PKCδ, these phosphorylation events require mTORC2; however, whether mTOR is the direct kinase for this site in cells remains controversial [6–9]. Phosphorylation at the hydrophobic motif controls the stability of the enzyme. Indeed, dephosphorylation of this site is the first step in the degradation of PKC as it destabilizes PKC and promotes its degradation. The PH domain leucine-rich repeat protein phosphatases (PHLPP) directly dephosphorylates the hydrophobic motif of PKCs, an event that requires the PH domain of PHLPP [10]. Thus, loss of PHLPP in the cell leads to an increase in steady state PKC levels. Conversely, PKC levels are low in cells in which any of the phosphorylation steps have been perburbed, including loss of TORC2 or loss of PDK-1 [7, 11]. Thus, priming phosphorylations regulate the steady state levels of PKC but not activity in an acute, agonist-dependent manner; instead, the spatial and temporal dynamics of PKC signaling are regulated by second messengers.
Regulation by second messengers
Although phosphorylated PKC is catalytically competent, an autoinhibitory pseudosubstrate the binds the substrate binding cavity maintains PKC inactive until the appropriate second messengers bind (Figure 2). Conventional PKCs are allosterically activated by binding to two second messengers: Ca2+ and diacylglycerol. Binding of the C2 domain to Ca2+ targets the kinase to the plasma membrane through hydrophobic interactions that drive binding to anionic phospholipids and through electrostatic interactions that contribute to retention of the C2 domain to membranes [12]. Once at the membrane, one of PKC’s C1 domains can then find and bind DAG, and event that provides the necessary energy to expel the pseudosubstrate and activate PKC [13]. Novel PKCs are activated solely by DAG, whereas atypical PKCs do not respond to either of these second messengers and are instead activated by protein-protein interactions.
Figure 2. Regulation of protein kinase C.
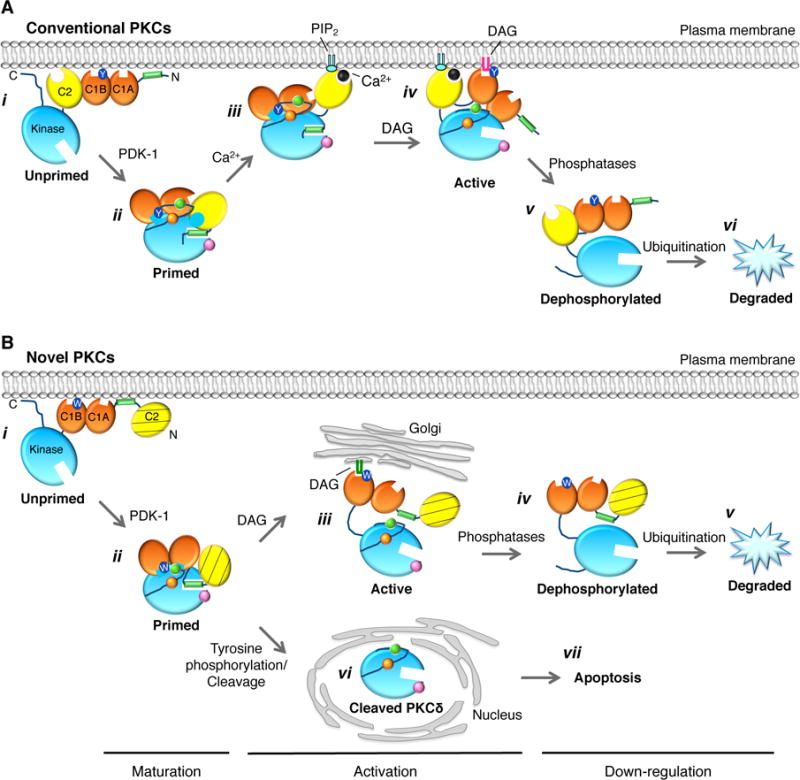
Model showing how maturation of cPKC masks C1 domains to increase the dynamic range of DAG sensing and thus PKC output. (A) Unprimed PKC is in an open conformation that associates with membranes via weak interactions from the C2 domain, both C1A and C1B domains, the exposed pseudosubstrate, and the C-terminal tail. In this conformation, both C1A and C1B domains are fully exposed. (B) Upon ordered phosphorylation of PKC at its activation loop (pink), turn motif (orange), and hydrophobic motif (green) sites, PKC matures into its closed conformation, in which both the C1A and C1B domains become masked, the pseudosubstrate binds the substrate binding site, and the enzyme localizes to the cytosol. This masking of the C1 domains prevents pretargeting of PKC to membranes in the absence of agonist-evoked increases in DAG, thus decreasing basal signaling. (C) In response to agonists that promote PIP2 hydrolysis, Ca2+-dependent binding of the C2 domain of cPKCs to the plasma membrane allows the low-affinity DAG sensor to find its membrane-embedded ligand, DAG. (D) Binding of DAG, predominantly to the C1B domain of PKCβII, expels the pseudosubstrate from the substrate-binding cavity and activates PKC. Use primarily of the lower-affinity C1B domain increases the dynamic range of PKC output as the signal does not saturate as readily using the lower affinity module and allows cPKCs to signal at the plasma membrane as opposed to the Golgi. (E) Dephosphorylation of activated PKC allows it to regain the exposed (open) conformation of unprimed PKC.
Differential binding of 2nd messengers between conventional and novel PKCs leads to substantial differences in their spatiotemporal dynamics of signaling. Firstly, conventional and novel PKCs predominantly translocate to different membranes. Conventional PKCs translocate to and are active at the plasma membrane because their Ca2+-binding C2 domain targets them there via PIP2, where they can then find diacylglycerol [12]. Novel PKCs do not have a functional C2 domain but have a C1A domain that has a 100-fold higher affinity for DAG [14] and, therefore, they translocate to DAG-rich endomembranes such as the Golgi. Indeed, impairing the ability of the C2 domain of PKCα to bind membranes forces it to the Golgi instead, as the C1 domain interaction becomes the dominant force [12]. Secondly, the kinetics of activation differ between conventional and novel PKCs. Conventional PKCs display rapid but transient activity at the plasma membrane that tracks with the initial Ca2+ release [15]. In fact, the kinetics of cPKC activation directly correlate with Ca2+ levels [16]. PKCβII exhibits oscillatory translocation to the plasma membrane in response to Ca2+ oscillations produced by histamine stimulation of HeLa cells. This in turn leads to oscillatory phosphorylation of membrane-localized substrates that tracks Ca2+ levels with a slight lag. Novel PKCs, on the other hand, are activated with slower kinetics because they do not respond to this fast Ca2+ release [15]. Thirdly, the duration of PKC activity, which is ultimately controlled by the persistence of the second messengers at a particular subcellular location, also differs among the PKCs. For example, DAG levels are more sustained at the Golgi than the plasma membrane, leading to prolonged novel PKC activity at the Golgi compared to the short-lived conventional PKC activity at the plasma membrane [15]. Thus, second messengers precisely dictate the kinetics, manganite, duration, and location of PKC activity and are responsible for the apparent differences between the three classes of PKCs.
PKC activity is exquisitely controlled by various mechanisms both under basal conditions and after agonist stimulation. For example, under basal conditions the E3 ligase for PKC, RING-finger protein that interacts with C kinase (RINCK), controls the amplitude of PKC signaling by regulating its levels [17]. RINCK interacts with the C1A domain of PKC and induces its ubiquitination and thus its degradation. Moreover, under basal conditions with limited DAG levels, PKCα interacts with diacylglycerol kinase ζ, which prevents its activation by locally metabolizing DAG [18]. Only under stimulated conditions in which enough DAG is locally produced to activate PKCα, allows PKCα to phosphorylate diacylglycerol kinase ζ leading to disassociation of the two proteins. However, agonist stimulation also ultimately leads to termination of PKC signaling through various mechanisms. PKC signaling is quickly terminated by the clearance of the respective 2nd messengers, but also by agonist-induced downregulation of the enzyme. Active PKC can be dephosphorylated by PHLPP or protein phosphatase 2A, shunting PKC to the detergent-insoluble fraction and inducing its ubiquitination and ultimately its proteasome-mediated degradation [19]. Agonist induced proteasome-mediated degradation of PKCα can, however, also occur via ubiquitination of plasma membrane-localized, fully primed PKC [20, 21]. Additionally, phosphorylated PKCα can also be internalized through lipid raft-mediated endocytic pathways and degraded by the lysosome [21, 22]. These mechanisms lead to desensitization of PKC signaling, thus maintaining its activity under exquisite control.
Regulation independent of 2nd messengers
Particular conventional and novel PKCs can also be activated independently of second messengers, adding to the complexity of PKC signaling. For example, certain PKCs can be activated by the accumulation of reactive oxygen species, which are often elevated in diseases such as cancer, cardiovascular disease, and neurodegeneration [23]. Specifically, H2O2 causes oxidation of cysteine residues within the C1B domain of PKCγ, inducing conformational changes that release PKCγ from its scaffold, leading to its translocation to the plasma membrane and subsequent DAG-independent activation [24, 25]. PKCδ is phosphorylated at multiple tyrosine residues by Src family kinases in response to acute stimulation of cells by H2O2, PMA, epidermal growth factors, or platelet-derived growth factor. Tyrosine phosphorylation can induce PKCδ’s DAG-independent activation without requiring membrane translocation and can alter its subcellular localization [26, 27]. For example, tyrosine phosphorylation of PKCδ at Tyr64 and Tyr155 in response to apoptotic stimuli induces a conformational change that exposes its nuclear localization sequence and Hsp90 binding site, allowing importin-α and Hsp90 to bind and import PKCδ into the nucleus where it can induce apoptosis [28]. H2O2-induced tyrosine phosphorylation at Tyr311 has also been proposed to activate PKCδ by inducing caspase-3 cleavage between its regulatory and catalytic domains [29]. Therefore, these agonist-induced phosphorylations and conformational changes can activate PKCs independently of 2nd messengers by either releasing PKCs from scaffolds or exposing binding sites for scaffolds to interact with.
Regulation by conformational changes
Another level of regulation of PKC activity is imparted by conformational changes, as PKCs are under precise conformation control, particularly during maturation. When first synthesized, PKC is in an open conformation that has both the C1A and C1B domains exposed (Figure 2). Upon phosphorylation at its 3 priming sites, PKC adapts a closed conformation that masks its C1 domains such that the lower affinity C1B domain is the predominant DAG binder. This conformational change optimizes PKC’s dynamic range of signaling such that it is not activated by basal diacylglycerol but can readily detect and be activated by a small, local increase in DAG [30]. Binding to their respective 2nd messengers leads to another conformational change that results in expulsion of the autoinhibitory pseudosubstrate and subsequent activation of PKC. Similarly to conventional PKCs, the pseudosubstrate and C1 domain of the atypical PKCζ also inhibit its activity through intramolecular interactions with the substrate-binding site and the αC helix, respectively [31, 32].
A recent study proposed that PKC is not only regulated by intramolecular interactions, but also by intermolecular interactions. Swanson et al. [33] showed evidence that PKCβII forms homo-dimers upon activation and that the dimers maintain PKC in an active conformation. Both C1 domains and the C2 domain contribute to this interaction, but the C-terminal tail is absolutely necessary for dimer formation. In fact, phosphorylation of the C-terminal tail is critical in maintaining PKC in a closed, inactive conformation, as lack of phosphorylation at these sites leads to PKC remaining in an open, exposed conformation [30] and increases basal dimerization [33]. Therefore, PKC undergoes conformational changes both during its maturation and during its activation in order to finely tune its activity.
Regulation by scaffolding
Scaffolding also plays an integral part in determining the precise location, duration, and amplitude of PKC activity, as well as in establishing substrate specificity (reviewed in [34, 35]). Considering that there are multiple PKC isozymes expressed in the same cell that are activated by the same stimuli, scaffolds provide a level of functional selectivity. Among the PKC scaffolds are receptors for activated C kinase (RACK), 14-3-3 proteins, and A-kinase anchoring proteins (AKAPs).
Particular scaffolds augment PKC signaling whereas others inhibit it. RACKs were the first scaffolds found to associate with active PKC. For PKCβII, this interaction occurs via its C2 domain and C-terminal tail and it stabilizes PKC’s active conformation, thus enhancing its activity towards co-scaffolded substrates [36–38]. The phosphoserine/threonine binding protein 14-3-3 binds to a pair of phosphoserines within the hinge region of PKCε, leading to its activation [39]. However, scaffolds can also be inhibitory towards PKC by sequestering it away from its substrates or maintaining it in an inactive conformation. For example, in lens epithelial cells, 14-3-3ε binds to PKCγ via its C1B domain and controls both its activity and localization, thus regulating gap junction activity [40]. AKAPs are another set of PKC scaffolds and were first identified to bind protein kinase A. AKAP12 binds to and attenuates PKCα and PKCδ signaling, thus preventing senescence and oncogenic transformation [41]. Similarly, AKAP5 inhibits PKC activity by binding to its catalytic pocket [42].
Scaffolds can also regulate the duration of PKC activity towards a substrate by co-scaffolding a phosphatase of the substrate. The phosphatase can thus rapidly dephosphorylate and attenuate signaling downstream of PKC substrates. Such an example is the coordination of PKC and protein phosphatase 2B/calcineurin on AKAP79 at the postsynaptic density in neurons [43]. Another AKAP (AKAP350/AKAP450/centrosome and Golgi localized PKN-associated protein or CG-NAP) was proposed to act as a scaffold for the maturation of PKC [44]. This AKAP was found to only associate with nascent PKCε at the Golgi/centrosome and this complex disassembled after PKCε maturation by phosphorylation. Interestingly, this complex also contains protein phosphatase 2A [45], which dephosphorylates PKC leading to its degradation. Perhaps PKC levels can be dynamically controlled on this scaffold through regulation of its phosphorylation. Scaffolding of PKC also has some clinical relevance because scaffolds have been shown to change the pharmacological prolife of PKC. Specifically, ATP-competitive inhibitors are ineffective against scaffolded PKC [42]. To explain how scaffolds amplify, accelerate, and insulate PKC signaling, Greenwald et al. [46] proposed a stochastic state-switching model. In this model, the complex containing PKC, its substrate, and its scaffold alters between inactive and active intermediate states, thus allowing phosphorylation of the substrate, even in the presence of active-site PKC inhibitors. Thus scaffolds are able to precisely control PKC activity and to confer functional selectivity.
Concluding Remarks
As PKC activity has to be precisely balanced at every subcellular location, its regulation is under intricate control. This control of the spatial and temporal dynamics of PKC signaling comes from regulation through various mechanisms, such as phosphorylation, binding to 2nd messengers, conformational changes, and binding to scaffolds. Perturbation of any of these mechanisms of control can lead to pathophysiological states. In fact, dysregulation of PKC activity has been observed in many diseases including cancer [47], diabetes [48], and neurodegenerative diseases such as Alzheimer’s [49]. Changes in expression of PKC scaffolds, such as RACK1, as opposed to PKC itself, have also been found to dysregulate PKC signaling in Alzhemier’s disease [50]. Therefore, understanding all mechanisms through which PKC is regulated is key to developing novel therapeutics to restore PKC signaling to physiological levels and subcellular locations.
Acknowledgments
We thank members of the lab for helpful suggestions.
Funding
This work was supported by the National Institutes of Health [grant number GM43154] to A.C.N. C.E.A. was supported in part by the National Science Foundation Graduate Research Fellowship [grant number DGE1144086] and by the UCSD Graduate Training Program in Cellular and Molecular Pharmacology through an institutional training grant from the National Institute of General Medical Sciences [grant number T32 GM007752].
Abbreviations used
- PKC
protein kinase C
- cPKC
conventional PKC
- DAG
diacylglycerol
- PS
phosphatidylserine
- PIP2
phosphatidylinositol 4,5-bisphosphate
- nPKC
novel PKC
- aPKC
atypical PKC
- PHLPP
PH domain leucine-rich repeat protein phosphatase
- RINCK
RING-finger protein that interacts with C kinase
- RACK
receptor for activated C kinase
- AKAP
A-kinase anchoring protein
References
- 1.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, Messing RO. Protein kinase C isozymes and the regulation of diverse cell responses. American journal of physiology. Lung cellular and molecular physiology. 2000;279:L429–438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 2.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. The Biochemical journal. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keranen LM, Dutil EM, Newton AC. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr Biol. 1995;5:1394–1403. doi: 10.1016/s0960-9822(95)00277-6. [DOI] [PubMed] [Google Scholar]
- 4.Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- 5.Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1) Curr Biol. 1998;8:1366–1375. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- 6.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. The EMBO journal. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. The EMBO journal. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Behn-Krappa A, Newton AC. The hydrophobic phosphorylation motif of conventional protein kinase C is regulated by autophosphorylation. Curr Biol. 1999;9:728–737. doi: 10.1016/s0960-9822(99)80332-7. [DOI] [PubMed] [Google Scholar]
- 10.Gao T, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. J Biol Chem. 2008;283:6300–6311. doi: 10.1074/jbc.M707319200. [DOI] [PubMed] [Google Scholar]
- 11.Balendran A, Hare GR, Kieloch A, Williams MR, Alessi DR. Further evidence that 3-phosphoinositide-dependent protein kinase-1 (PDK1) is required for the stability and phosphorylation of protein kinase C (PKC) isoforms. FEBS letters. 2000;484:217–223. doi: 10.1016/s0014-5793(00)02162-1. [DOI] [PubMed] [Google Scholar]
- 12.Scott AM, Antal CE, Newton AC. Electrostatic and hydrophobic interactions differentially tune membrane binding kinetics of the C2 domain of protein kinase Calpha. J Biol Chem. 2013;288:16905–16915. doi: 10.1074/jbc.M113.467456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orr JW, Keranen LM, Newton AC. Reversible exposure of the pseudosubstrate domain of protein kinase C by phosphatidylserine and diacylglycerol. J Biol Chem. 1992;267:15263–15266. [PubMed] [Google Scholar]
- 14.Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007;282:826–830. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 15.Gallegos LL, Kunkel MT, Newton AC. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J Biol Chem. 2006;281:30947–30956. doi: 10.1074/jbc.M603741200. [DOI] [PubMed] [Google Scholar]
- 16.Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen D, Gould C, Garza R, Gao T, Hampton RY, Newton AC. Amplitude control of protein kinase C by RINCK, a novel E3 ubiquitin ligase. J Biol Chem. 2007;282:33776–33787. doi: 10.1074/jbc.M703320200. [DOI] [PubMed] [Google Scholar]
- 18.Luo B, Prescott SM, Topham MK. Association of diacylglycerol kinase zeta with protein kinase C alpha: spatial regulation of diacylglycerol signaling. J Cell Biol. 2003;160:929–937. doi: 10.1083/jcb.200208120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Z, Liu D, Hornia A, Devonish W, Pagano M, Foster DA. Activation of protein kinase C triggers its ubiquitination and degradation. Mol Cell Biol. 1998;18:839–845. doi: 10.1128/mcb.18.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lum MA, Balaburski GM, Murphy ME, Black AR, Black JD. Heat shock proteins regulate activation-induced proteasomal degradation of the mature phosphorylated form of protein kinase C. J Biol Chem. 2013;288:27112–27127. doi: 10.1074/jbc.M112.437095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leontieva OV, Black JD. Identification of two distinct pathways of protein kinase Calpha down-regulation in intestinal epithelial cells. J Biol Chem. 2004;279:5788–5801. doi: 10.1074/jbc.M308375200. [DOI] [PubMed] [Google Scholar]
- 22.Lum MA, Pundt KE, Paluch BE, Black AR, Black JD. Agonist-induced down-regulation of endogenous protein kinase c alpha through an endolysosomal mechanism. J Biol Chem. 2013;288:13093–13109. doi: 10.1074/jbc.M112.437061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahman K. Studies on free radicals, antioxidants, and co-factors. Clinical interventions in aging. 2007;2:219–236. [PMC free article] [PubMed] [Google Scholar]
- 24.Lin D, Takemoto DJ. Oxidative activation of protein kinase Cgamma through the C1 domain. Effects on gap junctions. J Biol Chem. 2005;280:13682–13693. doi: 10.1074/jbc.M407762200. [DOI] [PubMed] [Google Scholar]
- 25.Lauer J, Banerjee D, Shanks D, Dai H, Gong YX, Prakash O, Takemoto D. NMR structure/function relationships of peptides corresponding to the C1B1 Region of PKC gamma. Protein and peptide letters. 2010;17:1–10. doi: 10.2174/092986610789909485. [DOI] [PubMed] [Google Scholar]
- 26.Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, Ohmae K, Kikkawa U, Nishizuka Y. Phosphorylation sites of protein kinase C delta in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6587–6592. doi: 10.1073/pnas.111158798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, Nishizuka Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci U S A. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adwan TS, Ohm AM, Jones DN, Humphries MJ, Reyland ME. Regulated binding of importin-alpha to protein kinase Cdelta in response to apoptotic signals facilitates nuclear import. J Biol Chem. 2011;286:35716–35724. doi: 10.1074/jbc.M111.255950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaul S, Anantharam V, Yang Y, Choi CJ, Kanthasamy A, Kanthasamy AG. Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cdelta in dopaminergic neuronal cells. J Biol Chem. 2005;280:28721–28730. doi: 10.1074/jbc.M501092200. [DOI] [PubMed] [Google Scholar]
- 30.Antal CE, Violin JD, Kunkel MT, Skovso S, Newton AC. Intramolecular conformational changes optimize protein kinase C signaling. Chemistry & biology. 2014;21:459–469. doi: 10.1016/j.chembiol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Neimanis S, Lopez-Garcia LA, Arencibia JM, Amon S, Stroba A, Zeuzem S, Proschak E, Stark H, Bauer AF, et al. Molecular Mechanism of Regulation of the Atypical Protein Kinase C by N-terminal Domains and an Allosteric Small Compound. Chemistry & biology. 2014 doi: 10.1016/j.chembiol.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 32.Lopez-Garcia LA, Schulze JO, Frohner W, Zhang H, Suss E, Weber N, Navratil J, Amon S, Hindie V, Zeuzem S, et al. Allosteric regulation of protein kinase PKCzeta by the N-terminal C1 domain and small compounds to the PIF-pocket. Chemistry & biology. 2011;18:1463–1473. doi: 10.1016/j.chembiol.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Swanson CJ, Ritt M, Wang W, Lang M, Narayan A, Tesmer J, Westfall M, Sivaramakrishnan S. Conserved Modular Domains Team Up to Latch-Open Active PKCalpha. J Biol Chem. 2014 doi: 10.1074/jbc.M113.534750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schechtman D, Mochly-Rosen D. Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene. 2001;20:6339–6347. doi: 10.1038/sj.onc.1204778. [DOI] [PubMed] [Google Scholar]
- 35.Antal CE, Newton AC. Spatiotemporal dynamics of phosphorylation in lipid second messenger signaling. Molecular & cellular proteomics : MCP. 2013;12:3498–3508. doi: 10.1074/mcp.R113.029819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:839–843. doi: 10.1073/pnas.91.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stebbins EG, Mochly-Rosen D. Binding specificity for RACK1 resides in the V5 region of beta II protein kinase C. J Biol Chem. 2001;276:29644–29650. doi: 10.1074/jbc.M101044200. [DOI] [PubMed] [Google Scholar]
- 38.Ron D, Luo J, Mochly-Rosen D. C2 region-derived peptides inhibit translocation and function of beta protein kinase C in vivo. J Biol Chem. 1995;270:24180–24187. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- 39.Kostelecky B, Saurin AT, Purkiss A, Parker PJ, McDonald NQ. Recognition of an intra-chain tandem 14-3-3 binding site within PKCepsilon. EMBO reports. 2009;10:983–989. doi: 10.1038/embor.2009.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen TA, Takemoto LJ, Takemoto DJ. Inhibition of gap junction activity through the release of the C1B domain of protein kinase Cgamma (PKCgamma) from 14-3-3: identification of PKCgamma-binding sites. J Biol Chem. 2004;279:52714–52725. doi: 10.1074/jbc.M403040200. [DOI] [PubMed] [Google Scholar]
- 41.Akakura S, Nochajski P, Gao L, Sotomayor P, Matsui S, Gelman IH. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle. 2010;9:4656–4665. doi: 10.4161/cc.9.23.13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoshi N, Langeberg LK, Gould CM, Newton AC, Scott JD. Interaction with AKAP79 Modifies the Cellular Pharmacology of PKC. Mol Cell. 2010;37:541–550. doi: 10.1016/j.molcel.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi M, Mukai H, Oishi K, Isagawa T, Ono Y. Association of Immature Hypophosphorylated Protein Kinase Cepsilon with an Anchoring Protein CG-NAP. J Biol Chem. 2000;275:34592–34596. doi: 10.1074/jbc.M005285200. [DOI] [PubMed] [Google Scholar]
- 45.Takahashi M, Shibata H, Shimakawa M, Miyamoto M, Mukai H, Ono Y. Characterization of a novel giant scaffolding protein, CG-NAP, that anchors multiple signaling enzymes to centrosome and the golgi apparatus. J Biol Chem. 1999;274:17267–17274. doi: 10.1074/jbc.274.24.17267. [DOI] [PubMed] [Google Scholar]
- 46.Greenwald EC, Redden JM, Dodge-Kafka KL, Saucerman JJ. Scaffold state switching amplifies, accelerates, and insulates protein kinase C signaling. J Biol Chem. 2014;289:2353–2360. doi: 10.1074/jbc.M113.497941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, Fields AP. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005;65:8905–8911. doi: 10.1158/0008-5472.CAN-05-2372. [DOI] [PubMed] [Google Scholar]
- 48.Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM, Gokce N, et al. Protein kinase C-beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95. doi: 10.1161/CIRCULATIONAHA.112.127514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim T, Hinton DJ, Choi DS. Protein kinase C-regulated abeta production and clearance. International journal of Alzheimer’s disease. 2011;2011:857368. doi: 10.4061/2011/857368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Battaini F, Pascale A, Lucchi L, Pasinetti GM, Govoni S. Protein kinase C anchoring deficit in postmortem brains of Alzheimer’s disease patients. Experimental neurology. 1999;159:559–564. doi: 10.1006/exnr.1999.7151. [DOI] [PubMed] [Google Scholar]